

Abstract
Because hyperpolarization-activated cyclic nucleotide-gated (HCN) ion channels modulate the excitability of cortical and hippocampal principal neurons, these channels play a key role in the hyperexcitability that occurs during the development of epilepsy after a brain insult, or epileptogenesis. In epileptic rats generated by pilocarpine-induced status epilepticus, HCN channel activity is downregulated by two main mechanisms: a hyperpolarizing shift in gating and a decrease in amplitude of the current mediated by HCN channels, Ih. Because these mechanisms are modulated by various phosphorylation signaling pathways, we hypothesized that phosphorylation changes occur at individual HCN channel amino acid residues (phosphosites) during epileptogenesis. We collected CA1 hippocampal tissue from male Sprague Dawley rats made epileptic by pilocarpine-induced status epilepticus, and age-matched naïve controls. We also included resected human brain tissue containing epileptogenic zones (EZs) where seizures arise for comparison to our chronically epileptic rats. After enrichment for HCN1 and HCN2 isoforms by immunoprecipitation and trypsin in-gel digestion, the samples were analyzed by mass spectrometry. We identified numerous phosphosites from HCN1 and HCN2 channels, representing a novel survey of phosphorylation sites within HCN channels. We found high levels of HCN channel phosphosite homology between humans and rats. We also identified a novel HCN1 channel phosphosite S791, which underwent significantly increased phosphorylation during the chronic epilepsy stage. Heterologous expression of a phosphomimetic mutant, S791D, replicated a hyperpolarizing shift in Ih gating seen in neurons from chronically epileptic rats. These results show that HCN1 channel phosphorylation is altered in epilepsy and may be of pathogenic importance.
Graphical Abstract
Introduction
Hyperpolarization-activated, cyclic nucleotide-gated (HCN) ion channels represent a unique family of voltage-gated channels both for their unusual biophysical properties and their effects on the excitability of pyramidal neurons within the cortex and hippocampus. Their biophysical properties have been a source of fascination and controversy for years, and their role in cortical and hippocampal excitability have made them an intriguing pharmacological target (Brennan et al., 2016; DiFrancesco et al., 2019; Santoro & Shah, 2020). Because they are localized to the distal dendrites of these neurons and are non-inactivated at resting potential, HCN channels produce a substantial resting conductance (Ih) that significantly diminishes summation of dendritic synaptic voltage transients and thus reduces pyramidal neuron excitability (Poolos et al., 2002; Poolos, 2012; Poolos & Johnston, 2012). Modulation of HCN channel voltage-dependent gating or dendritic expression thus can significantly affect pyramidal neuron excitability in cortex and hippocampus. Of the four isoforms of HCN channels, HCN1 comprises the majority of expression in these brain regions, with HCN2 comprising a minority of expression at about a 3:1 or 4:1 ratio of HCN1:HCN2. Also, HCN1 and HCN2 subunits can form functional HCN channels as tetramers of each isoform alone or as tetramers of mixed subunits (Santoro & Tibbs, 1999).
HCN channels have been implicated in human epilepsy and in animal models of epilepsy. Mutations of the HCN1 gene produce a severe early childhood epileptic encephalopathy consisting of frequent seizures, multifocal electroencephalogram (EEG) abnormalities, and developmental delay (Nava et al., 2014; Marini et al., 2018). A de novo loss-of-function HCN1 mutation that has been shown to increase neuronal excitability was also found in an epilepsy patient with generalized EEG abnormalities (Bonzanni et al., 2018). The HCN2 gene has not been clearly implicated in human epilepsy although case reports suggest a link to less severe epilepsy syndromes (Tang et al., 2008; DiFrancesco et al., 2011; Li et al., 2018). Genetic deletion of the Hcn1 gene in mice produces cortical hyperexcitability but not epilepsy (Nolan et al., 2004), while deletion of Hcn2 produces generalized, absence-like interictal discharges on EEG recordings (Ludwig et al., 2003). In animal models of acquired epilepsy such as post-status epilepticus (SE)-induced or febrile seizure-induced epilepsy, HCN channel gating, membrane surface expression, and protein expression are dysregulated in different phases of epileptogenesis and in isoform-specific ways.
In the acute phase immediately following the acute insult (SE, for example), HCN1 surface expression and maximal Ih amplitude are reduced and stay reduced into the chronic phase of epilepsy when spontaneous seizures occur. This is accompanied several days after the insult by long-lasting transcriptional downregulation of HCN1 expression (Jung et al., 2011). HCN2 expression, however, is transiently downregulated in the acute phase but returns to normal in chronic epilepsy (Brewster et al., 2002; Jung et al., 2011). HCN channels also undergo a downregulation in voltage-dependent gating which develops as seizures begin in the post-SE model and progressively worsen with increased seizure frequency as the animal develops chronic epilepsy (Jung et al., 2007). Both these changes in HCN1 expression and gating seen in chronic epilepsy downregulate Ih and contribute to neuronal hyperexcitability.
We previously published evidence that these changes in HCN1 biophysical properties during epileptogenesis were phosphorylation dependent. HCN1 voltage-dependent gating is downregulated by inhibition of p38 mitogen-activated protein kinase (p38 MAPK), and expression of this kinase is reduced in chronic epilepsy (Poolos et al., 2006; Jung et al., 2010). HCN1 channel surface membrane expression is acutely and irreversibly reduced by protein kinase C (PKC) activity following induction of epilepsy by SE (Williams et al., 2015). These studies demonstrated the modulation of HCN1 channel activity by phosphorylation in ways that were consistent with changes in phosphorylation signaling observed during epileptogenesis, but did not demonstrate that HCN1 channels themselves underwent phosphorylation changes in epilepsy.
In the present study we sought evidence that HCN channels undergo changes in phosphorylation during epileptogenesis and that these changes in phosphorylation modulate HCN channel activation. Because there was little existing knowledge about which residues are phosphorylated on HCN channels, we comprehensively mapped HCN1 and HCN2 channel phosphorylation sites (phosphosites) using mass spectrometry. We surveyed HCN channels in brain tissue from both naïve and epileptic animals, and in human brain tissue from medically refractory epilepsy patients. Our results demonstrate that HCN1 and HCN2 channels each have numerous phosphorylation sites, but that only one site, HCN1-S791, shows a change (an increase) in phosphorylation in epilepsy. Expression of mutant HCN1 channels in Xenopus oocytes replicating this phosphorylation change showed a downregulation of HCN1 channel gating, similar to that observed in native tissue from epileptic animals. In sum, this study demonstrates that changes in the phosphorylation of HCN1 channels may contribute to their loss of function in chronic epilepsy.
Experimental Procedures
Pilocarpine-induced status epilepticus (SE) in rats
Epileptic rats were generated using pilocarpine-induced SE as previously described (Jung et al., 2007). The University of Washington Institutional Animal Care and Use Committee approved all animal procedures before experimentation. To induce experimental SE, 6-week-old male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) were treated with scopolamine methylnitrate (1 mg/kg intraperitoneal [i.p]) 30 minutes before treating with pilocarpine hydrochloride (385 mg/kg i.p.). 60 minutes after observing convulsive SE, phenobarbital (PB 15 mg/kg i.p.) was administered every 30–45 minutes until observable seizures ceased. These rats, which become chronically epileptic after 3–4 weeks post-SE (Jung et al., 2007), were singly housed for 6–9 weeks before sacrifice. To generate 1 hr post-SE rats, animals were sacrificed after one hour of SE instead of administrating PB. As controls for chronically epileptic rats, 6-week-old naïve rats were singly housed for the same 6–9 week period. Naïve rats were sacrificed at 6 weeks old to serve as controls for the 1 hr post-SE rats. Rats were sacrificed with ketamine/xylazine (87/13 mg/kg i.p.), with the chronically epileptic animals and their age-matched controls exposed to 5% isoflurane beforehand. The procedure for brain slicing has been previously described (Williams et al., 2015). The tissue from brain slicing was frozen and stored at −80 °C.
Brain tissue samples from human epilepsy patients
Brain tissue from 10 human patients undergoing resective surgery at the University of Washington Regional Epilepsy Center for refractory epilepsy was collected at the time of operation. We received a waiver of consent from the University of Washington Institutional Review Board, and no protected health information was disclosed. These brain tissues were either of extratemporal neocortical origin (eight patients) or hippocampal (two patients) which after scalp and/or intracranial EEG studies were determined to contain the epileptogenic zone (EZ) where seizures arose. Each collected sample was immediately cut into smaller pieces, placed in microcentrifuge tubes, flash-frozen in liquid nitrogen, and stored at −80 °C. One sample per patient was used for mass spectrometry analysis.
Immunoprecipitation (IP)
We used immunoprecipitation (IP) to enrich for HCN channels. Starting with either pooled microdissected rat hippocampal CA1 regions or frozen resected human brain tissue from epilepsy patients, samples were homogenized in the following buffer: 50 mM tris base, 50 mM sodium chloride, 5 mM EDTA, 10 mM EGTA, 1 mM sodium orthovanadate, 2 mM sodium pyrophosphate, 4 ug/mL aprotinin, 4 mM para-nitrophenyl phosphate, 20 ug/mL leupeptin, 1 mM phenylmethylsulfonyl fluoride and 1% triton X-100 (Roberson et al., 1999). The homogenate was centrifuged at 20,800 × g at 4 °C for 15 mins, and the supernatant was transferred to a separate tube. After adding either anti-HCN1 antibody (Ab) (Antibodies Inc., Davis, CA; catalog #75–110) or anti-HCN2 Ab (Cell Signaling Technology, Danvers, MA; catalog #14957S), homogenates were incubated with gentle agitation overnight at 4 °C. Protein A/G UltraLink Resin (Thermo Fisher Scientific, Waltham, MA) was added, and the samples were incubated with gentle agitation at 4 °C for 4–6 hrs. After washing three times with homogenization buffer, we eluted with 1× Laemmli buffer with 5% sodium dodecyl sulfate and 25 mM dithiothreitol.
For HCN2 phosphosites in the human samples only, due to the limited amount of human tissue available, HCN2 was not enriched directly by the anti-HCN2 Ab (CST). Instead, because HCN2 subunits are known to colocalize with HCN1 subunits in the distal dendrites of hippocampal pyramidal cells (Notomi & Shigemoto, 2004), we were able to pull down HCN2 molecules that were bound to the targeted HCN1 subunits when immunoprecipitating with the anti-HCN1 Ab (Antibodies Inc.). Because of this co-immunoprecipitation of HCN2 molecules when targeting HCN1 proteins, we were able to identify spectra derived from HCN2 subunits and subsequently determine the HCN2 phosphosites and their phosphorylation levels within human brain tissues.
Western blotting
Western blotting was used to isolate HCN1 and HCN2 protein bands from the immunoprecipitated samples prepared above. Briefly, samples were run in a 4–15% polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA). Immunoprecipitation eluates as described in the previous section were incubated at 80 °C for 10 minutes and loaded in 4–15% acrylamide gels (Bio-Rad Laboratories). After transferring proteins to a nitrocellulose membrane, the following primary Abs were used: anti-HCN1 (Antibodies Inc., #75–110) or anti-HCN2 (CST, #14957S). Analysis of signals from fluorescent secondary antibodies (anti-rabbit IgG-Dylight800 and the anti-mouse-IgG-Dylight680; Thermo Fisher Scientific) were achieved with the Odyssey CLx Imaging system (Li-Cor Biosciences, Lincoln, NE).
In-gel digestion
Post-electrophoresis, gels were treated with Coomassie R-250 blue stain (Thermo Fisher Scientific) and washed to visualize the 110–120 kDa band, which was excised out from the gel and further cut into pieces about 1–2 mm3. After a series of contractions and expansions with acetonitrile and ammonium bicarbonate, respectively; reduction by dithiothreitol (DTT); and alkylation by iodoacetamide, samples were subjected to trypsin digestion. Afterwards, the peptides were extracted from the gel by 50% acetonitrile/5% formic acid solution and concentrated by vacuum desiccation.
Mass spectrometry analysis
All samples were analyzed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) equipped with a nanoACQUITY UPLC system (Waters, Milford, MA) and in-house-developed nano spray ionization source. Samples were loaded from the autosampler onto a 100 μm ID IntegraFrit trap (New Objective, Littleton, MA) packed with ReproSil-Pur C18-AQ 120 Å 5 μm material (Dr. Maisch, Entringen, Germany) to a bed length of 3 cm at a flow rate of 2 μL/min. After loading and desalting for 10 min with 0.1% formic acid plus 2% acetonitrile (LCMS grade from Thermo Fisher Scientific), the trap was brought in-line with a pulled fused-silica capillary tip (75-μm i.d.) packed with 35 cm of ReproSil-Pur C18-AQ 120 Å 5 μm (Dr. Maisch). Peptides were separated using a linear gradient, from 5–30% solvent B (LCMS grade 0.1 % formic acid in acetonitrile (Thermo Fisher Scientific)) in 90 min at a flow rate of 300 nL/min. Peptides were detected using a data-dependent (DDA) method. Survey scans of peptide precursors were performed in the Orbitrap mass analyzer from 400 to 1600 m/z at 120K resolution (at 200 m/z) with a 7e5 ion count target and a maximum injection time of 50 ms. The instrument was set to run in top speed mode with 3 sec cycle for the survey and the MS/MS scans. After a survey scan, tandem MS was performed on the most abundant precursors exhibiting a charge state from 2 to 5 of greater than 2e4 intensity by isolating them in the quadrupole with an isolation width of 1.6 m/z.
Higher-energy collisional dissociation (HCD) fragmentation was applied with a normalized collision energy of 30 %. Resulting fragments were detected in the Orbitrap mass analyzer at 30K resolution (at 200 m/z) with a 1e4 ion count target and a maximum injection time of 100 ms. The dynamic exclusion was set to 30 sec with a 20 ppm mass tolerance around the precursor and its isotopes. Monoisotopic precursor selection was enabled.
Data analysis from mass spectrometry
The raw instrument acquisition files were converted to the mzXML format using the ReAdW converter (Pedrioli et al., 2004). The peptide identification tool Comet (Eng et al., 2013) was used to search the mzXML files against a rat UniProt sequence database appended with sequences of common contaminants (UniProt Consortium, 2019). The relevant database search parameters include 20 ppm precursor tolerance, tryptic digest allowing two missed cleavages, variable modifications for oxidation of methionine and phosphorylation of serine, threonine, and tyrosine, static carbamidomethylation modification on cysteine, 0.02 fragment bin tolerance, and target-decoy search. The search results were further processed through the Trans-Proteomics Pipeline suite of tools for visualization and validation (Keller et al., 2005).
The following UniProt sequences were used to identify peptides: rat HCN1 – Q9JKB0 and F1LSH6; rat HCN2 – Q9JKA9; human HCN1 – O60741; and human HCN2 – Q9UL51. For the rat HCN1, Q9JKB0 and F1LSH6 are 98.6% homologous. Among the 13 differences, 10 are gaps, and 3 are variant amino acid residues: (A) Q9JKB0-T736 : F1LSH6-P736; (B) Q9JKB0-H799 : F1LSH6-N791; and (C) Q9JKB0-V840 : F1LSH6-M832. Of these three differences, only the H799:N791 variant was present in the regions “covered” by our mass spectrometric experiments involving the Sprague-Dawley rats used in this investigation. Furthermore, we only observed an asparagine (N) and never a histidine at this specific site (an example is displayed in Fig. 1D – the 4th to the last amino acid within the analyzed spectrum). Because Q9JKB0 is the “canonical” rat HCN1 sequence, we will refer to the Q9JKB0 sequence numbering in this study when discussing the rat HCN1 protein. However, as represented in Fig. 2A, at amino acid sequence #799, we have substituted the histidine for the empirically present asparagine.
Figure 1.

Identification of HCN1 and HCN2 phosphosites. (A) Flowchart of methods used for identifying phosphosites within HCN1 and HCN2. (B) Western blot demonstrating successful immunoprecipitation of HCN1 and HCN2. Black arrowheads indicate HCN bands. HCN1 from rat and human brain tissues and rat brain HCN2 migrate approximately between 110–120 kDa. Open arrowheads indicate artifacts from the HCN enrichment process, where the heavy- and light-chain IgGs (HC-IgG, LC-IgG) originating from the antibodies used in IP co-elute with the proteins of interest. (C) Sample spectra from mass spectrometry. The peak containing phosphorylated S791 (y12+) is marked by the red asterisk. (D) Example of spectral counting. The list consists of all spectra derived from peptides containing S791 from a single homogenate sample. Spectra that do not meet the threshold (“expect” value ≤ 0.01) are not included in the count and are labeled with a “#”. Spectra that contain phosphorylated S791 are labeled with a “p” with the encircled “p” denoting the specific spectrum displayed in (C). A red asterisk denotes phosphorylated S791 within the sequences shown in the “PEPTIDE” column.
Figure 2.

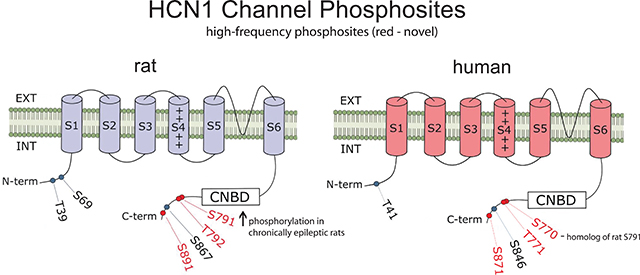
Comparison of rat and human HCN1 phosphosites. (A) Amino acid sequences of rat and human HCN1 are juxtaposed. Green highlights show regions of each HCN sequence that were observed by mass spectrometry. Ovals indicate detected phosphosites; red labels indicate novel phosphosites; bold labels indicate phosphosites observed in ≥ 50% of either species’ samples. Structural features of HCN channels are denoted: the intracellular N-terminal region, the six transmembrane domains (S1-S6), the pore-forming region, and an extensive intracellular C-terminal region incorporating the cyclic nucleotide binding domain (CNBD). (B) Representation of the rat HCN1 channel showing phosphosites that were phosphorylated in at least 50% of rat samples (with red labeling denoting novel phosphosites). (C) Representation of the human HCN1 channel showing phosphosites that are homologous to the prevalent rat phosphosites shown in (B). Novel phosphosites are labeled in red.
Spectral counting and determination of phosphorylation levels
From the data acquisition described above, identification and levels of phosphorylation of HCN1 and HCN2 channel phosphosites were achieved by spectral counting (Fig. 1D). Only those spectra whose “expected value” were ≤ 0.01 were treated as valid observations. The level of phosphorylation for a specific phosphosite within a given sample was calculated by:
(# of spectra where the specific phosphosite is phosphorylated) / (# of spectra containing that phosphosite regardless of phosphorylation state) X 100%.
Molecular biology
A full-length mouse Hcn1 cDNA cloned into a Xenopus oocyte expression plasmid (pGH19) was obtained (gift from Dr. Bina Santoro, Columbia University). To minimize frequent recombination events presumably due to poly-glutamine repeats in the carboxyl terminal of the channel subunit, this plasmid was propagated in a recA1-deficient bacterial strain (10-beta; New England Biolabs, Ipswich, MA). Site-directed mutagenesis of S791 was performed by standard overlap PCR with high-fidelity Taq Polymerase (Q5 Taq Polymerase; New England Biolabs), targeting a unique ~400 bp SexA1/XhoI cloning cassette with flanking primers. All WT and mutant clones were sequenced in entirety through their coding regions, using a commercial sequencing service (SimpleSeq; Eurofins Genomics, Louisville, KY). Large-scale plasmid preparations were generated with standard DNA purification kits (Maxiprep; QIAGEN, Redwood City, CA).
To generate capped cRNAs for Xenopus oocyte injections, plasmids were linearized with XhoI, for in vitro transcription by T7 RNA polymerase (mMessage mMachine T7 Transcription Kit; Ambion/Invitrogen, Waltham, MA), following manufacturer’s instructions. Defolliculated Xenopus oocytes (Stage IV-V) were obtained commercially (Xenocyte, Dexter, MI). Individual oocytes were injected with ~50 nL of cRNA (0.2 mg/mL) (Nanoject2, Drummond, Broomall, PA), and incubated in Barth’s solution (in mM: 88 NaCl, 1.0 KCl, 0.4 CaCl2, 0.33 Ca(NO3)2, 0.8 MgSO4, 5.0 Tris-HCl, 2.4 NaHCO3, pH 7.3, supplemented with 2.5 mM Na-Pyruvate, 100 U/mL Penicillin and 100 mg/mL Streptomycin) for 1–2 days at 19°C prior to recordings.
Electrophysiology
Two-electrode voltage clamp (TEVC) recordings were acquired on a TEVC workstation (TEV-700; Warner Instruments, Holliston, MA). Recording electrodes had resistances of 0.2–0.5 Mohm filled with 3.0 M KCl. Recordings were obtained in ND96 (in mM: 93.5 NaCl, 2.0 KCl, 1.8 CaCl2, 1.0 MgCl2, 5.0 HEPES, pH 7.5), at room temperature.
Oocytes were clamped at a holding potential of 0 mV. Current traces were evoked from a holding potential of 0 mV, cycled through a family of voltage steps from 10 mV to −120 mV (0.7 sec), returning to a 0 mV post-step, with a 10 sec inter-pulse interval to allow activated channels to fully equilibrate to the closed state. Conductance was measured by instantaneous tail currents at 0 mV, following activating hyperpolarizing voltage steps.
Oocytes were treated with the p38 MAPK inhibitor SB203780 (20 μM in ND96) (Tocris Bioscience, Minneapolis, MN) by soaking for ~20 min prior to recording.
Electrophysiology analysis
Uninjected oocytes exhibited an endogenous inward current activated by hyperpolarizing voltage steps negative to −90 mV (Supplemental Fig. S1), which varied in magnitude among batches of oocytes. Other investigators have reported similar induction of expression of endogenous ionic currents in oocytes injected with ion channel cDNA, including an inwardly-rectifying K+ current and a hyperpolarization-activated non-selective cation current (Tzounopoulos et al., 1995; Bauer et al., 1996; Weber, 1999). These endogenous inward currents complicate measurements of uncontaminated HCN1 currents at potentials negative to −90 mV. Therefore, derivations of conductance/voltage (G/V) plots, were performed in two steps similar to a procedure used by other investigators (Chen et al., 2001). Mean conductance values were first calculated for the voltage range 10 mV to −90 mV, then normalized to G at −90 mV. These data points were fitted to a single Boltzmann function (Origin8.5; OriginLab, Northampton, MA):
where: A1= maximal G/G−90mV
A2= minimal G/G−90mV
V1/2= voltage at half-maximal G/G−90mV
dx= slope factor
constraining only A2 to 0.
This procedure returned a fitted A1 value corresponding to an extrapolated Gmax, negative to −90 mV. This Gmax was used to normalize G/V plots a second time, and similarly refitted to single Boltzmann functions to allow normalized comparisons between WT and mutant HCN1 constructs.
Statistical analyses
Statistics are presented as means ± standard errors of mean (SEM). Because not all data sets were normally distributed, the Exact Two-Sample Fisher-Pitman Permutation test was used to determine the significance of results between the experimental and naïve groups. Additionally, because the same samples were used to investigate multiple phosphosites within a given HCN subunit, a Bonferroni correction factor was implemented to adjust the α for each HCN subunit (HCN1: α = 0.05/6; HCN2: α = 0.05/10). Graphically, data were presented as box plots.
Results
We previously reported that phosphorylation signaling modulates the biophysical properties of HCN channels. We showed that inhibition of p38 MAPK caused a hyperpolarizing shift in HCN channel voltage-dependent activation (or gating), and protein kinase C (PKC) activation caused a reduction of maximal amplitude of HCN channel-mediated current (Ih) as well as lowered surface expression of HCN channels (Poolos et al., 2006; Jung et al., 2010; Williams et al., 2015). During epileptogenesis both hyperpolarizing shifts in HCN channel activation and loss of maximal current occur, resulting in loss of function of HCN channels. We therefore hypothesized that one or more phosphorylation sites (serines, threonines, and tyrosine residues where phosphorylation occurs), or phosphosites, in HCN1 or HCN2 subunits undergo changes in phosphorylation level during epileptogenesis. Because there had been no prior studies describing the phosphosites of HCN subunits, which assemble as multimers to form functional channels, we sought to map HCN1 and HCN2 channel phosphosites using mass spectrometry in brain tissue from both human epilepsy patients and a rat model of temporal lobe epilepsy (TLE).
The flowchart (Fig. 1A) displays the process of identifying phosphosites in both HCN1 and HCN2 channels in brain tissue from both rats and human patients. Two stages of epileptogenesis (and age-matched controls) were studied in the animal model: the acute stage when rats are continuously seizing for 1 hour after onset of pilocarpine-induced SE (1 hr post-SE); and the chronically epileptic stage 6–9 weeks post-SE when spontaneous seizures average three/day (Jung et al., 2007). From each rat, hippocampal CA1 tissue was microdissected from rat brain slices and collected. Human brain tissue was obtained from 10 patients undergoing resection for medically refractory epilepsy, consisting of hippocampal tissue (two patients) or extratemporal neocortical tissue (eight patients). Each sample was enriched for either HCN1 or HCN2 subunits by immunoprecipitating with an anti-HCN isoform-specific antibody, which was confirmed by western blotting (Fig. 1B). (As noted in Methods, human HCN2 was not directly targeted during immunoprecipitation because of the limited amount of human brain tissue available from each patient. Instead, HCN2 that co-immunoprecipitated with human HCN1 subunit was analyzed.) Both HCN channel isoforms migrated at approximately 110–120 kDa due to similar numbers of total amino acids (rat HCN1 – 910 amino acids (a.a.) and human HCN1 – 890 a.a.; rat HCN2 – 863 a.a. and human HCN2 – 889 a.a.). All immunoprecipitated samples underwent mass spectrometry, with a representative spectrum shown in Fig. 1C. These spectra were used for HCN phosphosite identifications. Mapping HCN1 and HCN2 phosphosites in both humans and rats enabled us to assess whether HCN channel phosphorylation is conserved across species. Lastly, from these spectra, we determined the phosphorylation level at a specific phosphosite within a sample by spectral counting. An example of spectral counting, HCN1-S791 in a chronically epileptic rat, is shown in Fig. 1D. Phosphorylation level quantification via spectral counting with our rat model of TLE allowed us to uncover HCN phosphorylation changes related to epileptogenesis.
HCN1 phosphosites
Because HCN1 channels are the most abundant HCN channel in the hippocampus (Monteggia et al., 2000; Brewster et al., 2007) and most of the spontaneous seizures in an animal model of TLE originate in and around the hippocampus (Toyoda et al., 2013), we first focused on the HCN1 subunit. We successfully identified multiple phosphosites using mass spectrometry within HCN1 subunits in human epilepsy brain tissue and rat brain tissue from both the rat epilepsy model and age-matched controls (Table 1). We detected more phosphosites within the rat HCN1 subunit (14) than the human HCN1 subunit (10). Also, based on the Eukaryotic Phosphorylation Site Database (Lin et al., 2020) several of these HCN1 phosphosites have not been reported in either organism and, therefore, are novel: 7 in rats and 6 in humans. We found significant correspondence between human and rat HCN1 phosphosite patterns. Of the 14 identified rat phosphosites listed in Table 1, one phosphosite (S69) had a human homolog incapable of being phosphorylated (G80). Of the remaining 13 rat phosphosites, eight (62%) had a human homolog where phosphorylation was detected.
Table 1.
Sample-based frequency of phosphorylation at HCN1 phosphosites in rat and human brain tissues.
| HCN1 phosphosites | ||||
|---|---|---|---|---|
| HCN1 phosphosite (rat - Q9JKB0) | ratio (%) of samples found phosphorylated (rat, n = 42) | HCN1 phosphosite (human - O60741) | ratio (%) of samples found phosphorylated (human, n = 10) | species in which previously reported |
| T39 | 42/42 (100.0%) | T41 | 8/10 (80.0%) | rat/human |
| S53 | 5/42 (11.9%) | S56 | (0%) | |
| S69 | 40/42 (95.2%) | #(G80) | # | rat |
| T88 | 2/42 (4.8%) | T99 | (0%) | human |
| S105 | 17/42 (40.5) | S116 | (0%) | human |
| T461 | 9/42 (21.4%) | T472 | (0%) | |
| Y502 | 6/42 (14.3%) | Y513 | 1/10 (10.0%) | human |
| S588 | 6/42 (14.3%) | S599 | (0%) | |
| #(A635) | # | T646 | 1/10 (10.0%) | |
| S791 | 38/42 (90.5%) | S770 | 5/10 (50.0%) | |
| T792 | 33/42 (78.6%) | T771 | 3/10 (30.0%) | |
| S867 | 42/42 (100.0%) | S846 | 5/10 (50.0%) | rat/human |
| S868 | 7/42 (16.7%) | S847 | 1/10 (10.0%) | rat/human |
| S891 | 30/42 (71.4%) | S871 | 3/10 (30.0%) | |
| #(P892) | # | S872 | 4/10 (40.0%) | |
| S893 | 3/42 (7.1%) | S873 | 4/10 (40.0%) | |
Analogous HCN1 phosphosites between rat and human are displayed on the same line. (#) indicates corresponding amino acid residues where phosphorylation is not possible. Shown for each phosphosite is the ratio (%) of all samples analyzed where phosphorylation was detected, with boldface lettering indicating phosphorylation that occurred in at least 50% of samples within a species. Known HCN1 phosphosites in rats and humans are reported in the Eukaryotic Phosphorylation Site Database (Lin et al., 2020). Novel HCN1 phosphosites in which phosphorylation was detected by mass spectrometry are labeled in red.
There were five rat HCN1 phosphosites with human homologs where phosphorylation was possible but not detected. The lack of phosphorylation in some of these human HCN1 phosphosites possibly was due to a small number of samples. This supposition is supported by the relatively low frequency of phosphorylation at these rat HCN1 phosphosite homologs in the context of a larger rat sample size (n = 42).
Conversely, we detected ten human HCN1 phosphosites. Of these ten verified human HCN1 phosphosites, two of them (T646 and S872) did not have a homologous rat HCN1 phosphosite where phosphorylation was possible. Of the remaining human phosphosites, we observed that all eight (100%) phosphosites had a corresponding phosphorylatable rat residue where phosphorylation was detected. Taken together, cross comparisons between human and rat HCN1 phosphosites revealed similar patterns.
We next looked for high-frequency HCN1 phosphosites, in which phosphorylation was detected in at least 50% of samples within a species. Six of the 14 rat phosphosites (43%) showed phosphorylation in at least 50% of rat samples (bold-faced in Table 1), while three of 15 human phosphosites (20%) showed phosphorylation in at least 50% of human samples (bold-faced in Table 1). We observed phosphorylation in every homolog of these high-frequency phosphosites, except for the aforementioned rat HCN1-S69 phosphosite, where phosphorylation is impossible at the corresponding human site (G80). Therefore, we conclude that there is a conservation of high-frequency HCN1 phosphosites between humans and rats.
To further establish the similarity of HCN1 phosphosites between the two species, we performed an NCBI sequence BLAST between rat and human HCN1 proteins (Altschul et al., 1990). This comparison revealed robust sequence homology (91%), which explains the congruence of several structural features. Notably, all six transmembrane domains (S1-S6), the pore-forming regions, the cyclic nucleotide binding domains (CNBDs), and the SNL terminal tripeptide that is involved in the HCN1 channel’s interaction with the tetratricopeptide repeat-containing Rab8b interacting protein, TRIP8b, (Santoro et al., 2004; Santoro et al., 2009; Santoro et al., 2011) are aligned between the two homologous proteins (Fig. 2A). Moreover, comparison of regions covered by mass spectrometry showed similar coverage of the protein sequences (rat HCN1: 57.47%; human HCN1: 52.02%) and similar patterns (green highlights in Figs. 2A), with most of the covered regions known to be cytoplasmic/intracellular. These similar patterns can be directly attributed to the high homology between rat and human HCN1 subunits, including alignment of sections lacking arginines and lysines, the sites for trypsin digestion. These regions include rat sequences F132 – R184; I287 – K399; and M649 – K785, which contains a glutamine-rich region. This arrangement results in homologous segments being precluded from sequencing by mass spectrometry after the trypsin digestion, since peptides stemming from these regions are too large for mass spectrometry analyses. Most importantly, for HCN1 phosphosites that were observed to be phosphorylated by mass spectrometry (which are mapped out as oval-enclosed amino acid residues within the sequences in Fig. 2A), interspecies alignment was displayed for the majority of the identified phosphosites. Every identified phosphosite in both rats and humans resided outside of the S1-S6 transmembrane domains and was clustered within the terminal regions, especially the C-terminal end (Fig. 2A). Although much of the transmembrane domains were not covered by our mass spectrometry due to distribution of the lysines and arginines necessary for trypsin digestion, it is somewhat expected that no phosphosites were identified in amino acid residues embedded within the phospholipid bilayer. We also noted, however, that none of these phosphosites were localized in the CNBD, implying that none of the phosphosites directly affects the binding of cyclic nucleotides to the HCN1 channel.
The high degree of correspondence in HCN1 phosphosites between rats and humans supports the rationale of using the rat pilocarpine model of acquired epilepsy to study HCN1 phosphorylation mapping and to detect changes in phosphorylation levels at specific phosphosites during epilepsy. (Because we could not obtain control human brain tissues, we were unable to identify epilepsy-induced phosphorylation changes in epilepsy patients.) We analyzed only high-frequency rat HCN1 phosphosites where phosphorylation was detected in at least 50% of total rat samples (Table 1). These high-frequency rat HCN1 phosphosites also are depicted in Fig. 2B, and their corresponding human phosphosites shown in Fig. 2C. We investigated potential HCN1 phosphorylation changes at two stages of epileptogenesis in our rat model: the acute stage (1 hr post-SE), and the chronic epilepsy stage (6–9 weeks post-SE). These two stages have distinctive characteristics: the acute stage represents the induction of epilepsy by a prolonged seizure, while the chronic stage is characterized by spontaneous seizures. Therefore, we hypothesized that the molecular mechanisms responsible for these two different stages of epileptogenesis may differ and may manifest themselves as different phosphorylation patterns within HCN1 channels.
As shown in Fig. 3, we determined the average phosphorylation level at each of the six high-frequency rat phosphosites in each cohort. The phosphorylation level at a particular phosphosite in each sample was determined by spectral counting with the following calculation:
(# of spectra where the specific phosphosite is phosphorylated) / (# of spectra containing that phosphosite regardless of phosphorylation state) X 100%.
Figure 3.

HCN1 phosphorylation levels in epileptic vs naïve conditions in rats. Shown are the phosphorylation levels at six HCN1 phosphosites (where phosphorylation was detected in ≥ 50% of rat samples) between the chronic epilepsy group and their age-matched naïve group at two stages of epileptogenesis: acute (1 hour post-SE) and chronic (6–9 weeks post-SE). The limits of each box show the 25th and 75th percentiles of the data values; the median is depicted as the line within the box; and the whiskers show the 95th and 5th percentiles. (A) No phosphorylation changes were observed in 1 hr post-SE rats compared to age-matched naïve animals. (B) In chronic epileptic rats, S791 showed a significantly increased phosphorylation level compared to age-matched naïve animals, indicated by (**).
This calculation of phosphorylation levels is distinct from the prior calculation used in Table 1 that determined ratios of samples containing a specific phosphorylated phosphosite: Table 1 shows the number of samples within a species where we detected any phosphorylation at a specific phosphosite, while Fig. 3 shows the average ratio of individual spectra containing a specific phosphorylated phosphosite.
Except for N-terminally located T39, where the phosphorylation level consistently was over 65% for both cohorts, the phosphorylation levels of rat HCN1 phosphosites typically were relatively low, i.e., ranging from 8% to 33% (Fig. 3). When comparing each phosphosite between the 1 hr post-SE rats to their age-matched controls, there were no statistically significant phosphorylation changes at any of the six phosphosites (Fig. 3A). However, in chronically epileptic rats (Fig. 3B), the novel phosphosite S791 showed a 74% increase in phosphorylation level compared to age-matched controls [33.05 ± 3.51% in chronics (n = 8), 18.99 ± 2.65% in controls (n = 8); P = 0.0061] that was statistically significant after incorporating the Bonferroni correction factor for α (0.05/6 = 0.00833). This rat HCN1-S791 residue (and its human homolog, HCN1-S770) is situated within the C-terminal region of the HCN1 channel. Although S791 is not within the regions where HCN1 directly interacts with filamin A or TRIP8b (Gravante et al., 2004; Santoro et al., 2011; Noam et al., 2014), increased phosphorylation at this novel rat HCN1-S791 site may alter HCN1 channel association with other proteins.
HCN2 phosphosites
Although less abundant than HCN1 in the CA1 hippocampal region in adult rats, HCN2 does contribute to Ih (Bender et al., 2001; Brewster et al., 2007) and forms functional multimeric HCN channels with HCN1 (Much et al., 2003; Brewster et al., 2005). Therefore, we hypothesized that epilepsy also may induce phosphorylation changes in HCN2 channels. By employing the same experiments and techniques used in the HCN1 channel study, we sought to map out HCN2 phosphosites in both rats and human epilepsy patients.
We identified numerous phosphosites using mass spectrometry within HCN2 subunits in both rat and human brain tissues (Table 2). In these experiments, we directly enriched for HCN2 in rats but only indirectly via co-immunoprecipitation with HCN1 subunit in the human epilepsy patients. As with HCN1, we detected more phosphosites within the rat HCN2 subunit (16) than the human HCN2 subunit (14). However, based on the Eukaryotic Phosphorylation Site Database (Lin et al., 2020), only two novel HCN2 phosphosites, human S97 and rat T827, were detected. Further, both phosphosites had a low frequency of phosphorylation, and each lacked a homologous cross species phosphosite (Table 2).
Table 2.
Sample-based frequency of phosphorylation at HCN2 phosphosites in rat and human brain tissues.
| HCN2 phosphosites | ||||
|---|---|---|---|---|
| HCN2 phosphosite (rat - Q9JKA9) | ratio (%) of samples found phosphorylated (rat n = 19) | HCN2 phosphosite (human - Q9UL51) | ratio (%) of samples found phosphorylated (human, n = 10) | species in which previously reported |
| S67 | 3/19 (15.8%) | S80 | 3/10 (30.0%) | rat/human |
| S68 | 1/19 (5.3%) | S81 | (0%) | rat/human |
| T70 | (0%) | T87 | 2/10 (20.0%) | rat/human |
| #(G77) | # | S97 | 1/10 (10.0%) | |
| S90 | 18/19 (94.7%) | S110 | 4/10 (40.0%) | rat |
| S109 | 3/19 (15.8%) | S132 | 4/10 (40.0%) | human |
| S119 | 19/19 (100%) | S146 | 8/10 (80.0%) | rat/human |
| S743 | 18/19 (94.7%) | S771 | 6/10 (60.0%) | rat/human |
| S750 | 19/19 (100%) | S779 | 7/10 (70.0%) | rat/human |
| S756 | 19/19 (100%) | #(A785) | # | rat |
| S757 | 19/19 (100%) | S786 | 6/10 (60.0%) | rat/human |
| S764 | 9/19 (47.4%) | S793 | 8/10 (80.0%) | rat/human |
| Y766 | (0%) | Y795 | 6/10 (60.0%) | rat/human |
| S771 | 13/19 (68.4%) | #(A801) | # | rat |
| T827 | 1/19 (5.3%) | #(A853) | # | |
| S834 | 15/19 (79.0%) | S860 | 3/10 (30.0%) | rat/human |
| S840 | 15/19 (79.0%) | S866 | 6/10 (60.0%) | rat/human |
| S842 | 19/19 (100%) | S868 | 8/10 (80.0%) | rat/human |
| S847 | 9/19 (47.4%) | #(G873) | # | rat |
Analogous HCN2 phosphosites between rat and human are displayed on the same line. (#) indicates analogous amino acid residues where phosphorylation is not possible. Shown for each phosphosite is the ratio (%) of all samples analyzed where phosphorylation was detected, with boldface lettering indicating phosphorylation that occurred in at least 50% of samples within a species. Known HCN2 phosphosites in rat and humans are reported in the Eukaryotic Phosphorylation Site Database (Lin et al., 2020). Novel HCN2 phosphosites in which phosphorylation was detected by mass spectrometry are labeled in red.
We again focused on the correspondence between the HCN2 phosphosite patterns of the two species. Listed in Table 2, we identified 16 rat HCN2 phosphosites, 12 of which had a homologous human residue capable of being phosphorylated. Of these 12 rat phosphosites, 11 corresponding human HCN2 phosphosites (92%) were detected. Only rat HCN2-S68, which had relatively rare phosphorylation (5%), was the lone example of where no phosphorylation occurred in its homologous human HCN2 residue, S81. As before, the absence of phosphorylation at human HCN2-S81 possibly is due to a small human sample size (n = 10).
Conversely, of the 14 identified human HCN2 phosphosites listed in Table 2, one phosphosite (the novel S97) had a rat homolog incapable of being phosphorylated (G77). Of the remaining 13 human phosphosites, 11 (85%) had a rat homolog where phosphorylation was detected. Thus, we detected a high level of conservation of phosphosites between human and rat HCN2 channels.
As in the HCN1 study, we focused on the high-frequency HCN2 phosphosites, in which phosphorylation was detected in at least 50% of samples within a species. Ten of the 16 rat phosphosites (63%) showed phosphorylation in at least 50% of rat samples (bold-faced in Table 2), while eight of 14 human phosphosites (57%) showed phosphorylation in at least 50% of human samples (bold-faced in Table 1). We observed phosphorylation in both human and rat homologs of these high frequency phosphosites, except for the rat HCN2-Y766 phosphosite (see above) and human A785 and A801 residues, in which phosphorylation is not possible. Therefore, we conclude that there is a conservation of high-frequency HCN2 phosphosites between humans and rats.
To further illustrate the similarity of HCN2 phosphosites between the two species, we performed an NCBI sequence BLAST (Altschul et al., 1990) between rat and human HCN2 channel proteins. This comparison again revealed robust sequence homology (90%), which explains the congruence of several structural features. As with the HCN1 channels, all six transmembrane domains (S1-S6), the pore-forming region, the CNBD, and the C-terminal ending SNL tripeptide are aligned between the two homologous HCN2 proteins (Fig. 4A). Moreover, comparisons of regions covered by mass spectrometry showed somewhat similar coverage percentage of entire protein sequences (rat HCN2: 53.53%; human HCN2: 63.78%) and similar patterns (green highlights in Figs. 4A), with most of these covered regions known to be cytoplasmic/intracellular. These similar patterns can be attributed to the high homology between rat and human HCN1 subunits. One notable exception to coverage similarity is the N-terminal region. The presence of R57 in human HCN2 (that has no equivalent in the rat HCN2 sequence) allowed for generation of tryptic peptides suitable for sequencing in this region of human HCN2. Conversely, the absence of this arginine within this region of the rat HCN2 N-terminus would make the resultant peptide too large for mass spectrometry analysis.
Figure 4.

Comparison of rat and human HCN2 phosphosites. (A) Amino acid sequences of rat and human HCN2 are juxtaposed. Green highlights show regions of each HCN sequence that were observed by mass spectrometry. Ovals indicate detected phosphosites; red labels indicate novel phosphosites; bold labels indicate phosphosites observed in ≥ 50% of either species’ samples. Structural features of HCN channels are denoted: the intracellular N-terminal region, the six transmembrane domains (S1-S6), and an extensive intracellular C-terminal region incorporating the pore-forming region and the cyclic nucleotide binding domain (CNBD). (B) Representation of the rat HCN2 channel showing phosphosites that were phosphorylated in at least 50% of rat samples (with red labeling denoting novel phosphosites). (C) Representation of the human HCN2 channel showing phosphosites that are homologous to the prevalent rat phosphosites shown in (B).
Most importantly, for HCN2 phosphosites that were observed to be phosphorylated by mass spectrometry (which are mapped out as oval-enclosed amino acid residues within the sequences in Fig. 4A), we observed interspecies alignment for the vast majority of the identified phosphosites. As with HCN1 phosphosites, every identified HCN2 phosphosite in both rats and humans resides outside of the S1-S6 transmembrane domains. Specifically, the HCN2 phosphosites mainly were clustered within the terminal regions, especially the C-terminal end (Fig. 4A). Although most of the transmembrane domains were not covered by mass spectrometry, we again did not expect to identify phosphosites in these regions where amino acid residues are embedded within the phospholipid bilayer. We also noted that none of these phosphosites were resided within the CNBD, implying that none of the phosphosites sterically affects the binding of cyclic nucleotides to the HCN2 channel.
As with the HCN1 analysis, the high degree of correspondence in HCN2 phosphosites between rats and humans again corroborates using the rat pilocarpine model of acquired epilepsy to study HCN2 phosphorylation mapping and detect potential changes in phosphorylation levels at specific phosphosites during epilepsy. (As before, negative control human brain tissues were not available.) We analyzed only high-frequency rat HCN2 phosphosites where phosphorylation was detected in at least 50% of total rat samples (Table 2). These prevalent rat HCN2 phosphosites also are represented in Fig. 4B, and their homologous human phosphosites are shown in Fig. 4C. Because we detected the sole significant phosphorylation change in HCN1 channels in the chronic epilepsy stage (6–9 weeks post-SE), we focused our investigation of HCN2 phosphorylation changes only at this stage.
As shown in Fig. 5, we determined the average phosphorylation level at each of the 10 rat phosphosites in each cohort by spectral counting. Rat HCN2 phosphosite S750, which is within the C-terminal region, was the only phosphosite where the phosphorylation level was greater than 50%, averaging 70.1% for all rats involved. The phosphorylation levels of the other rat HCN2 phosphosites typically were moderately low, i.e., ranging from 14% to 47% (Fig. 5). Interestingly, when quantifying the overall phosphorylation level of high-frequency phosphosites in each rat (total # of phosphorylated phosphosites / total # of available phosphosites), the average was greater for these rat HCN2 phosphosites (39.26%) than for the high-frequency rat HCN1 phosphosites (23.65%). When comparing each phosphosite between the chronically epileptic rats to their age-matched controls, only one rat HCN2 phosphosite, S834, showed increased phosphorylation [41.36 ± 7.29% in chronic epileptic rats (n = 11), 17.50 ± 6.12% in controls (n = 8); p = 0.024]. However, after incorporating the Bonferroni correction factor for α (0.05/10 = 0.005), the increased HCN2-S834 phosphorylation was not statistically significant.
Figure 5.

HCN2 phosphorylation levels in epilepsy vs naïve conditions in rat. Shown are the phosphorylation levels of ten HCN2 phosphosites (where phosphorylation was detected in ≥ 50% of rat samples) between chronically epileptic rats (6–9 weeks post-SE) versus age-matched naïve animals. The limits of each box show the 25th and 75th percentiles of the data values; the median is depicted as the line within the box; and the whiskers show the 95th and 5th percentiles. No significant phosphorylation changes were observed between chronically epileptic and naïve animals.
Taking the HCN1 and HCN2 studies together, of the frequently observed phosphorylated rat HCN phosphosites (six in HCN1 and 10 in HCN2), only the novel rat HCN1-S791 phosphosite underwent a significant increase in phosphorylation levels, and only in the condition of chronic epilepsy.
Functional effects of S791 phosphorylation
The functional consequences of HCN1 phosphorylation at S791 on channel gating properties was examined by heterologous expression in Xenopus oocytes. To mimic constitutive S791 phosphorylation, the phosphomimetic mutation S791D was introduced by site-directed mutagenesis, along with the phosphoablative mutation S791A. The phosphomimetic mutation S791D replaces serine with the charged amino acid aspartate to mimic the biophysical effect of the addition of a charged phosphate group to the serine residue. The phosphoablative mutation S791A replaces serine with the hydrophobic uncharged alanine to simulate the absence of phosphorylation. All three HCN1 constructs (WT, S791D, S791A) were expressed in Xenopus oocytes, and currents were measured by two-electrode voltage clamp (TEVC) recordings. All three constructs expressed slowly gating inward currents activated by hyperpolarization, typical of HCN channels (Fig. 6C, 6D). At command potentials hyperpolarized to −90 mV, activation of contaminating endogenous inward currents was seen (described in Experimental Procedures and shown in Supplemental Fig. S1). We therefore generated conductance/voltage (G/V) plots based on tail current measurements by fitting data points obtained only from +10 mV to −90 mV. These G/V curves revealed a voltage of half-maximal activation (V1/2) for WT channels of −72.0 mV. V1/2 for S791D was shifted to −75.9 mV, indicating a modest but significant hyperpolarizing shift in the voltage-dependent activation of HCN1 channels when S791 is phosphorylated (G/Gmax at V= −70 mV, WT: 0.480 ± 0.021, n=20; S791D: 0.422 ± 0.017, n=22, p<0.05; Fig. 6A). Consistent with the low level of constitutive phosphorylation of S791 measured under naïve conditions in rat hippocampal tissue (~20%, Fig. 3B), the phosphoablative mutation S791A did not produce a significant change in HCN1 voltage-dependent activation, with V1/2 = −70.7 mV (G/Gmax at V= −70 mV: 0.491 ± 0.015, n=12, p>0.05). Together, these results show that phosphorylation of HCN1 channels at S791D produces a hyperpolarized shift in channel activation, similarly to what is seen in HCN channels from native hippocampal tissue taken from epileptic animals.
Figure 6.

Phosphomimetic mutation of S791 (S791D) negatively shifts HCN1 voltage-dependent activation. (A) Conductance/voltage (G/V) plots of WT HCN1, HCN1 with phosphomimetic mutation S791D, and HCN1 with phosphoablative mutation S791A are shown. Phosphomimetic mutation at S791 (S791D) produces an ~4 mV hyperpolarizing shift in HCN1 voltage-dependent activation, while ablation of phosphorylation with S791A mutation produces HCN1 activation that resembles WT. (B) Oocytes pretreated with the p38 MAPK inhibitor SB203780 (20 μM) exhibit negatively shifted voltage-dependent activation compared to WT. (C) Example WT HCN1 currents evoked by voltage steps from 10 mV to −120 mV, with tail currents measured at 0 mV. (D) Example HCN1-S791D currents evoked by identical voltage steps.
Phosphorylation of HCN1 channels by p38 MAPK has been shown to activate HCN channels by shifting their voltage-dependent activation towards depolarized potentials; conversely, application of p38 MAPK inhibitors shifts HCN channel activation in a hyperpolarized direction (Poolos et al., 2006). We sought to validate that HCN1 channels expressed in Xenopus oocytes retained the modulation by p38 MAPK that is exhibited in native hippocampal tissue. The p38 MAPK-specific inhibitor SB203780 (20 μM) was applied to oocytes for 20 mins prior to obtaining voltage-clamp recordings. As expected, inhibition of p38 MAPK shifted the voltage-dependent activation of WT HCN1 channels in a hyperpolarized direction relative to untreated WT controls (V1/2= −77.6 mV; G/Gmax at V= −70 mV: 0.388 ± 0.023, n=6, p<0.05). This validates that that HCN1 channels expressed in Xenopus oocytes can be modulated by phosphorylation and serves as a comparator for the change in V1/2 seen with the S791D mutation.
Discussion
Significant progress has been made in understanding the modulation of HCN channel biophysical properties by phosphorylation signaling pathways and in establishing an association between induced changes in HCN channel properties in epilepsy and dysregulation of phosphorylation signaling. Previous studies have demonstrated that several phosphorylation signaling pathways (p38 MAPK, calcineurin, and PKC) modulate HCN channel expression and function, and that epilepsy is known to induce changes in these phosphorylation pathways (Poolos et al., 2006; Jung et al., 2010; Williams et al., 2015). In this study we sought to identify for the first time the phosphosites within HCN channels; to detect any HCN channel phosphorylation changes associated with epilepsy; and to investigate if such epilepsy-related phosphorylation changes modulate HCN channel activity. Here, we focused on the two most abundant HCN channels in the brain, HCN1 and HCN2 (Biel et al., 2009). By mass spectrometry we detected in both human and rat brain homogenates numerous HCN1 and HCN2 phosphosites, some of which are novel. Most importantly, we identified epilepsy-associated increased phosphorylation at one of these novel phosphosites (HCN1-S791) in chronically epileptic rats. When expressing a phosphomimetic mutant that is analogous to constitutively phosphorylated S791 (rat HCN1-S791D) in Xenopus oocytes, we observed a hyperpolarizing shift in HCN1 channel voltage-dependent activation, similar to that seen in our rat model of acquired epilepsy (Jung et al., 2007; Jung et al., 2010). These findings represent the first survey of HCN channel phosphosites and also a demonstration that an epilepsy-induced phosphorylation change modulates HCN1 channel activity.
Although phosphorylation databases such as the Eukaryotic Phosphorylation Site Database catalog HCN1 and HCN2 phosphosites for both rats and humans (Lin et al., 2020), most of these listings derive from high-throughput shotgun phosphoproteomic data where HCN channels were not purified (Dammer et al., 2015; Nirujogi et al., 2015). In contrast, our investigation is the first to specifically enrich for HCN1 and HCN2 channels and identify their phosphosites via mass spectrometry. This mass spectrometry approach, which is similar to an earlier study that identified acute phosphorylation changes in the sodium channel Nav1.2 after kainic acid-induced seizures (Baek et al., 2014), yielded numerous phosphosites within HCN1 and HCN2 channels from both humans and rats. By mapping out identified HCN phosphosites, we found a high degree of cross-species similarity in phosphosites. This demonstrates that upstream phosphorylation signaling is largely conserved between rats and humans and suggests that a phosphorylation change seen in a rat model of epilepsy may occur in humans as well.
In this study we identified significantly increased phosphorylation at a specific HCN1 phosphosite, S791, in chronically epileptic rats compared to age-matched naïves. Supporting the relevance of this finding to epilepsy was that the phosphomimetic mutant S791D, when heterologously expressed in Xenopus oocytes, produced hyperpolarized change in Ih gating just as was seen in the chronic epilepsy phase of an animal model of temporal lobe epilepsy (Poolos et al., 2006; Jung et al., 2007; Jung et al., 2010). The magnitude of the expressed S791D mutant channel gating change seen (~4 mV) was smaller than that seen in native hippocampal tissue from chronically epileptic animals (~12 mV) reflecting either the possibility that modulation of other phosphorylation sites not sampled in our mass spectrometry analysis may contribute, or that other mechanisms modulating HCN channel biophysical properties are not preserved in the Xenopus expression system. Supporting this latter idea are the results of p38 MAPK inhibition of heterologously expressed WT HCN channels. p38 MAPK inhibition produced an ~6 mV hyperpolarized shift in expressed HCN1 channels, validating that the gating of expressed HCN channels can be modulated by phosphorylation in a similar fashion as is seen in native hippocampal neurons (Poolos et al., 2006). However, the much larger magnitude of the hyperpolarized gating change seen with p38 MAPK inhibition in native tissue (~25 mV) suggests that other factors in native neurons influence the effects of phosphorylation on HCN channels that are not present in heterologous expression by Xenopus oocytes. Thus our electrophysiology results and their relevance to HCN1 biophysical changes seen in animal models or human epilepsy should be interpreted with caution.
The identity of upstream signaling pathways responsible for increased phosphorylation of S791 in chronic epilepsy is unknown. In our prior work examining phosphorylation changes in an animal model of epilepsy, we found downregulation of p38 MAPK signaling—which is not consistent with increased S791 phosphorylation seen here (Jung et al., 2010). In another study we found acute, one hour post-SE changes in HCN channel surface expression putatively attributed to PKC activation (Williams et al., 2015). However, our mass spectrometry analysis failed to demonstrate any acute changes in HCN1 phosphorylation in the acute phase post-SE. Prior use of a prediction algorithm to identify putative HCN1 or HCN2 phosphorylation sites, while predicting phosphorylation of HCN1 by p38 MAPK (at T778, in a region not covered by our mass spectrometry analysis), did not identify S791 as residing in a consensus sequence phosphorylated by protein kinases (Poolos et al., 2006). Thus, the kinase or kinases responsible for phosphorylation of S791 remain unclear.
Recent studies have mapped out HCN1 channel mutations found in human epilepsy patients (Nava et al., 2014; Bonzanni et al., 2018; Marini et al., 2018). Their general consensus is that mutations affecting the pore-forming structures, including the transmembrane domains, are associated with severe forms of epilepsy, while the mutations located in the N- and C- terminals of the proteins are linked with milder phenotypes of the disease. Interestingly, none of the human mutations are located in the last 120 amino acids of the C-terminus, the region of highest clustering of phosphosites for both the rat and human HCN1 channel. In our study, the lone phosphosite that demonstrated increased phosphorylation in our rat model of epilepsy, HCN1-S791, is within the C-terminal. Because this phosphorylation change was detected within the chronic stage of epilepsy and not in the acute stage one hour post-SE, we conclude that this increased phosphorylation does not play a role in the initiation of the epileptic state. However, because numerous studies have shown that downregulation of HCN channels yields hyperexcitability in pyramidal neurons, and our phosphomimetic experiments show that increased phosphorylation at S791 downregulates HCN1 channel activity by inducing a hyperpolarizing shift in gating, we hypothesize that elevated phosphorylation at HCN1-S791 likely increases the excitability of pyramidal neurons and contributes to the maintenance of epilepsy in the chronic stage.
Despite the greater number of phosphosites and a greater degree of overall phosphorylation in HCN2 channels compared to HCN1 channels, no significant HCN2 phosphorylation changes were detected in our rat model of epilepsy. This may be consistent with prior studies that found only transient changes in mRNA or protein expression of HCN2 channels following induction of epilepsy which were not observed in chronically epileptic animals (Brewster et al., 2002; Jung et al., 2007).
Another limitation was the lack of human control tissue as a comparator for that from our human epilepsy patients. We believe that this is a difficult problem to solve: human autopsy tissue generally does not adequately preserve phosphorylation state so is not a useful comparator (Matsuo et al., 1994; Li et al., 2003; Wang et al., 2015); human epilepsy resection tissue taken at a distance from the most active seizure onset zone in extratemporal resection, while a useful comparator for changes produced by a spectrum of epileptic activity, cannot truly said to be from seizure-free brain (Beaumont et al., 2012). Because of the absence of these controls, we could not investigate any phosphorylation changes in human HCN channels that are associated with epilepsy. However, the high level of HCN1 phosphosite conservation between humans and rats, and the fact that we observed a high frequency of phosphorylation at S791 in both rat and at its human homolog S770, is suggestive that modulation of S770 may be occurring in human epilepsy as well. Interestingly, in both human epilepsy patient samples where the resected tissue originated from the hippocampal region, phosphorylation at S770 was detected.
Although most study of HCN channelopathy-related epilepsy has focused on HCN1 and HCN2 channels, recent data have also implicated HCN4 channels in the development of epilepsy (Rivolta et al., 2020). Loss-of-function mutations in HCN4 have been shown to underlie a benign form of generalized infantile epilepsy in one family (Campostrini et al., 2018) as well as contributing to seizures in cases of genetic generalized epilepsy (Becker et al., 2017). Additionally, ectopic HCN4 expression driven by hyperactive mechanistic target of rapamycin (mTOR) pathway in pyramidal neurons caused epilepsy in mice (Hsieh et al., 2020). However, given that HCN4 expression mainly in the thalamus in adults and in the cerebral cortex occurs only in infancy (Poolos, 2012; Santoro & Shah, 2020; Rivolta et al., 2020), this study, involving the hippocampal CA1 region in adult rats and either cortical or hippocampal tissues in adult human epilepsy patients, did not analyze phosphorylation within HCN4 channels.
This study joins prior work identifying changes in ion channel phosphorylation at the individual phosphosite level that may be important in the pathogenesis of epilepsy. Kv2.1 was demonstrated to be phosphorylated in a graded fashion and activity-dependent fashion, with dephosphorylation causing a downregulation of Kv2.1 function (Park et al., 2006). Nav1.2 is highly phosphorylated on multiple phosphosites, and induced seizures caused net dephosphorylation of these channels, along with a reciprocal methylation of other amino acid residues (Baek et al., 2014). KCNQ2/KCNQ3, an ion channel mutated in genetic forms of human epilepsy, is regulated by phosphorylation, with specific phosphosites involved in either tetramerization or channel inhibition (Surti et al., 2005).
Furthermore, of direct relevance to HCN channels, phosphorylation of TRIP8b (which regulates HCN channel trafficking and gating) was shown to augment its binding to HCN channels, and loss of TRIP8b phosphorylation at a single serine residue in experimental epilepsy diminished HCN channel expression (Foote et al., 2019). These studies all demonstrated that seizure- or activity-induced modulation of phosphorylation at even individual phosphosites can have a significant effect on ion channel biophysical properties. Because phosphorylation signaling—particularly when increased in a disease state—is potentially targetable by specific kinase inhibitors, further understanding of changes in phosphorylation signaling in epilepsy may lead to future therapeutic interventions. Perhaps such future novel therapeutic approaches may lead to treatments for the refractory fraction that makes up as much as one-third of all epilepsy patients (Kalilani et al., 2018).
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Neurological Disorders and Stroke: NIH R01 NS050229. We thank Priska von Haller from the University of Washington’s Proteomics Resource (UWPR) for her invaluable guidance and contributions to this mass spectrometry study. We also thank Bina Santoro for providing HCN1 cDNA for use in heterologous expression experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, Gish W, Miller W, Myers EW, & Lipman DJ (1990). Basic local alignment search tool. J Mol Biol, 215(3), 403–410. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Baek JH, Rubinstein M, Scheuer T, & Trimmer JS (2014). Reciprocal changes in phosphorylation and methylation of mammalian brain sodium channels in response to seizures. J Biol Chem, 289, 15363–15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CK, Falk T, & Schwarz JR (1996). An endogenous inactivating inward-rectifying potassium current in oocytes of Xenopus laevis. Pflugers Arch, 432(5), 812–820. doi: 10.1007/s004240050203 [DOI] [PubMed] [Google Scholar]
- Beaumont TL, Yao B, Shah A, Kapatos G, & Loeb JA (2012). Layer-specific CREB target gene induction in human neocortical epilepsy. J Neurosci, 32(41), 14389–14401. doi: 10.1523/JNEUROSCI.3408-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker F, Reid CA, Hallmann K, Tae HS, Phillips AM, Teodorescu G, Weber YG, Kleefuss-Lie A, Elger C, Perez-Reyes E, Petrou S, Kunz WS, Lerche H, & Maljevic S (2017). Functional variants in HCN4 and CACNA1H may contribute to genetic generalized epilepsy. Epilepsia Open, 2(3), 334–342. doi: 10.1002/epi4.12068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Brewster A, Santoro B, Ludwig A, Hofmann F, Biel M, & Baram TZ (2001). Differential and age-dependent expression of hyperpolarization-activated, cyclic nucleotide-gated cation channel isoforms 1–4 suggests evolving roles in the developing rat hippocampus. Neuroscience, 106(4), 689–698. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11682156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, & Zong X (2009). Hyperpolarization-activated cation channels: from genes to function. Physiol Rev, 89, 847–885. [DOI] [PubMed] [Google Scholar]
- Bonzanni M, DiFrancesco JC, Milanesi R, Campostrini G, Castellotti B, Bucchi A, Baruscotti M, Ferrarese C, Franceschetti S, Canafoglia L, Ragona F, Freri E, Labate A, Gambardella A, Costa C, Rivolta I, Gellera C, Granata T, Barbuti A, & DiFrancesco D (2018). A novel de novo HCN1 loss-of-function mutation in genetic generalized epilepsy causing increased neuronal excitability. Neurobiol Dis, 118, 55–63. doi: 10.1016/j.nbd.2018.06.012 [DOI] [PubMed] [Google Scholar]
- Brennan GP, Baram TZ, & Poolos NP (2016). Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channels in Epilepsy. Cold Spring Harb Perspect Med, 6(3), a022384. doi: 10.1101/cshperspect.a022384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster A, Bender RA, Chen Y, Dube C, Eghbal-Ahmadi M, & Baram TZ (2002). Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci, 22(11), 4591–4599. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12040066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster AL, Bernard JA, Gall CM, & Baram TZ (2005). Formation of heteromeric hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in the hippocampus is regulated by developmental seizures. Neurobiol Dis, 19(1–2), 200–207. doi: 10.1016/j.nbd.2004.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster AL, Chen Y, Bender RA, Yeh A, Shigemoto R, & Baram TZ (2007). Quantitative analysis and subcellular distribution of mRNA and protein expression of the hyperpolarization-activated cyclic nucleotide-gated channels throughout development in rat hippocampus. Cereb Cortex, 17(3), 702–712. doi: 10.1093/cercor/bhk021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campostrini G, DiFrancesco JC, Castellotti B, Milanesi R, Gnecchi-Ruscone T, Bonzanni M, Bucchi A, Baruscotti M, Ferrarese C, Franceschetti S, Canafoglia L, Ragona F, Freri E, Labate A, Gambardella A, Costa C, Gellera C, Granata T, Barbuti A, & DiFrancesco D (2018). A Loss-of Function HCN4 Mutation Associated With Familial Benign Myoclonic Epilepsy in Infancy Causes Increased Neuronal Excitability. Front Mol Neurosci, 11, 269. doi: 10.3389/fnmol.2018.00269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang J, & Siegelbaum SA (2001). Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol, 117(5), 491–504. doi: 10.1085/jgp.117.5.491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammer EB, Lee AK, Duong DM, Gearing M, Lah JJ, Levey AI, & Seyfried NT (2015). Quantitative phosphoproteomics of Alzheimer’s disease reveals cross-talk between kinases and small heat shock proteins. Proteomics, 15(2–3), 508–519. doi: 10.1002/pmic.201400189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco JC, Barbuti A, Milanesi R, Coco S, Bucchi A, Bottelli G, Ferrarese C, Franceschetti S, Terragni B, Baruscotti M, & DiFrancesco D (2011). Recessive loss-of-function mutation in the pacemaker HCN2 channel causing increased neuronal excitability in a patient with idiopathic generalized epilepsy. J Neurosci, 31(48), 17327–17337. doi: 10.1523/JNEUROSCI.3727-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco JC, Castellotti B, Milanesi R, Ragona F, Freri E, Canafoglia L, Franceschetti S, Ferrarese C, Magri S, Taroni F, Costa C, Labate A, Gambardella A, Solazzi R, Binda A, Rivolta I, Di Gennaro G, Casciato S, D’Incerti L, Barbuti A, DiFrancesco D, Granata T, & Gellera C (2019). HCN ion channels and accessory proteins in epilepsy: genetic analysis of a large cohort of patients and review of the literature. Epilepsy Res, 153, 49–58. doi: 10.1016/j.eplepsyres.2019.04.004 [DOI] [PubMed] [Google Scholar]
- Eng JK, Jahan TA, & Hoopmann MR (2013). Comet: an open-source MS/MS sequence database search tool. Proteomics, 13(1), 22–24. doi: 10.1002/pmic.201200439 [DOI] [PubMed] [Google Scholar]
- Foote KM, Lyman KA, Han Y, Michailidis IE, Heuermann RJ, Mandikian D, Trimmer JS, Swanson GT, & Chetkovich DM (2019). Phosphorylation of the HCN channel auxiliary subunit TRIP8b is altered in an animal model of temporal lobe epilepsy and modulates channel function. J Biol Chem, 294(43), 15743–15758. doi: 10.1074/jbc.RA119.010027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravante B, Barbuti A, Milanesi R, Zappi I, Viscomi C, & DiFrancesco D (2004). Interaction of the pacemaker channel HCN1 with filamin A. J Biol Chem, 279(42), 43847–43853. doi: 10.1074/jbc.M401598200 [DOI] [PubMed] [Google Scholar]
- Hsieh LS, Wen JH, Nguyen LH, Zhang L, Getz SA, Torres-Reveron J, Wang Y, Spencer DD, & Bordey A (2020). Ectopic HCN4 expression drives mTOR-dependent epilepsy in mice. Sci Transl Med, 12(570). doi: 10.1126/scitranslmed.abc1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Jones TD, Lugo JN, Sheerin AH, Miller JW, D’Ambrosio R, Anderson AE, & Poolos NP (2007). Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci, 27(47), 13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Warner LN, Pitsch J, Becker AJ, & Poolos NP (2011). Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. J Neurosci, 31(40), 14291–14295. doi: 10.1523/JNEUROSCI.1148-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Bullis JB, Lau IH, Jones TD, Warner LN, & Poolos NP (2010). Downregulation of dendritic HCN channel gating in epilepsy is mediated by altered phosphorylation signalling. J Neurosci, 30, 6678–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalilani L, Sun X, Pelgrims B, Noack-Rink M, & Villanueva V (2018). The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia, 59(12), 2179–2193. doi: 10.1111/epi.14596 [DOI] [PubMed] [Google Scholar]
- Keller A, Eng J, Zhang N, Li XJ, & Aebersold R (2005). A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol, 1, 2005.0017. doi: 10.1038/msb4100024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Gould TD, Yuan P, Manji HK, & Chen G (2003). Post-mortem interval effects on the phosphorylation of signaling proteins. Neuropsychopharmacology, 28(6), 1017–1025. doi: 10.1038/sj.npp.1300112 [DOI] [PubMed] [Google Scholar]
- Li M, Maljevic S, Phillips AM, Petrovski S, Hildebrand MS, Burgess R, Mount T, Zara F, Striano P, Schubert J, Thiele H, Nürnberg P, Wong M, Weisenberg JL, Thio LL, Lerche H, Scheffer IE, Berkovic SF, Petrou S, & Reid CA (2018). Gain-of-function HCN2 variants in genetic epilepsy. Hum Mutat, 39(2), 202–209. doi: 10.1002/humu.23357 [DOI] [PubMed] [Google Scholar]
- Lin S, Wang C, Zhou J, Shi Y, Ruan C, Tu Y, Yao L, Peng D, & Xue Y (2020). EPSD: a well-annotated data resource of protein phosphorylation sites in eukaryotes. Brief Bioinform. doi: 10.1093/bib/bbz169 [DOI] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, & Hofmann F (2003). Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. Embo J, 22(2), 216–224. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12514127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini C, Porro A, Rastetter A, Dalle C, Rivolta I, Bauer D, Oegema R, Nava C, Parrini E, Mei D, Mercer C, Dhamija R, Chambers C, Coubes C, Thévenon J, Kuentz P, Julia S, Pasquier L, Dubourg C, Carré W, Rosati A, Melani F, Pisano T, Giardino M, Innes AM, Alembik Y, Scheidecker S, Santos M, Figueiroa S, Garrido C, Fusco C, Frattini D, Spagnoli C, Binda A, Granata T, Ragona F, Freri E, Franceschetti S, Canafoglia L, Castellotti B, Gellera C, Milanesi R, Mancardi MM, Clark DR, Kok F, Helbig KL, Ichikawa S, Sadler L, Neupauerová J, Laššuthova P, Šterbová K, Laridon A, Brilstra E, Koeleman B, Lemke JR, Zara F, Striano P, Soblet J, Smits G, Deconinck N, Barbuti A, DiFrancesco D, LeGuern E, Guerrini R, Santoro B, Hamacher K, Thiel G, Moroni A, DiFrancesco JC, & Depienne C (2018). HCN1 mutation spectrum: from neonatal epileptic encephalopathy to benign generalized epilepsy and beyond. Brain, 141(11), 3160–3178. doi: 10.1093/brain/awy263 [DOI] [PubMed] [Google Scholar]
- Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O’Connor M, Trojanowski JQ, & Lee VM (1994). Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron, 13(4), 989–1002. doi: 10.1016/0896-6273(94)90264-x [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, & Nestler EJ (2000). Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain Res Mol Brain Res, 81(1–2). Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11000485 [DOI] [PubMed] [Google Scholar]
- Much B, Wahl-Schott C, Zong X, Schneider A, Baumann L, Moosmang S, Ludwig A, & Biel M (2003). Role of subunit heteromerization and N-linked glycosylation in the formation of functional hyperpolarization-activated cyclic nucleotide-gated channels. J Biol Chem, 278(44), 43781–43786. doi: 10.1074/jbc.M306958200 [DOI] [PubMed] [Google Scholar]
- Nava C, Dalle C, Rastetter A, Striano P, de Kovel CG, Nabbout R, Cances C, Ville D, Brilstra EH, Gobbi G, Raffo E, Bouteiller D, Marie Y, Trouillard O, Robbiano A, Keren B, Agher D, Roze E, Lesage S, Nicolas A, Brice A, Baulac M, Vogt C, El Hajj N, Schneider E, Suls A, Weckhuysen S, Gormley P, Lehesjoki AE, De Jonghe P, Helbig I, Baulac S, Zara F, Koeleman BP, Haaf T, LeGuern E, & Depienne C (2014). De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet, 46(6), 640–645. doi: 10.1038/ng.2952 [DOI] [PubMed] [Google Scholar]
- Nirujogi RS, Wright JD, Manda SS, Zhong J, Na CH, Meyerhoff J, Benton B, Jabbour R, Willis K, Kim MS, Pandey A, & Sekowski JW (2015). Phosphoproteomic analysis reveals compensatory effects in the piriform cortex of VX nerve agent exposed rats. Proteomics, 15(2–3), 487–499. doi: 10.1002/pmic.201400371 [DOI] [PubMed] [Google Scholar]
- Noam Y, Ehrengruber MU, Koh A, Feyen P, Manders EM, Abbott GW, Wadman WJ, & Baram TZ (2014). Filamin A promotes dynamin-dependent internalization of hyperpolarization-activated cyclic nucleotide-gated type 1 (HCN1) channels and restricts Ih in hippocampal neurons. J Biol Chem, 289(9), 5889–5903. doi: 10.1074/jbc.M113.522060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsaki G, Siegelbaum SA, Kandel ER, & Morozov A (2004). A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell, 119(5), 719–732. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15550252 [DOI] [PubMed] [Google Scholar]
- Notomi T, & Shigemoto R (2004). Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol, 471(3), 241–276. doi: 10.1002/cne.11039 [DOI] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Misonou H, & Trimmer JS (2006). Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science, 313(5789), 976–979. doi: 10.1126/science.1124254 [DOI] [PubMed] [Google Scholar]
- Pedrioli PG, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, Cheung K, Costello CE, Hermjakob H, Huang S, Julian RK, Kapp E, McComb ME, Oliver SG, Omenn G, Paton NW, Simpson R, Smith R, Taylor CF, Zhu W, & Aebersold R (2004). A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol, 22(11), 1459–1466. doi: 10.1038/nbt1031 [DOI] [PubMed] [Google Scholar]
- Poolos NP (2012). Hyperpolarization-activated Cyclic Nucleotide-Gated (HCN) Ion Channelopathy in Epilepsy. In Noebels JL, Avoli M, Rogawski MA, Olsen RW, & Delgado-Escueta AV (Eds.), Jasper’s Basic Mechanisms of the Epilepsies (4th ed.). [PubMed] [Google Scholar]
- Poolos NP, Bullis JB, & Roth MK (2006). Modulation of h-channels in hippocampal pyramidal neurons by p38 mitogen-activated protein kinase. J Neurosci, 26(30), 7995–8003. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16870744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, & Johnston D (2002). Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci, 5, 767–774. [DOI] [PubMed] [Google Scholar]
- Poolos NP, & Johnston D (2012). Dendritic ion channelopathy in acquired epilepsy. Epilepsia, 53 Suppl 9, 32–40. doi: 10.1111/epi.12033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivolta I, Binda A, Masi A, & DiFrancesco JC (2020). Cardiac and neuronal HCN channelopathies. Pflugers Arch, 472(7), 931–951. doi: 10.1007/s00424-020-02384-3 [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, & Sweatt JD (1999). The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci, 19(11), 4337–4348. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10341237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Hu L, Liu H, Saponaro A, Pian P, Piskorowski RA, Moroni A, & Siegelbaum SA (2011). TRIP8b regulates HCN1 channel trafficking and gating through two distinct C-terminal interaction sites. J Neurosci, 31(11), 4074–4086. doi: 10.1523/JNEUROSCI.5707-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Piskorowski RA, Pian P, Hu L, Liu H, & Siegelbaum SA (2009). TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron, 62(6), 802–813. doi: 10.1016/j.neuron.2009.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, & Shah MM (2020). Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels as Drug Targets for Neurological Disorders. Annu Rev Pharmacol Toxicol, 60, 109–131. doi: 10.1146/annurev-pharmtox-010919-023356 [DOI] [PubMed] [Google Scholar]
- Santoro B, Wainger BJ, & Siegelbaum SA (2004). Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J Neurosci, 24(47), 10750–10762. doi: 10.1523/JNEUROSCI.3300-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, & Tibbs GR (1999). The HCN gene family: molecular basis of the hyperpolarization-activated pacemaker channels. Ann N Y Acad Sci, 868, 741–764. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=10414361 [DOI] [PubMed] [Google Scholar]
- Surti TS, Huang L, Jan YN, Jan LY, & Cooper EC (2005). Identification by mass spectrometry and functional characterization of two phosphorylation sites of KCNQ2/KCNQ3 channels. Proc Natl Acad Sci U S A, 102(49), 17828–17833. doi: 10.1073/pnas.0509122102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Sander T, Craven KB, Hempelmann A, & Escayg A (2008). Mutation analysis of the hyperpolarization-activated cyclic nucleotide-gated channels HCN1 and HCN2 in idiopathic generalized epilepsy. Neurobiol Dis, 29(1), 59–70. doi: 10.1016/j.nbd.2007.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda I, Bower MR, Leyva F, & Buckmaster PS (2013). Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J Neurosci, 33(27), 11100–11115. doi: 10.1523/JNEUROSCI.0472-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Maylie J, & Adelman JP (1995). Induction of endogenous channels by high levels of heterologous membrane proteins in Xenopus oocytes. Biophys J, 69(3), 904–908. doi: 10.1016/S0006-3495(95)79964-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium T (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res, 47(D1), D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Hu W, Xie S, Gong CX, Iqbal K, & Liu F (2015). Rapid alteration of protein phosphorylation during postmortem: implication in the study of protein phosphorylation. Sci Rep, 5, 15709. doi: 10.1038/srep15709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber WM (1999). Endogenous ion channels in oocytes of xenopus laevis: recent developments. J Membr Biol, 170(1), 1–12. doi: 10.1007/s002329900532 [DOI] [PubMed] [Google Scholar]
- Williams AD, Jung S, & Poolos NP (2015). Protein kinase C bidirectionally modulates Ih and hyperpolarization-activated cyclic nucleotide-gated (HCN) channel surface expression in hippocampal pyramidal neurons. J Physiol, 593(13), 2779–2792. doi: 10.1113/JP270453 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.