

Abstract
The nuclear localized protein deacetylase, SIRT6, has been identified as a crucial regulator of biological processes that drive aging. Among these processes, SIRT6 can promote resistance to oxidative stress conditions, but the precise mechanisms remain unclear. The objectives of this study were to examine the regulation of SIRT6 activity by age and oxidative stress and define the role of SIRT6 in maintaining redox homeostasis in articular chondrocytes. Although SIRT6 levels did not change with age, SIRT6 activity was significantly reduced in chondrocytes isolated from older adults. Using dimedone-based chemical probes that detect oxidized cysteines, we identified that SIRT6 is oxidized in response to oxidative stress conditions, an effect that was associated with reduced SIRT6 activity. Enhancement of SIRT6 activity through adenoviral SIRT6 overexpression specifically increased the basal levels of two antioxidant proteins, peroxiredoxin 1 (Prx1) and sulfiredoxin (Srx) and decreased the levels of an inhibitor of antioxidant activity, thioredoxin interacting protein (TXNIP). Conversely, in chondrocytes derived from mice with cartilage specific Sirt6 knockout, Sirt6 loss decreased Prx1 levels and increased TXNIP levels. SIRT6 overexpression decreased nuclear-generated H2O2 levels and oxidative stress-induced accumulation of nuclear phosphorylated p65. Our data demonstrate that SIRT6 activity is altered with age and oxidative stress conditions associated with aging. SIRT6 contributes to chondrocyte redox homeostasis by regulating specific members of the Prx catalytic cycle. Targeted therapies aimed at preventing the age-related decline in SIRT6 activity may represent a novel strategy to maintain redox balance in joint tissues and decrease catabolic signaling events implicated in osteoarthritis (OA).
Keywords: Sirtuin, aging, oxidative stress, antioxidant, redox signaling, osteoarthritis
Graphical Abstract
Introduction
The sirtuins (SIRTs) represent a family of nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase enzymes that regulate multiple pathways required for cellular homeostasis via posttranslational modifications of target proteins (1). Of the seven mammalian sirtuins (SIRT1-7), key findings support a role for SIRT6 as a crucial regulator of processes that modulate longevity and drive aging (2,3). Transgenic overexpression of Sirt6 has been shown to improve metabolic function and extend lifespan in mice (4), whereas Sirt6 global knockout mice display a premature aging phenotype, metabolic dysfunction and a severely shortened lifespan (5). SIRT6 functions through deacetylating histone 3 at acetylated lysine residues at positions 9 and 56 (H3K9ac, H3K56ac) (6,7) and regulates an array of cellular processes including DNA repair (8,9), genomic stability (5), telomere integrity (6,7,10,11), metabolism (12), cellular senescence (13,14) and inflammation (15,16).
SIRT6 is emerging as a promising target for therapies aimed at preventing age-related diseases. (3,17). Current evidence also suggests that SIRT6 may play a crucial role in joint tissue homeostasis and regulate pathways relevant to osteoarthritis (OA) (18). OA represents the most common form of joint disease and is a primary cause of disability in the elderly (19). Intra-articular injections of lentiviral Sirt6 have been shown to reduce cartilage degradation in a surgery-induced OA mouse model (20), whereas Sirt6 haploinsufficiency increases joint inflammation, osteophyte growth, and chondrocyte hypertrophy in a diet-induced obesity mouse model of OA (21).
Accumulating evidence suggests that SIRT6 is intricately linked to the redox status of the cell (8,22-24). Oxidative stress conditions arise when the production of reactive oxygen species (ROS) exceeds the antioxidant capacity of the cell (25,26). In addition to inducing indiscriminate cellular damage, oxidative stress can posttranslationally modify redox sensitive proteins (via sulfenylation, nitrosation, hyperoxidation, disulfide bond formation, and glutathionylation) which can profoundly alter physiological cell signaling events to promote various age-associated diseases, including OA (19,25,26). SIRT6 contains at least two reactive cysteines that can undergo oxidation to form a sulfenic acid intermediate (termed sulfenylation), which represents a posttranslational modification that can profoundly alter the function of SIRT6 and its downstream targets (27,28). In response to oxidative stress or inflammatory stimuli, sulfenylation of SIRT6 was found to regulate its ability to bind to transcription factors (27) and regulate metabolic homeostasis (28). Alongside its redox sensitivity, SIRT6 also appears to be able to modulate redox balance, govern redox pathways and promote resistance to oxidative stress conditions (8,23)
Despite these promising studies there remains a significant lack of data defining the mechanisms that link SIRT6 to articular joint aging and no studies have investigated the role of SIRT6 to regulate redox homeostasis in joint tissues. As increases in ROS, including H2O2, are associated with joint aging and dysfunction (19), the ability of H2O2 to antagonize SIRT6-mediated cell signaling in joint tissues may be of fundamental importance and could contribute to the development and progression of OA. We have previously demonstrated that physiological and oxidative stress levels of H2O2 can oxidize and inhibit the activity of chondrocyte proteins implicated in cartilage homeostasis (29,30). Furthermore, our recent studies have established that targeted strategies aimed at enhancing antioxidant capacity can decrease oxidative stress levels, attenuate catabolic signaling events implicated in cartilage degradation and cell death, and promote pro-survival cell signaling pathways in human and mouse joint tissues (31,32). As such, the aims of this study were 1) to examine whether SIRT6 activity is regulated by age or oxidative stress conditions that are associated with aging and 2) to determine if SIRT6 is an important regulator of redox homeostasis and downstream cell signaling events in articular chondrocytes.
Materials and methods
Antibodies and reagents
Antibodies to phospho-p65 (ser536) (3033), total p65 (8242), Trx1 (2429), Trx2 (14907), HO-1 (5061), NQO1 (62262), TXNIP (c14715), SIRT6 (12486), TBP (8515), Histone 3 (9715) and β-tubulin (2146) were purchased from Cell Signaling Technology. Antibodies purchased from Abcam were to Prx1 (ab109506), Prx2 (ab109367, Prx3 (ab73349), Prx4 (ab59542), Prx6 (ab73350), PrxSO2/3 (ab16830), Nrf2 (ab62352), and β-actin (ab8226). The antibody to LDH was purchased from Fitzgerald (20-LR22) and the antibody to H3K9ac was from Millipore (06-942). The antibody to Srx was a kind gift from Dr. Sue Gho Rhee (Yonsei University, Seoul, South Korea). The TXNIP antibody used for IHC was from Proteintech (18243-1-AP). The antibody to AhpC was purified from rabbit serum (33). Salmonella typhimurium AhpC C165S protein was purified and expressed as previously described (Poole et al, 1996) and AhpC C165S was reduced and labeled with biotin maleimide as described (33). DCP-Bio1 was purchased from Kerafast. Menadione, H2O2, EX-527, N-ethylmaleimide (NEM), D-alanine, Flavin adenine dinucleotide disodium salt hydrate (FAD), iodoacetamide (IAM), and catalase were purchased from Sigma Aldrich. Dithiothreitol (DTT) was purchased from Life Technologies/Thermo Fisher Scientific. Endotoxin-free recombinant fibronectin fragment (FN-f) was expressed and purified as described (29).
Human primary chondrocyte isolation and culture
Primary human articular cartilage was obtained from the tali of deceased human tissue donors without known history of joint diseases through a collaboration between The Gift of Hope Organ and Tissue Donor Network (Itasca, IL) and Rush University Medical Center (Chicago, IL), with IRB approval. The donor cartilage was macroscopically inspected for damage or evidence of cartilage degeneration using a modified version of the 5-point Collins grading system (34) and only normal appearing cartilage was used. In total, normal cartilage was obtained from 139 donors (male, 108; Female, 31; age range 17-90 years). OA chondrocytes were isolated from cartilage removed from the knee joints of patients undergoing total knee arthroplasty through the Department of Orthopaedic Surgery at the University of North Carolina Medical Center (Chapel Hill, NC). Normal and OA chondrocytes were isolated by sequential digestion of cartilage with Pronase and Collagenase P and cells were seeded in monolayer in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) and antibiotics. The ages of tissue donors ranged from 17-90 years of age. For aging and OA studies, experiments were conducted on young (≤50 years), old (>50 years) and OA chondrocytes (age-independent). The rationale for young and old age cut-offs was chosen based on previous data showing that OA prevalence peaks at 50 years of age (35). In addition, previous publications demonstrate that chondrocytes isolated from cartilage donors aged ≥50 years of age display reduced sensitivity to IGF-1, increased sensitivity to Prx hyperoxidation (a marker of oxidative stress conditions) and altered redox signaling when compared to chondrocytes isolated from younger donors (31,36). In this study, intracellular generation of physiological and oxidative stress levels of H2O2 were induced by treatment of chondrocytes with fragments of fibronectin (FN-f) (1 μM), menadione (25 μM), DMNQ (25 μM), and varying concentrations of H2O2 (1-50 μM) as we have previously described (29-32), or by treatment with D-alanine (2-10 mM) in cells transduced with the NLS-HyPer-DAAO construct (described below).
Adenoviral transduction
Upon 60% confluency, chondrocyte monolayers were incubated in serum-free media containing adenovirus (4x108 viral particles/ml) and 1M CaCl (25μl/ml) for 2 hours (37°C). Adenovirus-containing media was aspirated, and chondrocytes were cultured in serum-containing media at 37°C for 48 hours. Chondrocytes were cultured in serum-free media prior to experimental incubations. An empty vector control (ad-Null) (Cell Biolabs) was used to test for non-specific effects of viral transduction. For co-transduction experiments (ad-SIRT6 + NLS-HyPer-DAAO or ad-Null + NLS-HyPer-DAAO), 4x108 viral particles/ml of each adenovirus were added in serum free media (with 25μl/ml 1M CaCl2) for 2 hours prior to incubation in serum-containing media at 37°C for 48 hours. Confirmation of co-transduction was confirmed at 48 hours by fluorescence detection using an EVOS m5000 imaging system (Thermo Fischer Scientific) and by immunoblotting.
Recombinant SIRT6 activity assay
The deacetylase activity of human recombinant SIRT6 was measured using the fluorometric SIRT6 activity assay kit (Abcam) in a 96 well plate format following manufacturer’s instructions. Briefly, duplicate wells contained ddH2O (25 μl), SIRT6 assay buffer (5 μl), Fluoro-Substrate peptide (5 μl), NAD+ (5 μl) and developer (5 μl). Reactions were initiated by addition of recombinant SIRT6 alone, recombinant SIRT6 in the presence of 50 μM H2O2 or recombinant SIRT6 in the presence of the SIRT6 inhibitor, EX527 (100 μM). Addition of 50 μM H2O2 in the absence of recombinant SIRT6 was also included to assess the effects of H2O2 alone on the assay system. Deacetylation of the Fluoro-Substrate peptide was measured at 15 mins (Ex/Em=480-500/520-540) on a Spectramax M2e multi-mode plate reader (Molecular Devices). Experiments were repeated on 3 separate occasions (n=3). Data are expressed as percentage SIRT6 activity at 15 mins compared to control (recombinant SIRT6 activity alone).
Detection of oxidized protein thiols using the biotinylated iodoacetamide (BIAM) enrichment method
Human articular chondrocytes were cultured in monolayer and treated with varying concentrations of exogenous H2O2 (0-50 μM) for 30 mins. The detection of oxidized thiols was analyzed using the BIAM labeling method as previously described (30). Briefly, cells were lysed in 1X Cell Signaling lysis buffer containing phosphatase inhibitor cocktail and 10 mM NEM in order to alkylate free thiols. Cell lysates were incubated for 30 mins under gentle agitation at 4°C and excess NEM was removed by desalting the samples using Zeba columns (Thermo Scientific). Desalted samples were harvested and treated with 5mM DTT for 1 hr at 4°C and incubated with 0.1 mM BIAM overnight under gentle agitation (4°C). BIAM selectively reacts with thiols that are susceptible to oxidation, allowing for identification of thiols in the sulfenic, disulfide, glutathionylated and nitrosated state. Centrifugation to remove insoluble proteins preceded protein quantification using the BCA assay. Streptavidin beads (Thermo Scientific) (120 μl) were loaded onto Pierce spin columns and washed twice in PBS and then in 0.1% SDS. Protein samples (200μg) were loaded into Pierce Spin columns and equal amounts (20μl 0.5ng) of pre-biotinylated AhpC was added into each sample as an internal standard that served as a loading control. Spin columns were rotated overnight at 4°C and then rinsed with PBS (3x). Samples were boiled in laemmli sample buffer to elute the beads, allowed to cool, centrifuged (3,200 rpm, 4 mins), and then equal amounts of supernatant were loaded onto 12% SDS-PAGE gels and immunoblotted under reducing conditions with antibodies to SIRT6 and AhpC.
Detection of sulfenylated thiols using the DCP-Bio1 enrichment method
Identification of DCP-Bio1 labeled lysates for detection of S-sulfenylated cysteine intermediates was performed as recently described (29,33,37). Human articular chondrocytes were cultured in monolayer and treated with 1 μM FN-f, 25 μM menadione or 25 μM DMNQ for 30 mins and lysed in standard lysis buffer containing 200 units/ml catalase and 5 mM DCP-Bio1. Samples were incubated at 4°C under gentle agitation for 30 mins and 20 mM IAM was added to each sample to block free sulfhydryls. Samples were incubated at 4°C under gentle agitation for an additional 30 mins. Cell lysates were centrifuged, and the soluble protein fraction was harvested. DCP-Bio1 labeled lysates were loaded onto Bio Gel P6 (Bio-Rad) columns and centrifuged at 1000g (4 min) to remove unbound DCP-Bio1. Protein content of DCP-Bio1 labeled lysates were quantified using the BCA assay. Streptavidin beads (120 μl) were loaded onto Pierce spin columns and washed twice in PBS and then in 0.1% SDS. Protein samples (200μg) were loaded into Pierce Spin columns and equal amounts (20μl 0.5ng) of pre-biotinylated AhpC was added into each sample as an internal standard that served as a loading control. Spin columns were rotated overnight at 4°C and then rinsed with TBST (3x), 2M urea (1x), 1M NaCl (1x), 0.1% SDS + 10mM DTT (2x) and PBS (2x). Samples were boiled in laemmli sample buffer to elute the beads, allowed to cool, centrifuged (3,200 rpm, 4 mins), and then equal amounts of supernatant were loaded onto 12% SDS-PAGE gels and immunoblotted under reducing conditions with antibodies to SIRT6 and AhpC. Due to our previous findings demonstrating the sensitivity of chondrocyte Prx2 to H2O2-induced oxidation (31,32), DCP-Bio1 labeled lysates were also immunoblotted for Prx2 as a positive control.
Induction and measurement of nuclear-generated H2O2 using the Nuclear Localization Signal-HyPer-D-amino acid oxidase (NLS-HyPer-DAAO) chemogenetic approach
To generate nuclear-specific H2O2, chondrocytes were transduced with NLS-HyPer-DAAO. This genetically engineered construct is contained in an adenoviral vector that is comprised of DAAO fused to the H2O2 indicator HyPer and targeted to the nucleus (NLS) (38). DAAO oxidizes exogenously added D-alanine (D-ala) to produce H2O2 specifically in the nucleus, which can then be directly visualized and monitored using HyPer. Preliminary experiments using exogenously added H2O2 (50μM) confirmed the sensitivity of HyPer in this construct (Supplementary Figure 1). To validate the effect of NLS-HyPer-DAAO to generate and measure nuclear H2O2, human chondrocytes were transduced with the NLS-HyPer-DAAO construct for 48 hours and chondrocytes were treated with various concentrations of D-ala (2-10 mM) for 15 mins. These concentrations of D-ala have previously been shown to generate varying levels of H2O2 in other cell types using HyPer-DAAO constructs (38-42). To test the effect of SIRT6 overexpression to regulate nuclear H2O2 generation, chondrocytes were co-transduced with the NLS-Hyper-DAAO expressing vector and either an empty vector control (ad-Null) or an adenoviral vector encoding SIRT6 (ad-SIRT6) and treated with 6mM D-ala for 15 mins. For each experiment, the cofactor, flavin adenine dinucleotide (FAD) (2.5 μM) was added to cell cultures during experimental exposures to maintain H2O2 generation beyond 10 mins (39). Differential Interference Contrast (DIC) and epifluorescence imaging was carried out on an Olympus IX81 widefield inverted microscope equipped with a motorized filter wheel and 20x/ 0.7NA objective. All experimental incubations were carried out in an Environmental Control Chamber (Tokai Hit) to ensure that conditions were maintained at 37°C in a humidified atmosphere containing 5% CO2. The fluorescent signal was excited sequentially using CFP (434/17) and FITC (475/35) bandpass excitation filters. The emission was collected using a FITC (530/43) bandpass emission filter for both excitations. Fluorescence images were recorded every 10 seconds. After at least 5 mins of background recording, D-ala was diluted in 50 μl of warm culture media and added to cell cultures without interruption of the recording. DIC images were taken prior to, and after, experimental manipulations. Ratiometric analysis was performed using a pipeline developed in CellProfiler software (43). Nuclear position was determined using the fluorescence signal from CFP excitation images (Supplementary Figure 2). The outlines of nuclei used as ROI for intensity calculations were documented for visual verification. For each ROI the ratio between integrated fluorescence intensity for CFP and FITC excitation was calculated and averaged across the frame for each time point of recording.
Analysis of chondrocyte signaling by immunoblotting
Chondrocyte monolayers were incubated overnight in serum-free conditions prior to experimental incubations. For analysis of basal protein levels, total cell lysates were prepared as previously described (31) and immunoblotting under reducing conditions was performed with the specified antibodies. In addition, chondrocytes were treated with 25 μM menadione to induce oxidative stress, for the indicated times. Nuclear and cytoplasmic (non-nuclear) chondrocyte fractions were extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) following manufacturer’s instructions and immunoblotted under reducing conditions with an antibody to PrxSO2/3 or phosphorylated p65. Immunoblots were stripped and reprobed with an antibody to total-p65 as a loading control. Immunoblotting with antibodies to the nuclear proteins lamin B or TBP and with an antibody to LDH as a cytoplasmic marker were used as loading controls and to confirm nuclear and cytoplasmic separation. The cytoplasmic (non-nuclear) fraction includes the contents of all organelles other than the nucleus.
SIRT6 functions as a specific H3 deacetylase that targets lys-9 (H3K9) and immunoblotting for the acetylated form of H3K9 (H3K9ac) can be used as an inverse marker of SIRT6 deacetylase activity (6). Thus, an increase in H3K9ac is regarded as a marker of decreased SIRT6 activity. Total chondrocyte histone fractions were isolated using the Histone Extraction kit (Abcam) and were immunoblotted with an antibody to H3K9ac. Histone 3 served as a loading control. In many cases, total protein levels were also analyzed using the Pierce™ Reversible Protein Stain Kit for Nitrocellulose Membranes (Thermo Fisher Scientific) to further confirm equal loading. In all cases, antibodies were diluted in TBST containing 5% blotting-grade blocker. All antibodies were used at a 1:1000 dilution, with the exception of the antibody to PrxSO2/3 (1:2000), the antibody to TXNIP (1:300) and the antibody to AhpC (1:500).
Prx redox status in response to treatment with D-alanine in cells transduced with NLS-HyPer-DAAO was assessed by visualizing the reduced and oxidized forms of the Prxs by non-reducing immunoblotting, as we (31,32), and others (44,45), have previously described. Briefly, chondrocytes were transduced with NLS-HyPer-DAAO and treated with 6mM D-alanine for 0-60 mins. In order to alkylate reduced thiols and prevent lysis-induced artificial oxidation of the Prxs, cell monolayers were rinsed in PBS and incubated for 10 mins in an NEM-containing alkylating buffer (40 mM HEPES, 50 mM NaCl, 1mM EGTA, 200 units/ml catalase, 100 mM NEM and phosphatase inhibitor cocktail 2 (pH7.4)). Cells were then lysed for 30 mins under gentle agitation at 4°C in lysis buffer containing 200 units/ml catalase, 100 mM NEM and phosphatase inhibitor cocktail (pH 7.4). Cell lysates were centrifuged, and the soluble protein fraction was harvested. Total protein concentrations were determined using the Pierce Micro BCA kit (Thermo Scientific). Protein lysates were boiled for 5 mins and immunoblotted under reducing or non-reducing conditions (presence or absence of 10% β-mercaptoethanol) as described (45).
RNA isolation and RT-qPCR from human chondrocytes
RT-qPCR data shown in Figure 1C was generated alongside a previously published study (46) (Sirt6 Taqman Assay ID: HS00966002_m1). For other data, RNA was isolated from human chondrocytes derived from younger and older adults using the RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions. Reverse transcription was performed using ImProm-II™ Reverse Transcriptase system (Promega) and PCR performed using SYBR Green Supermix (BioRad) and gene-specific oligos purchased from Qiagen on a QuantStudio™ 6 Flex machine (Applied Biosystems). Primer sequences used were: TXNIP; F: GGTCTTTAACGACCCTGAAAAGG; R: ACACGAGTAACTTCACACACCT; Prdx1 F: CATTCCTTTGGTATCAGACCCG; Prdx1 R: CCCTGAACGAGATGCCTTCAT; Srx1 F: CAGGGAGGTGACTACTTCTACTC; Srx1 R: CAGGTACACCCTTAGGTCTGA). Gene expression was normalized to TBP as housekeeping control.
Generation and validation of inducible cartilage specific Sirt6 deficient mice
All animal experiments were conducted in accordance with the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill. Acantm1(IRES-cre/ERT2)Crm (Aggrecan CreERT2) mice (47) were a kind gift from Dr. Benoit De Crombrugghe and were crossed to the conditional Sirt6tm1.1Cxd mouse (12) (Jackson Labs, stock number 017334, Bar Harbor, ME, USA) to generate Aggrecan-CreERT2; Sirt6fl/fl mice. Littermate Sirt6fl/fl (in the absence of Aggrecan-CreERT2) mice were used as Sirt6 controls. For in vitro femoral cap explant studies, 4-8-week-old mice were euthanized by CO2 asphyxiation and cervical dislocation. Femoral head cartilage explants from the hip joints of these mice were harvested by removal of the capital femoral epiphysis from the underlying bone. Femoral cap explants were cultured in serum-containing media for two days to allow for recovery post removal. Femoral cap explants were then cultured in media containing 5 μM 4-Hydroxytamoxifen (Sigma-Aldrich) for 2 media changes (total of 96 hours) to activate Cre recombinase activity of Aggrecan-CreERT2 and induce Sirt6 loss. Explants were maintained in serum-containing media for 1 week with media changes every 48 hours. Femoral caps were harvested and then homogenized in ceramic bead tubes (MOBIO) in lysis buffer (200 μl) using a Precellys homogenizer (Bertin Technologies) as previously described (31). Cell lysates were collected and centrifuged to remove non-soluble proteins. Protein concentrations were determined using the BCA assay prior to immunoblotting for proteins of interest.
Immunohistochemistry (IHC) on human knee joint cartilage sections
Human medial femoral condyle cartilage sections were a kind gift from Dr. Martin Lotz (Scripps Research Institute, La Jolla, CA). Cartilage surfaces were macroscopically graded using a modified Outerbridge scoring system and knee maps from the International Cartilage Repair Society (ICRS) as previously reported (48). Cartilage from younger adults had an average age of 43.0±6.2 yrs. (range, 36–48) and total knee macroscopic grade corresponded to grade II (mild changes, 59.0±1.73). Cartilage from older adults had an average age of 73.3±4.6 yrs. (range, 68–76) and total knee macroscopic grade corresponded to grade II (mild changes, 63.0±5.5). Despite no change in macroscopic grades, cartilage sections derived from older adults did show some superficial changes including cartilage thinning and cartilage surface fibrillation, when compared to younger cartilage when we performed IHC. IHC was performed as previously described (49) with modifications using the VECTASTAIN™ Elite™ ABC Kit. Sections were incubated in 3% H2O2 for 10 mins, followed by incubation in 10 mM sodium citrate buffer (pH 6.0, 90°C) for 30 mins. Sections were blocked in 5% donkey serum, rinsed in PBS and then incubated overnight with an antibody to TXNIP (Proteintech, 1:300 in 2% donkey serum) at room temperature. Sections were rinsed in PBS and then incubated in biotinylated secondary antibody (1 hr) prior to incubation in VECTASTAIN™ Elite™ Avidin-Biotin Complex (ABC) solution (30 mins). Slides were washed and incubated in Vector™ DAB peroxidase substrate and counterstained with hematoxylin. Images were taken with an Olympus BX60 microscope and analyzed with ImageJ.
Statistical analysis
Band intensity of immunoblots were quantified by densitometric analysis using ImageJ software and all statistical analysis was carried out in GraphPad Prism version 8. All data are presented as mean values±SD. In all cases, independent experiments were conducted on cells isolated from different tissues donors. Individual data points showing the responses from individual donors are also presented and exact biological replicates (from independent donors) are indicated in figure legends. Results were analyzed by paired or unpaired t-tests when comparing two groups, or one or two-way ANOVA when analyzing multiple groups. Post-hoc corrections were applied as appropriate. Specific p-values are presented in graphs. A p-value of <0.05 was deemed significant.
Results
Effects of age and oxidative stress on SIRT6 activity.
SIRT6 protein levels, as assessed by immunoblotting chondrocyte lysates derived from younger (29±7.7 yrs), older (66±3.9 yrs) or OA (62.7±3.4yrs) human cartilage did not reveal age or OA related differences (Figure 1A, B). Similarly, we did not observe an age-related difference in SIRT6 gene expression when we performed quantitative RT-qPCR on normal human chondrocytes isolated from adults ranging from 17-72 years of age (Figure 1C).
Figure 1. SIRT6 protein levels and gene expression are not altered by age or OA in primary human chondrocytes.
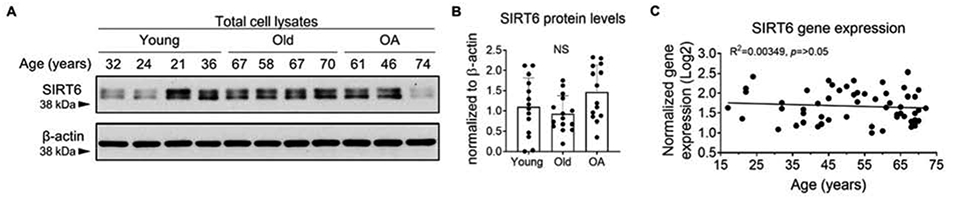
(A) Unstimulated primary human articular chondrocytes isolated from young (av age 29±7.7 yrs), old (av age 66±3.9 yrs) and OA (av age 61±11.8 yrs) cartilage were subjected to immunoblotting for total SIRT6. Representative immunoblots from young, old and OA samples are shown. (B) Densitometric analysis showing chondrocyte SIRT6 protein levels from young (n=14), old (n=15) and OA (n=14) cartilage tissue. SIRT6 was normalized to β-actin as a loading control. Quantified data is mean±SD showing individual data points from independent samples. Two-way ANOVA was used to test for differences between groups (NS = non-significant). (C) Basal SIRT6 gene expression measured by RT-qPCR in unstimulated articular chondrocytes derived from normal cartilage from adults aged 17-72 yrs. (n=57). The lowest value was set to 1 and SIRT6 levels were normalized to YWHAZ as a housekeeping control. R2 and p-values from linear regression analysis are presented.
We did however observe an age-related change in SIRT6 activity. As SIRT6 is a specific histone 3 (H3) deacetylase that targets lysine 9 (K9) (6), we used normalized H3K9 acetylation (H3K9ac) as an inverse marker of SIRT6 activity. Immunoblotting of chondrocyte histones with an antibody that detects H3K9ac revealed that chondrocytes derived from older adults (70.85±5.6 yrs.) displayed a significant increase in basal H3K9ac levels when compared to chondrocytes derived from younger adults (42.14±2.8 yrs.) (Figure 2A, B), an effect consistent with an age-related decline in SIRT6 activity. Since oxidative stress conditions are implicated in the aging phenotype (19,25,26), we next tested the effect of oxidative stress levels of H2O2 to regulate SIRT6 activity. Incubation of recombinant SIRT6 with 50 μM H2O2 significantly decreased SIRT6 activity by 27.5±2.8% within 15 mins (Fig 2C). Incubating SIRT6 with EX527, a SIRT6 chemical inhibitor also significantly decreased SIRT6 activity within 15 mins (67.1±11.0%) (Fig 2C).
Figure 2. SIRT6 activity is altered by age and oxidative stress.
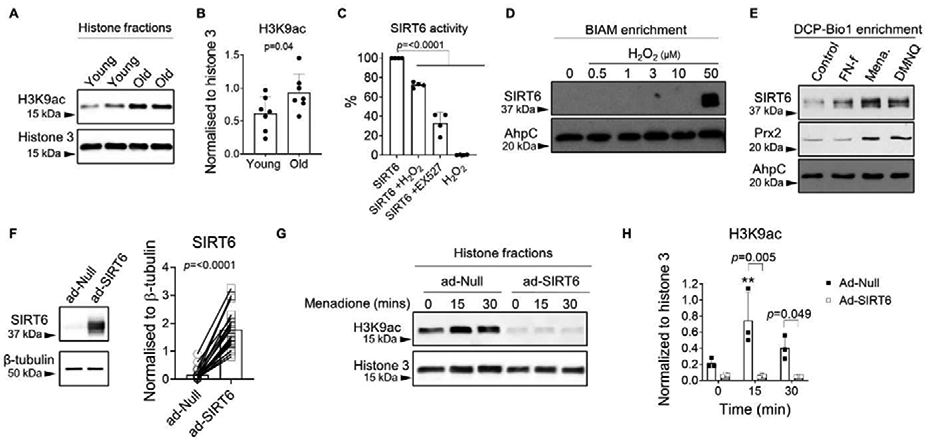
(A) Unstimulated confluent primary human chondrocytes isolated from young (42.14±2.8 years) and old (70.85±5.6 years) cartilage were lysed and histones were extracted prior to immunoblotting for the acetylated form of H3K9 (H3K9ac) as an inverse marker of basal SIRT6 activity. Representative immunoblots from young and old chondrocytes are shown. (B) Densitometric analysis showing basal H3K9ac protein levels in young and old chondrocytes (n=7). H3K9ac was normalized to total histone 3 as a loading control. Unpaired t-test was used to test for statistical differences between age groups. (C) Recombinant human SIRT6 was treated in the presence or absence of 50 μM H2O2 or the SIRT6 inhibitor, EX527 (100 μM), for 15 mins and a deacetylation-based assay was used to measure recombinant SIRT6 activity. Data are expressed as percentage SIRT6 activity compared to control (recombinant SIRT6 alone) at 15 mins (n=3 independent experiments). One-way ANOVA was used to test for differences compared to control. (D) Chondrocytes were treated with varying concentrations of exogenous H2O2 (0-50 μM) for 30 mins and biotinylated IAM (BIAM) was used to affinity tag proteins containing a variety of thiol oxidative modifications (R-SOH, R-SS-R, R-SSG, R-SNO) followed by pull-down with streptavidin beads and immunoblotting for SIRT6. (E) Chondrocytes were treated with FN-f (1μM), menadione (25 μM) or DMNQ (25 μM) for 30 mins, and DCP-Bio1 was used to specifically affinity tag proteins containing R-SOH (cysteine sulfenic acid intermediate) followed by pull-down with streptavidin beads and immunoblotting for SIRT6 and Prx2. Data shown are representative blots from n=3 independent experiments. AhpC was used as a loading control in BIAM and DCP-Bio1 experiments. (Mena.; menadione). (F) Human chondrocytes were transduced with an empty vector control (ad-Null) or an adenoviral vector encoding SIRT6 (ad-SIRT6) for 48 hours. A representative immunoblot showing typical levels of SIRT6 overexpression from total cell lysates is shown along with quantification from n=27 donors. (G) Chondrocytes were treated with 25 μM menadione for 0-30 mins to induce oxidative stress. Histones were extracted and immunoblotting for H3K9ac was used to assess SIRT6 activity levels. Representative immunoblots are shown. (H) Densitometric analysis from n=3 independent samples. H3K9ac protein levels were normalized to total histone 3 as a loading control. Asterisks represent significant differences compared to controls (**, p<0.01). Two-way ANOVA was used to test for differences between highlighted groups.
Based on this finding and published work that demonstrated the redox sensitivity of SIRT6 in other cell types (27,28), we hypothesized that SIRT6 is oxidized in chondrocytes under conditions of oxidative stress. To assess the redox sensitivity of SIRT6, we first treated human chondrocytes with various concentrations of H2O2 and captured reversibly modified thiols using biotinylated iodoacetamide (BIAM) labeling and affinity capture (30). At higher concentrations of H2O2, as would be associated with oxidative stress conditions in chondrocytes (30), we observed increased BIAM labeling of SIRT6 (Fig 2. D), suggesting that SIRT6 can be oxidatively modified in human chondrocytes. The BIAM method enriches for proteins containing a variety of thiol oxidative modifications, including glutathionylation (R-SSG), nitrosation (R-SNO) and sulfenylation (R-SOH). As H2O2-mediated sulfenylation of protein thiols is regarded as a key mechanism by which H2O2 regulates intracellular cell signaling events, we used an alternative method based on DCP-Bio1 labeling and affinity capture to specifically enrich for sulfenylated proteins (30). For this experiment, chondrocytes were incubated with fibronectin fragments (FN-f), a stimulus we have shown generates physiological levels of H2O2 relevant to chondrocyte signaling (30). This was compared to treatment with redox cycling oxidants, menadione and DMNQ, which we have shown generate oxidative stress levels of H2O2 in chondrocytes (31,32,50). We detected increased sulfenylation of SIRT6 in chondrocytes treated with both menadione and DMNQ (Figure 2E). Similarly, we also detected menadione and DMNQ-induced sulfenylation of peroxiredoxin 2 (Prx2), an intracellular thiol peroxidase that is highly sensitive to H2O2, and thus served as a positive control (Figure 2E). In contrast, treatment of chondrocytes with FN-f, which generates lower levels of H2O2 (30), did not yield detectable sulfenylation of SIRT6 or Prx2 (Figure 2E). Collectively, these findings demonstrate that SIRT6 is redox sensitive in chondrocytes and undergoes post-translational oxidative modifications, including sulfenylation, in response to oxidative stress levels of H2O2.
Since post-translational oxidative modifications including sulfenylation can activate or inhibit the function of redox sensitive proteins, and since we showed above that treatment of recombinant SIRT6 with excessive H2O2 inhibited its activity, we next investigated if oxidation inhibits SIRT6 activity in cells and if overexpression of SIRT6 could overcome this effect. To this end, we transduced human chondrocytes with either an adenoviral empty vector control (ad-Null) or an adenoviral vector encoding SIRT6 (ad-SIRT6) (Figure 2F) and treated cells with menadione to generate oxidative stress levels of H2O2 (Figure 2G). After treatment, chondrocyte histones were extracted and immunoblotted for H3K9ac in order to measure SIRT6 activity. In cells transduced with the control vector, menadione treatment led to a significant increase in H3K9ac within 15 mins (Figure 2G, H) suggesting that menadione-induced oxidative stress can rapidly inhibit endogenous SIRT6 activity, consistent with our results using recombinant SIRT6 and H2O2 (Figure 2C). Importantly, overexpression of SIRT6 significantly abrogated the menadione-induced increase in H3K9ac protein levels observed in null empty vector controls (Figure 2G, H), indicating that SIRT6 overexpression can increase intracellular SIRT6 activity. As such, we used the adenoviral vector to SIRT6 as a tool to overexpress SIRT6 and increase SIRT6 activity in further experiments.
SIRT6 regulates TXNIP, Prx1 and Srx levels in normal and OA chondrocytes.
To address the impact of SIRT6 on antioxidant function in chondrocytes, we transduced cells with the adenoviral vector to express SIRT6 or the null empty vector control and assessed basal protein levels of a plethora of antioxidant and redox-regulatory proteins. In total cell lysates, SIRT6 overexpression did not affect basal protein levels of the antioxidant proteins Prx2, Prx3, Prx4, Prx6, Trx2, HO-1 or NQO1 (Figure 3A, B). However, SIRT6 overexpression significantly reduced the basal protein levels of the thioredoxin inhibitor, TXNIP, while significantly increasing the basal protein levels of the antioxidant proteins Prx1 and Srx (Figure 4A, B). In agreement with these findings, SIRT6 overexpression significantly decreased TXNIP gene expression and increased Srx gene expression (Figure 4C) suggesting that SIRT6 modulates these factors at the level of transcription. Although Prx1 gene expression was increased in SIRT6 overexpressing cells when compared to controls, statistical significance was not reached (Figure 4C). The effect of SIRT6 overexpression to decrease basal TXNIP protein levels and increase basal Prx1 and Srx protein levels was also observed when we performed immunoblotting on chondrocyte nuclear fractions (Figure 4D, E) and cytoplasmic fractions (Figure 4F,G). In addition to Prx1 and Srx, SIRT6 overexpression significantly increased Nrf2 protein levels in normal chondrocytes (p=<0.0001, Figure 3A, B) but we were unable to consistently visualize Nrf2 protein in cytoplasmic and nuclear fractions (data not shown), likely due to its degradation during fractionation. SIRT6 overexpression also modestly increased Trx1 protein levels in total cell lysates (p=0.048, Figure 3A, B) but we did not observe a change in Trx1 protein levels when we immunoblotted for Trx1 in cytoplasmic and nuclear lysates (data not shown). Overexpression of SIRT6 in chondrocytes derived from OA tissue showed the same response as chondrocytes from normal tissue in terms of decreased TXNIP and increased Prx1 and Srx (Figure 5A, B).
Figure 3. Effect of adenoviral overexpression of SIRT6 on basal antioxidant protein levels.
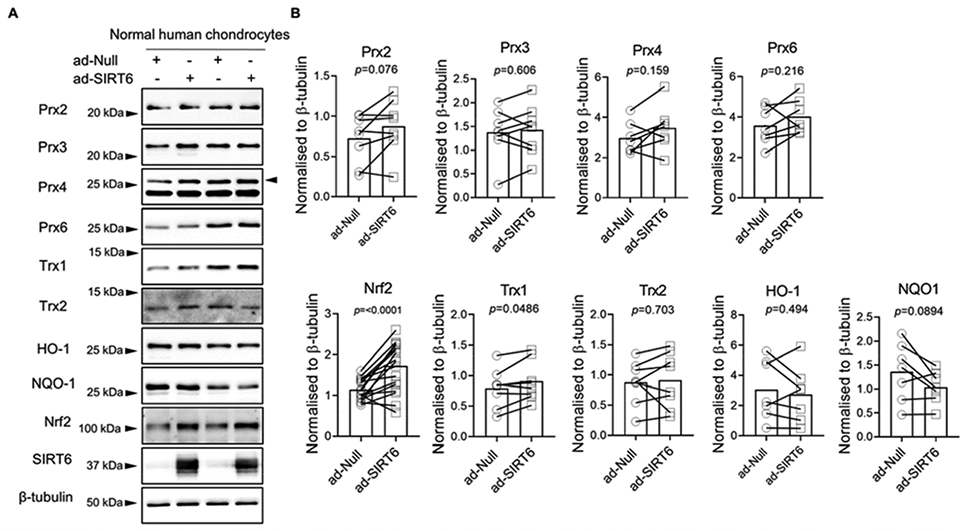
(A) Human articular chondrocytes were transduced with an empty adenoviral vector control (ad-Null) or an adenoviral vector encoding SIRT6 (ad-SIRT6) for 48 hours. Basal protein levels of Prx2, Prx3, Prx4, Prx6, Trx1, Trx2, HO-1, NQO1 and Nrf2 were assessed by immunoblotting. (B) Densitometric analysis from Prx2 (n=8), Prx3 (n=8), Prx4 (n=7), Prx6 (n=7), Nrf2 (n=15), Trx1 (n=8), Trx2 (n=8), HO-1 (n=7), NQO1 (n=7), and Nrf2 (n=15) immunoblots. Representative immunoblots are shown. Total protein levels were normalized to β-tubulin as a loading control. SIRT6 immunoblots confirmed SIRT6 overexpression. Quantified data is mean±SD showing individual data points from independent samples. Paired t-tests were used to test for statistical differences between group
Figure 4. Effect of adenoviral overexpression of SIRT6 on basal TXNIP, Prx1 and Srx levels in normal human articular chondrocytes.
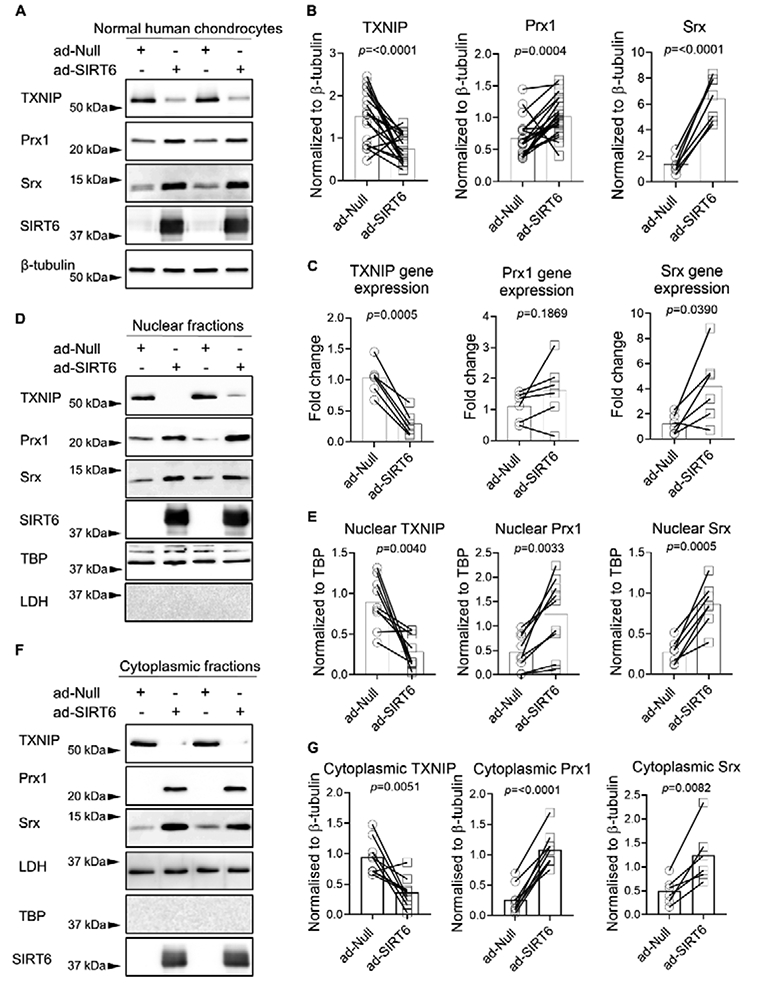
(A) Human articular chondrocytes were transduced with an empty adenoviral vector control (ad-Null) or an adenoviral vector encoding SIRT6 (ad-SIRT6) for 48 hours. Basal protein levels of TXNIP, Prx1 and Srx from total cell lysates were assessed by immunoblotting. Representative results from two separate individuals are shown in panels A, D, and F. (B) Densitometric analysis from TXNIP (n=18), Prx1 (n=21) and Srx (n=7) immunoblots. Total protein levels were normalized to β-tubulin as a loading control. (C) Chondrocytes were transduced with ad-Null or ad-SIRT6 for 48 hours and basal TXNIP, Prx1 and Srx mRNA expression were quantified by RT-qPCR (n=6 independent samples). Human articular chondrocytes were transduced with ad-SIRT6 or ad-Null for 48 hours and nuclear fractions (D,E) and cytoplasmic fractions (F,G) were isolated. Basal TXNIP (n=9), Prx1 (n=9), and Srx (n=6) were assessed by immunoblot. Nuclear protein levels were normalized to TBP and cytoplasmic protein levels were normalized to LDH. In all cases, representative immunoblots are shown. SIRT6 immunoblots confirmed SIRT6 overexpression. Paired t-tests were used to test for statistical differences between groups.
Figure 5. Effect of adenoviral overexpression of SIRT6 on basal TXNIP, Prx1 and Srx levels in OA human articular chondrocytes.
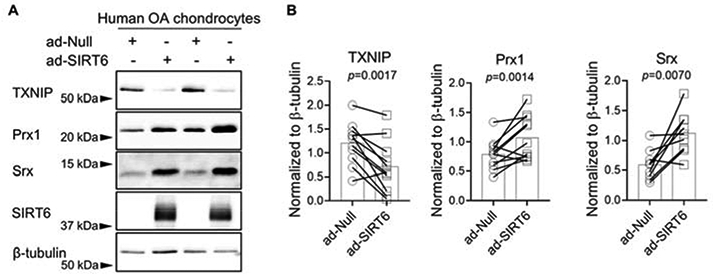
(A) Human OA articular chondrocytes were transduced with ad-Null or ad-SIRT6 for 48 hours. Basal protein levels of TXNIP, Prx1 and Srx from total cell lysates were assessed by immunoblotting. (B) Densitometric analysis from TXNIP (n=11), Prx1 (n=11) and Srx (n=9) immunoblots. Total protein levels were normalized to β-tubulin as a loading control. In all cases, representative immunoblots are shown. SIRT6 immunoblots confirmed SIRT6 overexpression. Paired t-tests were used to test for statistical differences between groups.
To further investigate the roles of SIRT6 on antioxidant and redox-regulatory pathways in chondrocytes, we crossed existing Aggrecan-CreERT2 and Sirt6fl/fl mice to generate inducible, cartilage-specific Sirt6 deficient mice. Femoral cap explants derived from mice with Sirt6 loss displayed a significant increase in basal TXNIP levels, when compared to chondrocytes derived from Sirt6 intact mice (Figure 6A, B). In addition, Sirt6 loss significantly decreased basal Prx1 protein levels when compared to controls (Figure 6A, B).
Figure 6. Effect of Sirt6 deficiency on chondrocyte TXNIP and Prx1 protein levels.
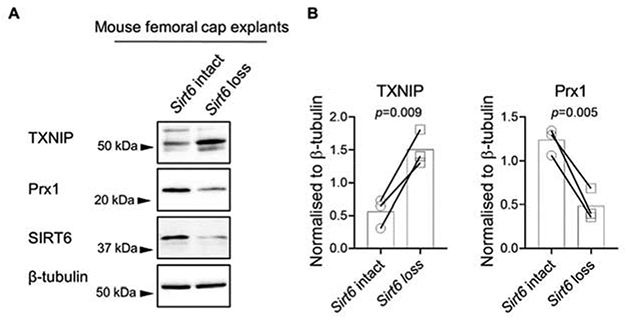
(A) Paired femoral caps from Aggrecan-CreERT2; Sirt6fl/fl (Sirt6 loss) or Sirt6fl/fl (Sirt6 intact) mice were treated with 4-Hydroxytamoxifen to activate Cre-mediated recombination ex vivo. Cell lysates were prepared and basal protein levels of TXNIP and Prx1 were assessed by immunoblotting. (B) Densitometric analysis from basal TXNIP and Prx1 immunoblots (n=3). In all cases, total protein levels were normalized to β-tubulin as a loading control. SIRT6 immunoblots confirmed Sirt6 loss. Unpaired t-test was used to test for statistical differences between groups.
TXNIP levels are increased in human knee joint cartilage sections derived from older adults.
Having found that 1) active SIRT6 can suppress TXNIP levels and 2) SIRT6 activity declines with age in chondrocytes, we hypothesized that TXNIP levels would exhibit an age-related increase in cartilage. We performed immunohistochemistry (IHC) with an antibody to TXNIP on normal human medial femoral condyle cartilage sections obtained from younger (43.0±6.2 yrs.) and older (73.3±4.6 yrs.) adults. Younger cartilage had a lower percentage of TXNIP-positive cells (23%±6.2%), which likely reflects basal levels of TXNIP in cartilage, whereas cartilage from older adults had a significantly higher percentage of TXNIP-positive stained cells (73.3±12.9%), consistent with an age-associated increase in cartilage TXNIP levels (Figure 7A, B).
Figure 7. TXNIP levels are increased in older human cartilage sections.

TXNIP levels in normal younger and normal older human knee cartilage tissue sections were evaluated by immunohistochemistry. Tissue sections were counterstained with hematoxylin. Human cartilage sections (n=3 independent samples) were macroscopically graded as described in ‘Experimental Procedures’. Younger tissues were from adults <50 years old (mean age, 43.0±6.2 yrs.), and older tissues were from adults >65 years old (mean age, 73.3±4.6 yrs.). Scale bar, 100μM. Data are presented as percentage TXNIP stained cells for young and old cartilage sections. Unpaired t-test was used to test for statistical differences between groups.
SIRT6 overexpression decreases menadione-induced cytoplasmic and nuclear Prx hyperoxidation and p65 phosphorylation in human chondrocytes.
We next tested the ability of SIRT6 to modulate oxidative stress in menadione treated chondrocytes using Prx hyperoxidation as a marker. For this, cytoplasmic (non-nuclear) and nuclear protein fractions were immunoblotted using an antibody that detects Prx 1-3 when it is in the sulfinic (SO2) or sulfonic forms (SO3). In control transduced cells, treatment with menadione led to a time dependent increase in cytoplasmic Prx hyperoxidation at 15 and 30 mins (Figure 8A, B), which is consistent with our previous findings in total cell lysates (30-32,50). In the cytoplasmic fractions, menadione-induced Prx hyperoxidation generated a familiar banding pattern that we have previously observed in total cell lysates and which we have shown to correspond with hyperoxidized forms of Prx1, 2 and 3 (Figure 8A). In the nuclear fractions, we found a similar, time-dependent increase in menadione-induced Prx1 and Prx2 hyperoxidation. Bands corresponding to hyperoxidized Prx3 were not observed in nuclear fractions as Prx3 is localized to the mitochondria. Overexpression of SIRT6 significantly decreased menadione-induced Prx hyperoxidation in both the cytoplasmic and nuclear fractions at 15 and 30 mins (Figure 8A, B, C), indicating a decrease in oxidative stress levels in cells overexpressing SIRT6. This finding encouraged us to assess the effects of SIRT6 overexpression on redox mediated, catabolic cell signaling events in chondrocytes by measuring phosphorylation of the NF-κB subunit p65 in nuclear and cytoplasmic chondrocyte fractions. In cells transduced with the empty vector, menadione-induced oxidative stress led to a significant increase in p65 phosphorylation at 30 mins in both the cytoplasmic and nuclear compartments (Figure 8D, E, F), an effect which was significantly abrogated by overexpression of SIRT6 (Figure 8D, E, F).
Figure 8. Adenoviral overexpression of SIRT6 attenuates menadione-induced Prx hyperoxidation and nuclear accumulation of phosphorylated p65 in human articular chondrocytes.

Human articular chondrocytes were transduced with an adenoviral vector encoding SIRT6 or an empty vector control for 48 hours and then treated with menadione (25 μM) for 0-30 mins. (A) Cytoplasmic and nuclear fractions were isolated and Prx hyperoxidation was detected by reducing immunoblots with an antibody to PrxSO2/3. Representative immunoblots are shown and specific Prxs are labelled. (B) Densitometric analysis from cytoplasmic and (C) nuclear PrxSO2/3 immunoblots (n=4). Data are expressed as relative intensity compared to untreated empty vector controls. (D) Cytoplasmic and nuclear lysates were also immunoblotted with antibodies to phosphorylated and total p65. LDH (non-nuclear protein) and TBP (nuclear protein) were immunoblotted for, in order to confirm successful cytoplasmic and nuclear separation. Immunoblots are representative results from n=6 independent samples. (E) Densitometric analysis from cytoplasmic and (F) nuclear phospho-p65 immunoblots. Asterisks represent significant differences compared to controls (**, p<0.01; ****, p<0.0001) (Two-way ANOVA). Two-way ANOVA was used to test for differences between time points.
SIRT6 overexpression decreases the NLS-HyPer-DAAO fluorescence ratio in human chondrocytes.
Since SIRT6 is a nuclear localized histone deacetylase enzyme and we have demonstrated it targets redox factors that are found in the nucleus, we speculated that SIRT6 may also directly modulate nuclear levels of H2O2. In order to generate and measure nuclear H2O2, we used a chemogenetic approach by transducing chondrocytes with a nuclear localized HyPer-DAAO construct (NLS-HyPer-DAAO) (38). Transduced cells showed high levels of expression and nuclear specificity (Supplementary Figure 3). Addition of 2 mM and 4 mM D-ala generated subtle increases in H2O2 levels whereas addition of 6-10 mM D-ala led to a consistent increase in H2O2 within 1 min, an effect which was maintained over the 15-min time course studied (Figure 9A, B).
Figure 9. Generation and measurement of nuclear-localized H2O2 and oxidation events using the NLS-HyPer-DAAO construct in human chondrocytes.

(A) An adenoviral vector encoding NLS-HyPer-DAAO was transduced into human articular chondrocytes and activated with varying concentrations of D-alanine (2-10 mM) for 15 mins to generate nuclear H2O2. Representative fluorescent images of the response to 6mM D-ala from n=3 independent samples. Images are shown in two different colors for presentation purposes. (B) Plot of calculated ratios between fluorescence intensities recorded using FITC and CFP excitation filters in response to varying concentrations of D-ala. Data was normalized to the average value prior to the addition of D-ala (representative data from n=3 independent samples). (C) Chondrocytes were treated with 6 mM D-ala for 0-60 mins prior to incubation in an NEM alkylating buffer to alkylate reduced thiols and cells were lysed in the presence of NEM. Under non-reducing conditions, immunoblotting for Prx1, Prx2 and Prx3 allowed for identification of Prx reduced monomers (labelled M on blots) and oxidized dimers (labelled D on blots). β-tubulin served as a loading control. (D) Densitometric analysis of D-alanine-induced Prx1, Prx2 and Prx3 dimer formation observed over the 60-min time course. Individual data points from n=5 independent samples are shown. Asterisks represent significant differences compared to untreated controls (*, p<0.05; **; p<0.01; ***; p<0.001) (Two-way ANOVA). (E) Human chondrocytes were co-transduced with ad-NLS-HyPer-DAAO and ad-Null or ad-SIRT6 vectors for 48 hrs. Plot of calculated ratios between fluorescence intensities recorded using FITC and CFP excitation filters in response to 6 mM D-alanine. Data was normalized to the average value prior to the addition of D-ala (representative data from n=3 independent samples).
To further validate this chemogenetic approach for generating nuclear H2O2 in human chondrocytes, we transduced cells with the NLS-HyPer-DAAO vector and assessed the redox status of Prx isoforms 1-3 in response to treatment with 6 mM D-ala by immunoblotting under non-reducing conditions. Based on our findings in cytoplasmic and nuclear fractions, we expected that nuclear generated H2O2 would oxidize nuclear and perhaps cytoplasmic Prx1 and Prx2, whereas Prx3, which is localized to the mitochondria, would be unaffected. Monomeric Prx1 was observed at around 22kDa and the oxidized, dimeric form was observed at around 38kDa, which is consistent with our previous report (32). In unstimulated conditions, Prx1 was mainly observed in the reduced monomeric form with low levels of Prx1 dimer formation (Figure 9C). Chondrocytes transduced with NLS-HyPer-DAAO and treated with 6 mM D-ala displayed significantly increased Prx1 dimer formation at 5 mins, when compared to untreated control conditions. This effect was maintained over the time course and was accompanied by the disappearance of monomeric Prx1, indicating H2O2 generated by NLS-HyPer-DAAO induced Prx1 oxidation (Figure 9C, D). Prx2 was observed in the reduced, monomeric form at approximately 21kDa and in the oxidized, dimeric form at approximately 44kDa, which agrees with our prior studies (31,32). The response of Prx2 to D-ala stimulation was similar to that observed for Prx1. A significant increase in Prx2 dimer formation was detected within 5 mins of treatment with D-ala, when compared to untreated conditions, and dimer formation was maintained over the time course (Figure 9C, D). Under oxidative stress conditions and at later time points (30-60 mins), we and others, have previously observed the disappearance of oxidized Prx dimers and the re-appearance of the Prx monomer, which indicates Prx hyperoxidation (31,32,44). For both Prx1 and Prx2, hyperoxidized monomer formation was not observed, suggesting that 6 mM D-ala activation of NLS-HyPer-DAAO led to physiological levels of ROS that oxidize the Prxs but do not cause Prx hyperoxidation (Figure 9C, D). As we have previously shown (31,32), monomeric Prx3 migrates at 24kDa with the dimeric form migrating at around 38kDa (Figure 9C). In response to treatment with 6 mM D-ala, we did not observe significant dimer formation or the appearance of a hyperoxidized monomer over our time course (Figure 9C, D), suggesting that mitochondrial Prx3 was not oxidized to form a disulfide, or hyperoxidized in this system.
To assess the effects of SIRT6 overexpression on nuclear H2O2, we co-transduced chondrocytes with the NLS-HyPer-DAAO construct and either the empty vector as control or the adenoviral vector encoding SIRT6 and stimulated cells with 6mM D-ala. D-alanine treatment of cells expressing the HyPer-DAAO construct led to a consistent increase in nuclear H2O2 generation within 1 min, an effect that was maintained over the 15 min time course studied. Importantly, overexpression of SIRT6 consistently decreased the NLS-HyPer-DAAO fluorescence ratio when compared to controls (Figure 9E), which we interpret as showing increased scavenging of H2O2, attributable to SIRT6-mediated alterations to Prx1, Srx, Nrf2, and TXNIP and enhancement of the antioxidant capacity of these cells.
Discussion and conclusions
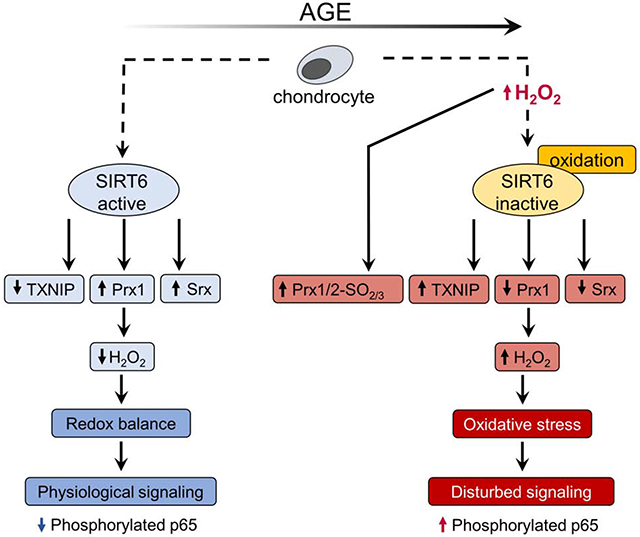
Emerging evidence suggests that SIRT6 is a crucial mediator of biological processes that can govern aging, longevity, and key pathways implicated in various stress responses, including resistance to oxidative stress conditions (3,8,22,23). Despite the purported roles of SIRT6 in attenuation of age-related disease processes, there is a lack of studies that address the role of SIRT6 on joint tissue homeostasis. As such, the current study was undertaken to examine the role of SIRT6 in aging, in response to oxidative stress conditions associated with aging, and on regulation of redox homeostasis in primary human chondrocytes. Our key findings demonstrate that chondrocyte SIRT6 activity declines in older, when compared to younger chondrocytes. We also show that SIRT6 is redox sensitive, being oxidized in response to H2O2, an effect that is associated with a decrease in SIRT6 activity. This is of particular importance as we show that active SIRT6 is an important regulator of chondrocyte redox balance by specifically increasing Prx1 and Srx antioxidant levels, and decreasing TXNIP levels. These events are linked to a decrease in nuclear generated H2O2 and a decrease in nuclear accumulation of phosphorylated p65, the catalytic isoform of NF-κB which is implicated in cartilage degradation and inflammation.
Our finding that chondrocyte SIRT6 protein levels do not change with age or OA is in contrast to the findings reported by Wu et al (20) who demonstrated an OA-related decrease in chondrocyte SIRT6 when compared to non-OA chondrocytes. Our study however, benefits from a much larger sample size, which was a strategic effort to overcome the heterogeneity commonly observed in chondrocytes derived from cartilage samples from adult humans. Rather than a change in SIRT6 levels, in a separate experiment, our data are the first to show that chondrocyte SIRT6 activity declines with age in chondrocytes. In addition to the age effect, we also demonstrate that SIRT6 is redox sensitive in chondrocytes and that SIRT6 oxidation leads to a decrease in SIRT6 deacetylase activity. In this study we assessed acetylation of H3K9 as an inverse marker of SIRT6 activity. SIRT6 has been well characterized as a highly specific H3 deacetylase that targets Lys9 and as a result, the acetylation/deacetylation status of H3K9 is commonly used to measure SIRT6 activity (6,8,12,23,51-55). However, we acknowledge that deacetylation of H3K9 could also have been modulated by other deacetylases that have been shown to target H3K9, including SIRT1, and to a lesser extent, HDAC3 and HDAC11 (56,57). Our observed redox-related change in SIRT6 activity is supported by the work of Hu et al (51), that demonstrated in HEK293 cells that ROS produced by SIN-1, which include nitric oxide, lead to posttranslational oxidative modifications of SIRT6 (namely nitrosation) and a decrease in SIRT6 catalytic activity. Posttranslational oxidative modifications of redox sensitive proteins are regarded as an important mechanism that can regulate cell signaling events (37). Given the plethora of SIRT6 targets, an age-associated or oxidative stress-induced decline in SIRT6 activity would likely lead to disturbances in physiological SIRT6 signaling that could contribute to age related pathology. Indeed, recent reports in other cell types demonstrate that SIRT6 sulfenylation leads to alterations in SIRT6-mediated regulation of glucose homeostasis (28) and binding to the transcription factor, HIF-1α (27). Further examination of mechanisms that regulate SIRT6 activity in primary human cells are required in order to fully understand the complex roles of SIRT6 in cellular homeostasis.
In the current study, we used an adenoviral vector to overexpress SIRT6 as a tool to maintain SIRT6 activity and we examined its ability to regulate redox status in chondrocytes. Although we acknowledge that SIRT6 levels are high in our overexpressing samples and should be interpreted with caution, our data suggests that SIRT6 can regulate specific chondrocyte antioxidant protein levels. We observed that SIRT6 overexpression specifically regulated members of the Prx catalytic cycle, namely Prx1 and Srx. The Prxs display high reactivity with H2O2 and are regarded as the primary antioxidant system that contributes to cellular redox homeostasis (58-60). During Prx-mediated removal of H2O2, the peroxidatic cysteine of Prx undergoes oxidation to the sulfenylated form (Prx−SOH). Ordinarily, the peroxidatic cysteine condenses with a resolving Prx cysteine, forming a disulfide which can be reduced, allowing the Prx to return to the active state. This reduction step is mediated by the thioredoxin reductase system which is NADPH dependent and comprised of thioredoxin (Trx) and thioredoxin reductase (TrxR) (61). If H2O2 levels are excessive, the Prxs can proceed to the sulfinic (Prx SO2−) and sulfonic (Prx-SO3−) forms, termed hyperoxidation, which can potentially inhibit Prx peroxidase activity and exacerbate oxidative stress conditions. Hyperoxidized Prxs in the sulfinic acid state can be reduced back to the active form by sulfiredoxin (Srx) (62). SIRT6 mediated enhancement of Prx1 and Srx levels supports our hypothesis that SIRT6 is an important mediator of redox balance and could contribute to maintaining the cellular antioxidant capacity through upregulating specific members of the Prx catalytic cycle. Additionally, our previous research has demonstrated that increasing intracellular levels of Prx1 can enhance basal levels of Akt and its downstream marker of activity, PRAS40 (32). Augmented Akt signaling is implicated in chondrocyte survival and cartilage matrix homeostasis (36,63); thus, it is conceivable that a SIRT6-mediated increase in Prx1 levels could positively modulate pro-anabolic pathways pertinent to joint homeostasis and may represent another layer of SIRT6-mediated regulation.
In addition to our findings for Prx1 and Srx, basal TXNIP levels were significantly decreased when we overexpressed SIRT6 in primary human chondrocytes, and significantly increased in our in vitro SIRT6 knockdown model. TXNIP can regulate redox homeostasis by binding to and inhibiting Trx, thereby preventing the regeneration of oxidized Prxs. Through this mechanism, TXNIP is associated with increased ROS generation and exacerbation of oxidative stress conditions (64). As such, a SIRT6-regulated reduction in TXNIP levels would likely contribute to more efficient elimination of ROS generation, especially in aged cells where an increase in basal levels of ROS are postulated to contribute to cellular dysfunction (26). In agreement with our findings, Qin et al (52) have demonstrated that conditional knockout of Sirt6 in beta cells increases TXNIP levels, due to an increase in the acetylation of H3K9 at the transcriptional start site of TXNIP (52). Such direct transcriptional suppression of TXNIP demonstrates the epigenetic control that SIRT6 exerts on redox-regulating genes and further highlights SIRT6 as a key modulator of redox balance. In addition to TXNIP, SIRT6 has been shown to transcriptionally regulate the expression of various genes by deacetylation of Lys9 on histone 3 (reviewed in (1)). Although beyond the scope of the current study, we anticipate that SIRT6 is regulating the expression of Prx1, Srx, Nrf2 and TXNIP through this mechanism, and experiments to this end represent an important avenue of our future research. In addition, SIRT6 has been shown to bind directly to the antioxidant transcription factor, Nrf2, and enhance the expression of Nrf2-regulated antioxidant genes through deacetylation of H3K56 at promoter regions (23). As Nrf2 has been shown to repress TXNIP (65) and our data showed a significant effect of SIRT6 to enhance Nrf2 protein levels, we hypothesize the existence of a SIRT6/Nrf2/TXNIP axis that may be an important mechanism in maintenance of redox regulation. Although our study did not examine the effect of SIRT6 to regulate Nrf2-mediated regulation of TXNIP, this represents an important avenue of future research. In addition, further experiments are needed to demonstrate that our observed effects are directly mediated by SIRT6 and not an indirect or correlative effect of SIRT6 adenoviral overexpression or knockdown.
As SIRT6 activity declines with age, we anticipated that during aging, TXNIP levels would increase due to a dampening of SIRT6-mediated TXNIP repression. Indeed, our data in human cartilage sections shows that TXNIP was significantly increased in older cartilage when compared to younger cartilage. Such an age-related increase in TXNIP levels is in accordance with other observations in different tissue types (66). Furthermore, evidence suggests that TXNIP may be implicated in the pathogenesis of OA. For example, mice that received surgery to induce post-traumatic OA (via destabilization of the medial meniscus (DMM model)) displayed significantly enhanced levels of cellular TXNIP staining in cartilage when compared to sham controls (67), suggesting that TXNIP may play an important role in joint tissue homeostasis.
Importantly, overexpression of SIRT6 attenuated global Prx hyperoxidation in response to menadione treatment and decreased nuclear generated H2O2 generated chemogenetically. We hypothesize that these findings are due to SIRT6-mediated upregulation of nuclear Prx1 and Srx and subsequent detoxification of nuclear generated or non-nuclear generated ROS. Prior evidence strongly suggests that NAD(P)H oxidase-4 (Nox4) is, at least partly, localized to the nuclear membrane and is an important source of nuclear superoxide generation in vascular endothelial cells that would likely contribute to ROS-induced damage (68). Although identification of the specific sources of nuclear ROS in chondrocytes were beyond the scope of this study, our findings suggest that targeted strategies aimed at restoring nuclear redox balance may be important for protecting against nuclear damage and activation of pro-catabolic signaling pathways. Indeed, SIRT6 overexpression reduced menadione-induced phosphorylation of cytoplasmic p65 and nuclear accumulation of phosphorylated p65 which represents the catalytic subunit of NF-κB implicated in upregulation of inflammatory pathways and cartilage degradation (69). Elsewhere, SIRT6 has been shown to attenuate NF-κB-dependent gene expression in chondrocytes (20), although the specific mechanism(s) responsible have not been studied. However, active SIRT6 has been shown to exert dynamic and specific control over NF-κB activation in various cell types by upregulating the expression of the NF-κB inhibitor, IkBα (70) and by deacetylation of H3K9 at the promoter regions of NF-κB target genes (71).
Our studies were conducted in articular chondrocytes in order to investigate SIRT6 in joint dysfunction and OA. An age-related increase in oxidative stress is recognized as a contributing factor in the development and progression of OA. Our prior in vivo and in vitro studies have demonstrated the importance of maintaining redox balance in chondrocytes, in part through modifying Prx levels, in order to decrease oxidative stress and associated cellular damage that contributes to OA (31,32). Our current findings extend these observations and suggest that SIRT6 may act as a critical regulator of antioxidant function through specifically modulating Prx1, Srx and TXNIP levels in both normal and OA chondrocytes. As we show that SIRT6 activity declines with age and in response to oxidative stress conditions associated with aging, we suggest that targeted approaches aimed at maintaining SIRT6 activity may represent an important therapeutic strategy to enhance antioxidant status, attenuate oxidative stress, and reduce catabolic signaling events associated with aging joint tissues. This could be specifically beneficial in OA, where an age-associated increase in oxidative stress conditions is hypothesized to play a key role in OA development and progression.
Supplementary Material
Highlights.
Age and oxidative stress levels of H2O2 decrease chondrocyte SIRT6 activity
SIRT6 overexpression increases Prx1 and Srx levels, while decreasing TXNIP levels
An age-associated decline in cartilage SIRT6 correlates with enhanced TXNIP levels
SIRT6 overexpression decreases H2O2 generation and attenuates p65 phosphorylation
Maintenance of SIRT6 activity during aging may govern chondrocyte redox balance
Acknowledgements
We thank the Gift of Hope Organ and Tissue Donor Network and the donor families for provision of normal human donor tissue. We thank Dr. Arkady Margulis for tissue procurement and Mrs. Arnavaz Hakimiyan from Rush University Medical Center for technical assistance. We thank Dr. Chris Olcott, Dr. Dan Del-Gaizo and the Department of Orthopaedic Surgery at the University of North Carolina Medical Center for provision of OA cartilage. We thank Dr. Martin Lotz for providing human knee cartilage sections. We also thank Kathryn Kelley, Meredith Rowe, Waverly Leonard, Mary Zhou, Mikalyn DeFoor, and Helen Willcockson for technical assistance.
Funding and additional information
This research was conducted while JAC was a Glenn/AFAR Postdoctoral Fellow and an Irene Diamond Fund/AFAR Postdoctoral Transition Awardee in Aging. This study was also funded by a grant from the National Institute on Aging (AG044034, RFL) and in part by the Klaus Kuettner Endowed Chair for Osteoarthritis Research (SC). TM is supported by funds from the National Institute on Aging grant R21 AG063073, the National Heart, Lung & Blood Institute, RO1 HL152173, and by the Brigham and Women's Hospital Health and Technology Innovation Award. EE was supported by the Austrian science foundation grant FWF (J4113).
Abbreviations:
- BIAM
biotinylated iodoacetamide
- DAAO
D-amino acid oxidase
- D-ala
D-alanine
- DTT
dithiothreitol
- H3K9ac
acetylated histone 3 lysine 9
- FAD
flavin adenine dinucleotide disodium salt hydrate
- HO-1
heme oxygenase-1
- IAM
iodoacetamide
- LDH
lactate dehydrogenase
- menadione
2-methyl-1,4-naphthoquinone
- NEM
N-ethylmaleimide
- NLS
nuclear localization signal
- NQO1
NAD(P)H Quinone Dehydrogenase-1
- OA
osteoarthritis
- PBS
phosphate buffered saline
- Prx
peroxiredoxin
- Prx-SOH
sulfenic acid
- Prx-SO2H
sulfinic acid
- Prx-SO3H
sulfonic acid
- ROS
reactive oxygen species
- SIRT
sirtuin
- Srx
sulfiredoxin
- TBP
TATA box binding protein
- Trx
thioredoxin
- Trxr
thioredoxin reductase
- TXNIP
thioredoxin interacting protein
Footnotes
Declaration of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kugel S, and Mostoslavsky R (2014) Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem Sci 39, 72–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roichman A, Kanfi Y, Glazz R, Naiman S, Amit U, Landa N, Tinman S, Stein I, Pikarsky E, Leor J, and Cohen HY (2017) SIRT6 Overexpression Improves Various Aspects of Mouse Healthspan. J Gerontol A Biol Sci Med Sci 72, 603–615 [DOI] [PubMed] [Google Scholar]
- 3.Tasselli L, Zheng W, and Chua KF (2017) SIRT6: Novel Mechanisms and Links to Aging and Disease. Trends Endocrinol Metab 28, 168–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, and Cohen HY (2012) The sirtuin SIRT6 regulates lifespan in male mice. Nature 483, 218–221 [DOI] [PubMed] [Google Scholar]
- 5.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, and Alt FW (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329 [DOI] [PubMed] [Google Scholar]
- 6.Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, Chang HY, Bohr VA, Ried T, Gozani O, and Chua KF (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452, 492–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, and Chua KF (2009) Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8, 2664–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, and Gorbunova V (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lombard DB, Schwer B, Alt FW, and Mostoslavsky R (2008) SIRT6 in DNA repair, metabolism and ageing. J Intern Med 263, 128–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tennen RI, Bua DJ, Wright WE, and Chua KF (2011) SIRT6 is required for maintenance of telomere position effect in human cells. Nat Commun 2, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tennen RI, and Chua KF (2011) Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem Sci 36, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH, Gao B, and Deng CX (2010) Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab 12, 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Xie JJ, Jin MY, Gu YT, Wu CC, Guo WJ, Yan YZ, Zhang ZJ, Wang JL, Zhang XL, Lin Y, Sun JL, Zhu GH, Wang XY, and Wu YS (2018) Sirt6 overexpression suppresses senescence and apoptosis of nucleus pulposus cells by inducing autophagy in a model of intervertebral disc degeneration. Cell Death Dis 9, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao G, Wang H, Xu C, Wang P, Chen J, Wang P, Sun Z, Su Y, Wang Z, Han L, and Tong T (2016) SIRT6 delays cellular senescence by promoting p27Kip1 ubiquitin-proteasome degradation. Aging (Albany NY) 8, 2308–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woo SJ, Noh HS, Lee NY, Cheon YH, Yi SM, Jeon HM, Bae EJ, Lee SI, and Park BH (2018) Myeloid sirtuin 6 deficiency accelerates experimental rheumatoid arthritis by enhancing macrophage activation and infiltration into synovium. EBioMedicine 38, 228–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon M, Van Meter M, Ablaeva J, Ke Z, Gonzalez RS, Taguchi T, De Cecco M, Leonova KI, Kogan V, Helfand SL, Neretti N, Roichman A, Cohen HY, Meer MV, Gladyshev VN, Antoch MP, Gudkov AV, Sedivy JM, Seluanov A, and Gorbunova V (2019) LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab 29, 871–885 e875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan RI, Nirzhor SSR, and Akter R (2018) A Review of the Recent Advances Made with SIRT6 and its Implications on Aging Related Processes, Major Human Diseases, and Possible Therapeutic Targets. Biomolecules 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dvir-Ginzberg M, Mobasheri A, and Kumar A (2016) The Role of Sirtuins in Cartilage Homeostasis and Osteoarthritis. Curr Rheumatol Rep 18, 43. [DOI] [PubMed] [Google Scholar]
- 19.Loeser RF, Collins JA, and Diekman BO (2016) Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol 12, 412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Y, Chen L, Wang Y, Li W, Lin Y, Yu D, Zhang L, Li F, and Pan Z (2015) Overexpression of Sirtuin 6 suppresses cellular senescence and NF-kappaB mediated inflammatory responses in osteoarthritis development. Sci Rep 5, 17602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ailixiding M, Aibibula Z, Iwata M, Piao J, Hara Y, Koga D, Okawa A, Morita S, and Asou Y (2015) Pivotal role of Sirt6 in the crosstalk among ageing, metabolic syndrome and osteoarthritis. Biochem Biophys Res Commun 466, 319–326 [DOI] [PubMed] [Google Scholar]
- 22.Rezazadeh S, Yang D, Tombline G, Simon M, Regan SP, Seluanov A, and Gorbunova V (2019) SIRT6 promotes transcription of a subset of NRF2 targets by mono-ADP-ribosylating BAF170. Nucleic Acids Res 47, 7914–7928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan H, Guan D, Liu X, Li J, Wang L, Wu J, Zhou J, Zhang W, Ren R, Zhang W, Li Y, Yang J, Hao Y, Yuan T, Yuan G, Wang H, Ju Z, Mao Z, Li J, Qu J, Tang F, and Liu GH (2016) SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res 26, 190–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao CY, and Kennedy BK (2016) SIRT6, oxidative stress, and aging. Cell Res 26, 143–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Go YM, and Jones DP (2017) Redox theory of aging: implications for health and disease. Clin Sci (Lond) 131, 1669–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones DP (2015) Redox theory of aging. Redox Biol 5, 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Gupta V, Carroll KS, and Liebler DC (2014) Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat Commun 5, 4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Long D, Wu H, Tsang AW, Poole LB, Yoza BK, Wang X, Vachharajani V, Furdui CM, and McCall CE (2017) The Oxidative State of Cysteine Thiol 144 Regulates the SIRT6 Glucose Homeostat. Sci Rep 7, 11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wood ST, Long DL, Reisz JA, Yammani RR, Burke EA, Klomsiri C, Poole LB, Furdui CM, and Loeser RF (2016) Cysteine-Mediated Redox Regulation of Cell Signaling in Chondrocytes Stimulated With Fibronectin Fragments. Arthritis Rheumatol 68, 117–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson KJ, Bolduc JA, Wu H, Collins JA, Burke EA, Reisz JA, Klomsiri C, Wood ST, Yammani RR, Poole LB, Furdui CM, and Loeser RF (2018) H2O2 oxidation of cysteine residues in c-Jun N-terminal kinase 2 (JNK2) contributes to redox regulation in human articular chondrocytes. J Biol Chem 293, 16376–16389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins JA, Wood ST, Nelson KJ, Rowe MA, Carlson CS, Chubinskaya S, Poole LB, Furdui CM, and Loeser RF (2016) Oxidative Stress Promotes Peroxiredoxin Hyperoxidation and Attenuates Pro-survival Signaling in Aging Chondrocytes. J Biol Chem 291, 6641–6654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins JA, Wood ST, Bolduc JA, Nurmalasari NPD, Chubinskaya S, Poole LB, Furdui CM, Nelson KJ, and Loeser RF (2019) Differential peroxiredoxin hyperoxidation regulates MAP kinase signaling in human articular chondrocytes. Free Radic Biol Med 134, 139–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, Daniel LW, and Poole LB (2010) Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol 473, 95–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muehleman C, Bareither D, Huch K, Cole AA, and Kuettner KE (1997) Prevalence of degenerative morphological changes in the joints of the lower extremity. Osteoarthritis Cartilage 5, 23–37 [DOI] [PubMed] [Google Scholar]
- 35.Cross M, Smith E, Hoy D, Nolte S, Ackerman I, Fransen M, Bridgett L, Williams S, Guillemin F, Hill CL, Laslett LL, Jones G, Cicuttini F, Osborne R, Vos T, Buchbinder R, Woolf A, and March L (2014) The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis 73, 1323–1330 [DOI] [PubMed] [Google Scholar]
- 36.Loeser RF, Gandhi U, Long DL, Yin W, and Chubinskaya S (2014) Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol 66, 2201–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klomsiri C, Nelson KJ, Bechtold E, Soito L, Johnson LC, Lowther WT, Ryu SE, King SB, Furdui CM, and Poole LB (2010) Use of dimedone-based chemical probes for sulfenic acid detection evaluation of conditions affecting probe incorporation into redox-sensitive proteins. Methods Enzymol 473, 77–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matlashov ME, Belousov VV, and Enikolopov G (2014) How much H(2)O(2) is produced by recombinant D-amino acid oxidase in mammalian cells? Antioxid Redox Signal 20, 1039–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alim I, Haskew-Layton RE, Aleyasin H, Guo H, and Ratan RR (2014) Spatial, temporal, and quantitative manipulation of intracellular hydrogen peroxide in cultured cells. Methods Enzymol 547, 251–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bogdanova YA, Schultz C, and Belousov VV (2017) Local Generation and Imaging of Hydrogen Peroxide in Living Cells. Curr Protoc Chem Biol 9, 117–127 [DOI] [PubMed] [Google Scholar]
- 41.Steinhorn B, Sorrentino A, Badole S, Bogdanova Y, Belousov V, and Michel T (2018) Chemogenetic generation of hydrogen peroxide in the heart induces severe cardiac dysfunction. Nat Commun 9, 4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mishina NM, Bogdanova YA, Ermakova YG, Panova AS, Kotova DA, Bilan DS, Steinhorn B, Arner ESJ, Michel T, and Belousov VV (2019) Which Antioxidant System Shapes Intracellular H2O2 Gradients? Antioxid Redox Signal 31, 664–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, Wiegraebe W, Singh S, Becker T, Caicedo JC, and Carpenter AE (2018) CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cox AG, Winterbourn CC, and Hampton MB (2010) Measuring the redox state of cellular peroxiredoxins by immunoblotting. Methods Enzymol 474, 51–66 [DOI] [PubMed] [Google Scholar]
- 45.Del Carlo M Jr., and Loeser RF (2002) Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum 46, 394–403 [DOI] [PubMed] [Google Scholar]
- 46.Diekman BO, Sessions GA, Collins JA, Knecht AK, Strum SL, Mitin NK, Carlson CS, Loeser RF, and Sharpless NE (2018) Expression of p16(INK) (4a) is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging Cell 17, e12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR, and de Crombrugghe B (2009) Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis 47, 805–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levy YD, Hasegawa A, Patil S, Koziol JA, Lotz MK, and D'Lima DD (2013) Histopathological changes in the human posterior cruciate ligament during aging and osteoarthritis: correlations with anterior cruciate ligament and cartilage changes. Ann Rheum Dis 72, 271–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ulici V, James CG, Hoenselaar KD, and Beier F (2010) Regulation of gene expression by PI3K in mouse growth plate chondrocytes. PLoS One 5, e8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Collins JA, Arbeeva L, Chubinskaya S, and Loeser RF (2019) Articular chondrocytes isolated from the knee and ankle joints of human tissue donors demonstrate similar redox-regulated MAP kinase and Akt signaling. Osteoarthritis Cartilage 27, 703–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu S, Liu H, Ha Y, Luo X, Motamedi M, Gupta MP, Ma JX, Tilton RG, and Zhang W (2015) Posttranslational modification of Sirt6 activity by peroxynitrite. Free Radic Biol Med 79, 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qin K, Zhang N, Zhang Z, Nipper M, Zhu Z, Leighton J, Xu K, Musi N, and Wang P (2018) SIRT6-mediated transcriptional suppression of Txnip is critical for pancreatic beta cell function and survival in mice. Diabetologia 61, 906–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang Z, Zhao J, Deng W, Chen Y, Shang J, Song K, Zhang L, Wang C, Lu S, Yang X, He B, Min J, Hu H, Tan M, Xu J, Zhang Q, Zhong J, Sun X, Mao Z, Lin H, Xiao M, Chin YE, Jiang H, Xu Y, Chen G, and Zhang J (2018) Identification of a cellularly active SIRT6 allosteric activator. Nat Chem Biol 14, 1118–1126 [DOI] [PubMed] [Google Scholar]
- 54.Iachettini S, Trisciuoglio D, Rotili D, Lucidi A, Salvati E, Zizza P, Di Leo L, Del Bufalo D, Ciriolo MR, Leonetti C, Steegborn C, Mai A, Rizzo A, and Biroccio A (2018) Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis 9, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein MA, Liu C, Kuznetsov VI, Feltenberger JB, Tang W, and Denu JM (2020) Mechanism of activation for the sirtuin 6 protein deacylase. J Biol Chem 295, 1385–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seto E, and Yoshida M (2014) Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 6, a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, and Reinberg D (2004) Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell 16, 93–105 [DOI] [PubMed] [Google Scholar]
- 58.Adimora NJ, Jones DP, and Kemp ML (2010) A model of redox kinetics implicates the thiol proteome in cellular hydrogen peroxide responses. Antioxid Redox Signal 13, 731–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wood ZA, Poole LB, and Karplus PA (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653 [DOI] [PubMed] [Google Scholar]
- 60.Perkins A, Nelson KJ, Parsonage D, Poole LB, and Karplus PA (2015) Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem Sci 40, 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Netto LE, and Antunes F (2016) The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol Cells 39, 65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lowther WT, and Haynes AC (2011) Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxid Redox Signal 15, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yin W, Park JI, and Loeser RF (2009) Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK signaling pathways. J Biol Chem 284, 31972–31981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nagaraj K, Lapkina-Gendler L, Sarfstein R, Gurwitz D, Pasmanik-Chor M, Laron Z, Yakar S, and Werner H (2018) Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc Natl Acad Sci U S A 115, 1045–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.He X, and Ma Q (2012) Redox regulation by nuclear factor erythroid 2-related factor 2: gatekeeping for the basal and diabetes-induced expression of thioredoxin-interacting protein. Mol Pharmacol 82, 887–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oberacker T, Bajorat J, Ziola S, Schroeder A, Roth D, Kastl L, Edgar BA, Wagner W, Gulow K, and Krammer PH (2018) Enhanced expression of thioredoxin-interacting-protein regulates oxidative DNA damage and aging. FEBS Lett 592, 2297–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ratneswaran A, Sun MM, Dupuis H, Sawyez C, Borradaile N, and Beier F (2017) Nuclear receptors regulate lipid metabolism and oxidative stress markers in chondrocytes. J Mol Med (Berl) 95, 431–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, Kuribayashi F, Imajoh-Ohmi S, Igarashi K, Shibata Y, Sueishi K, and Sumimoto H (2005) The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 10, 1139–1151 [DOI] [PubMed] [Google Scholar]
- 69.Olivotto E, Otero M, Marcu KB, and Goldring MB (2015) Pathophysiology of osteoarthritis: canonical NF-kappaB/IKKbeta-dependent and kinase-independent effects of IKKalpha in cartilage degradation and chondrocyte differentiation. RMD Open 1, e000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Santos-Barriopedro I, Bosch-Presegue L, Marazuela-Duque A, de la Torre C, Colomer C, Vazquez BN, Fuhrmann T, Martinez-Pastor B, Lu W, Braun T, Bober E, Jenuwein T, Serrano L, Esteller M, Chen Z, Barcelo-Batllori S, Mostoslavsky R, Espinosa L, and Vaquero A (2018) SIRT6-dependent cysteine monoubiquitination in the PRE-SET domain of Suv39h1 regulates the NF-kappaB pathway. Nat Commun 9, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, and Chua KF (2009) SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.