

Abstract
The disulfide isomerase ERp57, originally found in the endoplasmic reticulum, is located in multiple cellular compartments, participates in diverse cell functions and interacts with a huge network of binding partners. It was recently suggested as an attractive new target for cancer therapy due to its critical role in tumor cell proliferation. Since a major bottleneck in cancer treatment is the occurrence of hypoxic areas in solid tumors, the role of ERp57 in cell growth was tested under oxygen depletion in the colorectal cancer cell line HCT116. We observed a severe growth inhibition when ERp57 was knocked down in hypoxia (1% O2) as a consequence of downregulated c-Myc, PLK1, PDPK1 (PDK1) and AKT (PKB). Further, irradiation experiments revealed also a radiosensitizing effect of ERp57 depletion under oxygen deprivation. Compared to ERp57, we do not favour PDPK1 as a suitable pharmaceutical target as its efficient knockdown/chemical inhibition did not show an inhibitory effect on proliferation.
Subject terms: Biochemistry, Cancer, Cell biology, Molecular biology
Introduction
Despite advances in surgical quality, chemotherapies and irradiation, colorectal cancer (CRC) is still a predominant cancer in western countries due to marginal improvements in long-term survival rates1,2. A typical characteristic of solid tumors are hypoxic regions that represent a negative prognostic factor for a patients outcome and are therefore of high clinical relevance. Many promising chemotherapeutics failed when exposed to a hypoxic environment3. Involved in chemoresistance are the hypoxia-inducible factors (HIFs) 1–3, transcription factors that are key mediators for the cellular response to hypoxia. Predominantly HIF1 and HIF2 regulate the expression of numerous genes that drive adaptation and progression of tumor cells4. Further, oxygen deprived tumor areas show decreased generation of reactive oxygen species during radiotherapy that causes radioresistance, recurrence and metastasis5,6.
To explore new targets for cancer treatment, the endoplasmic reticulum (ER) is a suitable cellular place to look for, since a general feature of cancer growth is the abnormal need for high protein levels. Exclusively in the ER, secretory proteins are synthesized, get folded and transported to their final destination in the cell membrane or outside the cell. This whole process underlies a complex quality control and is prone to ER stress which can easily lead to cell death if the ER homeostasis is irreversibly disturbed.
Originally, the thiol-oxidoreductase ERp57 (aka PDIA3, GRP58) was found as an ER-luminal protein that participates in the folding and quality control of glycoproteins, in particular in the assembly of the major histocompatibility complex class I7. It belongs to the family of protein disulfide isomerases (PDIs) and beside its role as a chaperone, it acts as a catalyst for disulfide bond formation in the rather oxidizing environment of the ER. Like its close homologue PDI, the ERp57 protein consists of four domains a, b, b' and a' that form an U shape structure. This arrangement brings the a and a’ domains with its thioredoxin-like active sites in close proximity to each other to facilitate the redox reactions with its client proteins. Although catalytically inactive, the b and b’ domains are mandatory for its chaperoning function8.
We were recently able to demonstrate in the CRC cell line HCT116 under normoxic conditions, that depletion of ERp57 alone activated exclusively the PERK branch of the Unfolded Protein Response (UPR)9. The UPR is an ER signaling pathway that is critical for cell survival and is initiated under stressful conditions to restore ER homeostasis10. As we found out later, the activation of the PERK pathway was not due to ER stress caused by an overload of misfolded proteins, but rather due to ERp57`s role as an ER redox sensor that keeps PDI in a reduced state to maintain PERK in an inactive form11. Further, we recognized a p53-independent impaired cancer cell growth after ERp57 deficiency. Since a proportion of ERp57 was also detected outside the ER in the cytosol9,12,13, a direct participation of ERp57 in cytoplasmic signal transductions was conceivable and could explain this drastic impact on cell proliferation. Although we and others noticed a positive effect of ERp57 on mTOR-complex1 phosphorylation status12, an overall picture of the interplay between ERp57 and growth-related pathways is still missing.
In consequence, here we intended to clarify the role of ERp57 for hypoxic cancer cell growth in combination with irradiation and tested it exemplarily in the CRC cell line HCT116. By the use of a genetically controlled ERp57 knockdown (KD) we were able to detect substantial changes in proliferation signal cascades that underline the critical part of ERp57 for tumor formation under normoxic and hypoxic condition.
Results
Deprivation of ERp57 induces moderate apoptosis under hypoxic conditions
In an earlier work, we demonstrated that apoptosis is elevated in HCT116 colon cancer cells when ERp57 is depleted9. Throughout this study, we used the same lentivirally-based, doxycycline (DOX)-inducible knockdown system. To cover growth conditions that are also found in solid tumors, we extended the experimental setup from normoxia (21% O2) to mild hypoxia (1% O2) and performed Annexin/ propodium iodide (Anx/PI) staining (Fig. 1A): Sole treatment of ERp57 KD or irradiation increased apoptosis albeit less pronounced than in normoxia. Combined treatment of ERp57 depletion with irradiation nearly doubled the apoptotic fraction compared to the untreated hypoxic sample. Similar results were obtained using caspase-3 activity as a second readout for apoptosis (Fig. 1B). Additional western blot analysis showed induced levels for p53 and PUMA after ERp57 KD supporting our apoptotic measurements (Fig. 1C). However, their protein levels were upregulated oxygen-independently while Anx/PI staining and capase-3 assay measured decreased apoptosis under hypoxia. This discrepancy is probably due to hypoxia-induced expression of anti-apoptotic factors like BCL2 that bind and inactivate PUMA, thereby reducing the apoptotic response without detectable differences in protein levels14,15.
Figure 1.

Depletion of ERp57 increases apoptosis in hypoxia. (A) Annexin/ propidium iodide staining was performed to detect early and progressed apoptosis after radiation with 10 Gy or ERp57 KD. Annexin was conjugated to Pacific Blue and thereby designated as „Pacific Blue-H “ in the charts. Propidium iodide as indicator for progressed apoptosis was designated as PE-H. The diagram shows one representative result out of three performed experiments. The induction of apoptosis due to the respective treatment was illustrated as n-fold induction to the untreated sample. Early and late apoptosis were summed as comprehensive apoptosis. (B) Caspase-3 activity was measured 96 h after KD and 48 h after radiation with 10 Gy. Cells were in hypoxia for 72 h. (C) After the same treatment as in (B) pro-apoptotic proteins p53 and PUMA were checked by Western blot.
ERp57 is crucial for hypoxic cell proliferation and does not influence the HIF pathway
To evaluate a long-term effect of the ERp57 KD in hypoxia, clonogenic assays were performed (Fig. 2A). As in normoxia, the depletion of ERp57 alone drastically reduced the survival fraction (SF) down to 10% in hypoxia and was thereby even more effective than irradiation with 3 Gy. The combination of KD and irradiation with 3 Gy revealed the strongest phenotype of 2.7% SF under hypoxic condition. Next, we performed MTT assays to examine cell viability (Fig. 2B). The lack of ERp57 compromised cellular energy levels independent of the oxygen level as indicated by a decrease of at least 50% in normoxia and hypoxia compared to the respective controls. We further investigated a potential influence of ERp57 KD on hypoxia-inducible factor (HIF), the master regulator of O2 homeostasis. As demonstrated by Western blot for HIF1-α (Fig. 2C) and by luciferase assay for total HIF activity (Fig. 2D), the function of HIF was not affected by deprivation of ERp57.
Figure 2.

ERp57 promotes hypoxic proliferation, is radioprotective and does not effect HIF signaling. (A) For colony formation assay, cells were seeded supplemented with doxycycline and incubated under normoxic or hypoxic conditions. After 24 h, cells were irradiated with 0, 1 and 3 Gy and further incubated for 10 d before formed colonies were counted. Survival fraction was presented in bar graphs and representative images of colonies formed in 6 well plates are shown. The seeded cell number is indicated for each well. (B) MTT assay 96 h after ERp57 KD (n = 10). (C) Protein analysis of HIF1α 72 h after ERp57 KD and 24 h of hypoxia by western blot. As a positive control for HIF1α expression, normoxic cells were treated with 2 mM of prolyl-4-hydroxylase inhibitor dimethyloxalylglycine (DMOG). (D) HIF reporter gene assay. After co-transfection with pGLHIF1.3 (firefly luciferase, 3 × HRE) and pGL4.74 (renilla luciferase), ERp57 KD was induced. After 24 h in normoxia, cells were incubated for 48 h in normoxia/ hypoxia before cell lysis (n = 4).
PDPK1 depends on ERp57 but is expendable for cancer cell growth
We detected ERp57, originally described as a typical ER-resident protein, also in the cytoplasm of different cancer cell lines where it drives cell growth9. Hence, we started examining the phosphatidylinositol 3-phosphate kinase (PI3K)/ AKT pathway, one of the most well-known signal pathways that controls proliferation, cell cycle progression and metabolism in cancer cells16. First of all, we noticed changes in expression levels of the master kinase 3-phosphoinositide-dependent protein kinase 1 (PDPK1, PDK1) after depletion of ERP57: Total amount as well as the phosphorylated/ activated form of PDPK1 (pPDPK1) were both downregulated under normoxic and hypoxic conditions (Fig. 3A). To sort out if the lack of PDPK1 after ERp57 KD is responsible for the cell growth inhibition, we generated HCT116 cells with a doxycycline inducible small hairpin (shPDPK1) for PDPK1 and verified a KD efficiency of more than 90% for normoxia and hypoxia via immunoblotting (Fig. 3B). These cells were tested for cell viability, apoptosis and clonogenic survival after KD of PDPK1 but showed no phenotype at all (Fig. 3C–E). Instead, we observed a minor but not significant survival advantage when PDPK1 was missing (Fig. 3E). We compared this genetic approach with clonogenic survival assays of HCT116-shERp57 cells treated with the PDPK1 inhibitor GSK 2334470. However, we observed no altered proliferation after administration of the inhibitor either, only after ERp57 KD alone or in combination (Fig. 4A). Western blot analysis of activated/phosphorylated p70S6K, a PDPK1 downstream target, verified a functional PDPK1 inhibition for at least 4 days (Fig. 4B). As an additional control, the PI3K inhibitor LY294002 was used over the same time period. Further, we combined PDPK1 KD and chemical inhibition, analyzed cell viability by MTT assay and found a decreased viability in normoxia by about 40% and in hypoxia by about 35% (Fig. 4C). This combined treatment was also analyzed on protein level. Here, we observed a decrease of the PDPK1 target pGSK3β, while pPLK1 was unaffected and c-Myc levels were even upregulated (Fig. 4D).
Figure 3.

PDPK1 KD does not effect cancer cell growth. (A) Western blot analysis of PDPK1 after 96 h KD of ERp57. (B) Western blot analysis of inducible PDPK1 KD in HCT116 cells. (C) MTT viability assay. Cells were incubated 96 h after PDPK1 KD before analysis. (D) Apoptosis was analysed by Caspase-3 activity assay 96 h after PDPK1 KD. (E) Colony formation assay after PDPK1 KD. Irradiation with 0, 1 and 3 Gy took place 24 h after seeding. Formed colonies were counted 10 d after seeding. Survival fraction is presented in bar graphs and representative images of colonies formed in 6-Well plates are shown.
Figure 4.

Combination of ERp57 KD and chemical inhibition of PDPK1 does not effect cancer cell growth. (A) Colony formation assay after ERp57 KD in combination with PDPK1 inhibitor GSK 2334470 (3 µM). KD induction and inhibitor treatment was carried out right after seeding. The cells were irradiated 24 h after seeding. Formed colonies were counted 10 d after seeding. Survival fraction is presented in bar graphs and representative images of colonies formed in 6-Well plates are shown. (B) Western blot analysis of PDPK1 target p70S6K after 96 h of ERp57 KD and chemical inhibition of PDPK1 (3 µM GSK 2334470) or PI3K (50 µM LY294002) in normoxia. PCNA was used as a loading control. (C) MTT viability assay after 96 h of PDPK1 KD in combination with PDPK1 inhibition. (D) Western blot analysis after 96 h of PDPK1 KD in combination with chemical PDPK1 inhibition (3 µM GSK 2334470). For additional analysis of pPLK1, cells were treated with 0.1 µg/ml nocodazole 16 h before lysis to achieve G2/M arrest. DMSO was used as a vehicle control.
Deprivation of ERp57 impairs proliferation factors AKT, PLK1, ERK1/2 and c-Myc independently of cellular oxygen levels
Since the decrease of PDPK1 protein after ERp57 KD did not cause the observed cell growth inhibition, we analyzed other PI3K downstream mediators by western blot: While depletion of PDPK1 triggered phosphorylation of AKT at Ser473 and did not change its phosphorylation status at Thr308, KD of ERp57 instead minimized active AKT (pAKT-Ser473/ Thr308) under normoxic and hypoxic conditions, however more severe for pAKT-Ser473. We further noticed a reduced phosphorylation of AKT-target GSK3β under hypoxia (Fig. 5A, B). In addition, we tested the MAPK/ ERK signal pathway and could show a moderate downregulation of total ERK1/2 and pERK1/2 after depletion of ERp57 (Fig. 5A). As we noticed in our previous study a G2 arrest after KD of ERp579, we also looked into polo-like kinase 1 (PLK1) and oncogene c-Myc, both important factors for cell cycle progression and cell growth17,18. Comparing KD of PDPK1 and ERp57, we detected considerable differences in their expression/ activation levels (Fig. 5C): The lack of ERp57 resulted in a downregulation of total PLK1 and pPLK1 independent of the oxygen level, whereas the KD of PDPK1 rather increased the total amount of PLK1 in hypoxia but had no effect on its phosphorylation status (compare nocodazole treated samples). As for PLK1, we noticed a similar downregulation for c-Myc / p–c-Myc when ERp57 was depleted, even though less pronounced in hypoxia due to lesser expression levels of c-Myc under oxygen deprivation. In contrast, KD of PDPK1 even triggered the upregulation of c-Myc / p–c-Myc (compare non-nocodazole treated samples).
Figure 5.

KD of ERp57, but not PDPK1, impairs multiple proliferation factors independently of cellular oxygen levels. (A)–(C) Comparison of different proliferation factors in ERp57 KD and PDPK1 KD cells 96 h after induction of KD. (B) Cells were treated with 1 mM insulin 3 h before lysis. (C) To analyze pPLK1 in the G2/M phase, cells were treated with 0.1 µg/ml nocodazole 16 h before lysis. DMSO was used as a vehicle control.
As the depletion of ERp57 had a strong impact on PLK1, PDPK1, AKT and c-Myc, we further checked whether these proteins show a protein complex formation with ERp57 by CO-IP experiments (Fig. 6). After co-overexpression in HEK293T cells, only c-Myc could be identified as a potential protein interaction partner of ERp57. For the other targets, either no signal in the IP lane was detectable (PDPK1 and AKT) or the signal did not exceed the background level visible in the control IP (PLK1).
Figure 6.
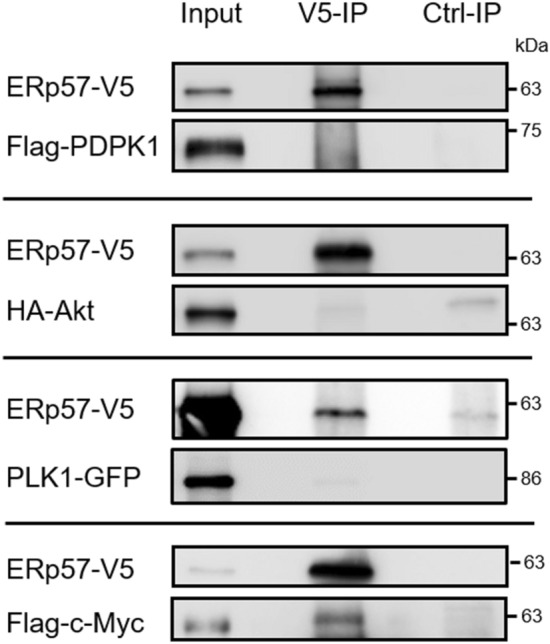
Analysis of protein–protein interaction for ERp57 with PDPK1, AKT, PLK1 and c-Myc. Plasmid pe-N1-ERp57-V5 was co-transfected with vectors CMV-3xFlag-hPDPK1, pCDNA3-HA-AKT1, pEYFP-PLK1 or pCMV4a-Flag-c-Myc into HEK293T cells. For all IP experiments, ERp57 was pulled with an anti-V5 antibody (V5-IP) and complex formation with PDPK1, AKT, PLK1 and c-Myc was determined by western blot analysis using anti-FLAG, anti-HA, anti-GFP and anti-c-Myc antibodies, respectively. 1–3% of total cell lysate was loaded as input.
Discussion
In previous work, we and others were able to detect a minor fraction of ERp57 in the cytosol9,12,13. Consequently, the impact of ERp57 depletion on the PI3K pathway and other proliferation factors described in this study suggested a direct or indirect protein interaction with cytoplasmic ERp57. Except for c-Myc, our Co-IPs could not confirm this for PDPK1, AKT or PLK1 although we cannot rule out protein complexes of ERp57 that were too weak or too transient to detect under the chosen conditions. Besides a direct involvement in proliferation from the cytoplasmic site, it was also conceivable that ERp57 promotes cell proliferation indirectly from the ER compartment: Since KD of ERp57 triggers ER stress via the PERK pathway and results in global protein shutdown9, this could also have caused attenuation of the affected growth factors. Notably, this hypothesis was tested recently in a study where the additional depletion of PDI inhibited the PERK-related UPR completely but did not alter the detrimental effect on proliferation after ERp57 KD alone11. Finally it is also possible that nuclear ERp57, acting as a transcriptional cofactor19, takes part in the expression of proliferation related pathways, as several studies could already show ERp57s influence on genes which are involved in intracellular traffic and stress responses19–21. However, since we have been not able to detect nuclear ERp57 in various cell lines9, we decided not to conduct gene expression analysis in this study.
PDPK1 is called a master kinase of the PI3K pathway for its outstanding role as an upstream kinase capable of phosphorylation and activation of a whole set of AGC kinases (e.g. AKT, S6K, RSK, PKC and SGK) that are involved in cell proliferation22. Beyond AKT, PDPK1 was further reported to trigger the well-studied oncogene c-Myc via phosphorylation of PLK123. Therefore, our initial findings of a severe growth inhibition pointed straight to PDPK1 as the main effector of the ERp57 KD since not only PDPK1, but also PLK1 and c-Myc were all depleted at the same time. The reduction in PLK1 could also explain the former observation of an increased G2 arrest after KD of ERp579. Unexpectedly, neither a chemical inhibition of PDPK1 with 3 µM compound GSK 2334470 (IC50 ≈ 10 nM) nor a generated cancer cell line with an inducible KD of PDPK1 (> 90% KD) did mimic the phenotype of ERp57 KD. Even though the combined treatment of inhibitor and KD finally effected tumor cell viability, the reduction did not exceed 40%. At first glance these results were hard to explain regarding the claimed central role for PDPK1. However, in line with our data is a former in vivo study reporting that an inducible PDPK1 KD of 90% efficiency failed to suppress tumor formation in three different mouse models of PTEN-deficient cancer24. The authors demonstrated that the residual PDPK1 protein was sufficient to uphold the PI3K pathway by continuous activation of AKT at Thr308. Furthermore, various cancer cell lines were able to bypass the PI3K pathway via phosphorylation of AKT by Ca2+/ calmodulin dependent protein kinase kinase (CAMKK2)25. Based on these and our results, it is at least questionable whether cancer patients would benefit from a monotherapy with a potent PDPK1 inhibitor molecule and it seems more advisable to target AKT directly.
In fact, numerous AKT inhibitors are in clinical development and passed phase 1/2 trials26. However, potent cancer drugs that only target one single pathway are likely to fail their clinical expectations due to the frequent occurrence of drug resistance. Redundant pathways can take over and bypass the blockade through rewiring the oncogenic cellular network27. Here, in search of new druggable targets, we tested the relevance of ERp57 for tumor growth in the context of oxygen deprivation. Insufficient oxygenation contributes to tumor progression and triggers stabilization of HIFs that mediate resistance to chemo- and radiotherapy4. Even though KD of ERp57 did not affect HIF activity, we presented compelling data that the depletion of ERp57 severely inhibits proliferation by downregulating a set of notorious oncogenes under moderate hypoxia (1% O2). Notably, we and others demonstrated earlier that ERp57 is also essential for sufficient mTORC1 activity9,12. Considering the size of the PDI family with more than 20 group members and a redundance in their substrate spectrum28, it was quite surprising that so many proliferation factors are compromised at once by the KD of one single family member. Although the number of ERp57`s interaction partners in various cell compartments continuously grow29, further research work is needed to reveal the complete network of ERp57 and the exact molecular mechanisms that explain such an impact on so many different cellular targets under normoxic and hypoxic conditions. In terms of ERp57 as a target for combination therapy, strong radio- and chemosensitizing effects were observed in cancer cells that were irradiated/ etoposide-treated under ERp57 KD in normoxia9. Here, these results were mirrored for irradiation treatment under oxygen deprivation indicating that inhibition of ERp57 has the potential to overcome hypoxic-induced tumor radioresistance. Interestingly, a role of nuclear ERp57 in DNA damage repair was already suggested in earlier studies30,31. PDI and ERp57 were also proposed as potential biomarker in ovarian cancer, since high expression correlates with poor patient outcome32. Depletion of ERp57 led to a severe impairment of the PI3K pathway which is frequently mutated and overactive in colorectal carcinoma, implying a rational strategy to overcome PI3K triggered, unregulated proliferation by inhibiting ERp5733. Therefore, and regarding numerous reports of ERp57′s involvement in other cancer models30,32,34–38, we postulate that this ERp57-dependend effect on cell growth, chemo- and radiosensitivity is not cell-type specific and affects cancer cells beyond colorectal and mammary origin used in our studies.
To our knowledge, no specific ERp57 inhibitor is available so far. But regarding the high structural similarity of ERp57 with its close homologue PDI and several reports of well tolerated PDI inhibitors in preclinical studies39–43, there is reasonable hope that a chemical (or genetic) inhibition of ERp57 could turn out to be more effective in mono- and combination therapy than single targeting inhibitors and could minimize toxicity in non-cancer tissue of patients during treatment.
Materials and methods
Antibodies and reagents
Anti-ERp57 (ab10287) and Anti-pAkt (Thr308) (ab194875) antibodies were from Abcam (Cambridge, UK). Anti-β-Actin (A2103) antibody, PDPK1 inhibitor GSK2334470 (SML0217), Nocodazole (M1404) and human Insulin (91077C) were from Sigma-Aldrich (Munich, Germany). Anti-p53 (GTX102965) antibody was from GeneTex (Hsinchu City, Taiwan). Anti-PUMA (ABC158) antibody was from Millipore (Burlington, MA, USA). Anti-HIF1α (610,959) was from BD Transduction Laboratories (San Jose, CA, USA). Anti-c-Myc (#13,987), Anti-ERK1/2 (#4695), Anti-GFP (#2956), Anti-HA-Tag (#3724), Anti-pAkt (Ser473) (#4060), Anti-p–c-Myc (#13,748), Anti-pERK1/2 (#4370), Anti-pGSK3β (#5558), Anti-PDPK1 (#3062), Anti-PLK1(#4513), Anti-p-p70S6K (#9205), Anti-pPDPK1 (#3438), Anti-pPLK1 (#9062), Anti-PCNA (#2586), Anti-V5-Tag (#13,202) antibodies and PI3K inhibitor LY294002 (#9901) were obtained from Cell Signaling Technology. DMOG (BML-EI347) was purchased from Enzo Life Sciences (Lörrach, Germany).
Cell lines, cell culture, hypoxic treatment and cell cycle synchronization
For all experiments colon carcinoma cell line HCT116 (ATCC, CCL-247) was used. Cells were cultured in McCoy’s 5A medium (Lonza, Basel, Switzerland) supplemented with 10% FBS and antibiotics. For normoxic conditions cells were incubated with 21% O2 and 5% CO2. For hypoxic conditions cells were kept in hypoxic incubator (Toepffer Lap Systems, Göppingen, Germany) with 1% O2 and 5% CO2. For cell cycle synchronization in G2/M phase cells were treated with 100 ng/ml nocodazole 16 h before lysis. Controls were treated with DMSO as vehicle. Treatment with 1 mM insulin took place 3 h before lysis. All experiments were performed with mycoplasma-free cells.
Plasmid construction, transfections, lentiviral production and knockdown induction
To introduce a C-terminal V5-tag into ERp57 (pe-N1-ERp57-V5), a modified QuickChange protocol was used44 to perform a PCR using pEGFP-N1-ERp57 as the template9 and primers ERp57-V5-for (5′-AAGGCACAGGAGGATCTC GGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGTAGCCACCGGTC GCCACCATGGTGAGC -3′) and ERp57-V5-rev (5′ GAGATCCTCCTGTGCCTT CTTCTTCTTCTTGGGTTTTTCTTCTTGAATTACAGGGGGG-3′). For transient transfection, ViaFect (Promega Corporations, Mannheim, Germany) was used as manufacture’s protocol suggests. Lentiviral production was conducted as described before11. Briefly, for transduction 2 × 105 HEK293T cells were incubated with 2 × 106 transduction units for 24 h in attendance of 8 µg/µl polybrene. Transduced HCT116 cells were selected by treatment with 2 µg/ml puromycin for 7 days. pLKO.1-shRNA-ERp57 tet-on (tetracycline inducible) contained the sequence 5′-GGAATAGTCCCATTAGCAAAG-3′ of ERp57 mRNA (GenBank acc. No. NM_005313). pLKO.1-shRNA-hPDPK1 tet-on contained the sequence 5′-CAAAGTTCTGAAAGGTGAAAT-3′ of hPDPK1 mRNA (GenBank acc. No. NM_001261816). Gene KD was induced by addition of 250 ng/ml doxycycline (DOX) to the medium.
Apoptosis analysis
Apoptosis were analyzed by caspase-3 enzyme activity assay and Annexin/ propidium iodide (Anx/PI) staining. Caspase-3 activity assay was performed as described previously45. AMC fluorescence was measured every 10 min at an excitation of 360 nm and emission of 460 nm over 2.5 h in a fluorescence reader (Synergy HT, Biotek, Bad Friedrichshall, Germany). For analysis, replicate values of a single timepoint in the linear range of the reaction were plotted. Measurement of apoptosis by flow cytometry with Anx/PI staining was performed as described previously9. Shortly, for staining 1 × 105 cells were incubated with 80 µg/ml PI and 9.6 µg/ml Annexin V conjugated to PacificBlue (No. 640917, Biolegend, San Diego, CA, USA) in Annexin V binding buffer (No. 422201, Biolegend) for 15 min at room temperature. 10 000 cells were analyzed on a FACS Canto II (BD Biosciences, Franklin Lakes, NJ, USA) per sample. Data were analyzed using FCS Express 4 Flow software (De Novo Software, Los Angeles, CA, USA).
Cell viability
Cell viability was analyzed by MTT assay. Therefore, 2 000 cells were plated in 96-well plates. After 96 h of knockdown induction cells were incubated with MTT for 4 h and afterwards lysed. Absorption was measured with fluorescence reader (Synergy HT, Biotek) at 540 nm.
Clonogenic survival assay
To check for long term cell survival colony formation assay (CFA) was performed as described previously46. To enable formation of colonies under irradiation, cells were irradiated with no more than 3 Gy. Briefly, 100 to 6400 cells were plated on coated 6-well plates. For coating 9.8 ml DPBS, 200 µl rat collagen (Biozol, Eching, Germany) and 5 µM NaOH were used. After 24 h cells were irradiated with 1 or 3 Gy. 9 d later cells were fixed using para-formaldehyde and stained with Coomassie brilliant blue (containing 0.1 Coomassie blue, 5% acetic acid, 45% methanol). For analysis, survival fraction (SF) was determined by dividing number of formed colonies by number of plated cells in respect of plating efficiency (PE) (SF = number of formed colonies/ number of plated cells/ PE). For PE following formula was used: PE = number of formed colonies/ number of plated cells).
Luciferase reporter gene assay
5 × 104 cells were seeded in 24-well plates. After 24 h, cells were transfected with 500 ng pGLHIF1.347 and 100 ng pGL4.74 renilla luciferase (#E6921, Promega) per well. After incubation for further 72 h, cells were lysed and prepared using the Dual Glow luciferase assay kit (Promega). Firefly and renilla luciferase were measured using the GloMax detection system (Promega) and normalized to renilla values to exclude variations in transfection.
SDS PAGE and Western blotting
Whole cell extracts were prepared using RIPA buffer. Proteins were separated in a 7.5% polyacrylamide gel and subsequently transferred onto a PVDF membrane. 5% skimmed milk in TBS-T (50 mM Tris/HCl, 150 mM NaCl, 0.5% Tween-20, pH 7.2) were used to block membrane against unspecific antibody binding. Incubation with antibodies were performed as manufactures recommended. To detect secondary antibodies, ECL kit (34,095, Thermo Fisher Scientific, Waltham, MA, USA) and FX7 chemo luminescence documentation system (Peqlap, Erlangen, Germany) were used. Densitometry was used to quantify protein band intensity using software IMAGEJ. After normalization to each loading control, samples were compared to the untreated sample (set to 1).
Co-immunoprecipitation
6 × 105 HEK293T cells were seeded in a 6 well plate and co-transfected with 2–3 µg plasmid of pe-N1-ERp57-V5 and 2–3 µg of plasmid pcDNA3-HA-AKT1 (Plasmid #73,408, Addgene), pEYFP-PLK1 (Plasmid #39,843, Addgene), pRP[Exp]mCherry-CMV > 3xFlag-hPDPK1 (= CMV-3xFlag-hPDPK1, #VB180820-1170jnv, VectorBuilder) or pCMV4a-Flag-c-Myc (Plasmid #102,625, Addgene). After 72 h of incubation, cells were lysed in caspase lysis buffer (Tris (pH 7.3) 50 mM, NaCl 150 mM, NP-40 1% (v/v)) and incubated for 4 h at 4 °C with V5 antibody. As a control IP, lysate without V5 antibody was used. Afterwards, the lysate was incubated for 1 h with protein S and G magnetic beads (Cell Signaling Technologies, Danvers, USA). Beads were washed 4 × in caspase lysis buffer and finally transferred to SDS-sample buffer containing DTT and were heated at 95 °C for 10 min before loading on a 7.5% polyacrylamide gel.
Statistical analysis
All experimental results were confirmed in at least 3 independent experiments except for Figs. 4B–D and 6 that were confirmed in two independent experiments. In graphs with bars at least three independent samples were taken unless it is otherwise described. Bars represent the mean of samples plus standard deviation (SD) of one representative experiment. For comparison two-way ANOVA by GraphPad Prism 7 (La Jolla, CA, USA) was applied with post hoc Bonferroni’s test. Specific P value is stated in the figures.
Supplementary Information
Acknowledgements
This study was funded by the research training group GRK1739 of the Deutsche Forschungs gemeinschaft. We are indebted to George Iliakis, Institut für Strahlenbiologie, Universität Duisburg-Essen, for providing radiation equipment.
Author contributions
T.O.: F.N., U.B., P.K., A.W., J.V., K.G., M.B., J.B. designed and performed experiments with contribution from E.M. T.O., F.N., P.K. and U.B. analyzed the data and wrote the manuscript with contributions from other authors.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Tobias Ocklenburg, Fabian Neumann, Eric Metzen and Ulf Brockmeier.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-86658-5.
References
- 1.De Rosa M, et al. Genetics, diagnosis and management of colorectal cancer (review) Oncol. Rep. 2015;34:1087–1096. doi: 10.3892/or.2015.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pandurangan AK, et al. Colorectal carcinogenesis: insights into the cell death and signal transduction pathways: a review. World J. Gastrointest. Oncol. 2018;10:244–259. doi: 10.4251/wjgo.v10.i9.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graham K, Unger E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018;13:6049–6058. doi: 10.2147/IJN.S140462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wigerup C, Pahlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016;164:152–169. doi: 10.1016/j.pharmthera.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Li F, et al. Radiation induces the generation of cancer stem cells: a novel mechanism for cancer radioresistance. Oncol. Lett. 2016;12:3059–3065. doi: 10.3892/ol.2016.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pettersen EO, et al. Targeting tumour hypoxia to prevent cancer metastasis: from biology, biosensing and technology to drug development. J. Enzyme Inhib. Med. Chem. 2015;30:689–721. doi: 10.3109/14756366.2014.966704. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, et al. ERp57 does not require interactions with calnexin and calreticulin to promote assembly of class I histocompatibility molecules, and it enhances peptide loading independently of its redox activity. J. Biol. Chem. 2009;284:10160–10173. doi: 10.1074/jbc.M808356200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozlov, G. et al. Crystal structure of the bb' domains of the protein disulfide isomerase ERp57. Structure (London, England : 1993)14, 1331–1339 (2006). [DOI] [PubMed]
- 9.Hussmann, M. et al. Depletion of the thiol oxidoreductase ERp57 in tumor cells inhibits proliferation and increases sensitivity to ionizing radiation and chemotherapeutics. Oncotarget6, 39247–39261. 10.18632/oncotarget.5746 (2015). [DOI] [PMC free article] [PubMed]
- 10.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 11.Kranz P, et al. PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR) Cell Death Dis. 2017;8:e2986. doi: 10.1038/cddis.2017.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramirez-Rangel I, et al. Regulation of mTORC1 complex assembly and signaling by GRp58/ERp57. Mol. Cell. Biol. 2011;31:1657–1671. doi: 10.1128/MCB.00824-10;10.1128/MCB.00824-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo, G. G. et al. Association of the chaperone glucose-regulated protein 58 (GRP58/ER-60/ERp57) with Stat3 in cytosol and plasma membrane complexes. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res.22, 555–563. 10.1089/10799900252982034 (2002). [DOI] [PubMed]
- 14.Kilic M, Kasperczyk H, Fulda S, Debatin KM. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene. 2007;26:2027–2038. doi: 10.1038/sj.onc.1210008. [DOI] [PubMed] [Google Scholar]
- 15.Kinoshita M, Johnson DL, Shatney CH, Lee YL, Mochizuki H. Cancer cells surviving hypoxia obtain hypoxia resistance and maintain anti-apoptotic potential under reoxygenation. Int. J. Cancer. 2001;91:322–326. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1064>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 16.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z, Sun Q, Wang X. PLK1, A potential target for cancer therapy. Transl. Oncol. 2017;10:22–32. doi: 10.1016/j.tranon.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grillo C, et al. Cooperative activity of Ref-1/APE and ERp57 in reductive activation of transcription factors. Free Radic. Biol. Med. 2006;41:1113–1123. doi: 10.1016/j.freeradbiomed.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 20.Chichiarelli S, et al. The stress protein ERp57/GRP58 binds specific DNA sequences in HeLa cells. J. Cell. Physiol. 2007;210:343–351. doi: 10.1002/jcp.20824. [DOI] [PubMed] [Google Scholar]
- 21.Aureli C, et al. ERp57/PDIA3 binds specific DNA fragments in a melanoma cell line. Gene. 2013;524:390–395. doi: 10.1016/j.gene.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Bayascas JR. PDK1: the major transducer of PI 3-kinase actions. Curr. Top. Microbiol. Immunol. 2010;346:9–29. doi: 10.1007/82_2010_43. [DOI] [PubMed] [Google Scholar]
- 23.Tan J, et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013;3:1156–1171. doi: 10.1158/2159-8290.cd-12-0595. [DOI] [PubMed] [Google Scholar]
- 24.Ellwood-Yen K, et al. PDK1 attenuation fails to prevent tumor formation in PTEN-deficient transgenic mouse models. Cancer Res. 2011;71:3052–3065. doi: 10.1158/0008-5472.can-10-2282. [DOI] [PubMed] [Google Scholar]
- 25.Gocher AM, et al. Akt activation by Ca(2+)/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J. Biol. Chem. 2017;292:14188–14204. doi: 10.1074/jbc.M117.778464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown JS, Banerji U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017;172:101–115. doi: 10.1016/j.pharmthera.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nussinov R, Jang H, Tsai CJ. The structural basis for cancer treatment decisions. Oncotarget. 2014;5:7285–7302. doi: 10.18632/oncotarget.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutkevich LA, Cohen-Doyle MF, Brockmeier U, Williams DB. Functional relationship between protein disulfide isomerase family members during the oxidative folding of human secretory proteins. Mol. Biol. Cell. 2010;21:3093–3105. doi: 10.1091/mbc.e10-04-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hettinghouse A, Liu R, Liu CJ. Multifunctional molecule ERp57: From cancer to neurodegenerative diseases. Pharmacol. Ther. 2018;181:34–48. doi: 10.1016/j.pharmthera.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chichiarelli S, et al. The stress protein ERp57/GRP58 binds specific DNA sequences in HeLa cells. J. Cell Physiol. 2007;210:343–351. doi: 10.1002/jcp.20824. [DOI] [PubMed] [Google Scholar]
- 31.Krynetskaia NF, Phadke MS, Jadhav SH, Krynetskiy EY. Chromatin-associated proteins HMGB1/2 and PDIA3 trigger cellular response to chemotherapy-induced DNA damage. Mol. Cancer Ther. 2009;8:864–872. doi: 10.1158/1535-7163.MCT-08-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samanta S, et al. Expression of protein disulfide isomerase family members correlates with tumor progression and patient survival in ovarian cancer. Oncotarget. 2017;8:103543–103556. doi: 10.18632/oncotarget.21569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vacante, M., Borzì, A. M., Basile, F. & Biondi, A. Biomarkers in colorectal cancer: current clinical utility and future perspectives. World J. Clin. Cases6, 869–881. 10.12998/wjcc.v6.i15.869 (2018). [DOI] [PMC free article] [PubMed]
- 34.Coe H, Jung J, Groenendyk J, Prins D, Michalak M. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem. 2010;285:6725–6738. doi: 10.1074/jbc.M109.054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leys CM, et al. Expression and prognostic significance of prothymosin-alpha and ERp57 in human gastric cancer. Surgery. 2007;141:41–50. doi: 10.1016/j.surg.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Liao CJ, et al. Glucose-regulated protein 58 modulates cell invasiveness and serves as a prognostic marker for cervical cancer. Cancer Sci. 2011;102:2255–2263. doi: 10.1111/j.1349-7006.2011.02102.x. [DOI] [PubMed] [Google Scholar]
- 37.Chichiarelli S, et al. Role of ERp57 in the signaling and transcriptional activity of STAT3 in a melanoma cell line. Arch. Biochem. Biophys. 2010;494:178–183. doi: 10.1016/j.abb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Cicchillitti L, et al. Characterisation of a multimeric protein complex associated with ERp57 within the nucleus in paclitaxel-sensitive and -resistant epithelial ovarian cancer cells: the involvement of specific conformational states of beta-actin. Int. J. Oncol. 2010;37:445–454. doi: 10.3892/ijo_00000693. [DOI] [PubMed] [Google Scholar]
- 39.Cole KS, et al. Characterization of an A-site selective protein disulfide isomerase A1 inhibitor. Biochemistry. 2018;57:2035–2043. doi: 10.1021/acs.biochem.8b00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kyani A, et al. Discovery and mechanistic elucidation of a class of protein disulfide isomerase inhibitors for the treatment of glioblastoma. ChemMedChem. 2018;13:164–177. doi: 10.1002/cmdc.201700629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Won JK, et al. Protein disulfide isomerase inhibition synergistically enhances the efficacy of sorafenib for hepatocellular carcinoma. Hepatology. 2017;66:855–868. doi: 10.1002/hep.29237. [DOI] [PubMed] [Google Scholar]
- 42.Vatolin S, et al. Novel protein disulfide isomerase inhibitor with anticancer activity in multiple myeloma. Cancer Res. 2016;76:3340–3350. doi: 10.1158/0008-5472.can-15-3099. [DOI] [PubMed] [Google Scholar]
- 43.Xu S, et al. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc. Natl. Acad. Sci. USA. 2012;109:16348–16353. doi: 10.1073/pnas.1205226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu H, Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janke K, et al. Factor inhibiting HIF-1 (FIH-1) modulates protein interactions of apoptosis-stimulating p53 binding protein 2 (ASPP2) J. Cell Sci. 2013;126:2629–2640. doi: 10.1242/jcs.117564. [DOI] [PubMed] [Google Scholar]
- 46.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat. Protocols. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 47.Kvietikova I, Wenger RH, Marti HH, Gassmann M. The transcription factors ATF-1 and CREB-1 bind constitutively to the hypoxia-inducible factor-1 (HIF-1) DNA recognition site. Nucleic Acids Res. 1995;23:4542–4550. doi: 10.1093/nar/23.22.4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.