

Abstract
Nuclear-lipid droplets (nLD)—a dynamic cellular organelle that stores neutral lipids, within the nucleus of eukaryotic cells—consists of a hydrophobic triacylglycerol –cholesterol-ester core enriched in oleic acid (OA) surrounded by a monolayer of polar lipids, cholesterol, and proteins. nLD are probably involved in nuclear-lipid homeostasis serving as an endonuclear buffer that provides or incorporates lipids and proteins participating in signaling pathways, as transcription factors and enzymes of lipid metabolism and nuclear processes. In the present work, we analyzed the nLD proteome and hypothesized that nLD-monolayer proteins could be involved in processes similar as the ones occurring in the cLD including lipid metabolism and other cellular functions. We evaluated the rat-liver–nLD proteome under physiological and nonpathological conditions by GeLC-MS2. Since isolated nLD are highly diluted, a protein-concentrating isolation protocol was designed. Thirty-five proteins were identified within the functional categories: cytoskeleton and structural, transcription and translation, histones, protein-folding and posttranslational modification, cellular proliferation and/or cancer, lipid metabolism, and transport. Purified nLD contained an enzyme from the lipid-metabolism pathway, carboxylesterase 1d (Ces1d/Ces3). Nuclear Carboxylesterase localization was confirmed by Western blotting. By in-silico analyses rat Ces1d/Ces3 secondary and tertiary structure predicted would be equivalent to human CES1. These results—the first nLD proteome—demonstrate that a tandem-GeLC-MS2-analysis protocol facilitates studies like these on rat-liver nuclei. A diversity of cellular-protein function was identified indicating the direct or indirect nLD participation and involving Ces1d/Ces3 in the LD-population homeostasis.
Keywords: Nuclear-lipid droplets, Proteomic, Nuclear proteins, Lipid metabolism, Lipase, Carboxylesterase, Triacylglyceride, Cholesterol-ester
Nuclear-lipid droplets; Proteomic; Nuclear proteins; Lipid metabolism; Lipase, Carboxylesterase; Triacylglyceride; Cholesterol-ester
1. Introduction
Nuclear-lipid droplets (nLD) are a dynamic cellular organelle that stores neutral lipids, within the nucleus of eukaryotic cells, as previously reported [1]. nLD are composed of a hydrophobic core of triacylglycerol (TAG) and cholesteryl ester (CE) enriched in oleic acid (OA) and surrounded by a monolayer of polar lipids, cholesterol (C), and proteins [1]. nLD are likely to be involved in nuclear-lipid homeostasis by serving as an endonuclear buffering system that provides or incorporates lipids and proteins participating in signaling pathways, such as transcription factors and enzymes of lipid metabolism and nuclear processes.
In liver cells, LDs are organized mainly within two intracellular compartments—the cLD in the cytoplasm, constituting the principal population at approximately 90%, and the nLD in the nucleus, corresponding to the remaining 10% [2]—though extremely small LDs have also been described in the lumen of the endoplasmic reticulum (ER) under particular conditions [3]. Both of the two major LD populations are reversibly altered by OA through a coordinated metabolism that determines size changes in the LD populations resulting from the ongoing biosynthesis of TAG, CE, C, and polar lipids (PLs), as already reported [2].
LDs are the cellular organelle that dynamically stores and compartmentalizes neutral lipids in eukaryotic and in at least certain prokaryotic organisms such as the oleaginous bacteria [4]. In general, when an organism contains TAGs or neutral lipids, LDs should also be present owing to their thermodynamically stable supramolecular structure. In this regard, it has been proposed the possibility that LDs could have been the earliest evolved organelle [4]. cLD coordinate cellular lipid metabolism in bacteria by direct protein interactions with the bacterial chromosome [4] and in eukaryotic organisms by association with almost all the cytoplasmic organelles by means of specific contact sites [5].
Accordingly, considerable cLD-related literature is found which indicates that monolayers proteins are involved in the LD structural organization and in the many cellular processes involving lipid metabolism [6, 7, 8, 9, 10], though only scarce information is available on the nLD. For example, under control conditions the cLD-associated proteins perilipin1 (PLIN1) and acetyl-CoA acetyltransferase1 (ACAT1) could not be identified in rat-liver nuclei [1], and only few proteins associated with nLD under stimulated conditions and ER stress have been described [11, 12].
The aim of the present work was to analyze nLD proteome by one-dimensional gel electrophoresis in combination with nanoliquid-chromatography and sequential tandem mass spectrometry (i.e., GeLC-MS2) in order to determine the structural organization and cellular functions of nLD. Our hypothesis was that the proteins in the nLD monolayer could be involved in processes similar to those of cLD—i.e., lipid metabolism including other functions.
Since nLD proteins correspond to a very low proportion of cellular proteins (0.0004%), as already reported [1]; we designed and applied a specific protocol to enrich the nLD sample in proteins and eliminate different LD components that might interfere (i.e., interferers) with the proteomic technique. Through this approach, a total of 35 different proteins in nLD were identified by GeLC-MS2 and classified in categories based on their tentatively assigned cellular function as follows: cytoskeleton and structural (31%), transcription and translation (23%), histones (20%), protein folding and posttranslational modifications (8.5%), cellular proliferation and cancer (8.5%), lipid metabolism (6%), and transport (3%). An enzyme from the lipid-metabolism pathway, the carboxylesterase 1d (Ces1d/Ces3), was identified in purified nLD and the carboxylesterase localization further confirmed by Western Blotting.
2. Materials and methods
2.1. Materials
Mouse monoclonal antibody against Ces1d/Ces3 (CES3(B-8): sc-374160); rabbit polyclonal antibody against ß-tubulin; horseradish-peroxidase-conjugated (HRPc) goat anti(mouse IgG) antibody; and HRPc goat anti(rabbit IgG) antibody were purchased from Santa Cruz, Biotechnology, Inc. (Santa Cruz, CA, USA) and LAP2β from BD Biosciences (San Jose, CA, USA). Alexafluor-594-labelled chicken anti(mouse IgG) and the boron-dipyrromethene dye BODIPY 493/503 were obtained from Invitrogen (Buenos Aires, Argentina) and 4′,6-diamidino-2-phenylindole (DAPI) from Sigma-Aldrich (St. Louis, MO, USA). Oleic acid sodium salt (≥99% purity) and fatty acid-free bovine-serum albumin (BSA) were acquired from Sigma-Aldrich (St. Louis, MO, USA); Amicon® ultracentrifugation filters from Millipore-Merck, Darmstadt, Germany); cOmplete™ EDTA-free protease-inhibitor cocktail from Roche Diagnostics-Roche Applied Science, Indianapolis, IN, USA; 3-[N,N-Dimethyl (3-myristoylaminopropyl)ammonium]propanesulfonate (ASB-14) from Sigma-Aldrich, St. Louis, MO, USA; and mass-spectrometry-grade trypsin from Promega Corporation, WI, USA. All the other reagents used in the assays were of standard laboratory grade.
2.2. Ethics statement
We conducted the study in strict accordance with standard procedures and in agreement with the local guidelines for vertebrate-animal welfare as well as with the United-States-Public-Health-Service and/or European-Union policies on the humane treatment of experimental animals (National Research Council, National Academy Press, Washington DC, 2010 and/or European Union Directive for Animal Experiments 2010/63/EU). The project was approved by the Institutional Animal Care and Use Committee (IACUC) of the Facultad de Ciencias Médicas, Universidad Nacional de La Plata, Argentina, and Protocol Number: P1-01-2017.
2.3. Animals
Experiments were performed on 180- to 200-g 60- to 70-day-old male Wistar rats. The animals were housed in rooms with 12:12 h light–dark diurnal cycle (midnight being the midpoint of the dark period), and the experiments were performed in accordance with the Guide for Care and Use of Laboratory Animals (1996, National Academy Press). The rats were maintained on a commercial standard-pellet diet (ACAI mouse and rat chow, San Nicolás, Buenos Aires, Argentina) plus tap water ad libitum. The diet contained (by weight) 20% proteins and 4% total lipids with a fatty-acid (FA) composition of 15.6% 16:0; 1.1% 16:1; 6.3% 18:0; 26.9% 18:1 (n-9); 1.6% 18:1 (n-7); 42.7% 18:2 (n-6); 5.7% 18:3 (n-3); and trace amounts of 14:0, 20:4 (n-6), and 22:6 (n-3).
2.4. Preparation of rat-liver homogenates and isolation of nuclei and nLD
Highly purified nuclei were isolated from rat-liver homogenates by sucrose-gradient ultracentrifugation after the method of Blobel and Potter [13], as modified initially by Kasper [14], and then slightly further in the laboratory [1]. The procedure stated in brief: Rats were euthanized by decapitation at 8 a.m. to standardize the effect of circadian rhythms [15] and the pooled livers from 10–12 animals homogenized at a ratio of 1:2 (w/v) in 0.25 M sucrose in Buffer A (50 mM Tris-HCl, 2.5 mM KCl, 5 mM MgCl2; pH 7.5) [15]. All steps were carried out at 4 °C. Morphological and biochemical criteria were used to assess the purity of the isolated nuclei [1].
nLD were isolated from fresh rat-liver nuclei (not previously frozen) and corresponded to the upper band (the α band) recovered from the sucrose gradient as previously reported [1]. Therefore, nuclei were first isolated from fresh liver (10–12 rats), then nLD were isolated from the nuclear fraction and finally, the newly isolated nLD fraction were frozen at -70 °C and stored. The nLD-isolation procedure was repeated ten times, thus involving a total of between 100 and 120 rats, in order to reach the amount of protein required for proteomic analysis.
The concentration of all fractions was determined by the protein content as measured by the method of Bradford [16] for the nLD and Lowry [17] for the homogenate and isolated nuclei with crystalline BSA as a standard. All fractions were stored at –70 °C until used.
2.5. Preparation of nLD-protein extracts
nLD proteins were extracted and concentrated from the α band of the sucrose-density gradient as described (Figure 1). All steps were done at 4 °C. The nLD fraction (α band) was filtered by centrifugation through a membrane of 3-kDa molecular-weight cutoff (Millipore Amicon® Ultra 0.5 ml 3K Centrifugal Filters). Then it was centrifuged in like manner through either the same membrane to produce the Sample A of Protocol A (Figure 1) containing the total proteins in the nLD fraction or a membrane of 100-kDa molecular-weight cutoff (Millipore Amicon® Ultra 0.5 ml 100K) to obtain Sample B in Protocol B constituting the total nLD-associated proteins (Figure 1). The nLD Sample A was then washed by centrifuging with Buffer B (50 mM Tris-HCl pH 8.3 with the protease-inhibitor cocktail). In addition, one aliquot of nLD Sample B was washed with Buffer B, whereas the other was washed with 0.5 M NaCl in the same buffer (Sample C in Protocol C for the nLD salt-resistant proteins). Then Buffer C—lysis Buffer-1 containing 7 M urea, 2 M thiourea, 4% [w/v] dimethyl[3-propyl azaniumyl]propane-1-sulfonate (CHAPS), 2% [w/v] of the zwitterionic-detergent ASB-14, and 30 mM Tris-HCl pH 8.3—was added to every sample in order to continue with the lipid removal step. The samples were left at 4 °C overnight and then kept on ice for sonication in a Branson 450 Sonifier with a tip of 1/8″ outer diameter (Danbury, CT, USA), first at 20% amplitude with cycles (on/off) of 0.5/0.5 s for 60 s and then at 35% with cycles of 0.5/0.5 s for 30 s. The sonicated samples were concentrated by filtration through nitrocellulose membranes of 10-kDa molecular-weight cutoff (Millipore Amicon® Ultra 0.5 ml 10K Centrifugal Filters).
Figure 1.
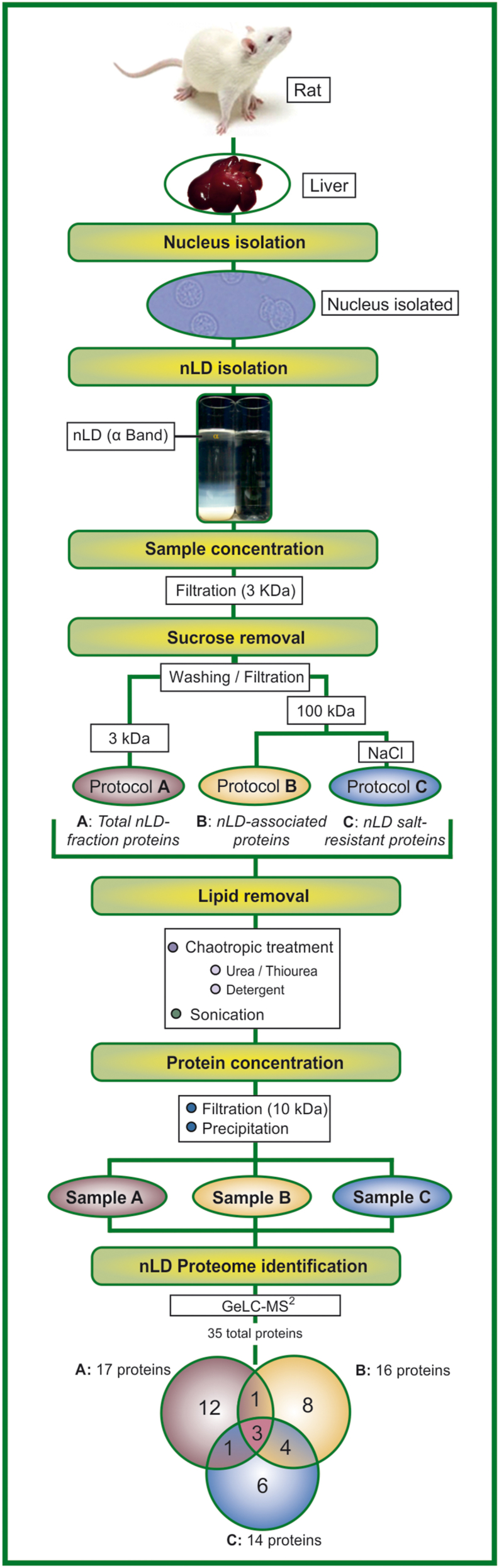
Scheme of nLD protein purification and identification. A schematic flowchart of nLD protein extraction, concentration, and identification is illustrated as explained in the Materials and Method section. nLD were isolated from rat-liver nuclei and corresponded to the sucrose-gradient upper band (α band). Three different subsets of nLD proteins were obtained: Sample A (A), the total proteins in the nLD fraction, comprising those from the nLD as well as the floating buoyant nuclear proteins; Sample B (B), the nLD-associated proteins; and Sample C (C), the nLD salt-resistant proteins. A total of 35 different proteins were identified among the three nLD samples; with 17, 16, and 14 total proteins comprising samples A, B, and C, respectively, as detailed in Supplementary Tables S1–S3. A total of 12, 8, and 6 unique proteins were identified in A (Only A), B (Only B), and C (Only C) samples, respectively; 1, 1, and 4 proteins were present in both samples A and B, A and C, and B and C, respectively; while 3 proteins were identified in all three nLD samples (A, B, and C) as detailed in Supplementary Table S4.
After protein precipitation from samples A, B, and C by means of a 2-D Clean-Up Kit (GE Healthcare Bio-Sciences Corp. NJ, USA); the pellet was suspended in Buffer D (lysis buffer-2 containing 8 M Urea, 2.5% [w/v] CHAPS, 2% [w/v] ASB-14, in Buffer E [30 mM Tris-HCl]; pH 8.3).
2.6. GeLC-MS2
2.6.1. Sodium-dodecyl-sulfate–polyacrylamide-gel electrophoresis (SDS-PAGE) and in-gel digestion
nLD-protein samples (A–C) were diluted for electrophoresis in Buffer F (0.35 M Tris-HCl, pH 6.8, 30% [w/v] glycerol, 10% [w/v] SDS, 0.012% bromphenol blue and 0.6 M dithiothreitol) and then incubated at room temperature for 30 min. The proteins were resolved by SDS-PAGE in 1-mm-thick 10% (w/v) uniform slab gels (Bio-Rad, USA) at 120 V in (25 mM Tris base, 192 mM glycine, 3.5 mM SDS; pH 8.3).
After electrophoresis, proteins were stained by Coomassie Colloidal stain (0.012% [w/v] Coomasie G-250, 10% [w/v] ammonium sulfate, 10% [w/v] phosphoric acid, 20% [v/v] methanol); [18]. Next, after separating entire gel lanes into slices, an in-gel–digestion protocol was carried out consisting of dithiothreitol reduction at room temperature (10 mM, 20 min) followed by iodoacetamide alkylation (25 mM, 20 min in the dark) and finally trypsin digestion (30 ng of trypsin in 25 mM ammonium bicarbonate) overnight at 37 °C. After proteolysis, the resulting peptides were extracted with 0.1% (v/v) formic acid in 25 mM ammonium bicarbonate and concentrated for vacuum evaporation. The peptide mixture was resuspended in 0.1% [v/v] formic acid.
2.6.2. Analysis by nanoliquid-chromatography–tandem-mass-spectrometry (nLC-MS2)
The tryptic peptides were separated by an initial nLC followed by tandem electrospray-MS analysis. The peptide separation was performed on an EASY-nLC–II system (Proxeon Biosystems, Denmark) connected to an amaZon-ETD ion-trap mass spectrometer (Bruker Daltonics, Bremen, Germany). The samples were loaded onto an EASY C18-A1 trap column, 100 μm × 20 mm, 5 μm (Thermo Fisher Scientific). The trap column was connected to an EASY C18-A2 analytical column, 75 μm × 100 mm, 3 μm (Thermo Fisher Scientific), equilibrated with 2% (v/v) aqueous acetonitrile with 0.1% (v/v) formic acid. The peptides were eluted at 250 nl/min, by means of a 100-min linear gradient of 3–35% acetonitrile. The mass spectrometer automatically switched between MS and the tandem MS2 second acquisition in positive mode. The full MS-survey spectra (m/z 100–3,000) were acquired in the amaZon-ETD ion trap in at an ultrascan-mode resolution. The ion-charge control target was set at 2 × 105 with a maximum ion-accumulation time of 170 ms. In the collision-induced dissociation–based procedures, the four most intense ions from each survey scan were subjected to fragmentation. The ions were obtained with an isolation window of 4 m/z units and provided in a dynamic-exclusion list for 60 s after being selected for a second sequential MS scan (MS2).
2.7. Electrophoresis and immunoblotting
Samples of liver homogenate and isolated whole nuclei were prepared for electrophoresis by first adding Laemmli sample Buffer H (125 mM Tris, 20% (v/v) glycerol, 4% (w/v) SDS, and 2% (v/v) 2-mercapthoethanol plus bromphenol blue; pH 6.8) and then boiling at 100 °C for 6 min. Samples (50–100 μg protein per well) were resolved by SDS-PAGE in 0.75-mm-thick 10% (w/v) uniform slab gels at 120 V for 100 min in Buffer G. The separated proteins were transferred to polyvinylidine-difluoride (PVDF) membranes (BioRad, Hercules, CA, USA) in the transfer Buffer I (25 mM Tris base, 192 mM glycine, 20% (v/v) methanol; pH 8.3). Nonspecific-binding sites on the membranes were blocked by an overnight incubation with 5% (v/v) nonfat milk in Buffer J (1.4 mM/l NaH2PO4, 8 mM Na2HPO4, 150 mM NaCl plus 0.1% [v/v] Tween-20; pH 7.4). The membrane strips were then incubated with the primary antibody diluted in 1% (v/v) nonfat milk in Buffer J (at 1:1,000 for anti-Ces1d/Ces3, 1:2000 for anti-LAP2β and 1:200 for anti-[ß-tubulin]) for 1 h at room temperature under gentle shaking. The immunoblots were developed through the use of secondary horse-radish-peroxidase–conjugated (HRPc) antibody. The membranes were washed 6 times for 5 min with Buffer J and then incubated for 1 h at room temperature under gentle shaking with the secondary HRPc-antibodies anti(mouse IgG) and anti(rabbit IgG) diluted 1:5,000 in Buffer J containing 1% (v/v) nonfat milk. Membranes were finally washed 6 times with Buffer J for 5 min each. The peroxidase activity was revealed by means of the Pierce enhanced-chemiluminescence (ECL) Western-blotting substrate (Pierce, Rockford, IL, USA) followed by chemiluminescence-image acquisition with an ImageQuant 350 digital-imaging system (GE Health Care, Buenos Aires, Argentina). Protein loading control was analyzed in the polyacrylamide gels and membranes after the transferring process by staining with Coomassie Blue and Ponceau S respectively [19].
2.8. MS-data analysis
Searches were performed by the Mascot search engine (Matrix Science, London, U.K.) with ProteinScape 3.0 software (Bruker Daltonics). Before the search, spectra were analyzed by means of the DataAnalysis software (Bruker Daltonics) with the default parameters set by the software developer. Carbamidomethyl (Cys) and oxidation (Met) were selected as variable modifications. Peptide-mass tolerances of 0.3 Da and 0.6 Da for the fragment masses were adopted as search parameters. Initially, it was considered each identified protein as an unambiguously assigned unique peptide that passed the identity score (Cutoff >35) in the Mascot searches; then a manual validation was performed on the MS2 spectra by fragment assignments on the b- and y-series ions.
2.9. In silico Ces1d/Ces3 analysis
The predicted model of the tertiary structure of rat Ces1d was obtained using the online server Phyre2 (Protein Homology/Analogy Recognition Engine) V 2.0 (http://www.sbg.bio.ic.ac.uk/phyre) [20]. Phyre2 uses different databases, such as Structural Classification of Proteins (SCOP) and Protein Data Bank (PDB). Through different sequential steps, such as profile construction, similarity analysis, and analysis of structural properties, Phyre2 selects the most appropriate template and, based on this, generates a protein model.
The sequences of rat Ces1d (ID: P16303) and human CES1 (ID: P23141) were extracted from the UniProt database and aligned using the ClustalW2 algorithm [21], with the following parameters: Gapopen 10.0; gapextend 0.5; endopen 10.0; endextend 0.5; Matrix: EBLOSUM62; Gap penalty: 10.0; Extend penalty: 0.5.
3. Results
3.1. nLD-protein samples
All biologic LDs contain a specific set of associated proteins that are considered to be located in the LD monolayer as integral or peripheral elements. Peripheral proteins, in particular, interact with the monolayer's hydrophobic or amphipathic domains that are composed of polar lipids, cholesterol, and/or proteins.
To determine the protein profile of the nLD, we employed proteomics. The nLD isolated from rat-liver nuclei became localized in a single, thin, whitish, practically translucent band (α band) recovered at the top of the initial sucrose gradient used. Since the nLD sample (α band) was very diluted in LDs [1], we designed a protocol specifically to enrich the nLD sample in proteins and eliminate different proteomics-interfering components (cf. the schematic flow chart Figure 1).
The proteins were obtained by three different extraction protocols in the initial concentration step. Our objective was to achieve the isolation of different subsets of the nLD proteins with restricted characteristics in order to reveal highly minor components as follows: Sample A comprised the total nLD-fraction proteins (A), consisting in the proteins collected at the top of the sucrose gradient in the α band—which fraction included the nLD proteins as well as the buoyant nuclear proteins. In contrast, samples B and C were freed from the nuclear buoyant proteins upon sequential filtering through Amicon® membranes of molecular-weight cutoff 100 kDa. Hence, samples B and C corresponded to the total nLD proteins (i. e., fraction B, the nLD-associated proteins) and nLD salt-resistant proteins (C), respectively.
One of the interfering components in the nLD sample (α band) was the sucrose carried over from the previous density gradients. This sucrose was removed, however, by the washing steps performed during the first sample concentration (Figure 1).
The lipids present in the nLD were also interfering components successfully removed by the treatments with chaotropic agents such as urea and thiourea and the nonionic detergents. Moreover, by a further sonication step, the size of the soluble lipid-detergent micelles formed was reduced, and finally removed from the protein sample by a filtration and a concentration step (Figure 1).
3.2. GeLC-MS2
The proteomics was performed by GeLC-MS2 to analyze all of the proteins present in each sample (i. e., A–C), with those first resolved by SDS-PAGE (Supplementary Figure S1). Those gels were sectioned, and each section was then digested with trypsin. The efficiency of the tryptic digestion of each of the samples was verified by matrix-assisted laser-desorption-ionization time-of-flight, i. e., MALDI-TOF MS (data not shown).
The peptides resulting from each digestion were resolved and registered by nLC–ion-trap-MS (nLC-IT), and the resulting data were analyzed by means of the MASCOT search tool in order to identify the greatest possible number of proteins present in each sample. The initial lists were then rigorously purged following the criteria set out in Materials and Methods. Once we obtained the lists of proteins, a comprehensive analysis was performed based on prior knowledge and the nature of the sample.
As a result of this study a total of 35 different proteins was identified in all the nLD samples (Figure 1). A list of 17 proteins in Sample A (A), 16 in Sample B (B), and 14 in Sample C (C) was identified as components of the nLD in each of the samples analyzed (Supplementary Tables S1–S3). It is possible that fraction A does not contain all of B and C proteins since those are very minor nLD components and/or were diminished during the given conditions. Moreover, is entirely possible that owing to the effect of signal suppression, the relatively abundant proteins in a sample prevent an identification of those less abundant.
3.3. Identification of nLD-associated proteins
The list of proteins identified in the three samples (A – C) corresponds to different protein subsets. It is interesting to point out that certain proteins were either found in only one sample or present in more than one (Supplementary Table S4).
We considered that the proteins only found in one type of sample corresponded to those exclusive to that fraction. In Only A, the 12 proteins would be buoyant nuclear species collected in the α band that would accordingly not correspond to the nLD monolayer proteins or be weakly associated with the nLD. In Only B, the 8 proteins would be peripheral species associated with the nLD through electrostatic forces that would be eliminated by salt washing. Finally, the 6 proteins exclusive to C would be nLD integral components that resisted salt washing.
The most common nLD proteins were those three found in all three samples (ABC: H33_RAT, K2C1_RAT, and ROA2_RAT). These proteins did not interact with the nLD by electrostatic forces, since those species were still present in samples obtained with or without the salt-washing step in the concentration protocol.
All the proteins identified in the three nLD samples (A - C) can be classified in the following functional categories: Lipid metabolism, Transcription and translation, Protein folding and posttranslational modification, Histones, Cytoskeletal and structural, Cellular proliferation and/or cancer, and Transport. On the basis of these tentatively assigned cellular functions, a total of 2, 8, 3, 7, 11, 3, and 1 protein were present in each of those respective categories (Figure 2).
Figure 2.

Distribution of the nLD proteins identified into functional categories. The 35 proteins identified in the nLD and listed in the figure were classified in the indicated categories based on their tentatively assigned cellular function as follows: cytoskeletal and structural, cellular proliferation and cancer, lipid metabolism, transcription and translation of proteins, histones, transport, and protein folding and posttranslational modification.
Only two enzymes involved in the Lipid metabolism category were identified in the nLD proteome. To the best of our knowledge, both enzymes have not been previously described in the nucleus: The ECHP_RAT (peroxisomal bifunctional enzyme) involved in lipid peroxidation and present in the Only-A fraction, would be a buoyant nuclear protein without an authentic nLD location, whereas CES1D_RAT, located in the Only-B fraction, participates in lipolysis and thus could be a peripheral nLD protein interacting by electrostatic forces with the LD monolayer (Supplementary Table S4). CES1D_RAT (name assigned by Mascot database nomenclature) would correspond to Ces3, so in the text it would be considered as Ces1d/Ces3 [3].
In the Cytoskeletal and structural category, several isoforms of keratin were identified distributed among all three samples, B, and C, in particular, 3 isoforms of K1(K1C10_RAT, K1C15_RAT, K1C19_RAT) and 7 isoforms of K2 (K22E_RAT, K2C1_RAT, K2C1B_RAT, K2C4_RAT, K2C5_RAT, K2C6A_RAT, K2C8_RAT) (Figure 2); but only the actin ACTB_RAT isoform was identified as a peripheral nLD protein, interacting with the LD monolayer by electrostatic forces (Supplementary Table S4, Only-B fraction). In particular, cytoskeletal proteins such as actin and myosin have already been described and characterized in the cell nucleus in normal tissues and under physiological conditions [22, 23], whereas keratins [24] and tubulin [25] have been identified in the nuclei of cancer and/or neoplastically transformed cells.
Nucleosomes contain an octameric core of the histones H2A, H2B, H3, and H4 that, along with the linker histone H1, comprises eukaryotic chromatin compacted into different domains. Histones were found in all three samples (A - C), and thus would not have interacted with the nLD monolayer simply by electrostatic forces (Supplementary Tables S1–S3). Several variants of core-nucleosome-histone subspecies of H2A and H2B were identified together with H33 (the canonical H3 variant) and H4 (the canonical core histone) (Figure 2); and although histones are nuclear proteins, these species have also been detected in the cLD of several cell types ranging from C. elegans, fly embryos, and moth cLD to the mammalian ß-cells of the islets of Langerhans and cells in mouse muscle and liver [26, 27, 28, 29, 30].
In particular, the histone H3.3 variant that was identified in all the nLD samples analyzed has been found to be enriched in the nucleosomes of active genes [31]. Considerable research is still required in order to clarify whether or not all histones (either canonical subspecies or specific variants) have an nLD location. In the present work, H1 was not identified by the nLD proteomics, but H1 had been previously detected in cLD [6].
The proteins involved in the Cellular proliferation and/or cancer category would be bona-fide nLD-associated species (i. e., not identified in Sample A). Namely, LAS2_RAT (Only B in Supplementary Table S4) would be a peripheral protein interacting by electrostatic forces with the nLD monolayer, whereas AKAP4_RAT and SNED1_RAT would be nLD integral proteins (Only C in Supplementary Table S4).
The importin beta subunit (IMB1_RAT; Supplementary Table S4) was identified in only Sample A and not in the purified nLD, probably because that protein weakly interacted with the nLD surface or belonged to the buoyant nuclear proteins. Moreover, Exportin 1 had been identified in the cLD proteome, and the amount of the protein was found to increase in TAG-enriched cLD [6]. Further research will be needed in order to prove whether or not LD trafficking truly occurs between the nucleus and the cytoplasm through the nuclear-pore complex.
The three proteins identified that are involved in Protein folding and posttranslational modification—GRP78, OGT1, and PDIA4—would either exert weak interactions with the nLD or belong to the buoyant proteins from the nucleus. These three are involved in processes such as protein folding, O-linked-N-acetylglucosamine (O-GlcNAc) glycosylation, and the rearrangement of disulfide bonds. O-GlcN acylation has already been documented in the nucleus, in mitochondria, and in the cytoplasm; but to the best of our knowledge not involving the cLD [32].
The eight proteins identified in nLD that were involved in transcription and posttranslation, those comprising heat-shock proteins, ribonucleoproteins, and transcription and splicing elements, would interact with the monolayer by electrostatic forces. nLD could store these proteins and/or interact with the specific chromatin domains where those processes take place. The heat-shock proteins, for their part, have also been identified in cLD [6].
3.4. Ces1d/Ces3
3.4.1. Carboxylesterase cellular topology
The approach undertaken in order to validate our hypothesis that Ces1d/Ces3 was a bona-fide nLD protein, since the enzyme identified in only Sample B, that is the peripheral proteins associated with the nLD through electrostatic forces, was to confirm a nuclear localization of the enzyme by Western blotting. This technique demonstrated Ces1d/Ces3 to be present in both the cell homogenates and the isolated nuclei from rat liver (Figure 3). The Ces1d/Ces3 immunoreactive band of the nuclear sample was much fainter than the corresponding band of the liver homogenate, these results would imply that Ces1d/Ces3 is present in a very small proportion in the nucleus, since all the samples were loaded with the same protein amount in each determination (Supplementary Figure S2). Moreover, as expected [1] LAP2β was enriched in the nuclear sample when compared with homogenate, and it can be considered as a nuclear loading control due to its nuclear localization. For the purpose of reference, ß-tubulin was used as a cellular-homogenate marker protein because of its cytoplasmic localization.
Figure 3.

Nuclear location of Ces1d/Ces3. The proteins from rat-liver homogenate and isolated nuclei were analyzed by Western blotting with antibodies against Ces1d/Ces3, LAP2β and ß-tubulin as described in Materials and Methods. Non-adjusted images and protein loading controls of samples are presented in Supplementary Figure S2.
3.4.2. In silico analysis of rat Ces1d/Ces3
Considering that up to now the available information on the rat Ces1d/Ces3 enzyme is scarce, and that its three-dimensional structure is not known since the protein has not been crystallized, the next objective was to perform a homology modeling of its tertiary structure. The human orthologue of rat Ces1d/Ces3 was used as the template, the enzyme CES1, which is a protein that is widely studied [3, 33].
In Figure 4 we can see that the tertiary structure predicted for rat Ces1d/Ces3 presents a great similarity with the tertiary structure of human CES1. On the other hand, in Supplementary Figure S3 we can see that the structure predicted for Ces1d/Ces3 correlates very well with the structure of the proposed protein that was used as a template (CES1 of human). 95% of the residues were modeled with more than 90% confidence (Figure 5). The amino acid sequence of rat Ces1d/Ces3 contains 565 residues, whereas human CES1 contains 567 residues. The amino acid sequences of both proteins share 80% identity. Previous studies in human CES1 have identified key residues that contribute to catalytic activity, subcellular localization, and enzyme structure. By modeling the tertiary structure and sequence alignment with human CES1 (Figure 5), we were able to predict such sites of interest in the rat Ces1d/Ces3 enzyme (Supplementary Figure S4).
Figure 4.

Modeling by homology of rat Ces1d/Ces3. Tertiary structure of human CES1 (right) and predicted model of the tertiary structure of rat Ces1d/Ces3 (left). The structure of human CES1 was determined by Bencharit et al [34]. The predicted tertiary structure for rat Ces1d/Ces3 was obtained using the Phyre2 web site. The rainbow color code describes the 3-D structures from the N-terminal (blue) to the C-terminal (red).
Figure 5.

Amino acid sequence alignments for rat Ces1d/Ces3 and human CES1. The EMBOSS Needle (Protein Alignment) server was used with the default parameters for the alignment. A sequence identity of 76.8% was obtained; with 2.4% of non-aligned regions (gaps). | shows identical residues between Ces1d/Ces3 and CES1;: shows residues with a high level of similarity; . shows residues with low level of similarity. Dashes indicate gaps. Red bold, the residues that make up the active site catalytic triad (Ser, Glu and His) are shown. Side door (green), Z-site (fuchsia) and Gate residues (olive green) are also highlighted in color. Violet highlight residues show potential N-glycosylation sites, and the yellow, the disulfide bond Cys residues. In red letters, N-terminal signal peptide and C-terminal residues are shown.
In human CES1 it has already been identified three main ligand binding sites: the catalytic triad (Ser221; Glu354; His468), the side door (Val424; Met425; Phe426), and the Z-site (Gly356), where substrates, fatty acids, and cholesterol analogs are bound, respectively; and the gate site (Phe551), which facilitates the release of products after catalysis [3, 34, 35, 36]. By modeling the tertiary structure and sequence alignment with human CES1 (Figure 5), we were able to predict such sites of interest in the rat Ces1d/Ces3 enzyme, Supplementary Figure S4. The catalytic triad is highly conserved among the family of carboxylesterases [3, 37, 38, 39, 40], and as shown in Supplementary Figure S4b in the predicted model of rat Ces1d/Ces3, the residues constituting the catalytic triad (Ser221; Glu353; His466) overlap almost perfectly with the corresponding residues belonging to human CES1. In the predicted structure of rat Ces1d/Ces3 the important domains: side door (Val422; Met423; Phe424) and Z-site (Gly355) (Figure 5) (Supplementary Figure S4c-d) overlap perfectly with that of the human CES1 protein that has been determined by crystallography [34, 35]. Meanwhile the gate residue corresponds to Phe551 in human CES1 [41] and according to Figure 5, in that position of the rat Ces1d/Ces3 sequence is the residue Arg549 (Supplementary Figure S4e). It is interesting to note that Arg is a basic and positively charged residue, while Phe is a non-polar residue. In human CES1, a single glycosylation site was detected (Asn79) [34,35], whereas in the rat Ces1d/Ces3 two potential sites (Asn79; Asn489) were identified [42] (Figure 5 and Supplementary Figure S4f). In rat Ces1d/Ces3, two disulfide bond forming residues were also detected [42] (Cys87/Cys-116 and Cys273/Cys284) (Figure 5 and Supplementary Figure S4g) and overlap with that observed in the crystallized human CES1 protein (Cys87/Cys116 and Cys274/Cys285) [35]. The N- and C-terminal sequences of both proteins are different. The N-terminal end possesses 18 residues [42, 43, 44], and the C-terminal has the retention sequence in the ER HIEL and HVEL, in CES1 from human and Ces1d/Ces3 from rat, respectively [37] (Figure 5 and Supplementary Figure S4h).
4. Discussion
The proteome of rat-liver nLD under physiological and nonpathological conditions was analyzed by GeLC-MS2. A specific protein-isolation protocol (Figure 1) was designed in order to effect a concentration of the proteins since nLD are isolated in a highly diluted sample that floats in a sucrose gradient (i. e., the α band). The protocol involved the isolation of the following three different subsets of nLD proteins, in order to be able to identify extremely minor cellular proteins with different physicochemical characteristics: the total proteins of the nLD fraction (Sample A) plus two additionally fractionated nLD samples, the nLD-associated proteins (Sample B) and the nLD salt-resistant proteins (Sample C). Under these experimental conditions, 35 proteins were identified in the three nLD samples (A–C) and classified into categories based on their tentatively assigned cellular functions as follows (Figure 2): cytoskeleton and structural (31%), transcription and translation (23%), histones (20%), protein folding and posttranslational modification (8.5%), cellular proliferation and/or cancer (8.5%), lipid metabolism (6%), and transport (3%).
Ces1d/Ces3 was the only protein of lipid metabolism that was identified by proteomics in the purified nLD protein sample and also confirmed by Western blotting. The enzyme presumably interacts with the LD monolayer by electrostatic forces (Supplementary Table S4). Under physiological conditions, Ces1d/Ces3 would be located mostly in the cytoplasm and to a lesser extent in the cell nucleus. In agreement with these results, as mentioned above, Ces1d/Ces3 has been described as having a cytoplasmic localization, in particular in association with the ER [3], the lumenal-ER LDs [3, 45], and cLD [43, 46, 47, 48] along with the analogous carboxylases Ces2a and Ces1c in the cLD [7].
Ces1d/Ces3 belongs to the large, highly conserved carboxylesterase multigene superfamily of the carboxyl-esterase hydrolases (EC 3.1.1.1) that can be found in virtually all species and with a great specificity [3]. These enzymes fall into the class of serine hydrolases that efficiently catalyze the hydrolysis of esters, thioesters, and amide bonds in a wide variety of molecules. Nevertheless, when CES1 amount/expression is genetically modify, the cellular derivations are complex and cannot be explained only by a lipolytic effect [33, 49, 50].
Carboxylesterase nomenclature has been standardized [3] and CES1D_RAT (name assigned by Mascot database nomenclature) would correspond to Ces3, so in the text it would be considered as Ces1d/Ces3. Ces1d/Ces3 has also been referred to under different aliases such as TGH (triacylglycerol hydrolase) and CEH (cholesteryl-ester hydrolase) [3]. Ces1d/Ces3 would be the rat functional ortholog to the human CES1, which protein has already been crystallized [3].
By in silico analyses, the three-dimensional structure predicted for rat Ces1d/Ces3 shows a high similarity with the 3-D structure of the human orthologous CES1, and we propose that the secondary and tertiary structures of rat Ces1d/Ces3 would be similar to that previously reported for human CES1. In this model of the tertiary structure of rat Ces1d/Ces3, potential key residues that would contribute to the catalytic activity, subcellular localization and structure of the protein were identified and localized. In particular, the spatial orientation of those residues that would participate in the catalytic activity (catalytic triad, side door and Z-site) of rat Ces1d/Ces3 correlates almost perfectly with those previously identified in human CES1.
Carboxylesterases play a critical role in cellular-lipid metabolism, energy homeostasis [51], and lipoprotein secretion as well as in drug and xenobiotic detoxification [3]. This enzyme family may also become involved in metabolic diseases (e. g., obesity, diabetes, and fatty liver), and Ces1d/Ces3 has been proposed as a possible preventive role in cancer development [48]. Therefore, we consider it highly relevant the development of a deeper understanding of the cellular role of the nuclear Ces1d/Ces3 both in the control physiological state and under deregulated or pathological conditions.
Carboxylesterase could be involved in nLD- and cLD-population homeostasis. In this regard, we have already demonstrated in liver cells that external OA induced an enhanced synthesis of TAGs, CEs, C, and polar lipids—which products were stored in the nLD and cLD causing both LD populations to increase markedly in size [2] and it would be important to determine the roll of carboxylesterase under these conditions. Moreover, previous studies in human macrophages demonstrated that under comparable conditions the carboxylesterase was found to translocate to the cLD in response to lipid overloads [52]. So, we can speculate that, a greater proportion of carboxylesterase close to the surfaces of LD, could facilitate the hydrolysis of TAG and CE stored in the droplets, in order to compensate for the synthesis of those lipids under the influence of high levels of external lipid load. Whereas, carboxylesterase also hydrolyzes MAGs to glycerol and FAs [53]. DAGs generated from TAG could be hydrolyzed by diacylglyceride lipase, an enzyme previously described in the cell nucleus that also is involved in phosphatidic-acid (PA) pathway to MAG [54, 55], and would participate in signaling mechanisms in the aging process [54].
The enzymatic activity of LD carboxylesterase could generate molecules in situ that in addition to being oxidized could constitute lipidic second messengers to regulate the nuclear- and/or cellular-lipid homeostasis. Within the nucleus, FAs released by carboxylesterase catalysis would be ligated to the nuclear liver fatty-acid–binding protein (L-FABP) [56], since that protein has been reported to play an active role in mobilizing FAs within the nuclear matrix [56]. These FAs could then bind to transcription factors such as the peroxisome proliferator-activated receptors (PPAR) [57], be activated to the corresponding CoA derivatives by the nuclear acyl-CoA synthetase (ACSL) [58] and then be reesterified to constitute nuclear-matrix lipids [56], and/or exit the nucleus into the cytoplasm (Figure 6).
Figure 6.

The involvement of nLD Ces1d/Ces3 in nuclear neutral-lipid catabolism.
In cultured Drosophila S2 cells loaded with lipids, the cLD lipase CG9186 was found to be elevated in comparison to the levels occurring under starvation conditions [59]. Moreover, the strength of interaction between proteins and the LD monolayer could determine their LD localization where weakly bound proteins would be displaced from the LD during lipolysis when the size of the LDs became reduced [59]. In this regard, we have proposed that Ces1d/Ces3 would be a peripheral LD protein interacting by electrostatic forces with the monolayer under control conditions but we do not know whether the carboxylesterase undergoes posttranslational modifications after external lipid load supplementation that could determine a translocation close to the LDs. For example, the LD-targeting mechanism of certain types of lipases and CTP: phosphocholine cytidylyltransferase α (CCT α) was reported to be mediated by phosphorylation [59, 60]. Moreover, in cLD, different mechanisms have been described and/or proposed to explain the targeting of proteins to the LD surface monolayer, among which models the palmitoylation of proteins would be included [9].
It would be of general interest the elucidation of the molecular mechanism involved in carboxylesterase LD interactions/translocation for its possible use as means of therapeutic targeting for the treatment of hepatic lipid-metabolism diseases in which neutral lipids such as TAG and/or CE become elevated along with a concomitant increase in the size of the cLD population. A LD localization of carboxylesterases could accordingly reduce the LD population size through the hydrolysis of those respective neutral lipids so as to restore the normal LD morphology.
The cLD marker proteins established previously were the classical PAT family proteins [61] —i. e., perilipin (PLIN 1), adipophilin (PLIN 2), TIP47: the tail-interacting protein of 47 kDa (PLIN 3), S3-12 (PLIN 4) and OXPAT, the oxidative tissue–enriched PAT protein (PLIN 5)—that are main cLD coat proteins, but none of those proteins were identified in nLD in the present experiments either by the proteomic protocols developed under physiological and nonpathological conditions or by Western blotting [1]. Moreover, the PLIN3 that had been previously identified by fluorescence microscopy in nLD from lipid-loaded Huh7 cells [11] was likewise not identified in rat liver nLD in the control physiological state. This difference could be due to the different cell type, experimental conditions and/or the possibility that the proteins could be at very low concentrations. Moreover, Huh7 cells have a different metabolic response to OA when compared with HepG2 cells [11, 12].
Under the influence of OA, CCTα —which enzyme participates in the synthesis of phosphatidyl choline— translocates to the surface of the cLD once the cells have been loaded with FAs and cLD population has increased in size as the result of an elevation in TAG synthesis [62]. It has also been demonstrated that CCTα is located in the nucleus and translocates between the inner nuclear membrane and the speckles [63]. By different types of ER stress conditions in Huh7 cell and in a high fat diet mouse model, it was observed that CCTα was located in nLD monolayer, under that conditions nLD would be dispersedly distributed within the nucleus and/or in association with the nucleoplamic reticulum [11, 12].
Considerable research has been reported on the proteomics of cLD in stimulated systems generally consisting in cultured cells exposed to lipid loading with mixtures of FAs (e. g., OA) or animals fed with high-fat diets in order to increase the cLD population and thus produce an elevated amount of LD proteins for the subsequent analyses. Moreover, many LD proteins have been reported to actively translocate to and/or from the LDs, and this possibility has to be considered when analyzing proteomics data. In this regard, proteomics has identified ACAT1 in cLD only in the livers of mice fed a high-fat diet and not in those livers under the control conditions [4]. In contrast, ACAT1 was not present in the nLD under control conditions in agreement with previous results since the enzyme could not be identified by Western blotting in isolated rat-liver nuclei [1].
On the basis of the cellular functions of the 35 proteins identified, we can propose the following characteristics and functions for the nLD in the cell, though further research is still needed:
-
•
nLD is a nuclear hydrophobic storage site for both lipids and proteins that would be delivered to specific locations within the nucleus, thus becoming available for different intranuclear processes. Actin would be involved in the nLD mobilization within the nucleus.
-
•
The hydrophobic nature of the nLD would also enable a three-dimensional space in the nucleus for the localization of nonspecific hydrophobic molecules (e. g., xenobiotics) in addition to the 2 dimensional nuclear-membrane surface.
-
•
nLD could be involved in epigenetic events (i. e., through the histones), in posttranslational modifications and/or the storage and release of specific transcription factors.
-
•
Carboxylesterase could play an important role in the cellular LD-population homeostasis.
5. Conclusions
In conclusion, the results reported here are the first describing the nLD proteome and accordingly demonstrate that the protocol developed that involves a tandem GeLC-MS2 analysis facilitates this type of study on the nuclei of rat liver. The approach revealed a wide diversity in cellular function of the proteins, what indicated that the nLD are directly or indirectly participating in those cellular processes; with the carboxylesterase Ces1d/Ces3 protein, for its part, being involved in cellular LD-population homeostasis.
Declarations
Author contribution statement
Lagrutta Lucía C, Trejo Sebastián A: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data.
Layerenza Juan P, Bronsoms Silvia: Performed the experiments.
Ves-Losada Ana: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Funding statement
This work was supported by CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas), PIP 2011 – 2013, N° 11220100100037 “Metabolismo lipídico en el núcleo celular”. UNLP (Universidad Nacional de La Plata): 11/M149 (2011-2013): “Metabolismo lipídico nuclear” (2011-2013), 11/M177 (2014-2017): “Bioquímica y Biología Celular de los Lípidos en Eukarya”, 11/M209: “Bioquímica y Biología Molecular de los Lípidos en Eukarya” (2018-2021). Boehringer Ingelheim Fonds Travel Grant: Proteomic analysis of nLD Setiembre de 2013 a Diciembre de 2013.
Data availability statement
Data included in article/supp. material/referenced in article.
Competing interest statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
Acknowledgements
The authors wish to acknowledge the valuable advice of Lic. Gerónimo Dubra in studies in silico, Lic. Romina V. Becerra for technical assistant and Dr Nicolás O Favale for his very valuable advice.
Contributor Information
Sebastián A. Trejo, Email: sebatrejo@gmail.com.
Ana Ves-Losada, Email: avlosada@biol.unlp.edu.ar.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Layerenza J.P., Gonzalez P., Garcia de Bravo M.M., Polo M.P., Sisti M.S., Ves-Losada A. Nuclear lipid droplets: a novel nuclear domain. Biochim. Biophys. Acta. 2013;1831(2):327–340. doi: 10.1016/j.bbalip.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Lagrutta L.C., Montero-Villegas S., Layerenza J.P., Sisti M.S., García de Bravo M.M., Ves-Losada A. Reversible nuclear-lipid-droplet morphology induced by oleic acid: a link to cellular-lipid metabolism. PloS One. 2017;12(1) doi: 10.1371/journal.pone.0170608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lian J., Nelson R., Lehner R. Carboxylesterases in lipid metabolism: from mouse to human. Protein Cell. 2018;9(2):178–195. doi: 10.1007/s13238-017-0437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang C., Yang L., Ding Y., Wang Y., Lan L., Ma Q. Bacterial lipid droplets bind to DNA via an intermediary protein that enhances survival under stress. Nat. Commun. 2017;8:15979. doi: 10.1038/ncomms15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuldiner M., Bohnert M. A different kind of love – lipid droplet contact sites. Biochim. Biophys. Acta (BBA) – Mol. Cell Biol. Lipids. 2017;1862(10, Part B):1188–1196. doi: 10.1016/j.bbalip.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Khor V.K., Ahrends R., Lin Y., Shen W.-J., Adams C.M., Roseman A.N. The proteome of cholesteryl-ester-enriched versus triacylglycerol-enriched lipid droplets. PloS One. 2014;9(8) doi: 10.1371/journal.pone.0105047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan S.A., Wollaston-Hayden E.E., Markowski T.W., Higgins L., Mashek D.G. Quantitative analysis of the murine lipid droplet-associated proteome during diet-induced hepatic steatosis. J. Lipid Res. 2015;56(12):2260–2272. doi: 10.1194/jlr.M056812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu M., Ge R., Liu W., Liu Q., Xia X., Lai M. Differential proteomics profiling identifies LDPs and biological functions in high-fat diet-induced fatty livers. J. Lipid Res. 2017;58(4):681–694. doi: 10.1194/jlr.M071407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bersuker K., Olzmann J.A. Establishing the lipid droplet proteome: mechanisms of lipid droplet protein targeting and degradation. Biochim. Biophys. Acta (BBA) – Mol. Cell Biol. Lipids. 2017;1862(10, Part B):1166–1177. doi: 10.1016/j.bbalip.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kramer D.A., Quiroga A.D., Lian J., Fahlman R.P., Lehner R. Fasting and refeeding induces changes in the mouse hepatic lipid droplet proteome. J. Proteomics. 2018;181:213–224. doi: 10.1016/j.jprot.2018.04.024. [DOI] [PubMed] [Google Scholar]
- 11.Ohsaki Y., Kawai T., Yoshikawa Y., Cheng J., Jokitalo E., Fujimoto T. PML isoform II plays a critical role in nuclear lipid droplet formation. J. Cell Biol. 2016;212(1):29–38. doi: 10.1083/jcb.201507122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sołtysik K., Ohsaki Y., Tatematsu T., Cheng J., Fujimoto T. Nuclear lipid droplets derive from a lipoprotein precursor and regulate phosphatidylcholine synthesis. Nat. Commun. 2019;10(1):473. doi: 10.1038/s41467-019-08411-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blobel G., Potter V.R. Nuclei from rat liver: isolation method that combines purity with high yield. Science. 1966;154(757):1662–1665. doi: 10.1126/science.154.3757.1662. [DOI] [PubMed] [Google Scholar]
- 14.Kasper C.B. Isolation and properties of the nuclear envelope. Methods Enzymol. 1974;31(Pt A):279–292. doi: 10.1016/0076-6879(74)31029-4. [DOI] [PubMed] [Google Scholar]
- 15.Actis Dato S.M., Catala A., Brenner R.R. Circadian rhythm of fatty acid desaturation in mouse liver. Lipids. 1973;8(1):1–6. doi: 10.1007/BF02533231. [DOI] [PubMed] [Google Scholar]
- 16.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72(1):248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 17.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 18.Neuhoff V., Arold N., Taube D., Ehrhardt W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988;9(6):255–262. doi: 10.1002/elps.1150090603. [DOI] [PubMed] [Google Scholar]
- 19.Goldman A., Harper S., Speicher D.W. Detection of proteins on blot membranes. Curr. Protoc. Protein. Sci. 2016;86 doi: 10.1002/cpps.15. 10.8.1-.8.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelley L.A., Sternberg M.J.E. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 21.Thompson J.D., Higgins D.G., Gibson T.J., Clustal W. Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiore A.P.Z.P., Spencer V.A., Mori H., Carvalho H.F., Bissell M.J., Bruni-Cardoso A. Laminin-111 and the level of nuclear actin regulate epithelial quiescence via exportin-6. Cell Rep. 2017;19(10):2102–2115. doi: 10.1016/j.celrep.2017.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Lanerolle P., Serebryannyy L. Nuclear actin and myosins: life without filaments. Nat. Cell Biol. 2011;13(11):1282–1288. doi: 10.1038/ncb2364. [DOI] [PubMed] [Google Scholar]
- 24.Hobbs R.P., Jacob J.T., Coulombe P.A. Keratins are going nuclear. Dev. Cell. 2016;38(3):227–233. doi: 10.1016/j.devcel.2016.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akoumianaki T., Kardassis D., Polioudaki H., Georgatos S.D., Theodoropoulos P.A. Nucleocytoplasmic shuttling of soluble tubulin in mammalian cells. J. Cell Sci. 2009;122(8):1111. doi: 10.1242/jcs.043034. [DOI] [PubMed] [Google Scholar]
- 26.Welte M.A., Gould A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta (BBA) - Mol. Cell Biol. Lipids. 2017;1862(10, Part B):1260–1272. doi: 10.1016/j.bbalip.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H., Zhou Z., Zhang H., Chen M., Li J., Ma Y. Shotgun proteomic analysis of the fat body during metamorphosis of domesticated silkworm (Bombyx mori) Amino Acids. 2010;38(5):1333–1342. doi: 10.1007/s00726-009-0342-8. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H., Wang Y., Li J., Yu J., Pu J., Li L. Proteome of skeletal muscle lipid droplet reveals association with mitochondria and apolipoprotein A-I. J. Proteome Res. 2011;10(10):4757–4768. doi: 10.1021/pr200553c. [DOI] [PubMed] [Google Scholar]
- 29.Zhang P., Na H., Liu Z., Zhang S., Xue P., Chen Y. Proteomic study and marker protein identification of Caenorhabditis elegans lipid droplets. Mol. Cell. Proteomics. 2012;11(8):317–328. doi: 10.1074/mcp.M111.016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsson S., Resjö S., Gomez M.F., James P., Holm C. Characterization of the lipid droplet proteome of a clonal insulin-producing β-cell line (INS-1 832/13) J. Proteome Res. 2012;11(2):1264–1273. doi: 10.1021/pr200957p. [DOI] [PubMed] [Google Scholar]
- 31.Kato S., Ishii T., Kouzmenko A. Point mutations in an epigenetic factor lead to multiple types of bone tumors: role of H3.3 histone variant in bone development and disease. BoneKEy Rep. 2015;4:715. doi: 10.1038/bonekey.2015.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X., Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017;18(7):452–465. doi: 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lian J., Bahitham W., Panigrahi R., Nelson R., Li L., Watts R. Genetic variation in human carboxylesterase CES1 confers resistance to hepatic steatosis. Biochim. Biophys. Acta (BBA) – Mol. Cell Biol. Lipids. 2018;1863(7):688–699. doi: 10.1016/j.bbalip.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 34.Bencharit S., Edwards C.C., Morton C.L., Howard-Williams E.L., Kuhn P., Potter P.M. Multisite promiscuity in the processing of endogenous substrates by human carboxylesterase 1. J. Mol. Biol. 2006;363(1):201–214. doi: 10.1016/j.jmb.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bencharit S., Morton C.L., Xue Y., Potter P.M., Redinbo M.R. Structural basis of heroin and cocaine metabolism by a promiscuous human drug-processing enzyme. Nat. Struct. Biol. 2003;10(5):349–356. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- 36.Fleming C.D., Bencharit S., Edwards C.C., Hyatt J.L., Tsurkan L., Bai F. Structural insights into drug processing by human carboxylesterase 1: tamoxifen, mevastatin, and inhibition by benzil. J. Mol. Biol. 2005;352(1):165–177. doi: 10.1016/j.jmb.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 37.Robbi M., Beaufay H. The COOH terminus of several liver carboxylesterases targets these enzymes to the lumen of the endoplasmic reticulum. J. Biol. Chem. 1991;266(30):20498–20503. [PubMed] [Google Scholar]
- 38.Satoh T., Hosokawa M. The mammalian carboxylesterases: from molecules to functions. Annu. Rev. Pharmacol. Toxicol. 1998;38(1):257–288. doi: 10.1146/annurev.pharmtox.38.1.257. [DOI] [PubMed] [Google Scholar]
- 39.Yan B., Yang D., Brady M., Parkinson A. Rat kidney carboxylesterase. Cloning, sequencing, cellular localization, and relationship to rat liver hydrolase. J. Biol. Chem. 1994;269(47):29688–29696. [PubMed] [Google Scholar]
- 40.Frey P.A., Whitt S.A., Tobin J.B. A low-barrier hydrogen bond in the catalytic triad of serine proteases. Science. 1994;264(5167):1927. doi: 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- 41.Holmes R., Cox L., VandeBerg J. Springer Netherlands; 2010. Mammalian Carboxylesterase 3: Comparative Genomics and Proteomics. Genetica; pp. 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dolinsky V.W., Gilham D., Alam M., Vance D.E., Lehner R. Triacylglycerol hydrolase: role in intracellular lipid metabolism. CMLS Cell. Mol. Life Sci. Cell. Mol. Life Sci. CMLS. 2004:1633–1651. doi: 10.1007/s00018-004-3426-3. Birkh+ñuser-Verlag. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehner R., Cui Z., Vance D.E. Subcellullar localization, developmental expression and characterization of a liver triacylglycerol hydrolase. Biochem. J. 1999;338(Pt 3):761–768. [PMC free article] [PubMed] [Google Scholar]
- 44.Lehner R., Verger R. Purification and characterization of a porcine liver microsomal triacylglycerol hydrolase. Biochemistry. 1997;36(7):1861–1868. doi: 10.1021/bi962186d. [DOI] [PubMed] [Google Scholar]
- 45.Wang H., Gilham D., Lehner R. Proteomic and lipid characterization of apolipoprotein B-free luminal lipid droplets from mouse liver microsomes: implications for very low density lipoprotein assembly. J. Biol. Chem. 2007;282(45):33218–33226. doi: 10.1074/jbc.M706841200. [DOI] [PubMed] [Google Scholar]
- 46.Gilham D., Alam M., Gao W., Vance D.E., Lehner R. Triacylglycerol hydrolase is localized to the endoplasmic reticulum by an unusual retrieval sequence where it participates in VLDL assembly without utilizing VLDL lipids as substrates. Mol. Biol. Cell. 2005;16(2):984–996. doi: 10.1091/mbc.E04-03-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang J., Oikawa S., Iwahashi H., Kitagawa E., Takeuchi I., Yuda M. Expression of proteins associated with adipocyte lipolysis was significantly changed in the adipose tissues of the obese spontaneously hypertensive/NDmcr-cp rat. Diabetol. Metab. Syndrome. 2014;6(1):8. doi: 10.1186/1758-5996-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quiroga A.D., Ceballos M.P., Parody J.P., Comanzo C.G., Lorenzetti F., Pisani G.B. Hepatic carboxylesterase 3 (Ces3/Tgh) is downregulated in the early stages of liver cancer development in the rat. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2016;1862(11):2043–2053. doi: 10.1016/j.bbadis.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 49.Blais D.R., Lyn R.K., Joyce M.A., Rouleau Y., Steenbergen R., Barsby N. Activity-based protein profiling identifies a host enzyme, carboxylesterase 1, which is differentially active during hepatitis C virus replication. J. Biol. Chem. 2010;285(33):25602–25612. doi: 10.1074/jbc.M110.135483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H., Wei E., Quiroga A.D., Sun X., Touret N., Lehner R. Altered lipid droplet dynamics in hepatocytes lacking triacylglycerol hydrolase expression. Mol. Biol. Cell. 2010;21(12):1991–2000. doi: 10.1091/mbc.E09-05-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quiroga A.D., Lehner R. Role of endoplasmic reticulum neutral lipid hydrolases. Trends Endocrinol. Metabol. 2011;22(6):218–225. doi: 10.1016/j.tem.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 52.Zhao B., Fisher B.J., Clair R.W.S., Rudel L.L., Ghosh S. Redistribution of macrophage cholesteryl ester hydrolase from cytoplasm to lipid droplets upon lipid loading. J. Lipid Res. 2005;46(10):2114–2121. doi: 10.1194/jlr.M500207-JLR200. [DOI] [PubMed] [Google Scholar]
- 53.Tsujita T., Okuda H. Fatty acid ethyl ester synthase in rat adipose tissue and its relationship to carboxylesterase. J. Biol. Chem. 1992;267(33):23489–23494. [PubMed] [Google Scholar]
- 54.Gaveglio V.L., Pasquar+® S.J., Giusto N.M. Metabolic pathways for the degradation of phosphatidic acid in isolated nuclei from cerebellar cells. Arch. Biochem. Biophys. 2011;507(2):271–280. doi: 10.1016/j.abb.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 55.Gaveglio V.L., Pasquar+® S.J., Giusto N.M. Phosphatidic acid metabolism in rat liver cell nuclei. FEBS Lett. 2013;587(7):950–956. doi: 10.1016/j.febslet.2013.01.074. [DOI] [PubMed] [Google Scholar]
- 56.Mate S.M., Layerenza J.P., Ves-Losada A. Incorporation of arachidonic and stearic acids bound to L-FABP into nuclear and endonuclear lipids from rat liver cells. Lipids. 2007;42(7):589–602. doi: 10.1007/s11745-007-3063-3. [DOI] [PubMed] [Google Scholar]
- 57.de la Rosa Rodriguez M.A., Kersten S. Regulation of lipid droplet-associated proteins by peroxisome proliferator-activated receptors. Biochim. Biophys. Acta (BBA) – Mol. Cell Biol. Lipids. 2017;1862(10, Part B):1212–1220. doi: 10.1016/j.bbalip.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 58.Ves-Losada A., Brenner R.R. Long-chain fatty Acyl-CoA synthetase enzymatic activity in rat liver cell nuclei. Mol. Cell. Biochem. 1996;159(1):1–6. doi: 10.1007/BF00226056. [DOI] [PubMed] [Google Scholar]
- 59.Kory N., Thiam A.R., Farese J., Walther T.-Á. Protein crowding is a determinant of lipid droplet protein composition. Dev. Cell. 2015;34(3):351–363. doi: 10.1016/j.devcel.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arnold R.S., DePaoli-Roach A.A., Cornell R.B. Binding of CTP:phosphocholine cytidylyltransferase to lipid Vesicles: diacylglycerol and enzyme dephosphorylation increase the affinity for negatively charged membranes. Biochemistry. 1997;36(20):6149–6156. doi: 10.1021/bi970023z. [DOI] [PubMed] [Google Scholar]
- 61.Sztalryd C., Brasaemle D.L. The perilipin family of lipid droplet proteins: gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta (BBA) - Mol. Cell Biol. Lipids. 2017;1862(10, Part B):1221–1232. doi: 10.1016/j.bbalip.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aitchison A.J., Arsenault D.J., Ridgway N.D. Nuclear-localized CTP:phosphocholine cytidylyltransferase +¦ regulates phosphatidylcholine synthesis required for lipid droplet biogenesis. Mol. Biol. Cell. 2015;26(16):2927–2938. doi: 10.1091/mbc.E15-03-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Favale N.O., Fernandez-Tome M.C., Pescio L.G., Sterin-Speziale N.B. The rate-limiting enzyme in phosphatidylcholine synthesis is associated with nuclear speckles under stress conditions. Biochim. Biophys. Acta. 2010;1801(11):1184–1194. doi: 10.1016/j.bbalip.2010.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data included in article/supp. material/referenced in article.