

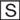
Abstract
Viruses require specific cellular receptors to infect their target cells. Angiotensin-converting enzyme 2 (ACE2) is a cellular receptor for two divergent coronaviruses, SARS coronavirus (SARS-CoV) and human coronavirus NL63 (HCoV-NL63). In addition to hostcell receptors, lysosomal cysteine proteases are required for productive infection by some viruses. Here we show that SARS-CoV, but not HCoV-NL63, utilizes the enzymatic activity of the cysteine protease cathepsin L to infect ACE2-expressing cells. Inhibitors of cathepsin L blocked infection by SARS-CoV and by a retrovirus pseudotyped with the SARS-CoV spike (S) protein but not infection by HCoV-NL63 or a retrovirus pseudotyped with the HCoV-NL63 S protein. Expression of exogenous cathepsin L substantially enhanced infection mediated by the SARS-CoV S protein and by filovirus GP proteins but not by the HCoV-NL63 S protein or the vesicular stomatitis virus G protein. Finally, an inhibitor of endosomal acidification had substantially less effect on infection mediated by the HCoV-NL63 S protein than on that mediated by the SARS-CoV S protein. Our data indicate that two coronaviruses that utilize a common receptor nonetheless enter cells through distinct mechanisms.
SARS coronavirus (SARS-CoV)4 is the etiological agent of severe acute respiratory syndrome (SARS). When a pathogenic form of SARS-CoV emerged in the winter of 2002–2003, it killed ∼800 people, ∼10% of those infected (1, 2, 3, 4). Three genetic and serologic groups of animal and human coronaviruses have been described. The five coronaviruses shown to cause human disease belong either to group 1 or group 2. Despite many unique features, SARS-CoV clusters phylogenetically with group 2 (5), a group that also includes the less pathogenic human coronaviruses OC43 and HKU1 (HCoV-OC43, HCoV-HKU1) (6, 7). Angiotensin-converting enzyme 2 is the cellular receptor for SARS-CoV (8, 9, 10, 11, 12). The cellular receptors for HCoV-OC43 and -HKU1 have not been identified. Group 1 coronaviruses include HCoV-229E, which, like many animal group 1 viruses, utilizes aminopeptidase N (CD13) as its cellular receptor (13, 14). Recently a novel group 1 coronavirus, HCoV-NL63 (HCoV-NL, HCoV-NH), has been described (15, 16, 17). HCoV-NL63 is a common, widely distributed pathogen associated with moderate respiratory illness in children (15, 16, 17, 18, 19, 20, 21, 22, 23). Despite its close similarity to other group 1 coronaviruses, HCoV-NL63 utilizes the SARS-CoV receptor ACE2 to infect cells (19).
The spike (S) proteins of coronaviruses mediate infection of receptor-bearing cells (24). Like many other class 1 fusion proteins such as those of HIV-1, influenza viruses, and filoviruses (ebola- and marburgviruses), some coronavirus S proteins are cleaved by a furin-like protease in virus-producing cells, generating two subunits (S1 and S2 in the case of coronaviruses). Producer-cell processing is necessary for infection mediated by the HIV-1 envelope glycoprotein and influenza virus hemagglutinin (25). The S protein of SARS-CoV is not processed in producer cells, but the S1 and S2 domains can be identified by their similarity with processed coronaviral S proteins (26). The S1 domain mediates an initial, high affinity association with the cellular receptor (27, 28, 29, 30). The C-terminal S2 domain mediates fusion of the viral and cellular membranes (26). The S2 domain contains a fusion peptide, a hydrophobic stretch of residues that associates with target cell membranes and facilitates mixing of viral and cellular bilayers. The coronavirus fusion peptide is located internally within S2. In contrast, the fusion peptides of influenza viruses and HIV-1, but not those of filoviruses, are immediately adjacent to the furin cleavage sites at the N termini of their S2-like domains (25).
Cathepsins are a diverse group of endosomal and lysosomal proteases that include aspartyl, serine, and cysteine proteases with both endo- and exopeptidase activities (31). The role of cathepsins in reovirus infection is well established (32, 33, 34, 35, 36). Following receptor-mediated endocytosis, the reovirus virion is converted to an infectious subvirion particle by partial proteolysis, mediated by cathepsins B, L, or S. One or more of these enzymes degrades the reovirus outer capsid protein σ3, exposing the underlying μ1 protein, which mediates penetration of endosomal membranes (37, 38, 39). Degradation of σ3 and autocatalytic cleavage of μ1 are essential for reovirus infection, and all known inhibitors of σ3 cleavage block reovirus replication (32, 34, 40). Recently, it has been demonstrated that infection mediated by the GP protein of the Zaire ebolavirus depends on cathepsin B and, to a lesser extent, on cathepsin L (41).
Here we show that cathepsin L plays an important role in SARS-CoV infection. In contrast, HCoV-NL63 infection is not dependent on cathepsin B, L, or S activities. Thus variations in the activity of cellular proteases can modulate the relative efficiencies with which these viruses enter ACE2-expressing cells.
EXPERIMENTAL PROCEDURES
Cells, Plasmids, and Inhibitors—The human embryonic kidney epithelial cell line 293T (CRL-11554), African green monkey kidney epithelial cell line Vero (CCL-81), and human bronchial epithelial cell line BEAS-2B (CRL-9609) were obtained from the American Tissue Type Culture Collection and maintained as recommended. The human endothelial cell line SLK (9402) was obtained from National Institutes of Health AIDS Research & Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health (SLK cell line was generated by Dr. Jay A. Levy and Dr. Sophie Leventon-Kriss) and maintained as recommended. Mouse embryonic fibroblasts derived from cathepsin L–/– mouse (MEFs L–/–) were a generous gift from Dr. Alexander Rudensky (42). The genes for SARS-CoV and HCoV-NL63 S proteins and the GP proteins of Lake Victoria marburg and Zaire ebolaviruses were generated by PCR amplification of overlapping oligonucleotides encoding codons optimized for mammalian expression and cloned into the pcDNA3.1 vector (Invitrogen) (43). Human cathepsin B, L, and S genes were PCR amplified from a human macrophage cDNA library and cloned into the pcDNA3.1 vector. A sequence encoding the c9 tag (GTETSQVAPA), an epitope of bovine rhodopsin recognized by the antibody 1D4, was added at the 3′-end of these genes to measure their expression (44). Inhibitor for serine protease AEBSF, aspartic protease inhibitor pepstatin A, and metalloprotease inhibitor phosphoramidon were purchased from Sigma. Cysteine protease inhibitor E64d, and inhibitors for cathepsins L (Z-FY(t-Bu)-DMK), cathepsin K (Z-l-NHNHCONHNH-LF-Boc), and cathepsin S (Z-FL-COCHO), were purchased from Calbiochem. Inhibitors for cathepsin B (CA-074 methyl ester) and B/L (Z-LLL-FMK) were purchased from Sigma.
Pseudotyped Virus Production and Infection—MLV-GFP virions pseudotyped with SARS-CoV S protein have been described previously (45). The experiments described here utilize S proteins of SARS-CoV and HCoV-NL63 with 27- and 24-residue deletions, respectively, in their cytoplasmic C termini. We have previously shown that such deletions enhance incorporation of the S proteins into the retroviral particle (45). Plasmids encoding the SARS-CoV or HCoV-NL63 S proteins were cotranfected with plasmid encoding the MLV gag and pol genes and with a construct bearing the MLV long terminal repeats, packaging signal, and the green fluorescent protein (GFP) gene (pQ-GFP), derived from the pQCXIX vector (Invitrogen). To generate pseudotyped MLV/GFP virions, HEK293T cells were transfected at a 5:5:2 ratio with gag/pol expressor, pQ-GFP, and plasmids encoding SARS-CoV or HCoV-NL63 S protein, ebola- or marburgvirus GP protein, HIV-1 envelope glycoprotein (HXB2 isolate), or vesicular stomatitis virus glycoprotein (VSV-G). Virus-containing cell culture supernatants were harvested 2 days later and filtered through a 0.45-μm filter. Target cells were prepared by transfecting HEK293T cells with expressor plasmids for ACE2 for SARS/MLV and NL63/MLV or CD4 and CXCR4 for HIV-1/MLV. Cells were replated onto polylysine-coated 24-well plates and infected ∼48 h after transfection. Level of infection was assessed 2 days later by measuring GFP expression by flow cytometry. For inhibitor studies, cells were preincubated for 3 h with medium containing the indicated inhibitors and infected for 5 h with virus suspension containing the same concentration of inhibitor. In experiments that included HCoV-NL63, to achieve similar infection levels among various pseudoviruses, SARS/MLV was diluted 5–7-fold, HIV-1/MLV by 2-fold, VSV-G/MLV by 20–30-fold, and NL63/MLV was not diluted. At these dilutions, mean fluorescence intensity of total cells incubated with these pseudoviruses ranged from 72 to 123 for NL63/MLV, from 63 to 151 for SARS/MLV, 46 to 96 for VSV-G/MLV, 90 to 151 for HIV/MLV, and 5 to 11 for uninfected cells.
Infection of Cells Expressing Exogenous Cathepsins—HEK293T cells grown on 6-well plates were transfected with 0.2–2 μg of plasmid expressing human ACE2 (9) and 2.5 μg of vector or plasmid expressing one of human cathepsins B, L, or S and replated the next day for flow cytometry, metabolic labeling, and infection. One aliquot of transfected cells was labeled with [35S]cysteine and [35S]methionine between 36 and 48 h following transfection to assess the expression of cathepsins in each transfection. Approximately 48 h after transfection, an additional aliquot of transfected cells was stained with goat anti-human-ACE2 antibody (R&D Systems) to assess ACE2 expression level, and a third aliquot of cells was infected with SARS/MLV, NL63/MLV, or VSV-G/MLV, as described above. The degree of infection is represented by GFP expression (mean fluorescence) in infected cells measured by flow cytometry 48–60 h after infection. Labeled cells were lysed in 0.5% Nonidet P-40/PBS containing protease inhibitor mixture (Sigma), and cathepsins were immunoprecipitated with anti-c9 tag antibody 1D4 (National Institutes of Health AIDS Research & Reference Reagent Program) and Protein A-Sepharose. Washed samples were analyzed by reducing SDS-PAGE and autoradiography.
Cathepsin Inhibitor Studies Using Infectious Virus—Vero cells were cultured at 34 °C in Iscove's modified Dulbecco's medium (BioWhittaker) supplemented with 5% fetal bovine serum. Cells in 96-well plates were pretreated with medium containing the indicated inhibitors and inoculated for 1 h with SARS-CoV (strain 5688, fourth passage) at a multiplicity of infection of 0.1 in the presence of the same concentration of inhibitors, and cells were washed twice with Iscove's modified Dulbecco's medium. Eight hours later, cells were fixed with 4% formaldehyde and 70% ethanol, 0.5% H2O2 successively, permeabilized with PBS/0.5% Tween 20, and incubated for 1 h at 37°C with 40-fold-diluted polyclonal antisera obtained from SARS-CoV-infected ferrets (Mustela furo). Horseradish peroxidase-conjugated goat-anti-ferret antibodies (Dako) were used at 1:50 dilution. The reaction was developed with 3-amino-9-ethylcarbazole (Sigma) according to the manufacturer's instruction, and infected cells were counted. LLC-MK2 cells were cultured at 34 °C in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. HCoV-NL63 (sixth passage) infection was carried out for 1 h at a multiplicity of infection of 0.01. After 24-h incubation, plates were processed as described above, and HCoV-NL63 infecton was detected by staining cells with 100-fold diluted human polyclonal serum, biotinylated anti-human antibody (1:250), and the VEC-TASTAIN ABC reagents (Vector Laboratories). All experiments were performed twice in duplicate. LLC-MK2 cells, HCoV-NL63, and human antiserum were generous gifts from Dr. Lia van der Hoek.
RESULTS AND DISCUSSION
We investigated the ability of inhibitors of several classes of proteases to modulate infection mediated by the SARS-CoV S protein, or by the HIV-1 envelope glycoprotein, included as a control. Murine leukemia viruses carrying the gene for GFP and pseudotyped with the SARS-CoV or HIV-1 fusion proteins (SARS/MLV and HIV-1/MLV, respectively) were incubated with 293T cells expressing ACE2 or the HIV-1 receptors CD4 and CXCR4. The infections were performed in the presence of the aspartic protease inhibitor pepstatin A, serine protease inhibitor AEBSF, cysteine protease inhibitor E64d, and metalloprotease inhibitor phosphoramidon. As shown in Fig. 1A , of four classes of protease inhibitors assayed, only E64d, a general inhibitor of cysteine proteases, blocked SARS/MLV infection. E64d had no significant effect on HIV-1/MLV infection.
FIGURE 1.
SARS-CoV S-protein-mediated pseudovirus entry is blocked by cathepsin L inhibitor. HEK293T cells were transfected with plasmids encoding human ACE2 or the HIV-1 receptors CD4 and CXCR4, replated, and infected with GFP-expressing MLV virus pseudotyped with the SARS-CoV S protein or HIV-1 envelope glycoprotein (SARS/MLV or HIV-1/MLV, respectively). Target cells were preincubated for 3 h with indicated concentrations of protease inhibitors (A) or cathepsin inhibitors (B) and incubated for 5 h with the indicated pseudoviruses and inhibitors. Infected cells were trypsinized 48–60 h later, and GFP expression was measured by flow cytometry. C, fluorescent micrographs of cells infected in the presence and absence of 10 μm cathepsin L inhibitor. Abbreviations used are: Phsdn, phosphoramidon; Pep A, pepstatin A; Cath L, cathepsin L.
SARS-CoV infection is sensitive to NH4Cl, an inhibitor of lysosomal acidification (45, 46), and roles for the lysosomal cysteine protease cathepsins in reovirus and ebolavirus infection have been described (32, 33, 34, 35, 36, 41). Accordingly, we investigated the ability of cathepsin inhibitors to block infection mediated by the SARS-CoV S protein. As shown in Fig. 1B, cathepsin L inhibitor (Z-FY(t-Bu)-DMK) potently blocked SARS/MLV infection, and cathepsin B inhibitor (CA-074 methyl ester) also showed consistent but less significant inhibition of infection. In contrast, HIV-1/MLV infection was enhanced by both of these inhibitors, consistent with other reports indicating that lysosomal degradation interferes with productive HIV-1 infection (47). Fig. 1C shows representative fields of SARS/MLV and HIV-1/MLV infection in the presence and absence of 10 μm cathepsin L inhibitor. These data imply that cathepsin L activity contributes substantially to the SARS-CoV S-protein-mediated infection.
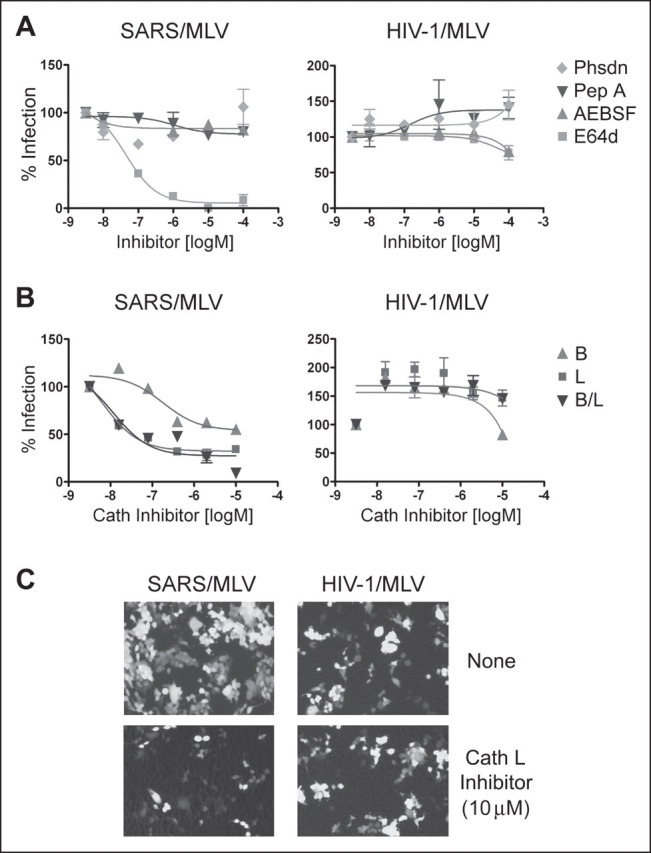
We then examined the ability of these protease inhibitors to block infection of replication-competent SARS-CoV and HCoV-NL63, a group 1 coronavirus that also utilizes ACE2 as its cellular receptor. Sindbis virus and VSV were also included in these studies as controls. Infection was assessed within 8 and 24 h of incubation with SARS-CoV and HCoV-NL63, respectively, thereby minimizing any potential effects of inhibitors on post-entry steps in viral replication. As shown in Fig. 2A , neither SARS-CoV nor HCoV-NL63 infection was inhibited by AEBSF or phosphoramidon. SARS-CoV infection was effectively blocked by cathepsin L inhibitor and less significantly by cathepsin B inhibitor (Fig. 2B) at concentrations comparable with those effective with pseudotyped virus. While cathepsin B inhibitor did not have any effect, cathepsin L inhibitor partially blocked HCoV-NL63 and Sindbis virus infection at the highest concentration in one of two cell lines assayed. This partial inhibition in LLK-MK2 cells may have been to due to cytotoxicity.
FIGURE 2.
Cathepsin (Cath) L inhibitor suppresses infectious SARS-CoV. Vero 118 cells (SARS-CoV, HCoV-NL63) and LLC-MK2 cells (HCoV-NL63) were infected with replication-competent SARS-CoV or HCoV-NL63 in the presence of indicated protease inhibitors (A) or cathepsin inhibitors (B) as described under”Experimental Procedures.“Sindbis virus and VSV are included as controls in both cell lines. Cells were fixed 8 h (for experiments with SARS-CoV, Sindbis virus, and VSV) or 24 h (for those with HCoV-NL63) post-infection, permeabilized and stained with ferret sera against SARS-CoV or with human serum against HCoV-NL63, as described. Positively stained cells were counted. Phsdn, phosphoramidon.
We further explored this difference in sensitivity to cathepsin inhibitors between HCoV-NL63 and SARS-CoV and sought to determine whether other cathepsin inhibitors could interfere with infection mediated by their respective S proteins. Inhibitors of cathepsin B, L, K, and S, the only cathepsins for which specific inhibitors are available, were examined. None of these inhibitors had a detectable effect on the infection of MLV pseudotyped with the HCoV-NL63 S protein (NL63/MLV) or with the fusion protein of vesicular stomatitis virus (VSV-G/MLV), whereas cathepsin L and cathepsin S inhibitors efficiently blocked SARS/MLV entry (Fig. 3A ). Again, cathepsin B inhibitor partially inhibited SARS/MLV infection. We also explored the ability of other broad protease inhibitors to block NL63/MLV infection. None of the protease inhibitors used in Fig. 1A had any effect on NL63/MLV infection (data not shown) nor did E64d, which again potently inhibited SARS/MLV (Fig. 3B). These data indicate that NL63/MLV infection is not dependent on the cysteine proteases that contribute to SARS/MLV infection nor on the protease activities blocked by the broader inhibitors assayed.
FIGURE 3.

HCoV-NL63 S-protein-mediated pseudovirus entry is not affected by cathepsin (Cath) inhibitors. Human ACE2-transfected HEK293T cells were preincubated for 3 h (A, B) or 30 min (C) with infection inhibitors and then incubated with SARS/MLV, NL63/MLV, HIV/MLV, or VSV-G/MLV in the presence of indicated cathepsin inhibitors (A), the broad cysteine protease inhibitor E64d (B), or NH4Cl, an inhibitor of endosomal acidification (C). Infectivity was assessed as described under”Experimental Procedures.“
The inability of E64d, a broad inhibitor of cyteine proteases, to block NL63/MLV infection raised the possibility that HCoV-NL63 and SARS-CoV enter cells via distinct mechanisms. Consistent with this possibility, NH4Cl, an inhibitor of acidification of lysosomal and endosomal compartments, had substantially less effect on NL63/MLV infection than on SARS/MLV (Fig. 3C). These data suggest that, in contrast with SARS-CoV, internalization of HCoV-NL63 to a low pH compartment may not be an essential step in its entry into the cell.
Because cathepsin inhibitors can cross-react, we investigated the consequences of exogenous cathepsin B, L, and S expression in 293T cells that express human ACE2. To ensure comparable ACE2 expression levels in cells transfected with various cathepsins, cells were transfected with various amounts of ACE2-expressing plasmid together with a fixed amount of cathepsin plasmid, and ACE2 cell-surface expression was assessed by flow cytometry following each transfection. As shown in Fig. 4A , exogenous cathepsin L markedly increased infection by SARS/MLV but had no effect on NL63/MLV or VSV-G/MLV. Exogenous cathepsin S also modestly enhanced SARS/MLV infection but, surprisingly, inhibited NL63/MLV infection. We speculate that overexpressed cathepsin S, which is secreted and active at neutral pH, may digest and inactivate the HCoV-NL63 S protein but not that of SARS-CoV. In parallel, epitope-tagged cathepsins were immunoprecipitated from an aliquot of the same cells and analyzed by SDS-PAGE. Fig. 4B shows that exogenous cathepsin L expression was lower than that of cathepsin B or S, despite its greater effect on SARS-CoV infection.
FIGURE 4.

Exogenous cathepsin (Cath) L enhances the entry of SARS/MLV, but not that of NL63/MLV.A, HEK293T cells were transfected with cathepsin B, L, or S expressor plasmids and various amounts of human ACE2 plasmid as described under”Experimental Procedures.“Cells were divided and replated for infection, metabolic labeling, or flow cytometry the following day. Two days post-transfection cells were incubated with SARS/MLV, NL63/MLV, or VSV-G/MLV. In parallel, cell surface ACE2 expression was assessed by flow cytometry with anti-human-ACE2 antibody. Infection by GFP-expressing pseudovirions, measured 2 days post-infection by flow cytometry (vertical axis), is plotted here against ACE2 expression (horizontal axis). B, approximately 36 h post-transfection, an aliquot of transfected cells used in A was labeled overnight with [35S]cysteine and [35S]methionine. Cells were lysed in 0.5% Nonidet P-40/PBS containing protease inhibitor mixture, immunoprecipitated with 1D4 antibody recognizing a C-terminal epitope present on the cathepsin protein, and analyzed by SDS-PAGE. Three lanes each for cathepsins B, L, and S represent three transfections (in the order of increasing amounts of ACE2 DNA). C, MEFs cultured from a cathepsin L–/– mouse were transduced with VSV-G/MLV carrying the human ACE2 gene and, 5 h later, with VSV-G/MLV carrying the human cathepsin L gene. Cells were split and infected with SARS/MLV, NL63/MLV, MARV/MLV, Ebola/MLV, or VSV-G/MLV. Infection efficiency was assessed 3 days later by measuring GFP expression by flow cytometry and is expressed as a percentage of GFP-expression observed with cells transfected with ACE2 alone.
Similarly, as shown in Fig. 4C, introduction of cathepsin L into mouse embryonic fibroblasts derived from mice lacking cathepsin L (MEFs L–/–) resulted in substantially enhanced infection by SARS/MLV and MLV pseudotyped with marburg- and ebolavirus GP proteins. Again, no enhancement of infection was observed with NL63/MLV or VSV-G/MLV. Additional experiments with similar results are shown in supplemental Fig. 1A. Collectively, these data show that introduction of cathepsin L into cells where this enzyme is limiting or absent can substantially boost infection mediated by the SARS-CoV but not the HCoV-NL63 S protein.
The role of cathepsins in the cellular entry of reoviruses has been appreciated for some time (34, 36). Recently, it has been demonstrated that infection mediated by the GP protein of Zaire ebolavirus also depends on cathepsin B and, to a lesser extent, on cathepsin L (41). Here we extend this observation to SARS-CoV, which appears to be more dependent on cathepsin L than on cathepsin B. Cathepsin S also appears to contribute modestly to SARS-CoV infection and may partially compensate for the absence of cathepsin L in some cells. We also show that infection mediated by the S protein of another coronavirus that utilizes ACE2 as a receptor, HCoV-NL63, is not dependent on any of these cathepsins. It remains to be investigated whether other cellular proteases contribute to HCoV-NL63 infection through a mechanism analogous to the role played by cathepsin L in SARS-CoV infection or whether HCoV-NL63, like HIV-1 and VSV, infects cells independently of target-cell proteases.
It is not yet clear how cathepsin L facilitates SARS-CoV infection. Several compatible possibilities exist. Cathepsin L may serve to nonspecifically degrade the S1 domain of SARS-CoV S protein, thereby permitting conformational transitions in the S2 domain necessary for fusion. ACE2 could also be a cathepsin target, thereby facilitating its dissociation with the S protein. Interestingly, we have observed that introduction of exogenous cathepsins reduces cell-surface expression of ACE2 (supplemental Fig. 1B). Cathepsin L could also cleave immediately upstream of the coronavirus fusion peptide in process equivalent to the furin cleavage of the HIV-1 and influenza fusion proteins.
Our studies show that SARS-CoV infection can be limited by low cathepsin L expression. Of note, mature endothelial cells express relatively low levels of cathepsin L (48), and despite their efficient expression of ACE2, little infection or replication of SARS-CoV has been observed in these cells (11, 12, 49). Although multiple factors may contribute to the infection efficiency of endothelial cells, one possibility is that low cathepsin L activity limits SARS replication in these cells. Studies determining whether HCoV-NL63 more efficiently infects these cells in vivo than SARS-CoV are warranted.
Finally our studies suggest that inhibitors specific to cathepsin L may be effective in treating SARS-CoV. However, they also raise the possibility that SARS-CoV can escape from a cathepsin L inhibitor by altering its S protein to function more like that of HCoV-NL63. Therefore, the means by which viruses escape from these inhibitors requires study.
Acknowledgments
We are grateful to Stefan Pohlman and Lia van der Hoek for kindly providing materials and to Bart Haagmans for help with the SARS-CoV infection experiments.
Footnotes
Note Added in Proof—While this manuscript was under review, Simmons et al. (Simmons, G., Gosalia, D. N., RenneKamp, A. J., Reeves, J. D., Diamond, S. L., and Bates, P. (2005) Proc. Natl. Acad. Sci. U. S. A.102, 11876–11882) published results simiar to those herein.
The abbreviations used are: SARS, severe acute respiratory syndrome; CoV, coronavirus; S, spike; ACE2, angiotensin-converting enzyme 2; MHV, mouse hepatitis virus; HCoV, human coronavirus; VSV-G, vesicular stomatitis virus glycoprotein; HIV, human immunodeficiency virus; MLV, murine leukemia virus; GFP, green fluorescent protein; PBS, phosphate-buffered saline; BSA, bovine serum albumin; AEBSF, 4-(2-aminoethyl)bezenesulfonyl fluoride.
Supplementary Material
REFERENCES
- 1.Fouchier R.A., Kuiken T., Schutten M., van Amerongen G., van Doornum G.J., van den Hoogen B.G., Peiris M., Lim W., Stohr K., Osterhaus A.D. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuiken T., Fouchier R.A., Schutten M., Rimmelzwaan G.F., van Amerongen G., van Riel D., Laman J.D., de Jong T., van Doornum G., Lim W., Ling A.E., Chan P.K., Tam J.S., Zambon M.C., Gopal R., Drosten C., van der Werf S., Escriou N., Manuguerra J.C., Stohr K., Peiris J.S., Osterhaus A.D. Lancet. 2003;362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 5.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guan Y., Peiris J.S., Zheng B., Poon L.L., Chan K.H., Zeng F.Y., Chan C.W., Chan M.N., Chen J.D., Chow K.Y., Hon C.C., Hui K.H., Li J., Li V.Y., Wang Y., Leung S.W., Yuen K.Y., Leung F.C. Lancet. 2004;363:99–104. doi: 10.1016/S0140-6736(03)15259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woo P.C., Lau S.K., Chu C.M., Chan K.H., Tsoi H.W., Huang Y., Wong B.H., Poon R.W., Cai J.J., Luk W.K., Poon L.L., Wong S.S., Guan Y., Peiris J.S., Yuen K.Y. J. Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W., Bao L., Zhang B., Liu G., Wang Z., Chappell M., Liu Y., Zheng D., Leibbrandt A., Wada T., Slutsky A.S., Liu D., Qin C., Jiang C., Penninger J.M. Nat. Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzeriaga C., Greenough T.C., Choe H., Farzan M. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S., Wong S.K., Huang I.C., Xu K., Vasilieva N., Murakami A., He Y., Marasco W.A., Guan Y., Choe H., Farzan M. EMBO J. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamming I., Timens W., Bulthuis M.L., Lely A.T., Navis G.J., van Goor H. J. Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding Y., He L., Zhang Q., Huang Z., Che X., Hou J., Wang H., Shen H., Qiu L., Li Z., Geng J., Cai J., Han H., Li X., Kang W., Weng D., Liang P., Jiang S. J. Pathol. 2004;203:622–630. doi: 10.1002/path.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeager C.L., Ashmun R.A., Williams R.K., Cardellichio C.B., Shapiro L.H., Look A.T., Holmes K.V. Nature. 1992;357:420–422. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tresnan D.B., Holmes K.V. Adv. Exp. Med. Biol. 1998;440:69–75. doi: 10.1007/978-1-4615-5331-1_9. [DOI] [PubMed] [Google Scholar]
- 15.van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J., Wolthers K.C., Wertheim-van Dillen P.M., Kaandorp J., Spaargaren J., Berkhout B. Nat. Med. 2004;10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fouchier R.A., Hartwig N.G., Bestebroer T.M., Niemeyer B., de Jong J.C., Simon J.H., Osterhaus A.D. Proc. Natl. Acad. Sci. U. S. A. 2004;101:6212–6216. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esper F., Weibel C., Ferguson D., Landry M.L., Kahn J.S. J. Infect. Dis. 2005;191:492–498. doi: 10.1086/428138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiu S.S., Chan K.H., Chu K.W., Kwan S.W., Guan Y., Poon L.L., Peiris J.S. Clin. Infect. Dis. 2005;40:1721–1729. doi: 10.1086/430301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofmann H., Pyrc K., van der Hoek L., Geier M., Berkhout B., Pohlmann S. Proc. Natl. Acad. Sci. U. S. A. 2005;102:7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moes E., Vijgen L., Keyaerts E., Zlateva K., Li S., Maes P., Pyrc K., Berkhout B., van der Hoek L., Van Ranst M. BMC Infect. Dis. 2005;5:6. doi: 10.1186/1471-2334-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bastien N., Anderson K., Hart L., Van Caeseele P., Brandt K., Milley D., Hatchette T., Weiss E.C., Li Y. J. Infect. Dis. 2005;191:503–506. doi: 10.1086/426869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arden K.E., Nissen M.D., Sloots T.P., Mackay I.M. J. Med. Virol. 2005;75:455–462. doi: 10.1002/jmv.20288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ebihara T., Endo R., Ma X., Ishiguro N., Kikuta H. J. Med. Virol. 2005;75:463–465. doi: 10.1002/jmv.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallagher T.M., Buchmeier M.J. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimitrov D.S. Nat. Rev. Microbiol. 2004;2:109–122. doi: 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao X., Dimitrov D.S. Cell. Mol. Life Sci. 2004;61:2428–2430. doi: 10.1007/s00018-004-4257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai J.C., Zelus B.D., Holmes K.V., Weiss S.R. J. Virol. 2003;77:841–850. doi: 10.1128/JVI.77.2.841-850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kubo H., Yamada Y.K., Taguchi F. J. Virol. 1994;68:5403–5410. doi: 10.1128/jvi.68.9.5403-5410.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonavia A., Zelus B.D., Wentworth D.E., Talbot P.J., Holmes K.V. J. Virol. 2003;77:2530–2538. doi: 10.1128/JVI.77.4.2530-2538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turk V., Turk B., Turk D. EMBO J. 2001;20:4629–4633. doi: 10.1093/emboj/20.17.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golden J.W., Bahe J.A., Lucas W.T., Nibert M.L., Schiff L.A. J. Biol. Chem. 2004;279:8547–8557. doi: 10.1074/jbc.M309758200. [DOI] [PubMed] [Google Scholar]
- 33.Golden J.W., Linke J., Schmechel S., Thoemke K., Schiff L.A. J. Virol. 2002;76:7430–7443. doi: 10.1128/JVI.76.15.7430-7443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebert D.H., Deussing J., Peters C., Dermody T.S. J. Biol. Chem. 2002;277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- 35.Jane-Valbuena J., Breun L.A., Schiff L.A., Nibert M.L. J. Virol. 2002;76:5184–5197. doi: 10.1128/JVI.76.10.5184-5197.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baer G.S., Ebert D.H., Chung C.J., Erickson A.H., Dermody T.S. J. Virol. 1999;73:9532–9543. doi: 10.1128/jvi.73.11.9532-9543.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandran K., Parker J.S., Ehrlich M., Kirchhausen T., Nibert M.L. J. Virol. 2003;77:13361–13375. doi: 10.1128/JVI.77.24.13361-13375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Odegard A.L., Chandran K., Liemann S., Harrison S.C., Nibert M.L. J. Virol. 2003;77:5389–5400. doi: 10.1128/JVI.77.9.5389-5400.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chandran K., Farsetta D.L., Nibert M.L. J. Virol. 2002;76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sturzenbecker L.J., Nibert M., Furlong D., Fields B.N. J. Virol. 1987;61:2351–2361. doi: 10.1128/jvi.61.8.2351-2361.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chandran K., Sullivan N.J., Felbor U., Whelan S.P., Cunningham J.M. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsieh C.S., deRoos P., Honey K., Beers C., Rudensky A.Y. J. Immunol. 2002;168:2618–2625. doi: 10.4049/jimmunol.168.6.2618. [DOI] [PubMed] [Google Scholar]
- 43.Holmes K.V., Dveksler G., Gagneten S., Yeager C., Lin S.H., Beauchemin N., Look A.T., Ashmun R., Dieffenbach C. Adv. Exp. Med. Biol. 1993;342:261–266. doi: 10.1007/978-1-4615-2996-5_40. [DOI] [PubMed] [Google Scholar]
- 44.Mirzabekov T., Bannert N., Farzan M., Hofmann W., Kolchinsky P., Wu L., Wyatt R., Sodroski J. J. Biol. Chem. 1999;274:28745–28750. doi: 10.1074/jbc.274.40.28745. [DOI] [PubMed] [Google Scholar]
- 45.Moore M.J., Dorfman T., Li W., Wong S.K., Li Y., Kuhn J.H., Coderre J., Vasilieva N., Han Z., Greenough T.C., Farzan M., Choe H. J. Virol. 2004;78:10628–10635. doi: 10.1128/JVI.78.19.10628-10635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J., Bates P. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei B.L., Denton P.W., O'Neill E., Luo T., Foster J.L., Garcia J.V. J. Virol. 2005;79:5705–5712. doi: 10.1128/JVI.79.9.5705-5712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urbich C., Heeschen C., Aicher A., Sasaki K., Bruhl T., Farhadi M.R., Vajkoczy P., Hofmann W.K., Peters C., Pennacchio L.A., Abolmaali N.D., Chavakis E., Reinheckel T., Zeiher A.M., Dimmeler S. Nat. Med. 2005;11:206–213. doi: 10.1038/nm1182. [DOI] [PubMed] [Google Scholar]
- 49.To K.F., Lo A.W. J. Pathol. 2004;203:740–743. doi: 10.1002/path.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.