

Abstract
Rapid decline of glomerular filtration rate estimated from creatinine (eGFRcrea) is associated with severe clinical endpoints. In contrast to cross-sectionally assessed eGFRcrea, the genetic basis for rapid eGFRcrea decline is largely unknown. To help define this, we meta-analyzed 42 genome-wide association studies from the Chronic Kidney Diseases Genetics Consortium and United Kingdom Biobank to identify genetic loci for rapid eGFRcrea decline. Two definitions of eGFRcrea decline were used: 3 mL/min/1.73m2/year or more (“Rapid3”; encompassing 34,874 cases, 107,090 controls) and eGFRcrea decline 25% or more and eGFRcrea under 60 mL/min/1.73m2 at follow-up among those with eGFRcrea 60 mL/min/1.73m2 or more at baseline (“CKDi25”; encompassing 19,901 cases, 175,244 controls). Seven independent variants were identified across six loci for Rapid3 and/or CKDi25: consisting of five variants at four loci with genome-wide significance (near UMOD-PDILT (2), PRKAG2, WDR72, OR2S2) and two variants among 265 known eGFRcrea variants (near GATM, LARP4B). All these loci were novel for Rapid3 and/or CKDi25 and our bioinformatic follow-up prioritized variants and genes underneath these loci. The OR2S2 locus is novel for any eGFRcrea trait including interesting candidates. For the five genome-wide significant lead variants, we found supporting effects for annual change in blood urea nitrogen or cystatin-based eGFR, but not for GATM or LARP4B. Individuals at high compared to those at low genetic risk (8–14 vs 0–5 adverse alleles) had a 1.20-fold increased risk of acute kidney injury (95% confidence interval 1.08–1.33). Thus, our identified loci for rapid kidney function decline may help prioritize therapeutic targets and identify mechanisms and individuals at risk for sustained deterioration of kidney function.
Keywords: Genome-wide association study, rapid eGFRcrea decline, end-stage kidney disease, acute kidney injury
Graphcal Abstract

Introduction
Rapid kidney function decline is an important risk factor for end-stage kidney disease (ESKD), cardiovascular events, and early mortality2,3. ESKD is a life-threatening condition with substantial individual and public health burden4–6 and a major endpoint in clinical nephrology trials. However, identifying and monitoring individuals at risk for ESKD is challenging. Two definitions of rapid decline in creatinine-based eGFR (eGFRcrea) are reported to increase ESKD risk 5- and 12-fold7,8, respectively, and thus recommended for clinical use: (i) rapid eGFRcrea decline of >5 mL/min/1.73m2/year and (ii) a ≥25% decline of eGFRcrea along with movement into a lower category of chronic kidney disease 8. Other surrogate endpoints of ESKD were implemented by interventional trials with follow-up duration of <5 years9,10, such as a doubling of creatinine levels (equivalent to a 57% eGFRcrea decline11) or an eGFRcrea decline of 30% or 40%.
Beside specific therapies in autoimmune driven glomerulopathies such as immunosuppressive agents12 or tolvaptan in polycystic kidney disease13, therapeutic options to slow down kidney function decline are largely limited to glycemic and blood pressure control as well as lipid-lowering drugs. Prior to the recent advent of SGLT2-inhibitors in large clinical trials14, these therapies had shown only moderate, if any, effect on clinically relevant renal endpoints15. Selecting genetically supported drug targets was estimated to double success rate in drug discovery1, in particular when the causal gene was suggested by Mendelian diseases or from genome-wide associations driven by coding variants16. This motivates genome-wide association studies (GWAS) for the identification and characterization of genetic variants associated with rapid kidney function decline.
A recent GWAS combining data from >1,000,000 individuals identified 264 loci associated with eGFRcrea based on one creatinine measurement (“cross-sectional eGFRcrea”)17. However, little is known about whether these or additional genetic factors are associated with rapid kidney function decline (“longitudinal kidney function traits”). Given the substantial organizational and temporal requirements of longitudinal studies, sample sizes for these studies are still limited compared to cross-sectional studies. Our previous longitudinal GWAS based on 61,078 individuals and ~3 million genetic variants did not identify any locus for rapid eGFRcrea decline18. New studies with longitudinal eGFRcrea measurements and new genomic reference panels enabling a denser and more precise genetic variant imputation now allow for a more powerful investigation.
We thus performed a GWAS meta-analysis across 42 longitudinal studies, consisting of 41 studies from the Chronic Kidney Disease Genetics (CKDGen) Consortium and UK Biobank, totaling >270.000 individuals with two eGFRcrea measurements across a time period of one to 15 years of follow-up. We implemented two definitions of rapid eGFRcrea decline that were feasible in population-based studies while preserving similarity to recommended surrogate clinical endpoints:
(1) “Rapid3” cases defined as eGFRcrea decline of >3 mL/min/1.73m2 per year compared to “no decline” (“Rapid3” controls, 1 to +1 mL/min/1.73m2 per year), (2) ”CKDi25” cases defined as ≥25% eGFRcrea decline during follow-up together with a movement from eGFRcrea≥60 mL/min/1.73m2 at baseline to eGFRcrea<60 mL/min/1.73m2 at follow-up compared to “CKDi25” controls defined as eGFRcrea≥60 mL/min/1.73m2 at baseline and follow-up (Figure 1).
Figure 1 |. Illustration of the case-control definitions of Rapid3 and CKDi25.
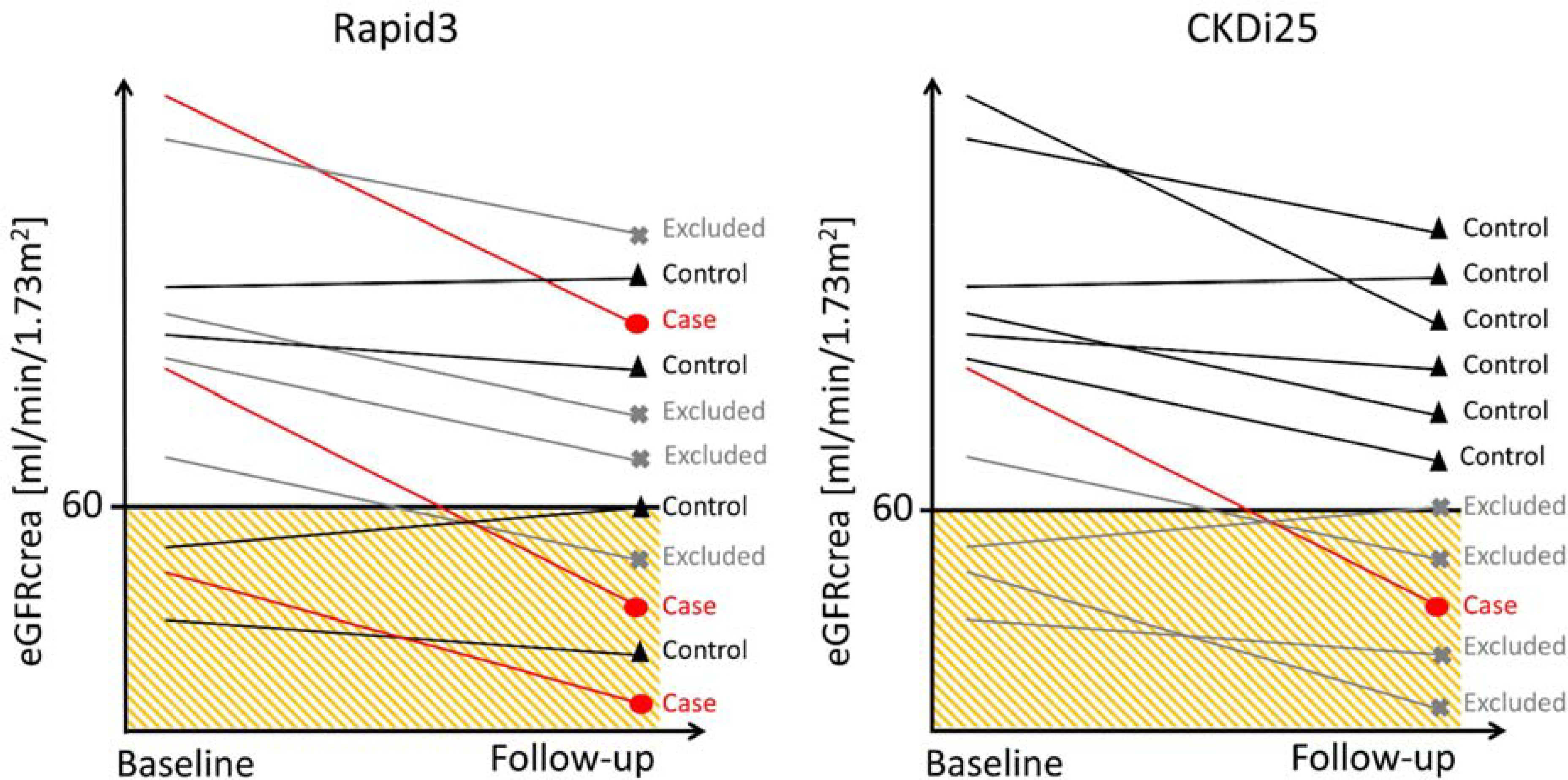
Rapid3 defines cases as individuals with an eGFRcrea decline>3 mL/min/1.73m2 per year and controls with an eGFRcrea decline between −1 and +1 mL/min/1.73m2 per year. CKDi25 defines cases as a ≥25% drop from baseline eGFRcrea≥60 mL/min/1.73m2 into eGFRcrea<60 mL/min/1.73m2 at follow-up and controls as an eGFRcrea≥60 mL/min/1.73m2 at baseline and follow-up. Shown are cases (red), controls (black) and excluded individuals (grey) according to the eGFRcrea values observed at baseline and follow-up.
RESULTS
Rapid eGFRcrea decline in 42 longitudinal studies
We collected phenotype summary statistics for Rapid3 and CKDi25 from 42 studies with genetic data and at least two measurements of creatinine (study-specific mean age of participants 33–68 years, study-specific median follow-up time 1–15 years; Methods, Supplementary Table 1A&B). Most studies were from European ancestry and population-based (32 European ancestry based, 34 population-based).
Several interesting aspects emerged: (i) as expected for studies covering general populations as well as elderly and patient populations, study-specific median baseline eGFRcrea ranged from 46.4 to 115.0 mL/min/1.73m2 (overall median=87.3 mL/min/1.73m2); (ii) case proportions ranged from 11% to 72% for Rapid3 and from 3% to 52% for CKDi25 (median=30% or 11%, respectively); (iii) there was no association of study-specific median age of participants or median follow-up time with Rapid3 or CKDi25 (Supplementary Figure 1A&B); (iv) most CKDi25 cases were a subgroup of Rapid3 cases in three example studies with different lengths of follow-up (Supplementary Table 2).
Four new genome-wide significant loci for rapid eGFRcrea decline
In each of the 42 studies, the >8 million genetic variants imputed via 1000 Genomes19 or Haplotype Reference Consortium (HRC)20 reference panels were tested for association with Rapid3 and CKDi25 using logistic regression adjusting for age, sex, baseline eGFRcrea (Supplementary Table 3, Methods). We meta-analyzed study-specific summary statistics by outcome (34,874 cases, 107,090 controls for Rapid3; 19,901 cases, 175,244 controls for CKDi25; Methods).
In our genome-wide approach, we selected genome-wide significant loci (i.e. ≥1 variant with P-value<5×10−8 within ±500kB; “lead variant” as the variant with the smallest P-value); within each locus, we searched for independently associated signals by conditional analyses (Methods). By this, we identified five lead variants across four loci (P-values=5.94×10−9 to 3.51×10−33, Figure 2, Table 1A): (i) the UMOD-PDILT locus was associated with Rapid3 and CKDi25 and showed a 2nd independent signal for CKDi25 (rs77924615; P-adjusted=2.98×10−10). For CKDi25, the independent odds ratios (OR) for the two UMOD-PDILT lead variants (rs12922822, rs77924615) were 1.06 per adverse allele per variant in a model containing both variants. (ii) One variant in each of the WDR72 and PRKAG2 loci was identified for CKDi25. (iii) A variant near OR2S2 was associated with Rapid3.
Figure 2 |. Four loci identified with genome-wide significance for Rapid3 or CKDi25.
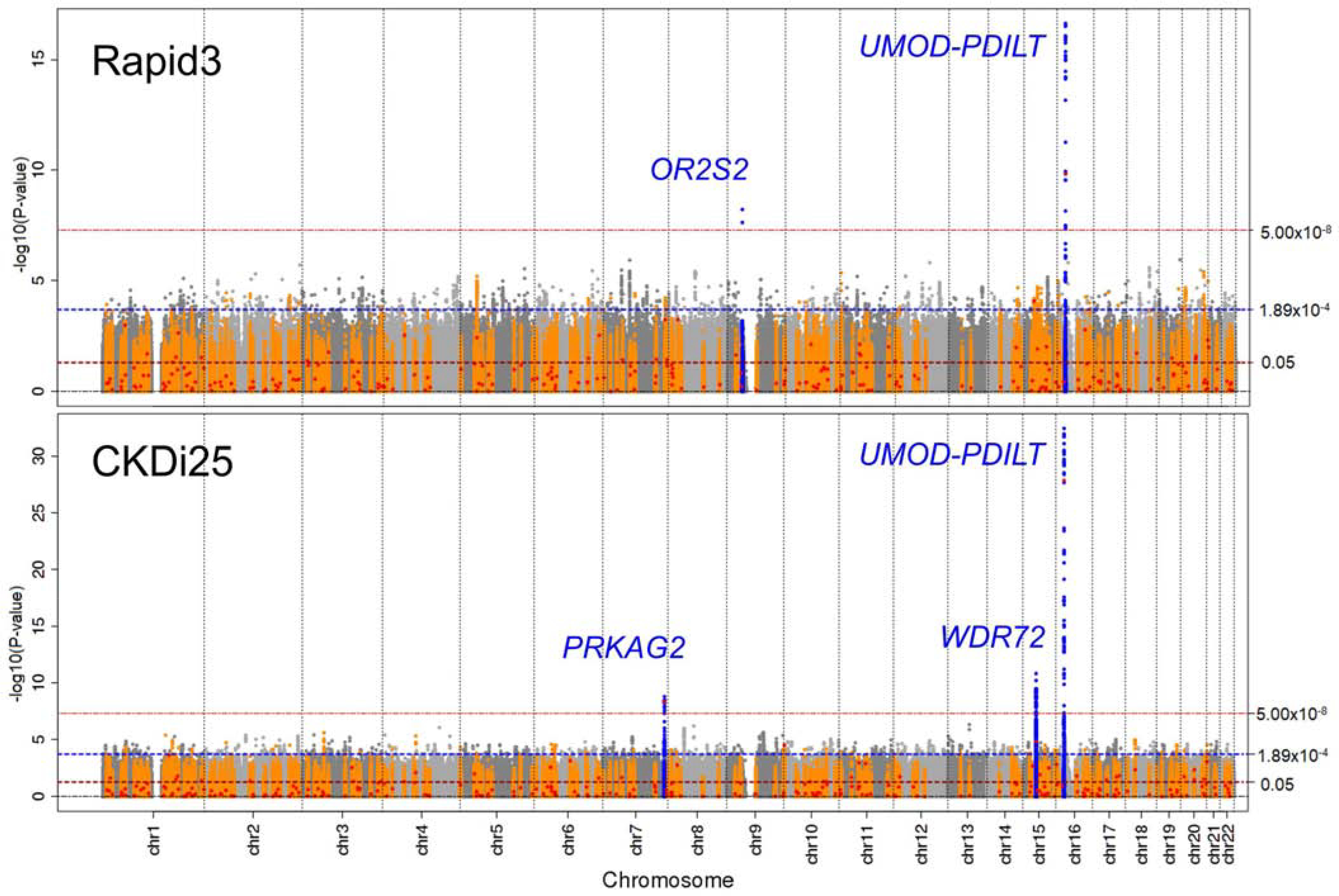
Shown are association P-values versus genomic position for Rapid3 (34,874 cases; 107,090 controls) and CKDi25 (19,901 cases; 175,244 controls). Horizontal dashed lines indicate genome-wide (5.00×10−8), Bonferroni-corrected (0.05/265≈1.89×10−4) and nominal (0.05) significance thresholds. The four identified genome-wide significant loci are annotated by the nearest genes (blue). The 264 loci reported previously for cross-sectional eGFRcrea17 are marked in orange and respective lead variants as red dots.
Table 1 |. Six loci from the genome-wide and candidate-based search for association with Rapid3 or CKDi25.
Shown are (A) the significant lead variants from the GWAS (genome-wide significance, P-value<5.0×10−8) and (B) the significant variants from the candidate-based approach inquiring the 265 variants reported for cross-sectional eGFRcrea17 (Bonferroni-corrected significance, P-value<0.05/265≈1.89×10−4).
| Rapid3 | CKDi25 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RSID | Chr:Position | Identifying analysis | Locus name | EA/OA | EAF | OR | P | OR | P | Locus/signal no. | Reference variant (R2) |
| (A) Genome-wide search (genome-wide significance, P-value<5×10−8) | |||||||||||
| rs13329952 | 16:20,366,507 | Rapid3 | [UMOD-PDILT] | t/c | 0.79 | 1.101 | 2.35×10−17 | 1.203 | 6.22×10−30 | 1.1 | |
| rs12922822 | 16:20,367,645 | CKDi25 | c/t | 0.81 | 1.103 | 1.13×10−16 | 1.224 | 3.51×10−33 | rs13329952 (0.91) | ||
| rs77924615 | 16:20,392,332 | CKDi25 2nd (a) | [UMOD-PDILT] | g/a | 0.79 | 1.023 | 0.0384 | 1.112 | 2.98×10−10 | 1.2 | |
| rs77593734 | 15:54,002,606 | CKDi25 | [WDR72] | t/c | 0.72 | 1.040 | 1.18×10−4 | 1.102 | 1.42×10−11 | 2 | |
| rs56012466 | 7:151,406,788 | CKDi25 | [PRKAG2] | a/g | 0.27 | 1.041 | 1.12×10−4 | 1.090 | 1.53×10−9 | 3 | |
| rs141809766 | 9:35,937,931 | Rapid3 | [OR2S2] | g/a | 0.02 | 1.222 | 5.94×10−9 | 1.065 | 0.252 | 4 | |
| (B) Candidate approach based on 265 (c) reported lead variants from cross-sectional eGFRcrea GWAS (significance P-value<0.05/265≈1.89×10−4) | |||||||||||
| rs34882080 (b) | 16:20,361,441 | CKDi25; Rapid3 | [UMOD-PDILT] | a/g | 0.81 | 1.100 | 1.11×10−15 | 1.216 | 2.98×10−31 | 1.1 | rs12922822 (0.99) |
| rs77924615 | 16:20,392,332 | CKDi25; Rapid3 | [UMOD-PDILT] | g/a | 0.79 | 1.084 | 1.40×10−10 | 1.256 | 1.29×10−28 | 1.2 | |
| rs690428 | 15:53,950,578 | CKDi25 | [WDR72] | a/c | 0.71 | 1.027 | 0.0117 | 1.078 | 1.46×10−5 | 2 | rs77593734 (0.42) |
| rs10254101 | 7:151,415,536 | CKDi25 | [PRKAG2] | t/c | 0.28 | 1.037 | 5.35×10−4 | 1.087 | 4.32×10−9 | 3 | rs56012466 (0.84) |
| rs80282103 | 10:899,071 | CKDi25 | [LARP4B] | t/a | 0.08 | 1.027 | 0.100 | 1.103 | 2.97×10−5 | 5 | |
| rs1145077 | 15:45,683,795 | Rapid3 | [GATM] | t/g | 0.40 | 1.038 | 7.94×10−5 | 1.042 | 1.93×10−3 | 6 | rs1145089 (0.99) |
RSID=Variant identifier on GRCh37, Chr:Position=Chromosome and Position on GRCh37, Identifying analysis=Trait und analysis for which the variant was identified with significant association (“2nd“ indicating the second signal analysis), Locus name=Nearest gene, stated in brackets to distinguish from gene and protein names, EA=Effect allele: cross-sectional eGFRcrea-lowering allele, OA=Other allele, EAF=Effect allele frequency, OR=Odds ratio, P=Genomic control corrected association P-value, Locus/ signal no.=Locus number and signal number highlighting that four of the six candidate-based identified variants capture the same locus/signal as the GWAS, Reference variant (R2)=Variant to which the identified variant is compared to in terms of correlation (spearman correlation coefficient squared). (a) Stated are OR and P-value for Rapid3 and CKDi25 adjusted for the lead variant of the respective primary GWAS (rs13329952 or rs12922822). Unadjusted OR=1.08 and 1.26 (P-value=1.40×10−10 and 1.29×10−28) for Rapid3 and CKDi25, respectively; (b) Lead variant of 2nd signal in [UMOD-PDILT] from cross-sectional eGFRcrea analysis in European ancestry17; (c) 264 reported lead variants plus the lead variant of the 2nd signal in [UMOD-PDILT] from cross-sectional eGFRcrea GWAS17.
For all variants and both outcomes, we observed no to moderate heterogeneity across studies (I2=0 to 43%). A sensitivity analysis restricted to European ancestry (31,101 cases, 102,485 controls for Rapid3; 19,419 cases, 169,087 controls for CKDi25) identified the same loci with the same or highly correlated lead variants (r2>0.84, Supplementary Table 4A). We also conducted a meta-analysis restricting to individuals of African ancestry (2,356 cases and 2,375 controls for Rapid3; 374 cases and 4,183 controls for CKDi25), but limited sample sizes prohibited an informative comparison with EUR results (Supplementary Table 4B, Supplementary Note 1).
Overall, we identified four loci associated at genome-wide significance for these binary rapid eGFRcrea decline traits.
Two additional loci for rapid eGFRcrea decline from a candidate-based search
Genetic variants with established association for cross-sectional eGFRcrea are candidates for association with rapid eGFRcrea decline. For our candidate-based approach, we selected the 264 lead variants and the 2nd signal lead variant in the UMOD-PDILT locus reported previously for eGFRcrea17 and tested these for association with Rapid3 and CKDi25 (judged at Bonferroni-corrected significance; 0.05/265=1.89×10−4). Among these, we found six variants in five loci significantly associated with Rapid3 and/or CKDi25 (Table 1B), yielding two variants that were associated with Rapid3 and/or CKDi25 independently from the five GWAS-identified variants, one each in LARP4B and GATM, were significantly associated with CKDi25 or Rapid3 (Supplementary Note 2, Supplementary Table 5, Supplementary Figure 2). Overall, our genome-wide and candidate-based approaches yielded seven independent variants in six loci associated with at least one of the rapid eGFRcrea decline traits.
Statistical evidence for the OR2S2 locus
For the OR2S2 locus, the only two genome-wide significant variants identified for Rapid3 were highly correlated and showed the largest odds ratio (OR) of all seven identified variants (rs141809766, rs56289282, r2=0.95; OR=1.22 and 1.21; P-value=5.94×10−9 and 2.11×10−8, respectively). Since these variants were not associated with cross-sectional eGFRcrea17 (P-value=0.16 or 0.18, n=542,354) and of low frequency in the general population (minor allele frequency, MAF=0.02), we evaluated the statistical robustness of this association: (i) the majority of studies showed consistent risk for rs141809766 (Supplementary Figure 3A); (ii) a leave-one-out sensitivity analysis showed no influential single study driving the signal (Supplementary Figure 3B); (iii) when focusing on European ancestry, we found similar results (Supplementary Table 4); (iv) the lack of association with cross-sectional eGFRcrea was confirmed in independent data (UK Biobank, n=364,686, e.g. rs141809766, P-value=0.65). In summary, these analyses supported this locus as a genuine finding.
Characterizing identified effects by alternative markers for kidney function
A challenge in using eGFRcrea to detect genetic variants for kidney function is the fact that it is influenced both by kidney function and creatinine production, the latter being linked to muscle mass21. Alternative biomarkers such as estimated GFR based on cystatin C22 (eGFRcys) and blood urea nitrogen17 (BUN) can be used to support eGFRcrea loci as kidney function loci. We thus evaluated the seven lead variants for their direction-consistent association with annual change in eGFRcys and BUN in UK Biobank (n=15,746 or 15,277, respectively; mean follow-up time=4.3 years): annual decline of eGFRcys and/or annual increase of BUN for the Rapid3/CKDi25-risk increasing allele. For completeness, we also present the seven variants’ association with cross-sectional eGFRcys and BUN (n=364,819 and 358,791). These analyses with alternative renal biomarkers supported UMOD-PDILT, WDR72, PRKAG2, and OR2S2, but not LARP4B or GATM loci (Table 2, Supplementary Note 3).
Table 2 |. Validation of the seven identified variants association with alternative renal biomarker in UK Biobank.
Shown are association results for annual change in estimated Glomerular Filtration Rate based on cystatin C (eGFRcys) and blood urea nitrogen (BUN) in UK Biobank (n up to 15,746 or 15,277, respectively). One-sided P-values are provided testing the allele that increased the risk of rapid eGFRcrea decline (usually the eGFRcrea-lowering allele, except for the OR2S2 lead variant) into the direction of annual eGFRcys decline and annual BUN increase. For completeness, also shown are association results for cross-sectional eGFRcys and BUN from UK Biobank (n up to 364,819 and 358,791) as well as previously reported BUN results from CKDGen17 (n=416,076), where one-sided P-values test the eGFRcrea-lowering allele into the direction of decreased eGFRcys and increased BUN levels.
| Locus/ signal no. [name] | RSID | eGFRcys change (a) UKBB | BUN change (a) UKBB | eGFRcys (b) UKBB | BUN (b) UKBB (CKDGen) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Effect | P | Effect | P | Effect | P | Effect | P | ||
| 1.1 [UMOD-PDILT] | rs13329952 | 0.0271 | 0.02 | −0.0036 | 0.45 | −0.0045 | 6.06×10−86 | 0.0024(0.0040) | 1.08×10−18 (1.62×10−22) |
| 1.1[UMOD-PDILT] | rs12922822 | 0.0289 | 0.01 | 0.0018 | 0.53 | −0.0046 | 2.17×10−85 | 0.0025 (0.0044) | 1.09×10−18 (8.79×10−21) |
| 1.2 [UMOD-PDILT] | rs77924615 | 0.0289 | 0.01 | −0.0519 | 0.03 | −0.0051 | 1.74×10−108 | 0.0029 (0.0053) | 2.38×10−26 (2.57×10−42) |
| 2 [WDR72] | rs77593734 | 0.0026 | 0.41 | −0.0429 | 0.03 | −0.0016 | 1.88×10−16 | 0.0014 (0.0026) | 1.59×10−9 (8.46×10−17) |
| 3 [PRKAG2] | rs56012466 | 0.0238 | 0.02 | −0.0652 | 2.75×10−3 | −0.0039 | 1.56×10−81 | 0.0046 (0.0057) | 8.73×10−80 (1.69×10−41) |
| 4 [OR2S2] | rs141809766 | 0.0537 | 0.04 | −0.1245 | 0.02 | 0.0005 | 0.80 | −0.00345 (−0.0018) | 0.70 (0.89) |
| 5 [LARP4B] | rs80282103 | 0.0241 | 0.10 | −0.0362 | 0.17 | −0.0037 | 4.87×10−29 | 0.0026 (0.0026) | 2.49×10−11 (4.90×10−7) |
| 6 [GATM] | rs1145077 | −0.0096 | 0.82 | 0.0150 | 0.75 | 0.0001 | 0.74 | −0.0004 (<0.0001) | 0.95 (0.46) |
Locus/signal no. [name]=Locus number and signal number [locus name], RSID=Variant identifier, Effect=Genetic effect, P=One-sided association P-value, UKBB=UK Biobank, (a) Annual change of eGFRcys and BUN was calculated as the baseline value minus the follow-up value divided by the years between baseline and follow-up. The age, sex and baseline eGFRcys/ BUN adjusted residuals were regressed on allele dosage, (b) The age and sex-adjusted residuals of the log eGFRcrea, eGFRcys and BUN were regressed on allele dosage.
From lead variants to the statistical signals
Each lead variant represents a signal consisting of correlated variants. Regional association plots (Supplementary Figure 4) illustrate that the seven rapid eGFRcrea decline signal mostly coincided with the cross-sectional eGFRcrea signal, except for a weaker signal in the WDR72 locus and no corresponding OR2S2 signal for cross-sectional eGFRcrea. Between the two traits, Rapid3 and CKDi25, the signals were mostly comparable, except for LARP4B and OR2S2.
To prioritize variants at identified signals, we ranked each signal variant by their posterior probability of driving the observed association and added them to the “99% credible set of variants” until the cumulative posterior probability was > 99% (Methods). Such a credible set is thus a parsimonious set of variants that most likely includes the causal variant, assuming that there is exactly one causal variant per signal and that this variant was analyzed23. When deriving the 99% credible sets of variants for each of the seven identified signals for Rapid3 and CKDi25 (Methods) wand comparing them with cross-sectional eGFRcrea credible sets17, we found the following (Table 3): (i) for most GWAS-derived signals, the credible sets coincided with those for cross-sectional eGFRcrea, except for the WDR72 locus; (ii) the credible set of the second UMOD-PDILT signal for CKDi25 consisted of precisely one variant, rs77924615, which was exactly the one credible set variant for eGFRcrea supporting this as the most likely causal variant for this association signal; (iii) the two correlated genome-wide significant variants in the OR2S2 locus for Rapid3 formed the credible set (posterior probability 77% and 23%, respectively); (iv) the credible sets for the two candidate-approach derived loci, LARP4B and GATM, included 1438 to 2955 variants for Rapid3 and CKDi25, which was due insufficiently strong associations resulting from the lack of genome-wide significance. We thus considered these credible sets unsuitable for in-silico follow-up and focused on further evaluation on the five genome-wide significant signals.
Table 3 |. Size of 99% credible sets of variants for the seven identified signals for Rapid3 or CKDi25.
Shown number of genes overlapping each of the six locus regions (lead variant +/−500kB) and the number of variants in the 99% credible set for each of the seven signals. The credible sets of variants were computed (i) for the two rapid eGFRcrea decline traits (Rapid3 and CKDi25) highlighting the set for the analysis that identified the locus/signal (signals 1.1 to 4 from genome-wide approach, signals 5 and 6 from candidate-based approach) and (ii) for cross-sectional eGFRcrea from CKDGen data as reported previously17.
| Locus/signal no. | Locus name (a) | Identifying trait (b) | Locus region (c) |
No. of variants in
99%credible set (overlap with eGFRcrea sets) |
No. of variants in 99% credible set
(overlap with CKDi25 sets) |
||||
|---|---|---|---|---|---|---|---|---|---|
| Chr | Start | Stop | No. of genes | Rapid3 | CKDi25 | eGFRcrea *** | |||
| 1.1 | [UMOD-PDILT] | Rapid3, CKDi25 | 16 | 19,866,507 | 20,867,645 | 13 | 14 (10) | 13 (11) | 16 (10) |
| 1.2 | [UMOD-PDILT] | CKDi25 2nd | 16 | 19,866,507 | 20,867,645 | s.a. | 1,059 | 1 (1) | 1 (1) |
| 2 | [WDR72] | CKDi25 | 15 | 53,502,606 | 54,502,606 | 1 | 2,931 | 37 (0) | 41 (0) |
| 3 | [PRKAG2] | CKDi25 | 7 | 150,906,788 | 151,906,788 | 14 | 2,671 | 16 (6) | 6 (6) |
| 4 | [OR2S2] | Rapid3 | 9 | 35,437,931 | 36,437,931 | 36 | 2 | 2,573 | NA |
| 5 | [LARP4B] | CKDi25 | 10 | 399,071 | 1,399,071 | 10 | 2,955 | 2,806 | 1 (d) |
| 6 | [GATM] | Rapid3 | 15 | 45,183,795 | 46,183,795 | 17 | 1,438 | 2,493 | 1 (d) |
Chr=Chromosome of locus region, Start/Stop=Start and stop of locus region on GRCh37, s.a.=see above, (a) Nearest gene(s), stated in brackets to distinguish from gene and protein names; (b) Indicates the trait for which the variant was identified with significant association (“CKDi25 2nd” indicating that this is the 2nd independent signal for the CKDi25 trait analysis); (c) Locus region defined as the region of the two lead variants identified for Rapid3 and CKDi25 in [UMOD-PDILT] or for the single lead variant identified for Rapid3 or CKDi25 in the other loci ±500 kB. The CKDi25 2nd signal (signal no. 1.2) is mapped to the [UMOD-PDILT] locus region from signal no. 1.1; (d) For the candidate-based identified loci [LARP4B] and [GATM], the statistics for the credible sets were instable due to the lack of genome-wide significance and yielded extremely wide credible set intervals. Since the CKDi25 or Rapid3 signal was very similar to the signal for cross-sectional eGFRcrea (Supplementary Figure 4E&F), we conducted the bioinformatic follow-up for the credible set variant derived from eGFRcrea previously.
From statistical evidence to biology
One of the key challenges in translating GWAS associations into an understanding of the underlying biology is the identification of variants and genes causing the statistical signal. It is unclear exactly what evidence to weigh in and how expansive the search for causal genes should be; ±500kB around the lead variant is often used (“locus region”). A variant is often considered more likely causal when it is in a credible set and predicted to have a relevant function, such as protein-altering (e.g. changing the peptide sequence, truncating, affecting RNA splicing) or modulating a gene’s expression24 (expression quantitative trait locus, eQTL). A gene is often considered more likely causal when it (i) contains a protein-altering credible set variant, (ii) is a target of an eQTL-variant, or (iii) has a kidney-related phenotype reported from animal models or monogenic disease. We annotated the credible set variants and the 64 genes across the five genome-wide significant signals accordingly (Methods, Supplementary Table 6A,B, 7A,B). We summarized the evidence per gene in a Gene PrioritiSation (GPS) Table and implemented a customizable score, where each category’s weight can be modified according to personal interest or preference (Supplementary Table 8).
By this, we identified eight genes with functional evidence (score ≥1; Table 4): two genes with protein-altering variant (WDR72, PRKAG2), four genes as target of a significant eQTL-variant (PDILT, WDR72, GALNTL5 and OR2S1P), and four genes with a phenotype in mice and/or human (UMOD, PRKAG2, GNE and CD72). Particularly interesting were the 36 genes in the OR2S2 locus (Supplementary Table 9) and the findings from in-silico follow-up in three of these genes: OR2S1P as an eQTL-target of the lead variant rs141809766 in lung tissue with a particularly high effect estimate also for kidney tissue (Supplementary Figure 5; no data available in NephQTL) and GNE as well as CD72 with abnormal morphology of podocytes or renal glomerulus in mice providing candidates for a potential kidney function biology.
Table 4 |. Gene Prioritization (GPS) for the genes across the four loci identified with genome-wide significance.
Shown are genes across the four loci, for which we found any relevant evidence: (i) blue: gene contains at least one credible set variant that was protein-altering (missense, non-mediated decay, NMD, or altered splicing; Supplementary Table 6A, information obtained from VEP49); (ii) orange: the gene’s expression shows a modulation by any of the signal’s credible set variant (expression quantitative trait loci, eQTL, in NephQTL50 or GTEx v851; Supplementary Table 6B), (iii) gene shows a kidney phenotype in mouse or human (MGI52, OMIM53; Supplementary Table 7A&B). The full GPS shows all genes overlapping the four loci (Supplementary Table 8) and the online version is searchable and customizable (i.e. the weights per column can be altered) to re-sort the table reflecting other preferences (www.genepi-regensburg.de/rapiddecline).
| Any credible set variants in gene | eQTL-modulated expression by any credible set variant | Evidenced kidney phenotype | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Weight | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||
| Locus name | Locus no. | Gene | Chromosome | Distance to 1st signal variant | #credible set variants in gene | Gene Priority Score | Missense | NMD | Altered splicing | NephQTL glomerulus | NephQTL tubulointerstitium | GTEx v8 kidney tissue | GTEx v8 any other tissue | In mice (MGI) | In human (OMIM) |
| [UMOD-PDILT] | 1 | UMOD | 16 | 0 | 10 | 2 | |||||||||
| [UMOD-PDILT] | 1 | PDILT | 16 | 2,846 | 1 | 1 | |||||||||
| [WDR72] | 2 | WDR72 | 15 | 0 | 37 | 2 | |||||||||
| [PRKAG2] | 3 | PRKAG2 | 7 | 0 | 16 | 2 | |||||||||
| [PRKAG2] | 3 | GALNTL5 | 7 | 246,675 | 0 | 1 | |||||||||
| [OR2S2] | 4 | OR2S1P | 9 | 75,251 | 0 | 1 | |||||||||
| [OR2S2] | 4 | GNE | 9 | 276,506 | 0 | 1 | |||||||||
| [OR2S2] | 4 | CD72 | 9 | −319,507 | 0 | 1 | |||||||||
Locus name=Nearest gene(s), stated in brackets to distinguish from gene or protein names, #credible set variants in gene region=#variants in the 99% credible set overlapping the gene’s region, Gene Priority Score=Cumulative score (here: weighing all categories equally; see Supplementary Table 8 for all genes in locus regions and online version for customization of weights), Blue section: gene contains ≥1 credible set variant overlapping the gene with relevant function (yes=blue/no=white); Orange section: locus/signal contains ≥1 credible set variant that modulates gene expression (yes=orange/no=white) in NephQTL glomerulus, NephQTL tubulointerstitium, GTEx v8 kidney tissue or GTEx v8 any tissue;Green section: gene shows a kidney-related phenotype (yes=green/no=white) in MGI Mouse kidney phenotype or OMIM Human kidney phenotype.
The cumulative genetic effect
A genetic risk score (GRS) is an approach to summarize the genetic profile of a person across the identified variants. We computed the GRS across the seven variants in four studies for Rapid3 and CKDi25 (overall 3,683 cases vs. 8,579 controls for Rapid3; 895 cases vs. 21,472 controls for CKDi25) and defined genetic high-risk and low-risk groups (individuals with 8–14 adverse alleles, ~30% in UK Biobank; 0–5 alleles, ~20%, respectively (Methods). In the meta-analysis of study-specific odds ratios, we found a 1.11-fold increased risk for Rapid3 (95%-confidence interval, CI, 0.99–1.24, P-value=0.07) and a 1.29-fold increased risk for CKDi25 (1.06–1.57, P-value=0.01, Table 5). The lower risk for Rapid3 compared to CKDi25 can be explained by the less pronounced effect sizes for Rapid3 for most variants in the GRS and by the fact that the only variant with a high effect for Rapid3 (near OR2S2) was rare and thus with little impact on the distribution of the GRS.
Table 5 |. Genetic Risk Score Analyses of Rapid3, CKDi25, End-stage Kidney Disease (ESKD) and Acute Kidney Injury (AKI).
Shown are the results of the unweighted Genetic Risk Score (GRS) across the seven variants identified for Rapid3 and/or CKDi25 counting Rapid3- or CKDi25-risk increasing alleles and its association with (A) Rapid3, (B) CKDi25, (C) ESKD, (D) and AKI. We show ORs for the comparison of genetic high-risk versus low-risk individuals (GRS≥7.5 versus GRS≤5.5). Associations are adjusted for age, sex and baseline eGFRcrea for Rapid3 and CKDi25 and adjusted for matching variables age-group and sex as well as quantitative age for ESKD and AKI.
| High versus low risk group: 8–14 adverse alleles versus 0–5 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| High risk group | Low risk group | |||||||||
| Study | Number of Cases | Number of Controls | OR | L95 | U95 | P | Number of Cases | Number of Controls | Number of Cases | Number of Controls |
| A) Rapid3 | ||||||||||
| UK Biobank | 2,416 | 5,828 | 1.05 | 0.92 | 1.20 | 0.49 | 488 | 1,205 | 721 | 1,840 |
| DIACORE | 705 | 532 | 0.95 | 0.70 | 1.31 | 0.77 | 169 | 136 | 189 | 147 |
| KORA-F3 | 321 | 851 | 1.85 | 1.26 | 2.72 | 0.00 | 85 | 184 | 69 | 250 |
| KORA-F4 | 241 | 1,368 | 1.34 | 0.88 | 2.03 | 0.17 | 52 | 314 | 61 | 388 |
| Meta-analysis | 3,683 | 8,579 | 1.11 | 0.99 | 1.24 | 0.07 | 794 | 1,839 | 1,040 | 2,625 |
| B) CKDi25 | ||||||||||
| UK Biobank | 518 | 14,518 | 1.19 | 0.92 | 1.53 | 0.18 | 113 | 2,972 | 142 | 4,514 |
| DIACORE | 124 | 1,584 | 1.22 | 0.72 | 2.05 | 0.46 | 34 | 359 | 32 | 449 |
| KORA-F3 | 168 | 2,651 | 1.68 | 1.03 | 2.74 | 0.04 | 49 | 592 | 32 | 735 |
| KORA-F4 | 85 | 2,719 | 1.50 < | 0.79 | 2.83 | 0.21 | 25 | 598 | 21 | 773 |
| Meta-analysis | 895 | 21,472 | 1.29 | 1.06 | 1.57 | 0.01 | 221 | 4,521 | 227 | 6,471 |
| C) ESKD (cases: ICD10 code N18.0 or N18.5; controls: no ICD10 code N18, eGFRcrea>60 mL/min/1,73m2, frequency-matched by age-group and sex) | ||||||||||
| 4D_KORA-F3 | 1,100 | 1,601 | 0.91 | 0.73 | 1.14 | 0.43 | 227 | 363 | 298 | 438 |
| GENDIAN_KORA-F4 | 470 | 1,545 | 1.11 | 0.82 | 1.50 | 0.50 | 103 | 345 | 124 | 455 |
| UKBBCaCo | 528 | 1,584 | 1.09 | 0.82 | 1.45 | 0.56 | 108 | 329 | 153 | 504 |
| Meta-analysis | 2,098 | 4,730 | 1.01 | 0.87 | 1.18 | 0.91 | 438 | 1,037 | 575 | 1,397 |
| D) AKI (cases: ICD 10 code N17; controls: no ICD10 code N17, frequency-matched by age-group and sex) | ||||||||||
| UKBBCaCo | 4,123 | 12,369 | 1.20 | 1.08 | 1.33 | 4.45×10−4 | 889 | 2,398 | 1,243 | 3,956 |
Study=Study name, OR=Odds Ratio, L95/ U95=Lower and Upper 95% confidence intervals, ESKD=End-stage Kidney Disease, UKBBCaCo=cases and controls from UK Biobank, AKI=Acute Kidney Injury.
Since rapid eGFRcrea decline is known to be associated with high ESKD risk, we were interested to see whether the genetic risk carried forward also to the severe renal endpoint further down the road. We gathered data on individuals with ESKD from three different sources (ICD-10 codes N18.5 and N18.6; UK Biobank, GENDIAN25 and 4D26, together 2,098 cases) and compared them to “healthy” individuals frequency-matched by age-groups and sex per case-source (eGFRcrea>60 mL/min/1.73 m2, no health record for chronic kidney impairment; UK Biobank, KORA-F3, KORA-F4, together 4,730 controls). When comparing the same GRS high-risk versus low-risk group as defined above, we found no association with ESKD risk (OR=1.01, 95% CI=0.87–1.18, P-value=0.91; Table 5).
When comparing the same GRS high-risk versus low-risk group for AKI risk in UK Biobank (ICD-10 code N17.0–17.9, 4,123 cases; 12,369 controls frequency matched on age-group and sex, eGFRcrea>60 mL/min/1.73m2, no record of AKI), we found a 1.20-fold statistically significant increased risk (95%CI: 1.08–1.33, P-value=4.45×10−4; Table 5). Thus, the derived GRS across the seven identified variants was associated with increased risk of AKI, but not ESKD.
DISCUSSION
Overall, we identified seven independent genetic variants across six loci that were significantly associated with two binary traits of rapid eGFRcrea decline, Rapid3 and/or CKDi25. In this GWAS meta-analysis of >40 studies with follow-up time of up to 15 years, we provide – to our knowledge - the first record of genome-wide significant variants for these traits. While there are several genetic studies for cross-sectional eGFRcrea (e.g.17,27, summarized in a review28) and some on annual eGFRcrea decline18,29,30, we adopted this extreme phenotype approach and focused on two binary traits for rapid eGFRcrea decline reported for increased ESKD risk7. Our work is unique in its large sample size for these two case-control definitions with ~35,000 Rapid3 cases and ~20,000 CKDi25 cases versus >100,000 controls. These trait definitions were based on precisely two creatinine measurements over time, which does not allow for a characterization of the slope, but for differentiating persons with rapid decline yes/no. Besides the fact that these traits require longitudinal data with all known challenges to maintain sample size, another challenge are the stringent case-control definitions as they exclude individuals with moderate decline or baseline eGFRcrea<60 mL/min/1.73m2 (neither a case, nor a control). To derive these case-control sample sizes, we had >270,000 individuals with at least two assessments of kidney function from population-based studies, exceeding previous work18 by >4- fold. Despite the relatively large sample size, we cannot exclude that the lack of association of an identified variant for one trait or the other as well as differences in effect sizes between traits might result from chance. We expect that the analysis of even larger samples in the future might increase the overlap of findings between the two traits and allow for a more formal comparison of effect sizes.
It might be considered a limitation that these binary traits were only similar, but not identical to KDIGO-recommended surrogate endpoints for ESKD. However, those endpoints would have limited the GWAS sample size even more. Our sample size is still much smaller than GWAS sample sizes for cross-sectional eGFRcrea, which might explain the relatively few identified loci for rapid decline, even with the candidate approach allowing for a less stringent threshold of significance, compared to the vast number of loci identified for cross-sectional eGFRcrea17. For example, our sample size for Rapid3 enabled a power of >80% to detect a variant with MAF=30% (2%) with 1.13-fold (1.28-fold) increased Rapid3 risk with genome-wide significance. There might be genetic variants with smaller MAF or smaller risk that have been missed. The sample size in Non-European ancestry individuals was too small for separate evaluation. There are current efforts to substantially enhance longitudinal studies and their molecular content31–33, also with Non-European ancestry, which will foster more GWAS on clinical endpoints in the future. Among the six identified loci for Rapid3 and/or CKDi25, four were identified with genome-wide significance (near UMOD-PDILT (2 signals), PRKAG2, WDR72 and OR2S2) and two among previously reported loci for cross-sectional eGFRcrea17 (LARP4B and GATM). Our in-silico follow-up highlighted the relevance of genome-wide significant associations for fine-mapping: credible sets identified via candidate-based approach contained >1000 variants, rendering the GPS unfeasible. For the four loci with genome-wide significance, the credible sets contained 1–40 variants, providing a more practical number of targets to turn the statistical signals into potentially relevant biological findings. For the four loci with genome-wide significance, our GPS helps prioritize genes for functional follow-up and provides the opportunity to customize the weighing of each piece of bioinformatic evidence. While some of the findings overlap with previous reports17 including functionally interesting variants mapping to the PRKAG2 and GALNTL5 gene both residing in the PRKAG2 locus, the WDR72 gene is supported with a missense variant that was not among credible set variants for cross-sectional eGFRcrea. Our data also highlights the two independent variants in the UMOD-PDILT locus known for large effects on eGFRcrea17 as the two strongest genetic risk factors for rapid eGFRcrea decline with each of the four adverse alleles increasing CKDi25 risk by 1.06-fold. One variant captures the signal in UMOD with unclear function and the other is the PDILT-residing variant rs77924615. The rs77924615 was reported as likely causal, modulating UMOD expression and urinary uromodulin concentrations17. The fact that this variant is the sole variant in the credible set for CKDi25 and for cross-sectional eGFRcrea17 provides a proof-of-concept that overlapping single-variant credible sets between cross-sectional and longitudinal traits may be indicative of the causal variant.
Particularly interesting is the OR2S2 locus, which was not identified by the previous GWAS of cross-sectional eGFRcrea17 and showed no association with cross-sectional eGFRcys or BUN here. In this locus, the genes OR2S1P, GNE, and CD72 were supported by our GPS: CD72 and GNE with evidence of abnormal morphology of podocytes or renal glomerulus, respectively, and by a link of CD72 molecules to systemic lupus erythematosus patients with renal involvement 34 or GNE mutation in mice as model for human glomerulopathy35. There is little published evidence on OR2S1P, but we find OR2S1P as target of an eQTL-variant that is a credible set variant and thus a likely variant to drive the association signal. We provide no independent replication for this locus association due to the lack of available comparable data for the low-frequency (MAF~2%) driver variants, but our sensitivity analyses supported the signal as genuine.
The genuineness of the OR2S2 locus for rapid kidney function decline was supported by consistent associationwith annual change in eGFRcys and BUN. These alternative biomarker results also supported five of the seven identified variants to be associated with kidney function (UMOD-PDILT (2 variants), WDR72, PRKAG2, OR2S2), but not the loci near GATM and LARP4B.
A challenge in clinical practice is the identification of individuals at increased risk of ESKD and little evidence on genetic factors for ESKD. Some GWAS including 500 to 4,000 ESKD cases reported genome-wide significant loci, but none of these overlap with the loci identified here29,36–44. Two genetic variants were identified in ~4,000 ESKD cases and equal number of controls36 testing 16 variants known for cross-sectional eGFRcrea. One variant, rs12918807, is highly correlated with our UMOD-PDILT lead variant rs12922822 (R2=1.00), but the other variant rs1260326, near GCKR, was not associated with rapid eGFRcrea decline (OR=1.01 and 1.00, P-value=0.396 and 0.757). Previous GWAS on ESKD may have been hampered by sample size: to detect a variant with MAF 30% (10%) and 1.1-fold increased disease risk at genome-wide significance with 80% power, the required sample size sizes is 13,500 (31,000) cases and similar number of controls; to detect such a variant with nominal significance, 2,700 (6,100) cases are needed. Therefore, ESKD case-control data with thousands of cases might work for candidate-based approaches, but will be underpowered for GWAS. While the genetic variants identified for rapid kidney function decline might be effective candidates, but we did not find increased ESKD risk comparing the high- versus low genetic profile in > 2100 ESKD patients and health controls. This could be due to insufficient power or survival bias on the adverse alleles45, but the data would also be in line with a lack of effect.
We did find a 1.20-fold increased risk for AKI comparing the genetic high-risk versus low-risk group in UK Biobank including 4000 individuals recorded for AKI. While AKI is defined as an acute event, AKI and particularly repeated episodes of AKI are known to deteriorate patients’ kidney function also chronically, at least for a subgroup46. Due to the nature of population-based studies in contrast to hospital-based studies, it is conceivable that some of the individuals in the GWAS studies had AKI between baseline and follow-up and that those with chronically rather than transiently reduced kidney function could have become cases for rapid decline. We assume it unlikely that persons in the acute phase of AKI come to the study center for a follow-up visit. While not each patient with AKI-episode will experience long-term and rapid deterioration of kidney function, individuals in the genetic high-risk group might include individuals at a higher risk of sustained deterioration of kidney function after AKI. Therefore, the genetic variants identified for rapid kidney function decline might capture mechanisms and individuals at increased risk for sustained kidney function deterioration after AKI.
METHODS
Overall 42 studies contributed GWAS results estimated via logistic regression on Rapid3 and CKDi25 with 1000 Genomes phase 3 v5 ALL47 or Haplotype Reference Consortium v.1.148 reference variants. After an inverse-variance weighted meta-analysis, genome-wide significantly associated loci including primary and secondary lead variants were identified. In addition, we identified loci among known loci for cross-sectional eGFRcrea17. We validated identified effects by alternative cross-sectional and longitudinal renal markers eGFRcys and BUN. We derived credible sets of variants for each identified signal and conducted a comprehensive in-silico follow-up for all genes underneath identified loci. Finally we estimated the cumulative genetic effect of the identified lead variants on rapid kidney function decline, ESKD, and AKI. A detailed description of the methods can be found in the Supplementary Material (Supplementary Methods).
Supplementary Material
Acknowledgements
We thank Daniele Di Domizio (Eurac Research) and Randy Rückner (University of Regensburg) for IT assistance. The University of Regensburg provided computing resources for the meta-analysis. We conducted this research using the UK Biobank resource under the application number 20272. General and study-specific acknowledgements and funding sources are provided in the Supplementary Online Material.
COMPETING INTERESTS
Dr. Biggs reports grants from National Heart Lung Blood Institute, during the conduct of the study. Dr. Boerwinkle reports grants from NIH, during the conduct of the study. Dr. Carroll reports grants from US National Institutes of Health, during the conduct of the study. Dr. Eckardt reports grants from Astra Zeneca, grants from Bayer, from FMC, grants from Vifor, during the conduct of the study; personal fees from Akebia, personal fees from Bayer, personal fees from Boehringer Ingelheim, personal fees from Vifor, grants from Amgen, outside the submitted work. Dr. Ho reports other from Sanofi Genzyme, other from Partners Healthcare, outside the submitted work. Dr. Kleber reports personal fees from Bayer, outside the submitted work. Dr. Koenig reports personal fees from AstraZeneca, personal fees from Novartis, personal fees from Pfizer, personal fees from The Medicines Company, personal fees from DalCor, personal fees from Kowa, personal fees from Amgen, personal fees from Corvidia, personal fees from Daiichi-Sankyo, personal fees from Berlin-Chemie, personal fees from Sanofi, personal fees from Bristol-Myers Squibb, grants and non-financial support from Singulex, grants and non-financial support from Abbott, grants and non-financial support from Roche Diagnostics, grants and non-financial support from Beckmann, outside the submitted work. Dr. Lukas reports to be employed by and stockholder of GlaxoSmithKline, outside the submitted work. Dr. Mychaleckyj reports grants from National Institutes of Health, during the conduct of the study. Dr. Nadkarni reports grants, personal fees and non-financial support from Renalytix AI, personal fees and non-financial support from Pensieve Health, personal fees from Reata, personal fees from AstraZeneca, personal fees from BioVie, personal fees from GLG Consulting, outside the submitted work. Dr. O’Donoghue reports grants from GlaxoSmithKline, during the conduct of the study; grants from Intarcia, grants and personal fees from Novartis, grants and personal fees from Amgen, grants from AstraZeneca, outside the submitted work. Dr. Psaty reports grants from the NIH during the conduct of the study; and Psaty serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. Dr. Rossing reports grants and other from Astra Zeneca, other from Astellas, other from Bayer, other from Boehringer Ingelheim, other from Gilead, other from Merck, grants and other from Novo Nordisk, other from Sanofi, other from Eli Lilly, other from Mundipharma, other from Vifor, outside the submitted work; all to Steno Diabetes Center Copenhagen. Dr. Rotter reports grants from NIH, during the conduct of the study. Dr. Scholz reports grants from Pfizer Inc., outside the submitted work. Dr. Verweij reports other from Regeneron Genetics Center, other from Genomics plc, outside the submitted work. Dr. Wallentin reports grants from AstraZeneca, grants from Boehringer Ingelheim, grants from Bristol-Myers Squibb/Pfizer, grants from GlaxoSmithKline, grants from Merck & Co., grants from Roche Diagnostics, other from Abbott, outside the submitted work. Dr. Wanner reports grants from Boehringer-Ingelheim, personal fees from Boehringer-Ingeheim, grants from Idorsia, grants from Sanofi-Genzyme, personal fees from Eli-Lilly, personal fees from Sanofi-Genzyme, personal fees from Akebia, personal fees from Mundipharma, personal fees from MSD, personal fees from AstraZeneca, personal fees from Bayer, from null, outside the submitted work. Dr. Waterworth reports at the time of contributing to this manuscript, I was a full time employee of GlaxoSmithKline. Dr. White reports grants and personal fees from Eli Lilly and Company, personal fees and non-financial support from AstraZeneca, grants and personal fees from Omthera Pharmaceuticals, grants and personal fees from Eisai Inc., grants and personal fees from DalCor Pharma UK Inc., grants and personal fees from CSL Behring LLC, grants and personal fees from American Regent, grants, personal fees and non-financial support from Sanofi-Aventis Australia Pty Ltd, grants, personal fees and non-financial support from Esperion Therapeutics Inc., personal fees from Genentech, Inc., grants, personal fees and non-financial support from Sanofi-Aventis, outside the submitted work. Dr. Woodward reports personal fees from Amgen, personal fees from Kirin, outside the submitted work. Dr. Yerges-Armstrong reports personal fees from GlaxoSmithKline, outside the submitted work. Dr. Chu reports being employed by Merck & Co during the conduct of the study and to be employed by GlaxoSmithKline, outside the submitted work. Dr. Wong reports grants from Allergan Bayer Boehringer-Ingelheim Genentech Merck Novartis Oxurion (formerly ThromboGenics) Roche Samsung Bioepis, personal fees from Allergan Bayer Boehringer-Ingelheim Genentech Merck Novartis Oxurion (formerly ThromboGenics) Roche Samsung Bioepis, personal fees from Allergan Bayer Boehringer-Ingelheim Genentech Merck Novartis Oxurion (formerly ThromboGenics) Roche Samsung Bioepis, personal fees from Allergan Bayer Boehringer-Ingelheim Genentech Merck Novartis Oxurion (formerly ThromboGenics) Roche Samsung Bioepis, grants from NMRC, grants from Novartis Singapore, during the conduct of the study; personal fees from Allergan Bayer Boehringer-Ingelheim Genentech Merck Novartis Oxurion (formerly ThromboGenics) Roche Samsung Bioepis, other from - Plano - EyRiS, outside the submitted work. Dr. Coresh reports grants from National Institute of Health, grants from National Kidney Foundation, during the conduct of the study; grants from National Institute of Health, grants from National Kidney Foundation, outside the submitted work. Dr. Nauck reports grants from Federal Ministry of Education and Research Germany, the Ministry of Cultural Affairs, the Social Ministry of the Federal State of Mecklenburg-West Pomerania, personal fees from Becton Dickinson (BD), grants from Federal Ministry of Education and Research, Germany, European Union Interreg IVa, personal fees from German Medical Association, German Centre for Cardiovascular Research (GCCR), National Cohort, outside the submitted work. Dr. Sieber reports being a full-time employee of GlaxoSmithKline plc. All other authors declare no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015. doi: 10.1038/ng.3314 [DOI] [PubMed] [Google Scholar]
- 2.Matsushita K, Selvin E, Bash LD, Franceschini N, Astor BC, Coresh J. Change in estimated GFR associates with coronary heart disease and mortality. J Am Soc Nephrol. 2009. doi: 10.1681/ASN.2009010025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coresh J, Turin TC, Matsushita K, et al. Decline in estimated glomerular filtration rate and subsequent risk of end-stage renal disease and mortality. JAMA - J Am Med Assoc. 2014. doi: 10.1001/jama.2014.6634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010. doi: 10.1016/S0140-6736(10)60674-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Astor BC, Matsushita K, Gansevoort RT, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int. 2011. doi: 10.1038/ki.2010.550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004. doi: 10.1056/NEJMoa041031 [DOI] [PubMed] [Google Scholar]
- 7.Turin TC, Coresh J, Tonelli M, et al. Short-term change in kidney function and risk of end-stage renal disease. Nephrol Dial Transplant. 2012. doi: 10.1093/ndt/gfs263 [DOI] [PubMed] [Google Scholar]
- 8.Andrassy KM. Comments on “KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease.” Kidney Int. 2013. doi: 10.1038/ki.2013.243 [DOI] [PubMed] [Google Scholar]
- 9.Levey AS, Inker LA, Matsushita K, et al. GFR decline as an end point for clinical trials in CKD: A scientific workshop sponsored by the national kidney foundation and the US food and drug administration. Am J Kidney Dis. 2014. doi: 10.1053/j.ajkd.2014.07.030 [DOI] [PubMed] [Google Scholar]
- 10.Levey AS, Gansevoort RT, Coresh J, et al. Change in Albuminuria and GFR as End Points for Clinical Trials in Early Stages of CKD: A Scientific Workshop Sponsored by the National Kidney Foundation in Collaboration With the US Food and Drug Administration and European Medicines Agency. In: American Journal of Kidney Diseases.; 2020. doi: 10.1053/j.ajkd.2019.06.009 [DOI] [PubMed] [Google Scholar]
- 11.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009. doi: 10.7326/0003-4819-150-9-200905050-00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cattran DC, Feehally J, Cook HT, et al. Kidney disease: Improving global outcomes (KDIGO) glomerulonephritis work group. KDIGO clinical practice guideline for glomerulonephritis. Kidney Int Suppl. 2012. doi: 10.1038/kisup.2012.9 [DOI] [Google Scholar]
- 13.Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012. doi: 10.1056/NEJMoa1205511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019. doi: 10.1016/S0140-6736(18)32590-X [DOI] [PubMed] [Google Scholar]
- 15.Taylor KS, McLellan J, Verbakel JY, et al. Effects of antihypertensives, lipid-modifying drugs, glycaemic control drugs and sodium bicarbonate on the progression of stages 3 and 4 chronic kidney disease in adults: A systematic review and meta-analysis. BMJ Open. 2019. doi: 10.1136/bmjopen-2019-030596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King EA, Wade Davis J, Degner JF. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019. doi: 10.1371/journal.pgen.1008489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wuttke M, Li Y, Li M, et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet. 2019. doi: 10.1038/s41588-019-0407-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorski M, Tin A, Garnaas M, et al. Genome-wide association study of kidney function decline in individuals of European descent. Kidney Int. 2015. doi: 10.1038/ki.2014.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Auton A, Abecasis GR, Altshuler DM, et al. A global reference for human genetic variation. Nature. 2015. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016. doi: 10.1038/ng.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schutte JE, Longhurst JC, Gaffney FA, Bastian BC, Blomqvist CG. Total plasma creatinine: An accurate measure of total striated muscle mass. J Appl Physiol Respir Environ Exerc Physiol. 1981. doi: 10.1152/jappl.1981.51.3.762 [DOI] [PubMed] [Google Scholar]
- 22.Köttgen A Genome-wide association studies in nephrology research. Am J Kidney Dis. 2010. doi: 10.1053/j.ajkd.2010.05.018 [DOI] [PubMed] [Google Scholar]
- 23.Maller JB, McVean G, Byrnes J, et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet. 2012. doi: 10.1038/ng.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet. 2018. doi: 10.1038/s41576-018-0016-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Böger CA, Haak T, Götz AK, et al. Effect of ACE and AT-2 inibitors on mortality and progression to microalbuminuria in a nested case-control study of diabetic nephropathy in diabetes mellitus type 2: Results from the GENDIAN study. Int J Clin Pharmacol Ther. 2006. doi: 10.5414/cpp44364 [DOI] [PubMed] [Google Scholar]
- 26.Wanner C, Krane V, März W, et al. Randomized controlled trial on the efficacy and safety of atorvastatin in patients with type 2 diabetes on hemodialysis (4D study): Demographic and baseline characteristics. Kidney Blood Press Res. 2004. doi: 10.1159/000080241 [DOI] [PubMed] [Google Scholar]
- 27.Hellwege JN, Velez Edwards DR, Giri A, et al. Mapping eGFR loci to the renal transcriptome and phenome in the VA Million Veteran Program. Nat Commun. 2019. doi: 10.1038/s41467-019-11704-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Köttgen A, Pattaro C. The CKDGen Consortium: ten years of insights into the genetic basis of kidney function. Kidney Int. 2020. doi: 10.1016/j.kint.2019.10.027 [DOI] [PubMed] [Google Scholar]
- 29.Parsa A, Kanetsky PA, Xiao R, et al. Genome-wide association of CKD progression: The chronic renal insufficiency cohort study. J Am Soc Nephrol. 2017. doi: 10.1681/ASN.2015101152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaewput W, Thongprayoon C, Chewcharat A, et al. Rate of Kidney Function Decline and Factors Predicting Progression of Kidney Disease in Type 2 Diabetes Mellitus Patients with Reduced Kidney Function: A Nationwide Retrospective Cohort Study. Ther Apher Dial. 2020. doi: 10.1111/1744-9987.13480 [DOI] [PubMed] [Google Scholar]
- 31.The German National Cohort: aims, study design and organization. Eur J Epidemiol. 2014. doi: 10.1007/s10654-014-9890-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sudlow C, Gallacher J, Allen N, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLoS Med. 2015. doi: 10.1371/journal.pmed.1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leitsalu L, Haller T, Esko T, et al. Cohort profile: Estonian biobank of the Estonian genome center, university of Tartu. Int J Epidemiol. 2015. doi: 10.1093/ije/dyt268 [DOI] [PubMed] [Google Scholar]
- 34.Vadasz Z, Goldeberg Y, Halasz K, et al. Increased soluble CD72 in systemic lupus erythematosus is in association with disease activity and lupus nephritis. Clin Immunol. 2016. doi: 10.1016/j.clim.2016.02.004 [DOI] [PubMed] [Google Scholar]
- 35.Kakani S, Yardeni T, Poling J, et al. The Gne M712T mouse as a model for human glomerulopathy. Am J Pathol. 2012. doi: 10.1016/j.ajpath.2011.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Böger CA, Gorski M, Li M, et al. Association of eGFR-related loci identified by GWAS with incident CKD and ESRD. PLoS Genet. 2011. doi: 10.1371/journal.pgen.1002292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sambo F, Malovini A, Sandholm N, et al. Novel genetic susceptibility loci for diabetic end-stage renal disease identified through robust naive Bayes classification. Diabetologia. 2014. doi: 10.1007/s00125-014-3256-2 [DOI] [PubMed] [Google Scholar]
- 38.Iyengar SK, Sedor JR, Freedman BI, et al. Genome-Wide Association and Trans-ethnic Meta-Analysis for Advanced Diabetic Kidney Disease: Family Investigation of Nephropathy and Diabetes (FIND). PLoS Genet. 2015. doi: 10.1371/journal.pgen.1005352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer ND, Ng MCY, Hicks PJ, et al. Evaluation of candidate nephropathy susceptibility genes in a genome-wide association study of African American diabetic kidney disease. PLoS One. 2014. doi: 10.1371/journal.pone.0088273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salem RM, Todd JN, Sandholm N, et al. Genome-Wide association study of diabetic kidney disease highlights biology involved in glomerular basement membrane collagen. J Am Soc Nephrol. 2019. doi: 10.1681/ASN.2019030218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sandholm N, Van Zuydam N, Ahlqvist E, et al. The genetic landscape of renal complications in type 1 diabetes. J Am Soc Nephrol. 2017. doi: 10.1681/ASN.2016020231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guan M, Ma J, Keaton JM, et al. Association of kidney structure-related gene variants with type 2 diabetes-attributed end-stage kidney disease in African Americans. Hum Genet. 2016. doi: 10.1007/s00439-016-1714-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guan M, Keaton JM, Dimitrov L, et al. An Exome-wide Association Study for Type 2 Diabetes–Attributed End-Stage Kidney Disease in African Americans. Kidney Int Reports. 2018. doi: 10.1016/j.ekir.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandholm N, Salem RM, McKnight AJ, et al. New Susceptibility Loci Associated with Kidney Disease in Type 1 Diabetes. PLoS Genet. 2012. doi: 10.1371/journal.pgen.1002921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reichel H, Zee J, Tu C, et al. Chronic kidney disease progression and mortality risk profiles in Germany: results from the Chronic Kidney Disease Outcomes and Practice Patterns Study. Nephrol Dial Transplant. 2020. doi: 10.1093/ndt/gfz260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.See EJ, Jayasinghe K, Glassford N, et al. Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019. doi: 10.1016/j.kint.2018.08.036 [DOI] [PubMed] [Google Scholar]
- 47.The 1000 Genomes Project Consortium. An integrated map of genetic variation. Nature. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prof Jonathan Marchini Prof Goncalo Abecasis Prof Richard Durbin. Haplotype Reference Consortium. http://www.haplotype-reference-consortium.org/. [Google Scholar]
- 49.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gillies CE, Putler R, Menon R, et al. An eQTL Landscape of Kidney Tissue in Human Nephrotic Syndrome. Am J Hum Genet. 2018. doi: 10.1016/j.ajhg.2018.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aguet F, Brown AA, Castel SE, et al. Genetic effects on gene expression across human tissues. Nature. 2017. doi: 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bult CJ, Blake JA, Smith CL, et al. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019. doi: 10.1093/nar/gky1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amberger JS, Hamosh A. Searching online mendelian inheritance in man (OMIM): A knowledgebase of human genes and genetic phenotypes. Curr Protoc Bioinforma. 2017. doi: 10.1002/cpbi.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.