

Summary
Voltage-gated sodium (NaV) channels, initially characterized in excitable cells, have been shown to be aberrantly expressed in non-excitable cancer tissues and cells from epithelial origins such as in breast, lung, prostate, colon, and cervix, whereas they are not expressed in cognate non-cancer tissues. Their activity was demonstrated to promote aggressive and invasive potencies of cancer cells, both in vitro and in vivo, whereas their deregulated expression in cancer tissues has been associated with metastatic progression and cancer-related death. This review proposes NaV channels as pharmacological targets for anticancer treatments providing opportunities for repurposing existing NaV-inhibitors or developing new pharmacological and nutritional interventions.
Subject areas: Cell Biology, Cancer
Graphical abstract
Cell Biology; Cancer
Introduction
Voltage-gated sodium (NaV) channels, composed of pore-forming NaVα and auxiliary NaVβ subunits, were initially characterized in excitable cells in which they are responsible for the triggering and the propagation of action potentials. Their physiological activity, through a transient depolarizing inward sodium current in cell types such as cardiomyocytes, skeletal muscle cells, or neurons, is well characterized as being responsible for the initiation of excitation-contraction, excitation-secretion, or excitation-expression couplings. As such, these ion channels are critical in numerous physiological functions and mutations in their encoding genes, as well as dysregulation of their activity may lead to serious pathologies called “sodium channelopathies.” NaV channels are targets for multiple inhibitory molecules that are FDA approved and clinically used in the treatment of pathologies such as cardiac angina or arrhythmias, epilepsies, chronic pain, or in anesthesiology.
Although the activity of NaV channels has been characterized about 70 years ago, recent data obtained in the past 5 years have shed light on the protein structure, arrangement, and functioning at the molecular level. Indeed, the activity of NaV channels, i.e. sodium currents (INa), was first recorded by Hodgkin and Huxley in 1952 from the squid giant axon, using the voltage-clamp technique. These pioneering experiments led to the ionic theory of membrane excitability (Hodgkin and Huxley, 1952). However, at that time the structure of NaV channels was not known and the first evidence of molecular properties came to light at the beginning of the 1980s with the identification of the channel proteins using radiolabeled-neurotoxins highly selective for NaV channels in combination with protein solubilization and purification methods (Agnew et al., 1980; Beneski and Catterall, 1980). Further structural insights into NaV channels were obtained by cloning and screening of cDNA libraries leading to the discovery of the amino acid sequence for these proteins and allowing for modeling of secondary structures based on aliphatic profiles (Noda et al., 1984, 1986). These seminal studies allowed development of a model in which the pore-forming NaV principal subunit in eukaryotes, later called NaVα-subunit, is composed of a single polypeptide chain of approximately 260 kDa containing four repeated homologous domains (I–IV) of six transmembrane segments (S1–S6). This protein was identified to interact with one or two single-span transmembrane auxiliary subunits, called NaVβ-subunits (30–40 kDa), initially characterized as bringing regulatory functions (Isom et al., 1992, 1994) to the macromolecular complex of eukaryotic NaV channels (Brackenbury and Isom, 2011; Catterall, 2000).
Interestingly, understanding of protein organization and function at the molecular level substantially progressed very recently with the use of X-ray protein crystallography and cryo-electron microscopy, first studying tetrameric prokaryotic channels sharing approximately 25–30% sequence identity with human channels. Thus information regarding the voltage-dependent gating; ion selectivity; drug binding; and open, closed, and inactivated states were acquired from the Arcobacter butzleri (NavAb) channel (Payandeh et al., 2011, 2012) (Lenaeus et al., 2017). The high-resolution crystal structure of the complete voltage-gated sodium channel (NaVMs) from Magnetococcus marinus was obtained in the activated open state, associated with electrophysiological recordings (Sula et al., 2017). Recently, structural data obtained for NaVAb provided a complete gating mechanism for voltage sensor function, pore opening, and the activation gate (Wisedchaisri et al., 2019). The first near-atomic resolution structure of a monomeric eukaryotic channel came from the recent study of the NaVPaS from the American cockroach, whereas structures of human voltage-gated sodium channel NaV1.2, NaV1.4, and NaV1.7, in complex with β-subunits were recently published (Pan et al., 2018, 2019; Shen et al., 2019). These structural data provide important insight into mechanisms underlying NaV channelopathies and for drug discovery. Briefly, each domain presents two functional modules: S1–S4 segments comprise the voltage-sensor module (VSM), whereas sections (S5 - P loop - S6) constitute the pore-forming module (PM). The positively charged arginine and lysine residues, positioned at every third residue within each S4 segment in the voltage-sensor module, sense changes in the transmembrane potential and transform this electrical stimulus into a fast conformational change of the pore-forming module, allowing the opening of the conductive pore, permitting Na+ influx. One to two milliseconds after pore opening, another fast voltage-dependent mechanism happens in the NaV channel protein, occluding Na+ influx, a process known as fast inactivation.
Another recent study has challenged the initial paradigms by which functional channels contain a monomeric NaVα subunit. Indeed, NaVα-subunits appear to assemble as dimers, and this physical interaction permits a coupled gating mechanism (Clatot et al., 2017). In humans, there are nine different genes encoding for NaVα-subunits, four of them clustered on chromosome 2: SCN1A (NaV1.1), SCN2A (NaV1.2), SCN3A (NaV1.3), and SCN9A (NaV1.7); three others located on chromosome 3: SCN5A (NaV1.5), SCN10A (NaV1.8), and SCN11A (NaV1.9); and two more located on chromosome 12 and 17: SCN8A (NaV1.6) and SCN4A (NaV1.4), respectively (Goldin, 2002). The amino acid sequence homology among NaV channels subtypes is higher than 70% in the transmembrane and extracellular motifs so that there are no distinct subfamilies; nonetheless, some of the isoforms are more closely related to each other, sharing chromosomal localization and sensitivity to tetrodotoxin (TTX), which has been explained by the early genomic duplication during the evolution of NaV channel genes. To date, four genes have been identified for NaVβ-subunits, one on chromosome 19: SCN1B (encoding for the two splicing variants, transmembrane NaVβ1 and soluble NaVβ1B) and the other three on chromosome 11: SCN2B (NaVβ2), SCN3B (NaVβ3), and SCN4B (NaVβ4). The structure of NaVβ-subunit is comprised of an N-terminal extracellular immunoglobulin-like domain, followed by an extracellular juxtamembrane region, a single transmembrane segment and a 34-44 amino-acid-length intracellular domain, except for the splice variant NaVβ1B, which is expressed as a soluble macromolecule (Brackenbury and Isom, 2011). Furthermore, it is worth noting that NaVβ-subunits not only influence NaVα-subunit trafficking and biophysical modulation but they have also been experimentally shown to act as cell adhesion molecules (CAMs), participating in both homophilic and heterophilic interactions, with contactin, N-cadherin, NrCAM, and several types of neurofascin and tenascin being the main binding proteins (Isom, 2002; Bouza and Isom, 2018).
NaVα and NaVβ subunits are differentially and developmentally expressed in several tissues and cell types (Black and Waxman, 2013; Roger et al., 2015). Initial studies identified NaV to be distributed in excitable tissues such as in mammalian central and peripheral nervous systems as well as in skeletal and cardiac muscle. The central nervous system channels mainly comprise the NaV1.1, NaV1.2, NaV1.3, and NaV1.6 isoforms, whereas the peripheral nervous system channels include NaV1.7, NaV1.8, and NaV1.9. NaV1.4 and NaV1.5 were characterized as being the main skeletal and heart muscle isoforms, respectively (Goldin, 2001), whereas they also have been identified to be expressed in the brain and in the dorsal root ganglion (Wang et al., 2008, 2009, 2018a, 2018b; Bergareche et al., 2015). Therefore, the expression of NaV has long been considered as the hallmark of excitable cells. Again, this paradigm has recently changed with the identification of expression (mRNA and protein) and sometimes activity at the plasma membrane (transient sodium currents) in non-excitable tissues and cells such as in chondrocytes, endothelial cells, microglia, astrocytes, fibroblasts, keratinocytes, islet β-cells, red blood cells, T-lymphocytes, dendritic cells, and macrophages, among others, in which the biological role and subcellular distribution of Navs is still elusive (Black and Waxman, 2013; Roger et al., 2015).
Recently, it has emerged that NaVα channels, as well as auxiliary NaVβ subunits, are aberrantly expressed in non-excitable cancer tissues and cells from different epithelial origins such as breast, lung, prostate, colon, and cervix, whereas they are not expressed in cognate non-cancer tissues. Their expression in carcinoma cells has been associated with cancer progression, suggesting that they could serve as cancer markers and prognostic factors. The expression and activity of NaV channels was shown to promote pro-cancerous properties and, importantly, to contribute to disease progression, in both in vitro and in vivo models. Recent studies shed light on the signaling pathways that are under the control of these channel proteins, coupling membrane activity to cellular properties. Furthermore, the inhibition of NaV channel activity, using small synthetic compounds and FDA-approved drugs as well as with natural dietary compounds potentially opens up new therapeutic strategies. In this review, we summarize current knowledge on the expression of NaV channels in cancers, highlight the signaling pathways involved, and discuss pharmacological and nutritional strategies that represent opportunities for novel anticancer treatments.
The aberrant expression of NaVα and NaVβ subunits in cancers
The expression of voltage-gated sodium channel subunits, both NaVα and NaV β, has been reported to be altered in several types of cancer (Table 1). The channel subunits have been detected by molecular biology and biochemical techniques, and in multiple cancer types NaV activity at the plasma membrane, i.e. INa, could be recorded. Most subunits have been shown to be upregulated in cancers, whereas some of them appear to be downregulated. This aberrant expression has been correlated with oncogenic properties in both in vitro and in vivo models of cancer, mostly in solid tumors including carcinomas (Figure 1).
Table 1.
Expression and identified roles of pore-forming NaVα and auxiliary NaVβ in cancer
| Isoform | Expression levels | Cancer type | Role in cancer, proposed mechanism | References |
|---|---|---|---|---|
| NaV1.1 | Upregulateda (mRNA, protein) | Lymph nodes from CRC | Unknown, unknown | Lin et al. (2019) |
| NaV1.2 | Upregulateda (Protein) | Liver | Unknown, unknown | The Human protein Atlas (www.proteinatlas.org) |
| NaV1.3 | Upregulateda (mRNA) | Ovarian | Unknown, unknown | Gao et al. (2010) |
| NaV1.4 | Upregulateda (mRNA) | Cervix | Unknown, unknown | Diaz et al. (2007) |
| NaV1.5 | Upregulateda,b (mRNA, protein, INa+) | Breast | ↑Invasionc,d ▪ Through an increased Src kinase activity promoting invadopodial formation and favoring an allosteric activation of NHE-1, extracellular acidification and enhanced activity of cysteine−cathepsins proteases ▪ Boosting the EMT transition via SIK1 ▪ Generating a sustained plasma membrane depolarization that leads to Rac1 activation and cytoskeleton reorganization ▪ Increasing MMP9 expression and reducing apoptosis |
Roger et al. (2003), Fraser et al. (2005), Brackenbury et al. (2007), Gillet et al. (2009), Brisson et al. (2011), Yang et al. (2012), Brisson et al. (2013), Driffort et al. (2014), Nelson et al. (2015a),b, Gradek et al. (2019), Yang et al. (2020), |
| Colorectal | ↑Invasionc Through the regulation of the transcriptional pathway PKA/ERK/c-jun/ELK-1/ETS-1 |
House et al. (2010), Baptista-Hon et al. (2014), House et al. (2015), Guzel et al. (2019), Poisson et al. (2020) | ||
| Ovarian | ↑Migrationc ↑Invasionc ↑Proliferationc ▪ By increasing the window current and depolarization of resting potential |
Gao et al. (2010), Liu et al. (2018) | ||
| Leukemia | ↑Invasionc ▪ Endosomal acidification and enhanced phagocytosis via calcium signaling |
Fraser et al. (2004), Carrithers et al. (2007), Carrithers et al., 2012, Rahgozar et al. (2013), Jones et al. (2014), Huang et al. (2015) | ||
| NaV1.6 | Upregulateda,b (mRNA, protein, INa+) | Cervix | ↑Invasionc ▪ Boosted activity of MMP2 and NHE-1 |
Diaz et al. (2007), Hernandez-Plata et al. (2012), Lopez-Charcas et al. (2018) |
| Leukemia | ↑Invasionc ▪ Invadopodial formation and enhanced activity |
Carrithers et al. (2009) | ||
| Melanoma | ↑Invasionc ▪ Invadopodial formation and enhanced activity |
Carrithers et al. (2009) | ||
| Lymph nodes from CRC | Unknown, unknown | Lin et al. (2019) | ||
| Downregulateda,b (mRNA, protein, INa+) | Erwin sarcoma | ↓Migrationc ↑Apoptosisc Through a repression of SCN8A by RING1B and maintaining low NF-κβ levels |
Hernandez-Munoz et al. (2016) | |
| Colorectal | Unknown, unknown | Igci et al. (2015) | ||
| NaV1.7 | Upregulateda,b (mRNA, protein, INa+) | Prostate | ↑Invasionc,d ▪ Cell motility increased via galvanotaxis |
Grimes et al. (1995), Diss et al. (2001), Diss et al. (2005), Yildirim et al. (2012), Bugan et al. (2019) |
| Lung (NSCLC) | ↑Invasionc ▪ Dysregulation of sodium homeostasis ▪ Increase of [NA+]i and depolarization of cell membrane |
Roger et al. (2007) Campbell et al. (2013) |
||
| Lung (SCLC) | Unknown Participation in the formation of a neuroendocrine-like tumor cell phenotype |
Pancrazio et al. (1989), Blandino et al. (1995), Onganer et al. (2005) | ||
| Stomach | ↑Invasionc,d ↑Tumor growthd ▪ Cancer progression through MACC1-mediated upregulation of NHE-1 |
Xia et al. (2016) | ||
| Endometrial | ↑Invasionc,d↑Tumor growthd↓Apoptosisd ▪Unknown | Liu et al. (2018), Liu et al. (2019) | ||
| Downregulateda (mRNA, protein) | Colorectal | Unknown, unknown ▪ Proposed within a risk score with high prognostic value for overall survival |
Pan et al. (2017) | |
| NaV1.8 | Upregulatedb (mRNA) | Lung (NSCLC) | Unknown, unknown | Roger et al. (2007) |
| NaV1.9 | Upregulatedb (mRNA) | Lung (NSCLC) | Unknown, unknown | Roger et al. (2007) |
| NaVβ1 | Upregulateda,b (mRNA, protein) | Prostate | Unknown, unknown | Diss et al. (2008) |
| Breast | ↑Tumor growthc,d ↑Vascularizationd ↑Invasionc ↓Apoptosis ▪ β1 trans-homophilic adhesion triggering of cell process outgrowth via Fyn kinase |
Nelson et al. (2014), Bon et al. (2016) | ||
| Downregulatedb (mRNA, protein) | Breast | ↑Invasionc By decreasing cell adhesion and facilitating cell migration |
Chioni et al. (2009) | |
| Lung | ↑Invasionc By decreasing cell adhesion |
Campbell et al. (2013) | ||
| Cervix | ↑Migrationc ▪Acting as a cell adhesion molecule ↓Proliferationc ▪By increasing the population of cells in the G0/G1 phase in cell cycle |
Sanchez-Sandoval and Gomora (2019) | ||
| NaVβ2 | Upregulatedb (mRNA, protein) | Prostate | ↑Invasionc ▪ Promotion of bipolar cell morphology, enhanced cell adhesion and motility through association with neural substrates. ↓Tumor volume in xenograft models‡ |
Jansson et al. (2012), Jansson et al. (2014) |
| NaVβ3 | Upregulatedb (mRNA, protein) | Bone | ↑Apoptosisc ▪ Increase of a p53-dependent apoptotic pathway |
Adachi et al. (2004) |
| NaVβ4 | Downregulateda,b (mRNA, protein) | Breast | ↑Invasionc,d ▪ Enhanced RhoA activity ▪ Mediated by the intracellular C-terminus motif ▪ Acquisition of a hybrid mesenchymal-amoeboid aggressive phenotype |
Bon et al. (2016) |
| Lung | Unknown, unknown | Bon et al. (2016) | ||
| Cervix | ↑Invasionc | Sanchez-Sandoval and Gomora (2019) | ||
| Thyroid | Unknown ▪ Its expression is an indicator of favorable recurrence-free survival |
Gong et al. (2018) |
Cancer tissue versus non-cancerous tissue.
Highly invasive cell line versus normal immortalized cell line.
In vitro.
In vivo.
Figure 1.

Expression of NaVα in carcinoma and role in invadopodial activity and invasion of extracellular matrices
Progression of precancerous into cancer cells is illustrated in the context in the malignant transformation of colon epithelium. Transformed cells have lost cell polarity, replication control, and cell-cell adherent junctions, and they acquired a mesenchymal pro-invasive phenotype. Migrating cancer cells develop a specialized actin-based membrane protrusions called “invadopodia” that facilitate cell invasion by providing a coupling of focal extracellular matrix (ECM) degradation together with a directional cell movement. NaV channels are expressed in invadopodial structures, co-localizing with the Na+/H+ exchanger type 1 (NHE1). Activity of NaV channels enhances the extrusion of protons by NHE1 and therefore the acidification of the peri-invadopodial microenvironment, thus favoring both secretion and activity of ECM proteases such as cysteine cathepsins and matrix metalloproteinases (MMPs). Cancer cell resting potential (Vm) is around −40 mV, in a window of voltage of NaV channels (overlap between activation and steady-state inactivation curves) in which a small proportion of channels are activated but non-inactivated, thus generating a small but continuous Na+ influx through a so-called “window sodium current.” NaV channels are also proposed to increase the intracellular levels of Ca2+ ions by the functioning of Na+/Ca2+ exchanger (NCX) in a “reverse mode.” Thus, the increase in the intracellular concentration of Na+ and Ca2+, sustains SRC kinase activity, leading to the polymerization of acting filaments and the formation of invadopodial structure.
NaVα and NaVβ subunits in prostate cancer
Although early works assessing ion channel activity identified sodium currents in small-cell lung cancer cells (Pancrazio et al., 1989), the first report of the direct contribution of NaV channels in cancer properties came from work performed in prostate cancer (PCa) cells by Prof. M. Djamgoz’ group almost 25 years ago. This first study was performed in two rat prostatic tumor cell lines in which they found a differential expression of voltage-activated Na+ currents: highly metastatic Mat Ly-Lu cells expressed Na+ currents, whereas weakly metastatic AT-2 cell did not express this type of current. For the first time, the functional relevance of the Na+ current was demonstrated: blocking INa with nanomolar concentrations of TTX reduced by ∼30% the invasiveness of the highly metastatic Mat-Ly-Lu cells (Grimes et al., 1995). Further evidence showed that the NaV1.7 pore-forming subunit, encoded by the SCN9A gene, was overexpressed in highly metastatic human and rat prostate cell lines in comparison with weakly metastatic cell lines (Diss et al., 2001). Later, this observation was confirmed in vivo by showing that NaV1.7 was overexpressed approximately 20 times in PCa biopsies versus non-cancer samples (Diss et al., 2005). Finally, in a rat model, inoculation with highly metastatic Mat-Ly-Lu cells promotes prostate cancer metastasis in vivo, and blockade of NaV in primary tumors with TTX (Yildirim et al., 2012) or ranolazine (Bugan et al., 2019) reduced lung metastases by 40% and 63%, respectively. However, most of these studies were performed in cell lines, and there are no systematic studies that show a positive correlation between mRNA/protein of NaV1.7 upregulation and human PCa patient samples.
Concerning NaVβ subunits, it was shown that NaVβ1 mRNA was the most highly expressed in strongly invasive compared with weakly invasive PCa cell lines (Diss et al., 2008). However, there was no significant difference in the expression of NaVβ1, or any other NaVβ subunits, between cancer and non-cancer human prostate specimens (Diss et al., 2008). NaVβ2 has been proposed to participate in prostate cancer biology. Indeed, the first experimental observation showed that the overexpression of NaVβ2, fused to GFP, was properly localized at the plasma membrane of weakly metastatic LNCaP cells inducing morphological changes consistent with a bipolar and elongated migratory phenotype (Jansson et al., 2012). These changes were accompanied by an increase in cell migration and Matrigel invasion in vitro, although there was no significant change in cell proliferation. However, the in vivo properties of LNCaP cells overexpressing the NaVβ2 subunit were quite unexpected, as subcutaneous tumor volume was drastically reduced compared with the control group (Jansson et al., 2012). Such contrasting effects could imply different mechanisms involved in tumor formation, growth, and metastatic behavior. A novel ex vivo organotypic spinal cord co-culture with LNCaP cells revealed that overexpression of NaVβ2 enhanced the association of PCa cells with nerve axons (Jansson et al., 2014). Furthermore, overexpression of NaVβ2 enhanced PCa cell migration, invasion, and growth, in the presence of the neuronal CAM laminin, suggesting that the NaVβ2 subunit may mediate metastatic behavior through association with neural substrates (Jansson et al., 2014).
NaVα and NaVβ subunits in breast and colorectal cancer
Breast cancer (BCa) is the most lethal female cancer worldwide (Bray et al., 2018), and colorectal cancer (CRCa) is the third most commonly diagnosed cancer (Arnold et al., 2017). The incidence of these cancers is gradually increasing, thus representing a serious global health problem. The main cause of patient mortality from these two types of cancers, as for the majority of carcinomas, is the development of metastases in distant organs, following the dissemination of cancer cells from the primary tumor (Figure 1).
Multiple studies have investigated the expression of NaV channels and their contribution to tumor progression and metastasis in BCa and CRCa. Knowledge about signaling pathways and cellular mechanisms induced by NaV channels have been mostly acquired from these cancers. In both cases, the major isoform identified was NaV1.5, encoded by the SCN5A gene. In BCa samples, NaV1.5 is overexpressed as compared with normal tissues (Fraser et al., 2005). A high expression was correlated with cancer recurrence, metastasis development, and reduced patient survival (Yang et al., 2012). Most of the mechanistic studies in BCa have been performed in human cancer cell lines such as MDA-MB-231 (highly metastatic) and compared with weakly metastatic cell lines such as MCF-7. It has been shown initially that MDA-MB-231 expresses a TTX-resistant Na+ current, lacking in MCF-7 cells (Roger et al., 2003), which is encoded by a neonatal splice variant of the SCN5A gene (Fraser et al., 2005). This neonatal variant is due to a switch from adult exon 6B to fetal exon 6A, which are mutually exclusive and encode for a part of the voltage sensor, segments 3 and 4 located in the domain I of the channel. Therefore, these two variants, called hNaV1.5 and hNaV1.5e for the adult and the neonatal channels respectively, show different electrophysiological properties in terms of voltage sensitivity and current kinetics (Murphy et al., 2012; Onkal et al., 2008). These changes result in a greater Na+ influx for neonatal hNaV1.5e (Onkal et al., 2008). In the heart, splicing of SCN5A is developmentally regulated, such that the neonatal exon 6A is rapidly replaced by the “adult” exon 6B after birth (Murphy et al., 2012) and molecular determinants explaining the abnormal expression of hNaV1.5e in cancer cells have not been identified so far. Nevertheless, the inhibition of channel activity, by either pharmacological (TTX, ranolazine and phenytoin) or molecular (siRNA and inhibitory antibody) approaches, has shown its contribution to migration and invasion of BCa cell lines (Roger et al., 2003; Fraser et al., 2005; Brackenbury et al., 2007; Driffort et al., 2014; Yang et al., 2012). There has also been some significant progress into uncovering the mechanisms underlying the promotion of invasiveness behavior by NaV1.5. Experimental evidence suggested that NaV1.5 induces pro-migratory and pro-invasive properties through a persistent activity at the membrane potential called “window current,” and a correlated depolarization of the membrane voltage of breast cancer cells. Particularly, NaV1.5 activity induced the allosteric modulation of the Na+-H+ exchanger type 1 (NHE-1), resulting in an increased activity, leading to the acidification of the extracellular space, thus favoring the pH-dependent activity of proteolytic cysteine cathepsins (Gillet et al., 2009; Brisson et al., 2011). In addition, NaV1.5 expression and activity were demonstrated to increase Src kinase activity, which promotes the acquisition of an invasive morphology (invadopodia) in MDA-MB-231 cells. Taken together, these observations indicate that NaV1.5 promotes invadopodia activity of breast cancer cells and the invasion of the surrounding ECM (Brisson et al., 2013) (Figure 1). Recently, NaV1.5 was identified as importantly promoting the epithelial-to-mesenchymal transition (EMT) and cancer cell invasiveness through the regulation of the salt-inducible kinase 1 (SIK1) (Gradek et al., 2019). Furthermore, NaV1.5 activates the small GTPase Rac1 by sustaining a plasma membrane depolarization, which as a regulator of activation, induces cytoskeletal reorganization and cellular migration (Yang et al., 2020). In addition, in vivo experiments have shown that NaV1.5 activity promotes metastasis in immunodeficient mice (Driffort et al., 2014; Nelson et al., 2015a, 2015b). NaV1.5 activity also increases MMP9 expression and reduces apoptosis in primary tumors in vivo (Nelson et al., 2015b).
The SCN5A gene and its protein product the NaV1.5 channel have also been shown to be overexpressed in colorectal cancer biopsies, as compared with non-cancer samples (House et al., 2010). NaV1.5 was found to be expressed at the plasma membrane of tumor cells, and its activity (INa) was recorded in several carcinoma cell lines (mainly SW-480, SW-620, and HT-29) (House et al., 2010). In colon cancer cells, NaV1.5 activity promotes cancer cell invasion in vitro, in both 2- and 3-dimensional models, and regulates a network of invasion-promoting genes via modulation of the PKA/ERK/c-JUN/ELK-1/ETS-1 transcriptional pathway (House et al., 2010, 2015; Poisson et al., 2020) (Figure 2). It was shown that, similar to BCa, the neonatal exon 6A splice variant of the NaV1.5 isoform has a predominant contribution to the invasiveness of CRCa cell lines (Guzel et al., 2019), even though both adult hNaV1.5 and neonatal hNaV1.5e splice variants could be detected (Baptista-Hon et al., 2014).
Figure 2.

Participation of NaVα and/or NaVβ in pro-metastatic signaling pathways
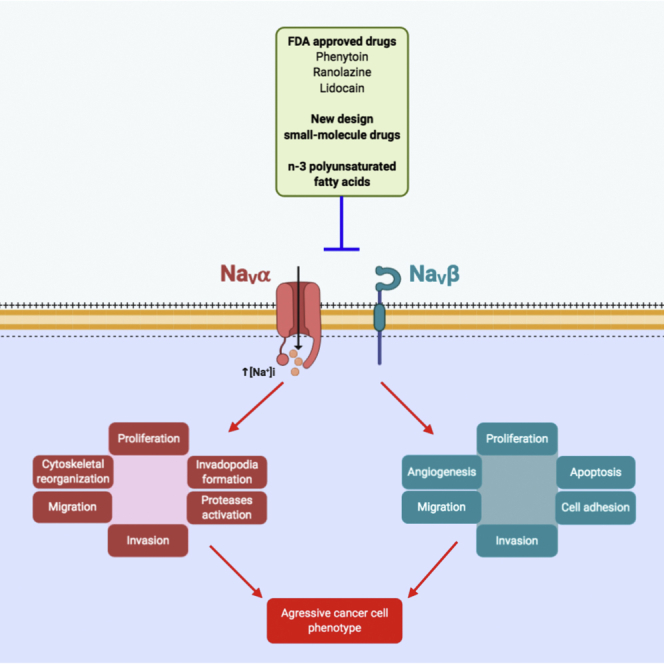
NaVα subunit overexpression and activity in cancer cells trigger biochemical or an electro-biochemical cascades, leading to the acquisition of a pro-invasive cell phenotype. NaV is co-localized with NHE1 in caveolin-1 (Cav-1)-containing lipid rafts and promotes the efflux of protons. NaV activity can be further stimulated by the use of pharmacological activators such as veratridine (inhibitor of the inactivation phase). Activity of NaVα subunits leads to a cAMP-independent activation of protein kinase A (PKA) that activates the cytosolic small GTPase Ras-related protein 1 (Rap1A/B) and the extracellular-signal-regulated kinases (ERK1/2). The transcription factor (TF) metastasis associated in colon cancer 1 (MACC1) is activated by the p38/NF-κβ signaling, whereas the TFs c-jun, ELK1, and ETS1 are activated by ERK1/2 and the zinc finger protein SNAI1 is activated through a NaVα-dependent mechanism regulating the expression of genes associated with cytoskeleton reorganization, cell motility, extracellular matrix degradation, and cell invasiveness. It has been demonstrated that MACC1 upregulates the expression of the SLC9A1 gene, encoding for NHE1, thus enhancing its activity at plasma membrane. On the other hand, the electro-biochemical triggering begins with a resting potential depolarization due to the activity of NaVα subunits promoting the activation and recruitment of the small GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1) at the leading edge of migrating cells. Transforming growth factor β 1 (TGF-β1) increases the expression levels of NaV channels genes (SCNxA), whereas ring finger protein 1 (RING1B), RE1 silencing transcription factor (REST), histone deacetylase 2 (HDAC2) and salt inducible kinase 1 (SIK-1), as well as the n-3 polyunsaturated fatty acids n-3 (PUFA) repress their expression. SIK-1 also impairs the functioning of NHE1 exchanger. The “auxiliary subunit” NaVβ4 is expressed in normal epithelial cells but is importantly downregulated in invasive cells and high-grade metastatic tumors. The absence of this protein, but specifically the lack of the intracellular C-terminus domain, triggers the acquisition of an amoeboid-mesenchymal hybrid phenotype dependent of the small GTPase Ras homolog family member A (RhoA). NaVβ1 proteins have a dual role in cancer cells acting as cell adhesion molecules (CAMs), reducing cell migration and proliferation. However, it has also been demonstrated that NaVβ1 promotes tumor growth, metastasis, and vascularization via the proto-oncogene tyrosine-protein kinase Fyn. The Rho-associated protein kinases (ROCK1/2) negatively regulate the expression of NaVα subunits, therefore, silencing or inhibition of these repressors restore NaV channels activity promoting an aggressive cell phenotype. Pharmacological intervention with FDA-approved drugs or new-design small-molecule lead compounds against NaV channels represents a promising strategy to decrease sodium-channel-associated metastases.
A study reported the downregulation of the SCN9A gene, encoding for NaV1.7, in CRCa (Pan et al., 2017). In this study, authors analyzed genes differentially expressed in CRCa utilizing three Gene Expression Omnibus (GEO) datasets. By screening 46 biomarkers associated with cancer proliferation, drug-resistance, and metastasis, i.e., genes closely associated to patient overall survival, they proposed a risk score with high prognostic value based on the expression of five genes: MET (MET proto-oncogene and receptor tyrosine kinase), CPM (carboxypeptidase M), SHMT2 (serine hydroxymethyltransferase 2), GUCA2B (guanylate cyclase activator 2B), and SCN9A. MET and SHMT2 were upregulated whereas CPM, GUCA2B, and SCN9A were downregulated. Interestingly, this observation was confirmed in the human protein atlas immunohistochemistry database (www.proteinatlas.org), as the staining for NaV1.7 was lower in some CRCa sample tissues (Pan et al., 2017). There are also reports indicating the downregulation of NaV1.6, encoded by the SCN8A gene, in CRCa (Igci et al., 2015). Tumor samples from CRCa patients exhibited reduced expression of NaV1.6 compared with paired tumour-surrounding normal tissues. SCN8A mRNA levels, analyzed by real-time qPCR, were significantly lower in tumor tissues and in patients younger than 45 years. Results also reveal a relationship between SCN8A expression, gender, grade of CRCa, tumor location, and histopathological classification (Igci et al., 2015). On the contrary, NaV1.6 protein was highly expressed in metastatic lymph nodes from CRCa patients (Lin et al., 2019). Although the reduced expression of SCN8A, encoding for NaV1.6, and SCN9A, encoding for NaV1.7, might harbor predictive values in CRCa, we are still missing clear information to assert whether they have a role, either causative or consecutive, in the carcinogenesis or whether their expression dysregulation is only correlative to cancer transformation or progression. Indeed, the functional activity of NaV1.6 and/or NaV1.7 at the plasma membrane of colorectal non-cancer or cancer cells has not been demonstrated so far. Furthermore, it cannot be excluded that these channels might be expressed in intracellular compartments, in which they might play diverse functions. Eventually, it is not clear at the moment whether these changes in expression levels concern epithelial cells, or non-epithelial cells in the colorectal tract, such as immune cells, which are key protagonists in colorectal carcinogenesis. As such, the participation of SCN8A and SCN9A in CRCa biology will require further studies.
The expression of NaVβ subunits has been studied in BCa and in CRCa. Some NaVβ have been shown to be upregulated, whereas others are downregulated in cancer tissues, and mostly these changes appear to correlate with the metastatic behavior of cancer cells, in particular with cell migration and invasion. Most research performed so far studying the role of NaVβ subunits in metastatic behavior has been undertaken in BCa. Originally, it was shown that NaVβ1 was more abundantly expressed in the weakly metastatic MCF-7 than in the highly metastatic MDA-MB-231 cell line. Interestingly, when MCF-7 cells were transfected with specific siRNA directed against NaVβ1, cell adhesion was reduced by 35%, whereas migration was increased by 121%. In contrast, stable expression of NaVβ1 in MDA-MB-231 cells increased process length and adhesion while reducing lateral motility and proliferation. Thus, NaVβ1 was proposed to act as a cell adhesion molecule in BCa cells, negatively controlling cellular migration (Chioni et al., 2009). Later, it was found that NaVβ1 was the NaVβ subunit most expressed in BCa and was upregulated (both mRNA and protein) in BCa biopsies, compared with normal breast tissue (Nelson et al., 2014; Bon et al., 2016). More importantly, by using a xenograft model of BCa, it was shown that NaVβ1 overexpression increased tumor growth, metastasis, and vascularization, while decreasing apoptosis in the primary tumors. Therefore, this study was the first showing the functional role for NaVβ1 in tumor growth and metastasis in vivo (Nelson et al., 2014). Consistent with these results, the use of siRNA to specifically target NaVβ1 expression in MDA-MB-231 cells inhibited cancer cell invasion (Bon et al., 2016).
The participation of NaVβ3 in tumorigenesis process is poorly understood. Two missense mutations have been identified in the SCN3B gene in high-grade metastatic colorectal cancer biopsies (Sjoblom et al., 2006). The first report suggested that non-mutated NaVβ3 mediates a p53-dependent apoptotic pathway in Saos-2, a bone osteosarcoma cell line, after DNA damage (Adachi et al., 2004). In agreement with this, the SCN3B gene is not expressed in highly invasive MDA-MB-231 breast cancer cells (Gillet et al., 2009) or weakly invasive MCF-7 cells (Chioni et al., 2009). In non-tumour breast samples, SCN3B expression was the lowest among all NaVβ encoding genes and was still significantly reduced in cancer samples (Bon et al., 2016).
The SCN4B gene was shown to play a critical role as a metastasis-suppressor gene in BCa (Bon et al., 2016). In this study SCN4B mRNA appeared to be significantly expressed in normal breast, colon, rectum, lung, and prostate but consistently downregulated in cancer samples. Furthermore, NaVβ4 protein was expressed in normal epithelial cells but significantly reduced in BCa biopsies, especially in high-grade primary and metastatic tumors. In vitro experiments showed that reducing NaVβ4 expression potentiates cell migration and invasiveness though an increase in RhoA activity and the acquisition of a hybrid mesenchymal-amoeboid aggressive phenotype. This effect was independent of NaVα channel activity and was prevented by overexpression of the intracellular C-terminus of NaVβ4. On the contrary, SCN4B overexpression reduced cancer cell invasiveness and tumor progression. The findings are in line with previous observations showing decreased levels of SCN4B in invasive versus non-invasive PCa cells (Diss et al., 2008). Interestingly, a recent study identified dysregulated miRNA in CRCa and reported an increased miR-424-5p expression in tumor samples that was associated with poor prognosis (Dai et al., 2020). miR-424-5p was found to be elevated in the peripheral blood of CRCa patients, most probably secreted in tumor exosomes. In this study, it was demonstrated that overexpression of SCN4B inhibited HT-29 CRCa cell proliferation, migration, and invasion, and expression of SCN4B was directly inhibited by miR-424-5p (Dai et al., 2020). These results support the tumour-suppressor role of SCN4B in CRCa and identified miR-424-5p as a regulator of its expression in tumors.
NaVα and NaVβ subunits in lung cancer
There are two subtypes of lung cancer, small-cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). Early works assessing ion channel activity have been undertaken in small-cell lung cancer cells. In these pioneering works, INa was initially recorded in human H146, H69, and H128 small-cell lung cancer cell lines (Pancrazio et al., 1989), although this study did not relate the presence of NaV currents to lung tumorigenesis process itself. These currents were most probably due to TTX-sensitive channels, because they were fully inhibited by 5 μM TTX. Although neither the molecular characterization of the channels nor their biological role was described, they were proposed to participate in a “neuroendocrine-like” tumor cell phenotype (Pancrazio et al., 1989). Later, it was shown that these sodium currents were actually able to participate in the generation of action potentials in H146 SCLC cells, thus supporting this idea. Interestingly, 5 μM TTX abolished these action potentials, which implies the contribution of a TTX-resistant current (Blandino et al., 1995). In fact, H146 cell sodium currents demonstrated an IC50 to TTX of 215 nM, leading the authors to indicate that NaV channels were weakly TTX-sensitive. However, this concentration is too high to consider the channels to belong to the TTX-S category but too low to belong to TTX-R isoforms. The most likely explanation for this would be the expression of a population of different isoforms of NaV at the plasma membrane of cells, thus leading to an apparent IC50 that would be intermediate between TTX-S and TTX-R. Nevertheless, the possibility that NaV channels expressed in these cells are variants (splice-variants or polymorphism variants) showing specific pharmacological properties cannot be excluded.
The hypothesis of the role of NaVα in the acquisition of a neuroendocrine phenotype was also proposed by M. Djamgoz's team (Onganer et al., 2005). Unexpectedly, in this later study, the endocytic activity of SCLC cells was inhibited by using lower nanomolar concentrations of TTX, suggesting the participation of TTX-sensitive sodium channels in these SCLC cells. In addition, they found mRNA encoding for NaV1.3, NaV1.5, and NaV1.6 in H69, H209, and H510 cell lines. The latter also showed the additional presence of NaV1.9 mRNA. Thus, it remains to be elucidated which NaV subunit is responsible for the generation of TTX-resistant action potentials in H146 cells (Blandino et al., 1995) and whether a TTX-resistant NaV channels contribute to migration, invasion, or some other metastatic component, other than endocytic activity (Onganer et al., 2005), in SCLC cell lines. More studies are needed investigating the expression profile and role of NaV channels in SCLC biopsy tissue.
Later analyses were also performed in non-small cell lung cancer (NSCLC) in which the NaV1.7 isoform was shown to potentiate cancer cell invasion (Roger et al., 2007; Campbell et al., 2013). Although different NSCLC cell lines (H23, H460, and Calu-1) express mRNA for several Na+ channel isoforms (Roger et al., 2007), the selective inhibition of NaV1.7 activity (using TTX) or reduction of expression (by using small interfering RNA) reduced H460 cell invasion by up to 50%. On the contrary, weakly invasive A549 cells showed no evidence of functional NaV channels (Roger et al., 2007). In addition, exogenous overexpression of the NaV1.7 subunit was sufficient to promote TTX-sensitive invasion of these cells. Interestingly, NaV1.7 protein expression was found to be higher in cancerous compared with normal-matched human lung tissue (Campbell et al., 2013). It is worth noting that at least in one NSCLC cell line (Calu-1), expression of a TTX-resistant NaV channel significantly contributes to the invasion capacity of this strongly metastatic cell line. However, the molecular identity of the molecular mediator of INa has not been fully characterized. Non-quantitative PCR results suggested that mRNAs encoding the three known TTX-resistant NaV channels (NaV1.5, NaV1.8, and NaV1.9) may be more abundantly expressed in Calu-1 than in H23 and H460 cells (Roger et al., 2007). Although kinetics and TTX-sensitivity of currents recorded in Calu-1 cells suggest NaV1.5 activity, more studies are needed to properly identify the molecular identity of the NaV channels mediating the Na+ current in these cells. In addition, there are currently no data correlating NaVα expression in lung cancer tissue with clinical outcome.
The expression of NaVβ in LCa has been assessed in several studies but so far it is difficult to conclude a general pattern. SCN1B mRNA was found to be expressed in H460, Calu-1, and A549 but not in the H23 NSCLC cell lines. It was also expressed in non-cancer NL-20 and BEAS-2B cells. SCN2B appeared to be weakly expressed in A549 cancer and NL-20 non-cancer cells, whereas not expressed at all in H23, H460, and Calu-1 cancer cell lines. SCN3B mRNA was found to be expressed in all these cell types with the only exception of H460. SCN4B mRNA was expressed in cancer H23 and non-cancer NL-20 and BEAS-2B but not in H460, Calu-1, and A549 (Roger et al., 2007). In patient samples, SCN4B expression levels were downregulated in lung cancer compared with normal lung tissue, and preliminary immunohistochemical analyses in lung cancer tissue microarrays showed a tendency toward decreased protein expression in high-grade primary lung tumors and metastases (Bon et al., 2016). The role of the NaVβ1 protein in cell adhesion was also proposed in human non-small cell lung cancer cell lines (Campbell et al., 2013). In this study, it was shown that the highly invasive H460 cells exhibited very low expression of all NaVβ subunit mRNAs, confirming previous results (Roger et al., 2007), whereas A549 cells expressed 8-fold higher levels of NaVβ1 mRNA. Accordingly, cell adhesion was 2-fold higher in A549 cells compared with H460 cells (Campbell et al., 2013). Moreover, manipulation of NaVβ1 mRNA expression by using siRNA or cDNA targeting SCN1B in these two cell lines confirmed the contribution of this subunit in the promotion of cell adhesion and reduced invasion (Campbell et al., 2013).
NaVα and NaVβ subunits in gastric cancer
In gastric cancer (GCa) tissue samples and in two human GCa cell lines (BGC-823 and MKN-28 cells), it was shown that the SCN9A gene, encoding NaV1.7, is the most abundantly expressed NaVα isoform (Xia et al., 2016). A systematic evaluation of 319 GCa tumor tissue samples by immunohistochemistry revealed a correlation of NaV1.7 expression with poor prognosis, as well as correlation with the expression of the NHE1 exchanger type 1 and the oncoprotein metastasis associated in colon cancer-1 (MACC1). In addition, NaV1.7 suppression resulted in reduced invasion and proliferation rates of GC cells and growth of GC xenografts in nude mice (Xia et al., 2016). In brief, results of this study indicate that NaV1.7 promotes GCa progression through MACC1-mediated upregulation of NHE1.
NaVα and NaVβ subunits in cervical cancer
The product of the SCN8A gene, the NaV1.6 channel, has been shown to be upregulated in cervical cancer (CeCa). In a study performed by using primary cultures derived from three different patient CeCa biopsies, the presence of functional NaV channels has been identified and INa recorded. Primary cells from CeCa biopsies expressed mRNA for different TTX-sensitive NaVα subunits: NaV1.1–1.4, NaV1.6, and NaV1.7 (Diaz et al., 2007). Among these, only the SCN8A gene encoding for NaV1.6 was shown to be overexpressed by about 40-fold at the mRNA level in CeCa primary cultures and biopsies in comparison with non-cancerous cervical tissue. The functional relevance of this NaV channel was demonstrated by blocking its activity with TTX as well as with the Cn2 specific toxin, which in both cases led to a significant decrease in the invasion capacity of CeCa primary culture cells, without affecting proliferative or migratory cell behavior (Hernandez-Plata et al., 2012). This suggested a role for NaV1.6 in extracellular matrix degradation, and indeed NaV1.6-mediated invasiveness of CeCa cells specifically involved MMP-2 activity along with increased expression of the NHE1 exchanger (Lopez-Charcas et al., 2018). In addition, CeCa cell lines more abundantly express the mRNA for the NaV1.6 variant, which has exon 18 deleted (Δ18 variant) rather than the neonatal and adult splice variants. This variant appeared to be distributed in intracellular compartments (Lopez-Charcas et al., 2018). However, the functional relevance of the Δ18 variant to the metastatic behavior of CeCa cells remains to be elucidated. Another interesting question regarding the expression of this Δ18 variant of NaV1.6 in CeCa cells is whether it has the same function in intracellular compartments as observed in macrophages and melanoma cells in which the channel has a role in podosome formation and activity (Carrithers et al., 2009).
The role of NaVβ1 as a migration suppressor gene was demonstrated in three different CeCa cell lines (HeLa, SiHa, and CaSki) in which SCN1B mRNA levels were around 3- to 6-fold higher than those of NaVβ2, NaVβ3, or NaVβ4. However, differences in protein levels among the four NaVβ subunits were more discrete; NaVβ1 was again the most highly expressed in HeLa and CaSki cells, whereas in SiHa cells, protein levels for all NaVβ were more uniform (Sanchez-Sandoval and Gomora, 2019). Previously, the same group had demonstrated that NaVβ1 mRNA levels were also slightly higher in CeCa biopsies than in non-CeCa tissue (Hernandez-Plata et al., 2012). In addition, it was demonstrated that NaVβ1 regulated SiHa cell proliferation, specifically by affecting the proportion of cells in the G0/G1 phase of cell cycle (Sanchez-Sandoval and Gomora, 2019). Because NaVβ3 was proposed to have anti-cancer properties (Adachi et al., 2004), the effect of its expression in CeCa cells was tested. However, neither its overexpression nor its downregulation affected proliferation in CeCa cell lines, suggesting that the likely pro-apoptotic activity of NaVβ3 might not be a generalized mechanism in all cancer types or cells. In this regard, it has been suggested that the p53 protein status in CeCa cell lines is under the control of the E6 protein, the main oncogene expressed as a result of human papillomavirus (HPV) infection (the most frequent risk factor for CeCa incidence) of cervical epithelial cells. The early expression of E6 protein leads to the specific ubiquitination and degradation of p53 (Scheffner et al., 1993), thereby inactivating any pro-apoptotic effect due to the NaVβ3 expression in basal conditions. In line with this interpretation, SCN3B expression was increased almost 2-fold in CeCa biopsies when compared with non-cancer samples (Hernandez-Plata et al., 2012). Further studies are needed to fully understand the potential role of SCN3B as well as the mechanism involved in the pro-apoptotic effect in cancer cells. More recent observations in CeCa cell lines confirm the contribution of NaVβ4 to cell invasive potential, as the downregulation of SCN4B leads to an increase in the percentage of invading cells in three CeCa cell lines (Sanchez-Sandoval and Gomora, 2019). However, a previous study indicated that mRNA levels for NaVβ4 were not significantly different between CeCa and non-CeCa biopsies tissues (Hernandez-Plata et al., 2012).
NaVα subunits in ovarian cancer and endometrial cancers
In ovarian cancer (OCa), the NaV1.5 isoform appears to be the main NaVα subunit expressed and contributing to the migration and invasion capabilities of cancer cells (Gao et al., 2010; Liu et al., 2018); however, the splicing status of NaV1.5 in this carcinoma is currently unknown.
In endometrial cancer tissues, a recent study identified the SCN9A gene, encoding for the NaV1.7 channel, as being the most highly expressed NaVα subunit. NaV1.7 expression level was associated with tumor size, local lymph node metastasis, and 5- and 10-year survival. Pharmacological inhibition using the PF-05089771 blocker selective for NaV1.7 and NaV1.8 induced cancer cell apoptosis and reduced cancer cell invasion (Liu et al., 2019).
NaVβ in papillary thyroid cancer
Recent results obtained in papillary thyroid cancer (PTC) show that SCN4B is downregulated at both RNA and protein level as compared with normal thyroid tissues (Gong et al., 2018). Importantly, by using databases such as the Gene Expression Omnibus (GEO) and the Cancer Genome Atlas (TCGA)-Thyroid Cancer (THCA), the authors found that SCN4B expression was an independent indicator of favorable recurrence-free survival (RFS) in patients with classical PTC, further contributing to the notion of the SCN4B as a metastases-suppressor gene (Gong et al., 2018). So far, nothing is known about the expression of NaVα subunits in PTC.
NaVα and NaVβ subunits in leukemia cells
Although most results related to NaV in cancer were obtained from solid tumors, predominantly carcinomas, there are also some indications that NaV expression might also be dysregulated in hematological disorders such as leukemia, in which they could bear oncogenic properties. In Jurkat leukemic T cell lymphoblasts, original evidence showed that a small fraction of ∼10% displayed INa and mRNA encoding for NaV1.5, NaV1.6, and to a lesser extent NaV1.7 and NaV1.9, were detected (Fraser et al., 2004). INa was likely carried mostly by a TTX-resistant NaV channel because an IC50 of ∼1 μM was measured. Importantly, invasion was reduced by 93% when the cells were treated with 10 μM TTX (Fraser et al., 2004). However, more recent data have shown that NaV1.6, NaV1.7, and NaV1.3 (in that order) are the most abundant NaV isoforms in three acute lymphocytic leukemia cell lines, including Jurkat, MOLT-4, and BALL-1 cells, as well as in peripheral blood mononuclear cells (PBMC). In this study, INa recorded from approximately 20% of MOLT-4 cells was completely abolished by 2 μM TTX, indicative of TTX-sensitive channels. The same concentration of TTX decreased the invasion of MOLT-4 and Jurkat cells by 90% (Huang et al., 2015).
Interestingly, semi-quantitative PCR results indicated the presence of both the neonatal (18N) and the Δ18 (exon 18 skipped) isoforms of NaV1.6 channel in the THP-1 monocytic leukemia cell line (Carrithers et al., 2009). Neither of these two variants form functional channels at the plasma membrane (Plummer et al., 1997). Instead, the Δ18 NaV1.6 channel isoform is expressed in vesicular intracellular compartments and crucially contributes in the control of podosome and invadopodia formation (Carrithers et al., 2009). In addition, SCN5A (NaV1.5) is expressed in the late endosome, rather than at the plasma membrane of the THP-1 cells. The intracellular NaV1.5 channel was shown to enhance endosomal acidification and phagocytosis (Carrithers et al., 2007), Ca2+ signaling, and phenotypic differentiation in human macrophages (Carrithers et al., 2011). The same group later demonstrated that SCN5A was expressed as a new splice variant lacking exon 25, resulting in a deletion of 18 amino acids in domain III (Rahgozar et al., 2013), generating non-selective outward currents and small inward currents in a heterologous expression system (Jones et al., 2014).
NaVα in Ewing sarcoma
The Ewing sarcoma (ES) is the second most common primary malignant bone tumor in children and adolescents, following osteosarcoma (Choi et al., 2014). RING1B, a member of the polycomb family of epigenetic regulators, is highly expressed in primary ES tumors. Depletion of RING1B with shRNA in ES cells enriched the expression of genes involved in hematological development, without affecting cellular differentiation (Hernandez-Munoz et al., 2016). Importantly, in ES cells, RING1B directly binds to the promoter of SCN8A, and its depletion results in enhanced NaV1.6 expression and function. In addition, the migratory speed of RING1B-depleted ES cells was attenuated, suggesting an inverse correlation between SCN8A expression and the migration capabilities of ES cells. Finally, reduced NaV1.6 function appeared to protect ES cells from apoptosis by a mechanism that maintains low NF-κB levels (Hernandez-Munoz et al., 2016). These findings revealed striking differences in the participation of SCN8A and its product, the NaV1.6 channel, in sarcomas compared with carcinomas and leukemia. Indeed, NaV1.6 appeared to have anti-cancer properties in ES, whereas it has pro-invasive functions in carcinomas and leukemia. Therefore, further studies are needed to fully understand the function of SCN8A across different types of cancer.
Conclusions on the roles of NaVα and NaVβ subunits in cancers
Pore-forming NaVα subunits
As previously indicated, the three main NaVα-encoding genes found to be upregulated in cancers are SCN5A, SCN8A, and SCN9A, which encode NaV1.5, NaV1.6, and NaV1.7, respectively. On the other hand, recent reports showed the downregulation of SCN8A and SCN9A genes in some cases. The molecular determinants explaining why these specific isoforms are overexpressed in cancers are not known and might be tissue specific. However, it is tempting to consider the deregulation in tumors of transcription factors that normally restrict the expression of a suite of genes associated with specific tissue functioning in adult tissues, such as the repressor element silencing transcription factor (REST) that restrict the expression of NaVα channels in excitable cells (Bruce et al., 2004; Chong et al., 1995) or other epigenetic regulations such as histone acetylation/deacetylation (performed by Histone Acetylases HAT and Histones Deacetylases HDAC, respectively), DNA, or histone methylation. Indeed, it was recently proposed that REST and HDAC2 play important role as epigenetic regulators and their inhibition in MCF-7 breast cancer cells enhanced the expression of NaV1.5 and promoted invasive capacities (Kamarulzaman et al., 2017). Nevertheless, it is interesting to notice that, when specifically studied in cancer cells, several neonatal splice variants of channels have been identified (Fraser et al., 2005; Lopez-Charcas et al., 2018; Baptista-Hon et al., 2014; Carrithers et al., 2009), thus supporting the common hypothesis of the re-expression of developmental genes in cancers.
These channels are present at the plasma membrane of cancer cells where sodium currents have been recorded. With the exception of NaV1.6 in Ewing sarcoma cells (Hernandez-Munoz et al., 2016), all NaVα isoforms have been shown to bear oncogenic properties, promoting cancer cell invasion in vitro, as well as other behaviors associated with metastasis, such as the acquisition of elongated and mesenchymal-like phenotypes, directed migration, proliferation, regulation of endocytosis, control of intracellular and perimembrane pH, and extracellular matrix degradation (Yang et al., 2012; Hernandez-Plata et al., 2012; Roger et al., 2003; Grimes et al., 1995; Fraser et al., 2005; Gillet et al., 2009; Mycielska et al., 2003; Djamgoz et al., 2001; Brisson et al., 2013). In addition, NaVα subunits promote tumor growth, invasion, and metastasis in in vivo rodent models (Nelson et al., 2015b; Driffort et al., 2014; Batcioglu et al., 2012; Yildirim et al., 2012). Comparative studies performed in different cancer types indicate the involvement of these NaVα isoforms in similar functional properties, arguing for isoform-independent signaling pathways. The activity of the channels at the plasma membrane appears to be critical. Indeed, Navα subunits are functionally active in cancer cell lines and primary tumor cells cultured in vitro, as well as in murine tumor xenograft tissue slices in vivo (Fraser et al., 2005; Roger et al., 2003; Hernandez-Plata et al., 2012; Nelson et al., 2015b), and their inhibition, using different drugs and small molecules such as TTX, ranolazine, phenytoin, Cn2 or PF-05089771, inhibits invasion (Nelson et al., 2015a, 2015b; Driffort et al., 2014; Batcioglu et al., 2012; Yildirim et al., 2012; Lopez-Charcas et al., 2018; Roger et al., 2003, 2007; Liu et al., 2019).
Importantly, the membrane potential (Vm) of cancer cells is typically relatively depolarized compared with terminally differentiated non-cancer cells (Yang and Brackenbury, 2013). At this range of Vm, NaVα channels would be expected to be predominant in the inactivated state. However, in cancer cells the Vm is generally between −40 and −30 mV and is situated in a window of voltage that provides a small non-inactivating persistent Na+ current flowing into the cell, locally increasing intracellular Na+ concentration (Yang et al., 2012; Roger et al., 2003; Campbell et al., 2013). Recently a Na+-dependent intracellular signaling pathway, involving Salt-inducible kinase 1, has been proposed to account for pro-invasive effects of NaVα (Gradek et al., 2019).
The main role attributed to Na+ is to serve as a mere mediator of the membrane potential, in excitable as well as in non-excitable cells. It is also characterized to support ion (among which Na+/K+, Na+/Ca2+, Na+/K+/Cl−, Na+/HCO3−) exchanges and nutrient/metabolite transports across membranes (Na+/glucose for example). The role of second messenger is mostly attributed to the Ca2+ ion, for which a lot of specific probes and tools have been developed over the last 20 years. In contrast, no direct and specific biological sensors for Na+ have been identified, and tools to study Na+ evolution still lack sensitivity or dynamics. Yet, there is some evidence suggesting that Na+ could act as a second messenger per se and might regulate several important signaling pathways in normal cells. Indeed, recent data support a direct role of Na+ in controlling kinases activity (Jaitovich and Bertorello, 2010), membrane fluidity, and protein diffusion through an interaction with phospholipids (Hernansanz-Agustin et al., 2020) or to induce inflammatory stress (Amara et al., 2016). Therefore, this raises the possibility that Na+ could also serve as a second messenger in cancer cells to activate signaling pathways promoting aggressiveness. To further support this hypothesis, it is worth mentioning that early studies questioned the involvement of intracellular Na+ content and the consequences on malignant cell proliferation, invasive capacities, and the development of metastases (Cone, 1974). Indeed, much higher Na+ concentrations have been recorded in tumor cells, as compared with non-cancer cells by energy-dispersive X-ray microanalyses (Cameron et al., 1980) as well as by 23Na-magnetic resonance imaging (Ouwerkerk et al., 2007; Jacobs et al., 2004; Zaric et al., 2016) and was proposed to serve as an indicator of malignancy.
The inward Na+ current may also further depolarize the Vm, which might also participate in promoting migration. In support of this hypothesis, it was demonstrated in breast cancer cells that NaV1.5 sustained Vm depolarization, which activated the RhoGTPase Rac1, subsequently inducing cytoskeletal reorganization and cellular migration (Yang et al., 2020) (Figure 2). Although there is clear evidence that NaVα channels expressed at the plasma membrane are critical in the acquisition of oncogenic properties, the discovery of splice variants with expression restricted to intracellular compartments, such as endosomes, phagosomes, or lysosomes (Lopez-Charcas et al., 2018; Carrithers et al., 2009), suggests a more complex role.
(More than) auxiliary NaVβ subunits
The main NaVβ-encoding gene found to be upregulated in cancers is SCN1B, encoding for the NaVβ1 subunit (Diss et al., 2008; Nelson et al., 2014; Bon et al., 2016; Sanchez-Sandoval and Gomora, 2019). On the other hand, there are multiple reports showing the downregulation of SCN4B (NaVβ4) (Hernandez-Plata et al., 2012; Bon et al., 2016; Diss et al., 2008; Gong et al., 2018; Sanchez-Sandoval and Gomora, 2019) and of SCN3B (NaVβ3) in some instances (Bon et al., 2016; Adachi et al., 2004).
The NaVβ1 subunit has been shown to increase cancer proliferation, cell adhesion, increase neurite-like process outgrowth formation, and promote cancer cell invasion, while slowing migration in vitro (Nelson et al., 2014; Chioni et al., 2009; Bon et al., 2016; Sanchez-Sandoval and Gomora, 2019). In vivo, NaVβ1 overexpression increases angiogenesis and reduces apoptosis, thus increasing tumor growth and metastasis (Nelson et al., 2014). Taken together, these results are in favor of a pro-cancerous role of NaVβ1, and the effects appear to be dependent in part on the regulation of the NaVα pore-forming subunit as well as on the extracellular CAM motif (Nelson et al., 2014). NaVβ2 expression in prostate cancer cells also increases process extension, adhesion, invasion, and migration in vitro but reduces tumor take in vivo (Jansson et al., 2012, 2014). Conversely, NaVβ3 and NaVβ4 may function as tumor suppressors. The SCN3B gene contains p53 response elements, and NaVβ3 suppresses colony formation and promotes chemotherapy-induced apoptosis in a p53-dependent manner (Adachi et al., 2004). NaV β4 expression is downregulated in breast, colorectal, lung, cervical, and prostate tumors and papillary thyroid cancer compared with normal tissue (Hernandez-Plata et al., 2012; Bon et al., 2016; Diss et al., 2008; Gong et al., 2018; Sanchez-Sandoval and Gomora, 2019). In addition, NaVβ4 functions as a tumor and metastasis suppressor gene in vivo (Bon et al., 2016). This tumor-suppressing function occurs via β4-mediated control of RhoA GTPase activation (Bon et al., 2016) (Figure 2).
NaVα as anticancer targets for repurposed drugs and new small inhibitory molecules
NaVα are attractive drug targets because of the broad therapeutic potential of their blockers. Considering the fact that NaVα are expressed in metastatic cells in various tumors, significant effort has been made to develop NaVα blockers as potential drugs for cancer treatment. This section of the review focuses on such efforts that took place in the past 10 years. These efforts for blocker development can broadly be classified into two sections: (1) repurposing drugs that are FDA approved for other clinical uses (local and general anesthetics, antiepileptic and anticonvulsant, antiarrhythmic drugs); (2) rational design and development of novel NaVα blockers for cancer treatment.
Repurposing of FDA-approved NaVα blockers
There are numerous existing NaVα inhibitors licensed for clinical use. In several cases NaVα inhibition is considered an off-target effect of these drugs. For example, tricyclic antidepressants, including amitriptyline, inhibit not only the serotonin transporter but also several neurotransmitter receptors and voltage-activated ion channels including NaVα. In other cases NaVα inhibition is considered the primary mechanism of the drug's intended therapeutic effect. This is true for several anti-seizure medications (e.g. phenytoin and carbamazepine) and all of the local anaesthetics, although these too have additional off-target actions. Regardless of whether it is a primary or secondary effect of a licensed medication, NaVα inhibition might be beneficial in patients with cancers associated with NaVα expression. This raises the intriguing possibility that approved NaVα-inhibiting drugs might be repurposed to treat cancer.
Benefits of repurposing approved medications include prior knowledge of their mechanisms of action and the availability of toxicology and safety data, thereby avoiding the need for drug discovery and early phase clinical trials. Drawbacks include limited potential for developing intellectual property, leading to a lack of both funding potential and industry involvement (Pushpakom et al., 2019). Nevertheless, despite the potential drawbacks, there are some notable successes, and a well-trodden pathway to repurposing is through the use of electronic health records to link prescribing data to potentially beneficial health outcomes. A good example of the impact of this type of retrospective clinical analysis is the identification of an association of aspirin use with reduced risk of colon cancer (Dube et al., 2007). Similar approaches are being used in studies exploring a possible relationship between NaVα inhibitors and outcomes in cancer patients.
Anti-seizure and class 1 antiarrhythmic NaVα inhibitors
A recent study, using retrospective clinical analysis to explore the possibility of a beneficial effect of NaV inhibiting medications, examined several class 1 antiarrhythmic and antiseizure medications in patients with breast, bowel, or prostate cancer (Fairhurst et al., 2015). The combined analysis revealed that these medications (including class I antiarrhythmic drugs, lamotrigine, carbamazepine phenytoin, and valproate) were collectively associated with decreased median time to death compared with the control patient group, with significantly increased mortality in the drug group. This study clearly does not support the idea of repurposing antiseizure NaVα inhibitors in the treatment of breast, bowel, or prostate these cancers. However, as the authors pointed out, the causes of death were not available in the large primary care dataset, and co-morbidities were among the likely confounding factors. In many cases, patients treated with NaVα-inhibiting drugs will be suffering from life-threatening disorders such as epilepsy, and it is difficult to completely accommodate this confound in retrospective analyses.
Analgesic NaVα inhibitors
There has been considerable recent interest in the idea that anesthetics and analgesics used during surgical tumor excision might influence subsequent cancer recurrence. Surgery can cause the release of tumor cells into the circulation, and the number of postsurgical circulating cancer cells is known to be a negative prognostic indicator of disease-free survival (Yu et al., 2018). The perioperative period, i.e. immediately before, during, and after surgery, may therefore be an opportune time for interventions that inhibit the potential for metastatic invasion. A variety of drugs are typically administered during surgery including general anesthetics, analgesics, and anti-muscarinic and neuromuscular blockers. Some of these, such as inhalational general anesthetics, may have the potential to worsen outcomes by suppressing the immune response (Stollings et al., 2016). By contrast, local anesthetics may provide more favorable outcomes. Local anesthetics are often administered regionally to provide blockade of afferent nociceptive fibers entering the spinal cord. Several retrospective clinical studies suggest that regional analgesia during breast and prostate cancer surgery increases disease-free survival (Forget et al., 2019). The use of regional anesthesia diminishes or abolishes the need for general anesthetic during surgery. It was therefore initially hypothesized that the general anesthetic sparing effect of regional analgesia with local anesthetics accounts for the apparent beneficial effect (Sessler et al., 2008). However, a recent large prospective multicenter trial comparing outcomes after breast cancer surgery under inhalational anesthesia with or without paravertebral analgesia by ropivacaine or levobupivacaine revealed no difference in disease-free survival (Sessler et al., 2019).
Most of the ongoing clinical trials exploring the impact of anesthetic technique on cancer outcomes are predicated on the idea that the potential benefit of local anesthetics is conferred indirectly through their inhalational anesthetic sparing effect. However, it is possible that local anesthetics such as lidocaine, ropivacaine, and levobupivacaine provide a direct beneficial effect through NaVα inhibition (Baptista-Hon et al., 2014; Elajnaf et al., 2018). If this is the case, then a more direct approach for administering these drugs directly onto the tumor may prove to be beneficial. Lidocaine, in addition to being a local anesthetic, is also used intravenously as a class 1b antiarrhythmic agent and a circulating analgesic. Ongoing clinical trials will test whether lidocaine delivered directly onto breast tumors prior to excision or intravenously during the perioperative period for colon cancer surgery will prolong postoperative disease-free survival (NCT01916317, R.A Badwe, 2013; NCT02786329, D. Ionescu, 2016). We await the outcome of these trials with interest and note that there are several other approved NaVα-inhibiting drugs that should be examined in retrospective clinical studies for potential beneficial effects in cancer outcomes.
Rational design of small molecule NaVα blockers
Rational designing of NaVα inhibitors has been difficult because detailed structural information of drug-binding sites for this integral membrane protein were lacking until very recently. Therefore, early effort for the discovery of NaVα blockers mainly relied on strategies such as ligand-based drug design, natural-product-based drug design, in silico screening, and similarity searches. However, recent reports on the structures of human and bacterial NaVα and bound ligands shed light on their binding site (Cervenka et al., 2018; Nguyen et al., 2019; Pan et al., 2018; Payandeh et al., 2011; Shen et al., 2017, 2018, 2019). These reports will certainly aid in the structure-based design and discovery of NaVα blockers. A summary of the available reports on the identification and evaluation of NaVα blockers for potential use in cancer therapy follows.
One such effort to identify NaVα blockers used a pyrrole-imidazole marine alkaloid, clathrodin (Hodnik et al., 2013). Clathrodin was originally isolated from the Agelas clathrodes sponge. Several conformationally restricted analogues of clathrodin containing a 4,5,6,7-tetrahydrobenzo [d] thiazol-2-amine moiety are blockers of NaV1.3, NaV1.4, NaV1.5, and NaV1.7 channels. These compounds display state-dependent inhibitory activity of these channels at low micromolar concentrations. The most active compound (4e, Figure 3) identified from this study represents a novel selective blocker of NaV1.4 channel with an IC50 value of 8 μM. The use of clathrodin analogues as a template for ligand-based virtual screening of commercially available ZINC library of compounds using ROCS software identified two potent lead compounds 2 and 16 (Tomasic et al., 2013) (Figure 3). These blocked INa produced by NaV1.7 with IC50 values of 7 μM and 9 μM, respectively.
Figure 3.

Chemical structures of known NaVα blockers with anticancer effects
Plant-derived polyphenolic natural products have also been reported as NaVα inhibitors. For example, the plant phenolic, resveratrol (Figure 3), found at high concentrations in red grapes inhibits NaVα with an IC50 value of 50 μM (Fraser et al., 2014). Resveratrol also suppresses lateral cell motility by up to 25%, transverse cell motility by 31%, and cell invasion by 37%, without affecting cellular proliferation or cell viability of MAT-LyLu cells. Another similar phenolic is caffeic acid phenethyl ester (CAPE, Figure 3) isolated from honeybee propolis. CAPE blocks NaV activity in several invasive cancer cell lines, including breast (MDA-MB-231 and MDA-MB-468), colon (SW620), and non-small cell lung cancer (H460). Motility and invasion of MDA-MB-231 cells were reduced by up to 14% and 51%, respectively by CAPE at 1 μM without affecting cell proliferative activity (Fraser et al., 2016).
Shaheen et al. used an in silico approach to identify NaVα blockers with superior pharmacological profile compared with phenytoin (PHT) and carbamazepine (CBZ) in 2015 (Shaheen et al., 2015). They conducted a similarity search in the PubChem database with PHT and CBZ as query molecules using the Tanimoto-based similarity search. The search was further refined by docking of these molecules into the binding site of the homology model of SCN1A using MolDock program. This study identified high-affinity compounds similar to PHT and CBZ. The lead compounds were further evaluated for toxicity profiles and biological activity. Two of the best compounds identified by this study, NSC403438 and AGN-PC-0BPCBP (Figure 3), demonstrated better binding affinity to NaVα compared with PHT and CBZ, with NSC403438 being a superior inhibitor of INa with lower toxicity, better IC50 value, and optimal bioactivity.
NaVα inhibitors were also derived from the natural product, crambescin (Nakazaki et al., 2016). Enantiomerically pure crambescin A, B, and C carboxylic acid derivatives were synthesized and evaluated for their ability to block NaVα. Structure activity relationship studies revealed that the natural enantiomer of crambescin B, carboxylic acid (Figure 3), is the most active compound with activity comparable to TTX. The cyclic guanidinium moiety present in this molecule is indispensable for its activity.
In 2018, Dutta et al. utilized a highly predictive, comprehensive 3D-QSAR model for the design of NaVα blockers (Dutta et al., 2018). The NaVα-binding data (IC50) for 67 compounds were used to train a comprehensive CoMFA model, which effectively covered 3D space and spanned over 4 orders of magnitude in biological activity. Potency predictions by this model have been highly accurate for more than 30 compounds that were synthesized and evaluated. Five compounds shown or predicted to have low nanomolar NaVα binding were further evaluated for the inhibition of hNaV1.5e currents in individual breast cancer MDA-MB-231 cells. Of these, two lead compounds, 1 and 4 (Figure 3), were found to be most effective in whole-cell patch-clamp studies and showed significant invasion inhibitory activities at concentrations as low as 1 μM without affecting cell viability.
Boezio et al. reported several sulfonamides with highly selective NaV1.7 blockade activity (Boezio et al., 2018). This novel series of blockers contained a triazole sulfone, which served as a bioisostere for the acyl sulfonamide group. This work resulted in the discovery of a series of potent NaV1.7 blockers with selectivity over NaV1.5 and favorable pharmacokinetic properties in rodents. An example of such a blocker is compound 35 (Figure 3). In the same year, Yapa et al. reported a known inhibitor of TRPM8, N-(3-aminopropyl)-2-{[(3-methylphenyl) methyl]oxy}-N-(2-thienyl methyl)benzamide (AMTB, Figure 3), as a NaVα blocker in breast cancer MDA-MB-231 cells (Yapa et al., 2018). AMTB decreased viable cell number in MDA-MB-231 and SK-BR-3 breast cancer cell lines and also reduced the migration of MDA-MB-231 cells. These studies provided the evidence that these effects are not related to TRPM8 inhibition but rather caused by the NaVα blockade caused by AMTB. Gumushan Aktas et al. investigated the potential effects of a natural flavanone, naringenin (Figure 3), on the motility of MAT-LyLu cells in 2018 (Gumushan Aktas and Akgun, 2018). This study revealed that naringenin inhibited cell proliferation at higher concentrations (75 μM), whereas it decreased the movement of MAT-LyLu cells at low concentrations (5 μM and 10 μM). Moreover, naringenin inhibited cell motility by reducing the expression of the SCN9A gene at the mRNA level. In conclusion, naringenin was found to have direct or indirect blocking activity on the SCN9A-encoded channel. Most recently, in 2019, Wang et al. has reported NaV1.7 channel blockers using a comparative molecular field analysis (CoMFA) model for the binding of ligand to NaVα, generated based on diverse set of compounds. No channel current blockade data were presented in the paper. However, there was an extensive anticancer evaluation of the identified lead compounds S0154 and S0161 (Wang et al., 2019). Both showed anticancer and anti-metastatic effects against PC3 prostate cancer cells and significantly inhibited cell viability, with IC50 values in the range of 5–26 μM. Both these compounds inhibited the expression of NaVα, increased the intracellular level of Na+, and caused cell-cycle arrest in G2/M phase. The compounds also inhibited the invasion of PC3 cells. Furthermore, S0161 inhibited the PC3 tumor growth by about 51% in an in vivo xenograft model (Wang et al., 2019).
In conclusion, there have been major advances in NaVα-targeted drug discovery over the last decade. Specific NaVα isoforms have been implicated in the metastasis development of a variety of tumors raising the possibility of developing tumor selective drugs. Recent advances in the discovery of high-resolution crystal and cryoEM structures of NaVα should further advance the field with structure-based drug design efforts.
NaVα as targets for nutritional management of cancers
Cancer is a metabolic disease that depends on bioenergetic parameters (Penkert et al., 2016). Cancer progression is generally associated with the survival of cancer cells under conditions of low oxygen levels and nutrient deprivation and relies on metabolic adaptations (Dumas et al., 2017; Hanahan and Weinberg, 2011). These metabolic adaptations allow cancer cells to survive the pressure of environmental conditions and to fulfill the high energy demands associated with their high anabolic activity (Payen et al., 2016; Porporato et al., 2016). Also, this metabolic switch toward an aerobic glycolysis brings selective advantages by promoting invasive activities and metastatic properties (Brisson et al., 2012; Webb et al., 2011). Furthermore, the metabolic reprogramming concerns not only tumor cells but also multiple cell types and organs of the host, thus leading to an overall deregulation of the energetic balance in patients, called tumor cachexia (Fearon et al., 2012). This devastating syndrome, initially triggered by the release of soluble tumor factors and the participation of a systemic inflammation, is characterized by anorexia, the loss of skeletal muscle mass, in some cases with the loss of adipose tissue mass, and a general weakening of patients, impeding their quality of life and decreasing the tolerance to antineoplastic therapies (Fonseca et al., 2020; Biswas and Acharyya, 2020). In this context, bringing a nutritional support to patients is required to allow holding the most efficient possible treatment. Nutritional interventions are mostly aimed at preventing the wasting of body compartments in patients but could also be a source of active anti-cancer molecules. Furthermore, diet represents a controllable component of the environment and brings promising strategies to increase treatment efficacy in combination with conventional chemotherapeutics. Some dietary compounds have also been shown to decrease the risk of carcinoma development and may prolong the survival of patients (Dumas et al., 2017).
As pointed out in the previous section, several natural compounds in the diet, such as resveratrol and caffeic acid phenethyl esters, are effective at inhibiting NaVα subunits (Fraser et al., 2014, 2016). Dietary lipids have also been proposed to inhibit NaVα and modulate cancer progression. Indeed, dietary lipids incorporate into cellular membrane and alter NaVα or their pharmacology (Agwa et al., 2018; D'avanzo, 2016). Among dietary lipids, long-chain n-3 polyunsaturated fatty acids (n-3 PUFA) have been described in epidemiological studies to delay or prevent the appearance of breast cancer (Rose et al., 1996; Bougnoux et al., 2010). From both in vivo and in vitro studies, n-3 PUFA have been reported to induce multiple anti-tumour effects, and their dietary consumption was associated with a lower risk of cancers, such as breast or colorectal cancers (Bougnoux, 1999; Bougnoux et al., 2010; Eltweri et al., 2016). Even though n-3 PUFA were suggested to prevent prostate cancer (Moreel et al., 2014; Li et al., 2017), their beneficial effect remains to be demonstrated through more intervention trials or observational studies (Aucoin et al., 2016). A pilot study showed that fatty acid composition of breast adipose tissue differed according to breast cancer focality: low levels of the two long-chain n-3 PUFA docosahexaenoic acid (DHA, 22:6n-3) and eicosapentaenoic acid (EPA, 20:5n-3) were associated with tumor multifocality, which is considered a marker of cancer aggressiveness (Ouldamer et al., 2016a). These results could indicate that differences in adipose tissue concentration, a surrogate of past dietary uptake, may contribute to mechanisms influencing cancer progression.
Long-chain n-3 PUFA have been proposed to increase tumor sensitivity to chemotherapeutic agents with no sensitization of normal tissues and no additional side effect (Bougnoux et al., 2010). As such, DHA and EPA have generated intense interest due to their ability to reduce resistance to anthracyclines, taxanes, or radiotherapy in mammary tumor models (Bougnoux et al., 2010; Hajjaji et al., 2011, 2012; Ouldamer et al., 2016b).
N-3 PUFA have also been shown to have anti-invasive and anti-metastatic properties (Blanckaert et al., 2010; Gillet et al., 2011; Rose et al., 1994, 1997; Bougnoux et al., 2010). On the other hand, they are capable of modulating the activity of NHE exchangers (Besson et al., 1996; Lacroix et al., 2008) and ion channels (Tillman and Cascio, 2003; Jude et al., 2003). Interestingly, in expression systems and in native rat cardiomyocytes, the activity of NaV1.5 was initially found to be inhibited by n-3 PUFA (Kang and Leaf, 1996; Kang et al., 1997) and in initial studies have proposed that these effects could be mediated by a direct binding of n-3 PUFA to specific residues of the channel (Xiao et al., 1998, 2001). Therefore, n-3 PUFA could also exert their beneficial effects on cancers through a reduction of NaV1.5 (Pignier et al., 2007; Gillet et al., 2011). However, contrasting results were obtained in human breast cancer cells in which INa was not inhibited by acute applications of n-3 PUFA, even at high concentrations (30–50 μM) (Wannous et al., 2015) at which they also have anti-proliferative effects (Barascu et al., 2005). This discrepancy might be due to the fact that cancer cells mostly express the hNaV1.5e neonatal splice variant (Fraser et al., 2005). However, growing breast cancer cells in the presence of low doses of DHA (0.5–10 μM) reduced SCN5A gene expression and levels of NaV1.5 proteins and INa (Wannous et al., 2015; Isbilen et al., 2006). This inhibition of SCN5A expression was mediated by the lipid-sensitive nuclear receptor peroxisome proliferator-activated receptor β (PPAR-β). Correlatively, the inhibition of NaV1.5 activity was also responsible for a reduced activity of the downstream protagonist NHE-1, thus decreasing H+ efflux, preventing extracellular matrix degradation proteolytic activity, and inhibiting breast cancer cell invasiveness (Wannous et al., 2015). A recent report also demonstrated the efficacy of EPA to reduce migration and proliferation of ovarian cancer cells by inhibiting NaV1.5 (Liu et al., 2018).
Such regulations concerning other NaVα involved in cancer properties, i.e. NaV1.6 and NaV1.7, should be investigated. It is also of interest to note that n-3 PUFA, through the activation of PPAR-γ, have been shown to downregulate the expression of NHE-1 and reduce cancer colony growth (Kumar et al., 2009). Furthermore, incorporation of n-3 PUFA into phospholipids induces changes in the physico-chemical properties of cell membranes (Yaqoob and Shaikh, 2010), which in turn affects NHE-1 activity (Dendele et al., 2014).
Further to these effects on NaV channels and downstream signaling pathways, it should be mentioned that n-3 PUFA might exert a multiplicity of actions by interfering with several signaling pathways, some of them being beneficial to the prevention or treatment of cancer. As such, a lack of specificity is not obligatorily detrimental, and pleiotropic effects might increase the efficacy of the anticancer treatment.
N-3 PUFA supplementations were proposed to have beneficial effects in reducing mammary tumor growth, by slowing down cancer cell proliferation (Barascu et al., 2005). N-3 PUFA treatment was demonstrated to inhibit cyclin B1 and the expression of the cell division cycle 25C phosphatase, which dephosphorylates cyclin-dependent kinase 1 (Barascu et al., 2005). In addition, the nuclear receptor PPARβ appeared to regulate the DHA-related inhibition of MDA-MB-231 and MCF-7 cells proliferation. This allowed identifying PPARβ as an important protagonist in the inhibition of breast cancer cell proliferation and mammary tumor growth under DHA-enriched diet (Wannous et al., 2013). N-3 PUFA have been proposed to regulate autophagy in cancer cells and as such could be involved in both survival and apoptosis, depending on the carcinogenetic phase and the treatment context (Ferro et al., 2020). DHA was found to induce apoptosis in cancer cells (Jing et al., 2011). DHA-induced autophagy was associated with p53 loss, with the activation of AMPK and the decrease in the activity of mTOR. Autophagy inhibition suppressed apoptosis, and autophagy induction further enhanced apoptosis in response to DHA treatment (Jing et al., 2011). There is evidence that n-3 PUFA may inhibit the expression of EMT markers and reduce associated invasive properties in cancer cells (D'eliseo et al., 2016; Yin et al., 2016). Altogether, these effects could bring beneficial values to delay the appearance/diagnosis of a primary tumor and as such be of interest in primary prevention.
N-3 PUFA, which are highly peroxidizable, were also proposed to improve the efficacy of anticancer treatments by amplifying oxidative stress generated by anthracyclines or radiotherapy (Bougnoux et al., 2009). The acquisition of resistance to chemotherapeutic agents represents an important limitation in cancer. Resistance to taxanes has been proposed to depend on the induction of signaling pathways such as PI3K/Akt and ERK1/2, which promote survival and cell growth in human cancer cells. In docetaxel-treated MDA-MB-231 cells, phosphorylated-ERK1/2 levels were increased by 60% in both membrane and nuclear compartments, compared with untreated cells, and ERK1/2 activation depended on PKCε and PKCδ activation. In comparison, in cells treated with DHA, docetaxel was unable to increase PKCε and PKCδ levels, thus resulting in the reduction of ERK1/2 phosphorylation and the increase in docetaxel efficacy (Chauvin et al., 2016). In addition to these effects, n-3 PUFA were proposed to increase the efficacy of the chemotherapeutic treatment by remodeling the tumor vascular network, thereby improving the delivery of anticancer drugs within the tumor (Kornfeld et al., 2012). These results support the hypothesis that n-3 PUFA, which do not induce any toxic effect, could be used as an adjuvant to cancer therapy.
Conclusions and perspective for the treatment of cancers
There is now clear evidence that the abnormal expression of NaV subunits occurs during carcinogenesis and is associated with cancer progression toward metastatic states. Several different NaVα play a role, including NaV1.5, NaV1.6, and NaV1.7, but the intracellular signaling pathways they regulate appear to be the same or very similar in all studied cancer types, leading to the induction of invasive properties. The activity of such channels at the plasma membrane, and consequent INa, appear critical. Therefore, the inhibition of NaVα in cancer cells represents a new anti-cancer strategy that could be achieved in several ways alone or in combination: repurposing existing NaVα-inhibitory drugs, developing new small inhibitory molecules, and through dietary interventions. As shown above, NaVβ subunits also have important roles in cancer cell biology and in cancer progression, acting both as auxiliary subunits of NaVα and as CAMs. However, they do not exert a recordable activity per se, and their direct “inhibition” or “activation” would be challenging. Therefore, dietary strategies aiming at controlling their expression, i.e. reducing the expression of SCN1B and SCN2B, while maintaining the expression of SCN3B and SCN4B, might represent powerful strategies. For this purpose, future studies aiming at unraveling transcriptional and epigenetic regulators will be of high interest.
Acknowledgments
Authors declare no conflict of Interest. This consortium was supported by a grant from LE STUDIUM, Loire Valley Institute for Advanced Studies, Orléans & Tours, France (“Pharmacological and nutritional targeting of voltage-gated sodium channels in the treatment of epithelial cancers”, Grant number Y17C3). Research performed in S.R. lab was supported by the University of Tours, the “Ligue Nationale Contre le Cancer—Interrégion Grand-Ouest”, the Région Centre-Val de Loire, the “Association CANCEN”. S.R. was recipient of a prize “Prix Ruban Rose Avenir 2017” from the Charity “Ruban Rose”. O.L.-C. was recipient of a funding from the “Fondation pour la Recherche Médicale (FRM)” (SPF201909009198). WJB acknowledges funding from Cancer Research UK (A25922) and Breast Cancer Now (2015NovPhD572). J.C.G. research has been supported by CONACYT-México grant A1-S-19171 and PAPIIT-DGAPA-UNAM grant IN209820. S.E.V. acknowledges the funding from US National Institutes of Health (R21CA226491 and R21DE028349). TGH acknowledges the Stewart family for their generous support of research investigating ion channels in colon cancer. Figures 1 and 2 were created using BioRender.com.
Author contributions
All authors participated to the writing of the review, which was directed by SR.
Declaration of interests
Authors declare no competing interest.
References
- Adachi K., Toyota M., Sasaki Y., Yamashita T., Ishida S., Ohe-Toyota M., Maruyama R., Hinoda Y., Saito T., Imai K. Identification of SCN3B as a novel p53-inducible proapoptotic gene. Oncogene. 2004;23:7791–7798. doi: 10.1038/sj.onc.1208067. [DOI] [PubMed] [Google Scholar]
- Agnew W.S., Moore A.C., Levinson S.R., Raftery M.A. Identification of a large molecular weight peptide associated with a tetrodotoxin binding protein from the electroplax of Electrophorus electricus. Biochem. Biophys. Res. Commun. 1980;92:860–866. doi: 10.1016/0006-291x(80)90782-2. [DOI] [PubMed] [Google Scholar]
- Agwa A.J., Peigneur S., Chow C.Y., Lawrence N., Craik D.J., Tytgat J., King G.F., Henriques S.T., Schroeder C.I. Gating modifier toxins isolated from spider venom: modulation of voltage-gated sodium channels and the role of lipid membranes. J. Biol. Chem. 2018;293:9041–9052. doi: 10.1074/jbc.RA118.002553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara S., Ivy M.T., Myles E.L., Tiriveedhi V. Sodium channel gammaENaC mediates IL-17 synergized high salt induced inflammatory stress in breast cancer cells. Cell. Immunol. 2016;302:1–10. doi: 10.1016/j.cellimm.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold M., Sierra M.S., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–691. doi: 10.1136/gutjnl-2015-310912. [DOI] [PubMed] [Google Scholar]
- Aucoin M., Cooley K., Knee C., Fritz H., Balneaves L.G., Breau R., Fergusson D., Skidmore B., Wong R., Seely D. Fish-derived omega-3 fatty acids and prostate cancer: a systematic review. Integr. Cancer Ther. 2016;16:32–62. doi: 10.1177/1534735416656052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista-Hon D.T., Robertson F.M., Robertson G.B., Owen S.J., Rogers G.W., Lydon E.L., Lee N.H., Hales T.G. Potent inhibition by ropivacaine of metastatic colon cancer SW620 cell invasion and NaV1.5 channel function. Br. J. Anaesth. 2014;113:i39–i48. doi: 10.1093/bja/aeu104. [DOI] [PubMed] [Google Scholar]
- Barascu A., Besson P., Le Floch O., Bougnoux P., Jourdan M.L. CDK1-cyclin B1 mediates the inhibition of proliferation induced by omega-3 fatty acids in MDA-MB-231 breast cancer cells. Int. J. Biochem. Cell Biol. 2005;38:196–208. doi: 10.1016/j.biocel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Batcioglu K., Uyumlu A.B., Satilmis B., Yildirim B., Yucel N., Demirtas H., Onkal R., Guzel R.M., Djamgoz M.B. Oxidative stress in the in vivo DMBA rat model of breast cancer: suppression by a voltage-gated sodium channel inhibitor (RS100642) Basic Clin. Pharmacol. Toxicol. 2012;111:137–141. doi: 10.1111/j.1742-7843.2012.00880.x. [DOI] [PubMed] [Google Scholar]
- Beneski D.A., Catterall W.A. Covalent labeling of protein components of the sodium channel with a photoactivable derivative of scorpion toxin. Proc. Natl. Acad. Sci. U S A. 1980;77:639–643. doi: 10.1073/pnas.77.1.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergareche A., Bednarz M., Sanchez E., Krebs C.E., Ruiz-Martinez J., De La Riva P., Makarov V., Gorostidi A., Jurkat-Rott K., Marti-Masso J.F., Paisan-Ruiz C. SCN4A pore mutation pathogenetically contributes to autosomal dominant essential tremor and may increase susceptibility to epilepsy. Hum. Mol. Genet. 2015;24:7111–7120. doi: 10.1093/hmg/ddv410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson P., Gore J., Vincent E., Hoinard C., Bougnoux P. Inhibition of Na+/H+ exchanger activity by an alkyl-lysophospholipid analogue in a human breast cancer cell line. Biochem. Pharmacol. 1996;51:1153–1158. doi: 10.1016/0006-2952(96)00029-9. [DOI] [PubMed] [Google Scholar]
- Biswas A.K., Acharyya S. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer. 2020;20:274–284. doi: 10.1038/s41568-020-0251-4. [DOI] [PubMed] [Google Scholar]
- Black J.A., Waxman S.G. Noncanonical roles of voltage-gated sodium channels. Neuron. 2013;80:280–291. doi: 10.1016/j.neuron.2013.09.012. [DOI] [PubMed] [Google Scholar]
- Blanckaert V., Ulmann L., Mimouni V., Antol J., Brancquart L., Chenais B. Docosahexaenoic acid intake decreases proliferation, increases apoptosis and decreases the invasive potential of the human breast carcinoma cell line MDA-MB-231. Int. J. Oncol. 2010;36:737–742. doi: 10.3892/ijo_00000549. [DOI] [PubMed] [Google Scholar]
- Blandino J.K., Viglione M.P., Bradley W.A., Oie H.K., Kim Y.I. Voltage-dependent sodium channels in human small-cell lung cancer cells: role in action potentials and inhibition by Lambert-Eaton syndrome IgG. J. Membr. Biol. 1995;143:153–163. doi: 10.1007/BF00234661. [DOI] [PubMed] [Google Scholar]
- Boezio A.A., Andrews K., Boezio C., Chu-Moyer M., Copeland K.W., Dimauro E.F., Foti R.S., Fremeau R.T., Jr., Gao H., Geuns-Meyer S. 1,2,4-Triazolsulfone: a novel isosteric replacement of acylsulfonamides in the context of NaV1.7 inhibition. Bioorg. Med. Chem. Lett. 2018;28:2103–2108. doi: 10.1016/j.bmcl.2018.04.035. [DOI] [PubMed] [Google Scholar]
- Bon E., Driffort V., Gradek F., Martinez-Caceres C., Anchelin M., Pelegrin P., Cayuela M.L., Marionneau-Lambot S., Oullier T., Guibon R. SCN4B acts as a metastasis-suppressor gene preventing hyperactivation of cell migration in breast cancer. Nat. Commun. 2016;7:13648. doi: 10.1038/ncomms13648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougnoux P. n-3 polyunsaturated fatty acids and cancer. Curr. Opin. Clin. Nutr. Metab. Care. 1999;2:121–126. doi: 10.1097/00075197-199903000-00005. [DOI] [PubMed] [Google Scholar]
- Bougnoux P., Hajjaji N., Ferrasson M.N., Giraudeau B., Couet C., Le Floch O. Improving outcome of chemotherapy of metastatic breast cancer by docosahexaenoic acid: a phase II trial. Br. J. Cancer. 2009;101:1978–1985. doi: 10.1038/sj.bjc.6605441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougnoux P., Hajjaji N., Maheo K., Couet C., Chevalier S. Fatty acids and breast cancer: sensitization to treatments and prevention of metastatic re-growth. Prog. Lipid Res. 2010;49:76–86. doi: 10.1016/j.plipres.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Bouza A.A., Isom L.L. Voltage-gated sodium channel beta subunits and their related diseases. Handb Exp. Pharmacol. 2018;246:423–450. doi: 10.1007/164_2017_48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury W.J., Chioni A.M., Diss J.K., Djamgoz M.B. The neonatal splice variant of Nav1.5 potentiates in vitro invasive behaviour of MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2007;101:149–160. doi: 10.1007/s10549-006-9281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury W.J., Isom L.L. Na channel beta subunits: overachievers of the ion channel family. Front. Pharmacol. 2011;2:53. doi: 10.3389/fphar.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Brisson L., Driffort V., Benoist L., Poet M., Counillon L., Antelmi E., Rubino R., Besson P., Labbal F., Chevalier S. NaV1.5 Na+ channels allosterically regulate the NHE-1 exchanger and promote the activity of breast cancer cell invadopodia. J. Cell Sci. 2013;126:4835–4842. doi: 10.1242/jcs.123901. [DOI] [PubMed] [Google Scholar]
- Brisson L., Gillet L., Calaghan S., Besson P., Le Guennec J.Y., Roger S., Gore J. Na(V)1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H(+) efflux in caveolae. Oncogene. 2011;30:2070–2076. doi: 10.1038/onc.2010.574. [DOI] [PubMed] [Google Scholar]
- Brisson L., Reshkin S.J., Gore J., Roger S. pH regulators in invadosomal functioning: proton delivery for matrix tasting. Eur. J. Cell Biol. 2012;91:847–860. doi: 10.1016/j.ejcb.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Bruce A.W., Donaldson I.J., Wood I.C., Yerbury S.A., Sadowski M.I., Chapman M., Gottgens B., Buckley N.J. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc. Natl. Acad. Sci. U S A. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugan I., Kucuk S., Karagoz Z., Fraser S.P., Kaya H., Dodson A., Foster C.S., Altun S., Djamgoz M.B.A. Anti-metastatic effect of ranolazine in an in vivo rat model of prostate cancer, and expression of voltage-gated sodium channel protein in human prostate. Prostate Cancer Prostatic Dis. 2019;22:569–579. doi: 10.1038/s41391-019-0128-3. [DOI] [PubMed] [Google Scholar]
- Cameron I.L., Smith N.K., Pool T.B., Sparks R.L. Intracellular concentration of sodium and other elements as related to mitogenesis and oncogenesis in vivo. Cancer Res. 1980;40:1493–1500. [PubMed] [Google Scholar]
- Campbell T.M., Main M.J., Fitzgerald E.M. Functional expression of the voltage-gated Na+-channel Nav1.7 is necessary for EGF-mediated invasion in human non-small cell lung cancer cells. J. Cell Sci. 2013;126:4939–4949. doi: 10.1242/jcs.130013. [DOI] [PubMed] [Google Scholar]
- Carrithers L.M., Hulseberg P., Sandor M., Carrithers M.D. The human macrophage sodium channel NaV1.5 regulates mycobacteria processing through organelle polarization and localized calcium oscillations. FEMS Immunol. Med. Microbiol. 2011;63:319–327. doi: 10.1111/j.1574-695X.2011.00853.x. [DOI] [PubMed] [Google Scholar]
- Carrithers M.D., Chatterjee G., Carrithers L.M., Offoha R., Iheagwara U., Rahner C., Graham M., Waxman S.G. Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A. J. Biol. Chem. 2009;284:8114–8126. doi: 10.1074/jbc.M801892200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrithers M.D., Dib-Hajj S., Carrithers L.M., Tokmoulina G., Pypaert M., Jonas E.A., Waxman S.G. Expression of the voltage-gated sodium channel NaV1.5 in the macrophage late endosome regulates endosomal acidification. J. Immunol. 2007;178:7822–7832. doi: 10.4049/jimmunol.178.12.7822. [DOI] [PubMed] [Google Scholar]
- Catterall W.A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Cervenka R., Lukacs P., Gawali V.S., Ke S., Koenig X., Rubi L., Zarrabi T., Hilber K., Sandtner W., Stary-Weinzinger A., Todt H. Distinct modulation of inactivation by a residue in the pore domain of voltage-gated Na(+) channels: mechanistic insights from recent crystal structures. Sci. Rep. 2018;8:631. doi: 10.1038/s41598-017-18919-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvin L., Goupille C., Blanc C., Pinault M., Domingo I., Guimaraes C., Bougnoux P., Chevalier S., Maheo K. Long chain n-3 polyunsaturated fatty acids increase the efficacy of docetaxel in mammary cancer cells by downregulating Akt and PKCepsilon/delta-induced ERK pathways. Biochim. Biophys. Acta. 2016;1861:380–390. doi: 10.1016/j.bbalip.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Chioni A.M., Brackenbury W.J., Calhoun J.D., Isom L.L., Djamgoz M.B. A novel adhesion molecule in human breast cancer cells: voltage-gated Na+ channel beta1 subunit. Int. J. Biochem. Cell Biol. 2009;41:1216–1227. doi: 10.1016/j.biocel.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E.Y., Gardner J.M., Lucas D.R., Mchugh J.B., Patel R.M. Ewing sarcoma. Semin. Diagn. Pathol. 2014;31:39–47. doi: 10.1053/j.semdp.2014.01.002. [DOI] [PubMed] [Google Scholar]
- Chong J.A., Tapia-Ramirez J., Kim S., Toledo-Aral J.J., Zheng Y., Boutros M.C., Altshuller Y.M., Frohman M.A., Kraner S.D., Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Clatot J., Hoshi M., Wan X., Liu H., Jain A., Shinlapawittayatorn K., Marionneau C., Ficker E., Ha T., Deschenes I. Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 2017;8:2077. doi: 10.1038/s41467-017-02262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone C.D., Jr. The role of the surface electrical transmembrane potential in normal and malignant mitogenesis. Ann. N. Y. Acad. Sci. 1974;238:420–435. doi: 10.1111/j.1749-6632.1974.tb26808.x. [DOI] [PubMed] [Google Scholar]
- D'avanzo N. Lipid regulation of sodium channels. Curr. Top Membr. 2016;78:353–407. doi: 10.1016/bs.ctm.2016.04.003. [DOI] [PubMed] [Google Scholar]
- D'eliseo D., Di Rocco G., Loria R., Soddu S., Santoni A., Velotti F. Epitelial-to-mesenchimal transition and invasion are upmodulated by tumor-expressed granzyme B and inhibited by docosahexaenoic acid in human colorectal cancer cells. J. Exp. Clin. Cancer Res. 2016;35:24. doi: 10.1186/s13046-016-0302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W., Zhou J., Wang H., Zhang M., Yang X., Song W. miR-424-5p promotes the proliferation and metastasis of colorectal cancer by directly targeting SCN4B. Pathol. Res. Pract. 2020;216:152731. doi: 10.1016/j.prp.2019.152731. [DOI] [PubMed] [Google Scholar]
- Dendele B., Tekpli X., Hardonniere K., Holme J.A., Debure L., Catheline D., Arlt V.M., Nagy E., Phillips D.H., Ovrebo S. Protective action of n-3 fatty acids on benzo[a]pyrene-induced apoptosis through the plasma membrane remodeling-dependent NHE1 pathway. Chem. Biol. Interact. 2014;207:41–51. doi: 10.1016/j.cbi.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Diaz D., Delgadillo D.M., Hernandez-Gallegos E., Ramirez-Dominguez M.E., Hinojosa L.M., Ortiz C.S., Berumen J., Camacho J., Gomora J.C. Functional expression of voltage-gated sodium channels in primary cultures of human cervical cancer. J. Cell Physiol. 2007;210:469–478. doi: 10.1002/jcp.20871. [DOI] [PubMed] [Google Scholar]
- Diss J.K., Archer S.N., Hirano J., Fraser S.P., Djamgoz M.B. Expression profiles of voltage-gated Na(+) channel alpha-subunit genes in rat and human prostate cancer cell lines. Prostate. 2001;48:165–178. doi: 10.1002/pros.1095. [DOI] [PubMed] [Google Scholar]
- Diss J.K., Fraser S.P., Walker M.M., Patel A., Latchman D.S., Djamgoz M.B. Beta-subunits of voltage-gated sodium channels in human prostate cancer: quantitative in vitro and in vivo analyses of mRNA expression. Prostate Cancer Prostatic Dis. 2008;11:325–333. doi: 10.1038/sj.pcan.4501012. [DOI] [PubMed] [Google Scholar]
- Diss J.K., Stewart D., Pani F., Foster C.S., Walker M.M., Patel A., Djamgoz M.B. A potential novel marker for human prostate cancer: voltage-gated sodium channel expression in vivo. Prostate Cancer Prostatic Dis. 2005;8:266–273. doi: 10.1038/sj.pcan.4500796. [DOI] [PubMed] [Google Scholar]
- Djamgoz M.B.A., Mycielska M., Madeja Z., Fraser S.P., Korohoda W. Directional movement of rat prostate cancer cells in direct-current electric field: involvement of voltagegated Na+ channel activity. J. Cell Sci. 2001;114:2697–2705. doi: 10.1242/jcs.114.14.2697. [DOI] [PubMed] [Google Scholar]
- Driffort V., Gillet L., Bon E., Marionneau-Lambot S., Oullier T., Joulin V., Collin C., Pages J.C., Jourdan M.L., Chevalier S. Ranolazine inhibits NaV1.5-mediated breast cancer cell invasiveness and lung colonization. Mol. Cancer. 2014;13:264. doi: 10.1186/1476-4598-13-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube C., Rostom A., Lewin G., Tsertsvadze A., Barrowman N., Code C., Sampson M., Moher D., Force U.S.P.S.T. The use of aspirin for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2007;146:365–375. doi: 10.7326/0003-4819-146-5-200703060-00009. [DOI] [PubMed] [Google Scholar]
- Dumas J.F., Brisson L., Chevalier S., Maheo K., Fromont G., Moussata D., Besson P., Roger S. Metabolic reprogramming in cancer cells, consequences on pH and tumour progression: integrated therapeutic perspectives with dietary lipids as adjuvant to anticancer treatment. Semin. Cancer Biol. 2017;43:90–110. doi: 10.1016/j.semcancer.2017.03.004. [DOI] [PubMed] [Google Scholar]
- Dutta S., Lopez Charcas O., Tanner S., Gradek F., Driffort V., Roger S., Selander K., Velu S.E., Brouillette W. Discovery and evaluation of nNav1.5 sodium channel blockers with potent cell invasion inhibitory activity in breast cancer cells. Bioorg. Med. Chem. 2018;26:2428–2436. doi: 10.1016/j.bmc.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elajnaf T., Baptista-Hon D.T., Hales T.G. Potent inactivation-dependent inhibition of adult and neonatal NaV1.5 channels by lidocaine and levobupivacaine. Anesth. Analg. 2018;127:650–660. doi: 10.1213/ANE.0000000000003597. [DOI] [PubMed] [Google Scholar]
- Eltweri A.M., Thomas A.L., Metcalfe M., Calder P.C., Dennison A.R., Bowrey D.J. Potential applications of fish oils rich in omega-3 polyunsaturated fatty acids in the management of gastrointestinal cancer. Clin. Nutr. 2016;36:65–78. doi: 10.1016/j.clnu.2016.01.007. [DOI] [PubMed] [Google Scholar]
- Fairhurst C., Watt I., Martin F., Bland M., Brackenbury W.J. Sodium channel-inhibiting drugs and survival of breast, colon and prostate cancer: a population-based study. Sci. Rep. 2015;5:16758. doi: 10.1038/srep16758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon K.C., Glass D.J., Guttridge D.C. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 2012;16:153–166. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Ferro F., Servais S., Besson P., Roger S., Dumas J.F., Brisson L. Autophagy and mitophagy in cancer metabolic remodelling. Semin. Cell Dev. Biol. 2020;98:129–138. doi: 10.1016/j.semcdb.2019.05.029. [DOI] [PubMed] [Google Scholar]
- Fonseca G., Farkas J., Dora E., Von Haehling S., Lainscak M. Cancer cachexia and related metabolic dysfunction. Int. J. Mol. Sci. 2020;21:2321. doi: 10.3390/ijms21072321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forget P., Aguirre J.A., Bencic I., Borgeat A., Cama A., Condron C., Eintrei C., Eroles P., Gupta A., Hales T.G. How anesthetic, analgesic and other non-surgical techniques during cancer surgery might affect postoperative oncologic outcomes: a summary of current state of evidence. Cancers (Basel) 2019;11:592. doi: 10.3390/cancers11050592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser S.P., Diss J.K., Chioni A.M., Mycielska M.E., Pan H., Yamaci R.F., Pani F., Siwy Z., Krasowska M., Grzywna Z. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin. Cancer Res. 2005;11:5381–5389. doi: 10.1158/1078-0432.CCR-05-0327. [DOI] [PubMed] [Google Scholar]
- Fraser S.P., Diss J.K., Lloyd L.J., Pani F., Chioni A.M., George A.J., Djamgoz M.B. T-lymphocyte invasiveness: control by voltage-gated Na+ channel activity. FEBS Lett. 2004;569:191–194. doi: 10.1016/j.febslet.2004.05.063. [DOI] [PubMed] [Google Scholar]
- Fraser S.P., Hemsley F., Djamgoz M.B.A. Caffeic acid phenethyl ester: inhibition of metastatic cell behaviours via voltage-gated sodium channel in human breast cancer in vitro. Int. J. Biochem. Cell Biol. 2016;71:111–118. doi: 10.1016/j.biocel.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Fraser S.P., Peters A., Fleming-Jones S., Mukhey D., Djamgoz M.B. Resveratrol: inhibitory effects on metastatic cell behaviors and voltage-gated Na(+) channel activity in rat prostate cancer in vitro. Nutr. Cancer. 2014;66:1047–1058. doi: 10.1080/01635581.2014.939291. [DOI] [PubMed] [Google Scholar]
- Gao R., Shen Y., Cai J., Lei M., Wang Z. Expression of voltage-gated sodium channel alpha subunit in human ovarian cancer. Oncol. Rep. 2010;23:1293–1299. doi: 10.3892/or_00000763. [DOI] [PubMed] [Google Scholar]
- Gillet L., Roger S., Besson P., Lecaille F., Gore J., Bougnoux P., Lalmanach G., Le Guennec J.Y. Voltage-gated sodium channel activity promotes cysteine cathepsin-dependent invasiveness and colony growth of human cancer cells. J. Biol. Chem. 2009;284:8680–8691. doi: 10.1074/jbc.M806891200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet L., Roger S., Bougnoux P., Le Guennec J.Y., Besson P. Beneficial effects of omega-3 long-chain fatty acids in breast cancer and cardiovascular diseases: voltage-gated sodium channels as a common feature? Biochimie. 2011;93:4–6. doi: 10.1016/j.biochi.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Goldin A.L. Resurgence of sodium channel research. Annu. Rev. Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- Goldin A.L. Evolution of voltage-gated Na(+) channels. J. Exp. Biol. 2002;205:575–584. doi: 10.1242/jeb.205.5.575. [DOI] [PubMed] [Google Scholar]
- Gong Y., Yang J., Wu W., Liu F., Su A., Li Z., Zhu J., Wei T. Preserved SCN4B expression is an independent indicator of favorable recurrence-free survival in classical papillary thyroid cancer. PLoS One. 2018;13:e0197007. doi: 10.1371/journal.pone.0197007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradek F., Lopez-Charcas O., Chadet S., Poisson L., Ouldamer L., Goupille C., Jourdan M.L., Chevalier S., Moussata D., Besson P., Roger S. sodium channel Nav1.5 controls epithelial-to-mesenchymal transition and invasiveness in breast cancer cells through its regulation by the salt-inducible kinase-1. Sci. Rep. 2019;9:18652. doi: 10.1038/s41598-019-55197-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes J.A., Fraser S.P., Stephens G.J., Downing J.E., Laniado M.E., Foster C.S., Abel P.D., Djamgoz M.B. Differential expression of voltage-activated Na+ currents in two prostatic tumour cell lines: contribution to invasiveness in vitro. FEBS Lett. 1995;369:290–294. doi: 10.1016/0014-5793(95)00772-2. [DOI] [PubMed] [Google Scholar]
- Gumushan Aktas H., Akgun T. Naringenin inhibits prostate cancer metastasis by blocking voltage-gated sodium channels. Biomed. Pharmacother. 2018;106:770–775. doi: 10.1016/j.biopha.2018.07.008. [DOI] [PubMed] [Google Scholar]
- Guzel R.M., Ogmen K., Ilieva K.M., Fraser S.P., Djamgoz M.B.A. Colorectal cancer invasiveness in vitro: predominant contribution of neonatal Nav1.5 under normoxia and hypoxia. J. Cell. Physiol. 2019;234:6582–6593. doi: 10.1002/jcp.27399. [DOI] [PubMed] [Google Scholar]
- Hajjaji N., Besson P., Bougnoux P. Tumor and non-tumor tissues differential oxidative stress response to supplemental DHA and chemotherapy in rats. Cancer Chemother. Pharmacol. 2012;70:17–23. doi: 10.1007/s00280-012-1884-0. [DOI] [PubMed] [Google Scholar]
- Hajjaji N., Schubnel V., Bougnoux P. Determinants of DHA incorporation into tumor tissue during dietary DHA supplementation. Lipids. 2011;46:1063–1069. doi: 10.1007/s11745-011-3573-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hernandez-Munoz I., Figuerola E., Sanchez-Molina S., Rodriguez E., Fernandez-Marino A.I., Pardo-Pastor C., Bahamonde M.I., Fernandez-Fernandez J.M., Garcia-Dominguez D.J., Hontecillas-Prieto L. RING1B contributes to Ewing sarcoma development by repressing the NaV1.6 sodium channel and the NF-kappaB pathway, independently of the fusion oncoprotein. Oncotarget. 2016;7:46283–46300. doi: 10.18632/oncotarget.10092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Plata E., Ortiz C.S., Marquina-Castillo B., Medina-Martinez I., Alfaro A., Berumen J., Rivera M., Gomora J.C. Overexpression of NaV 1.6 channels is associated with the invasion capacity of human cervical cancer. Int. J. Cancer. 2012;130:2013–2023. doi: 10.1002/ijc.26210. [DOI] [PubMed] [Google Scholar]
- Hernansanz-Agustin P., Choya-Foces C., Carregal-Romero S., Ramos E., Oliva T., Villa-Pina T., Moreno L., Izquierdo-Alvarez A., Cabrera-Garcia J.D., Cortes A. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature. 2020;586:287–291. doi: 10.1038/s41586-020-2551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin A.L., Huxley A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodnik Z., Tomasic T., Masic L.P., Chan F., Kirby R.W., Madge D.J., Kikelj D. Novel state-dependent voltage-gated sodium channel modulators, based on marine alkaloids from Agelas sponges. Eur. J. Med. Chem. 2013;70:154–164. doi: 10.1016/j.ejmech.2013.07.034. [DOI] [PubMed] [Google Scholar]
- House C.D., Vaske C.J., Schwartz A.M., Obias V., Frank B., Luu T., Sarvazyan N., Irby R., Strausberg R.L., Hales T.G. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 2010;70:6957–6967. doi: 10.1158/0008-5472.CAN-10-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House C.D., Wang B.D., Ceniccola K., Williams R., Simaan M., Olender J., Patel V., Baptista-Hon D.T., Annunziata C.M., Gutkind J.S. Voltage-gated Na+ channel activity increases colon cancer transcriptional activity and invasion via persistent MAPK signaling. Sci. Rep. 2015;5:11541. doi: 10.1038/srep11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Lu C., Wu Y., Ouyang S., Chen Y. Identification and functional characterization of voltage-gated sodium channels in lymphocytes. Biochem. Biophys. Res. Commun. 2015;458:294–299. doi: 10.1016/j.bbrc.2015.01.103. [DOI] [PubMed] [Google Scholar]
- Igci Y.Z., Bozgeyik E., Borazan E., Pala E., Suner A., Ulasli M., Gurses S.A., Yumrutas O., Balik A.A., Igci M. Expression profiling of SCN8A and NDUFC2 genes in colorectal carcinoma. Exp. Oncol. 2015;37:77–80. [PubMed] [Google Scholar]
- Isbilen B., Fraser S.P., Djamgoz M.B. Docosahexaenoic acid (omega-3) blocks voltage-gated sodium channel activity and migration of MDA-MB-231 human breast cancer cells. Int. J. Biochem. Cell Biol. 2006;38:2173–2182. doi: 10.1016/j.biocel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Isom L.L. The role of sodium channels in cell adhesion. Front. Biosci. 2002;7:12–23. doi: 10.2741/isom. [DOI] [PubMed] [Google Scholar]
- Isom L.L., De Jongh K.S., Catterall W.A. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- Isom L.L., De Jongh K.S., Patton D.E., Reber B.F., Offord J., Charbonneau H., Walsh K., Goldin A.L., Catterall W.A. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- Jacobs M.A., Ouwerkerk R., Wolff A.C., Stearns V., Bottomley P.A., Barker P.B., Argani P., Khouri N., Davidson N.E., Bhujwalla Z.M., Bluemke D.A. Multiparametric and multinuclear magnetic resonance imaging of human breast cancer: current applications. Technol. Cancer Res. Treat. 2004;3:543–550. doi: 10.1177/153303460400300603. [DOI] [PubMed] [Google Scholar]
- Jaitovich A., Bertorello A.M. Intracellular sodium sensing: SIK1 network, hormone action and high blood pressure. Biochim. Biophys. Acta. 2010;1802:1140–1149. doi: 10.1016/j.bbadis.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Jansson K.H., Castillo D.G., Morris J.W., Boggs M.E., Czymmek K.J., Adams E.L., Schramm L.P., Sikes R.A. Identification of beta-2 as a key cell adhesion molecule in PCa cell neurotropic behavior: a novel ex vivo and biophysical approach. PLoS One. 2014;9:e98408. doi: 10.1371/journal.pone.0098408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson K.H., Lynch J.E., Lepori-Bui N., Czymmek K.J., Duncan R.L., Sikes R.A. Overexpression of the VSSC-associated CAM, beta-2, enhances LNCaP cell metastasis associated behavior. Prostate. 2012;72:1080–1092. doi: 10.1002/pros.21512. [DOI] [PubMed] [Google Scholar]
- Jing K., Song K.S., Shin S., Kim N., Jeong S., Oh H.R., Park J.H., Seo K.S., Heo J.Y., Han J. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy. 2011;7:1348–1358. doi: 10.4161/auto.7.11.16658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A., Kainz D., Khan F., Lee C., Carrithers M.D. Human macrophage SCN5A activates an innate immune signaling pathway for antiviral host defense. J. Biol. Chem. 2014;289:35326–35340. doi: 10.1074/jbc.M114.611962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jude S., Bedut S., Roger S., Pinault M., Champeroux P., White E., Le Guennec J.Y. Peroxidation of docosahexaenoic acid is responsible for its effects on I TO and I SS in rat ventricular myocytes. Br. J. Pharmacol. 2003;139:816–822. doi: 10.1038/sj.bjp.0705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarulzaman N.S., Dewadas H.D., Leow C.Y., Yaacob N.S., Mokhtar N.F. The role of REST and HDAC2 in epigenetic dysregulation of Nav1.5 and nNav1.5 expression in breast cancer. Cancer Cell Int. 2017;17:74. doi: 10.1186/s12935-017-0442-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.X., Leaf A. Evidence that free polyunsaturated fatty acids modify Na+ channels by directly binding to the channel proteins. Proc. Natl. Acad. Sci. U S A. 1996;93:3542–3546. doi: 10.1073/pnas.93.8.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.X., Li Y., Leaf A. Regulation of sodium channel gene expression by class I antiarrhythmic drugs and n - 3 polyunsaturated fatty acids in cultured neonatal rat cardiac myocytes. Proc. Natl. Acad. Sci. U S A. 1997;94:2724–2728. doi: 10.1073/pnas.94.6.2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld S., Goupille C., Vibet S., Chevalier S., Pinet A., Lebeau J., Tranquart F., Bougnoux P., Martel E., Maurin A. Reducing endothelial NOS activation and interstitial fluid pressure with n-3 PUFA offset tumor chemoresistance. Carcinogenesis. 2012;33:260–267. doi: 10.1093/carcin/bgr274. [DOI] [PubMed] [Google Scholar]
- Kumar A.P., Quake A.L., Chang M.K., Zhou T., Lim K.S., Singh R., Hewitt R.E., Salto-Tellez M., Pervaiz S., Clement M.V. Repression of NHE1 expression by PPARgamma activation is a potential new approach for specific inhibition of the growth of tumor cells in vitro and in vivo. Cancer Res. 2009;69:8636–8644. doi: 10.1158/0008-5472.CAN-09-0219. [DOI] [PubMed] [Google Scholar]
- Lacroix J., Poet M., Huc L., Morello V., Djerbi N., Ragno M., Rissel M., Tekpli X., Gounon P., Lagadic-Gossmann D., Counillon L. Kinetic analysis of the regulation of the Na+/H+ exchanger NHE-1 by osmotic shocks. Biochemistry. 2008;47:13674–13685. doi: 10.1021/bi801368n. [DOI] [PubMed] [Google Scholar]
- Lenaeus M.J., Gamal El-Din T.M., Ing C., Ramanadane K., Pomes R., Zheng N., Catterall W.A. Structures of closed and open states of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. U S A. 2017;114:E3051–E3060. doi: 10.1073/pnas.1700761114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Gu Z., Pan Y., Wang S., Chen H., Zhang H., Chen W., Chen Y.Q. Dietary supplementation of alpha-linolenic acid induced conversion of n-3 LCPUFAs and reduced prostate cancer growth in a mouse model. Lipids Health Dis. 2017;16:136. doi: 10.1186/s12944-017-0529-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Lv Y., Xu J., Mao X., Chen Z., Lu W. Over-expression of Nav1.6 channels is associated with lymph node metastases in colorectal cancer. World J. Surg. Oncol. 2019;17:175. doi: 10.1186/s12957-019-1715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Liu D., Liu J.J., Zhao C., Yao S., Hong L. Blocking the Nav1.5 channel using eicosapentaenoic acid reduces migration and proliferation of ovarian cancer cells. Int. J. Oncol. 2018;53:855–865. doi: 10.3892/ijo.2018.4437. [DOI] [PubMed] [Google Scholar]
- Liu J., Tan H., Yang W., Yao S., Hong L. The voltage-gated sodium channel Nav1.7 associated with endometrial cancer. J. Cancer. 2019;10:4954–4960. doi: 10.7150/jca.31544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Charcas O., Espinosa A.M., Alfaro A., Herrera-Carrillo Z., Ramirez-Cordero B.E., Cortes-Reynosa P., Perez Salazar E., Berumen J., Gomora J.C. The invasiveness of human cervical cancer associated to the function of NaV1.6 channels is mediated by MMP-2 activity. Sci. Rep. 2018;8:12995. doi: 10.1038/s41598-018-31364-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreel X., Allaire J., Leger C., Caron A., Labonte M.E., Lamarche B., Julien P., Desmeules P., Tetu B., Fradet V. Prostatic and dietary omega-3 fatty acids and prostate cancer progression during active surveillance. Cancer Prev. Res. (Phila) 2014;7:766–776. doi: 10.1158/1940-6207.CAPR-13-0349. [DOI] [PubMed] [Google Scholar]
- Murphy L.L., Moon-Grady A.J., Cuneo B.F., Wakai R.T., Yu S., Kunic J.D., Benson D.W., George A.L., Jr. Developmentally regulated SCN5A splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia. Heart Rhythm. 2012;9:590–597. doi: 10.1016/j.hrthm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mycielska M.E., Fraser S.P., Szatkowski M., Djamgoz M.B. Contribution of functional voltage-gated Na+ channel expression to cell behaviors involved in the metastatic cascade in rat prostate cancer: II. Secretory membrane activity. J. Cell. Physiol. 2003;195:461–469. doi: 10.1002/jcp.10265. [DOI] [PubMed] [Google Scholar]
- Nakazaki A., Nakane Y., Ishikawa Y., Yotsu-Yamashita M., Nishikawa T. Asymmetric synthesis of crambescin A-C carboxylic acids and their inhibitory activity on voltage-gated sodium channels. Org. Biomol. Chem. 2016;14:5304–5309. doi: 10.1039/c6ob00914j. [DOI] [PubMed] [Google Scholar]
- Nelson M., Millican-Slater R., Forrest L.C., Brackenbury W.J. The sodium channel beta1 subunit mediates outgrowth of neurite-like processes on breast cancer cells and promotes tumour growth and metastasis. Int. J. Cancer. 2014;135:2338–2351. doi: 10.1002/ijc.28890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M., Yang M., Dowle A.A., Thomas J.R., Brackenbury W.J. The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol. Cancer. 2015;14:13. doi: 10.1186/s12943-014-0277-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M., Yang M., Millican-Slater R., Brackenbury W.J. Nav1.5 regulates breast tumor growth and metastatic dissemination in vivo. Oncotarget. 2015;6:32914–32929. doi: 10.18632/oncotarget.5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P.T., Demarco K.R., Vorobyov I., Clancy C.E., Yarov-Yarovoy V. Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel. Proc. Natl. Acad. Sci. U S A. 2019;116:2945–2954. doi: 10.1073/pnas.1817446116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M., Ikeda T., Kayano T., Suzuki H., Takeshima H., Kurasaki M., Takahashi H., Numa S. Existence of distinct sodium channel messenger RNAs in rat brain. Nature. 1986;320:188–192. doi: 10.1038/320188a0. [DOI] [PubMed] [Google Scholar]
- Noda M., Shimizu S., Tanabe T., Takai T., Kayano T., Ikeda T., Takahashi H., Nakayama H., Kanaoka Y., Minamino N. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- Onganer P.U., Seckl M.J., Djamgoz M.B. Neuronal characteristics of small-cell lung cancer. Br. J. Cancer. 2005;93:1197–1201. doi: 10.1038/sj.bjc.6602857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onkal R., Mattis J.H., Fraser S.P., Diss J.K., Shao D., Okuse K., Djamgoz M.B. Alternative splicing of Nav1.5: an electrophysiological comparison of 'neonatal' and 'adult' isoforms and critical involvement of a lysine residue. J. Cell. Physiol. 2008;216:716–726. doi: 10.1002/jcp.21451. [DOI] [PubMed] [Google Scholar]
- Ouldamer L., Goupille C., Vilde A., Arbion F., Body G., Chevalier S., Cottier J.P., Bougnoux P. N-3 polyunsaturated fatty acids of marine origin and multifocality in human breast cancer. PLoS One. 2016;11:e0147148. doi: 10.1371/journal.pone.0147148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouldamer L., Nadal-Desbarats L., Chevalier S., Body G., Goupille C., Bougnoux P. NMR-based lipidomic approach to evaluate controlled dietary intake of lipids in adipose tissue of a rat mammary tumor model. J. Proteome Res. 2016;15:868–878. doi: 10.1021/acs.jproteome.5b00788. [DOI] [PubMed] [Google Scholar]
- Ouwerkerk R., Jacobs M.A., Macura K.J., Wolff A.C., Stearns V., Mezban S.D., Khouri N.F., Bluemke D.A., Bottomley P.A. Elevated tissue sodium concentration in malignant breast lesions detected with non-invasive 23Na MRI. Breast Cancer Res. Treat. 2007;106:151–160. doi: 10.1007/s10549-006-9485-4. [DOI] [PubMed] [Google Scholar]
- Pan X., Li Z., Huang X., Huang G., Gao S., Shen H., Liu L., Lei J., Yan N. Molecular basis for pore blockade of human Na(+) channel Nav1.2 by the mu-conotoxin KIIIA. Science. 2019;363:1309–1313. doi: 10.1126/science.aaw2999. [DOI] [PubMed] [Google Scholar]
- Pan X., Li Z., Zhou Q., Shen H., Wu K., Huang X., Chen J., Zhang J., Zhu X., Lei J. Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science. 2018;362:eaau2486. doi: 10.1126/science.aau2486. [DOI] [PubMed] [Google Scholar]
- Pan Y., Zhang H., Zhang M., Zhu J., Yu J., Wang B., Qiu J., Zhang J. A five-gene based risk score with high prognostic value in colorectal cancer. Oncol. Lett. 2017;14:6724–6734. doi: 10.3892/ol.2017.7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancrazio J.J., Viglione M.P., Tabbara I.A., Kim Y.I. Voltage-dependent ion channels in small-cell lung cancer cells. Cancer Res. 1989;49:5901–5906. [PubMed] [Google Scholar]
- Payandeh J., Gamal El-Din T.M., Scheuer T., Zheng N., Catterall W.A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012;486:135–139. doi: 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J., Scheuer T., Zheng N., Catterall W.A. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen V.L., Porporato P.E., Baselet B., Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell. Mol. Life Sci. 2016;73:1333–1348. doi: 10.1007/s00018-015-2098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkert J., Ripperger T., Schieck M., Schlegelberger B., Steinemann D., Illig T. On metabolic reprogramming and tumor biology: a comprehensive survey of metabolism in breast cancer. Oncotarget. 2016;7:67626–67649. doi: 10.18632/oncotarget.11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignier C., Revenaz C., Rauly-Lestienne I., Cussac D., Delhon A., Gardette J., Le Grand B. Direct protective effects of poly-unsaturated fatty acids, DHA and EPA, against activation of cardiac late sodium current: a mechanism for ischemia selectivity. Basic Res. Cardiol. 2007;102:553–564. doi: 10.1007/s00395-007-0676-x. [DOI] [PubMed] [Google Scholar]
- Plummer N.W., Mcburney M.W., Meisler M.H. Alternative splicing of the sodium channel SCN8A predicts a truncated two-domain protein in fetal brain and non-neuronal cells. J. Biol. Chem. 1997;272:24008–24015. doi: 10.1074/jbc.272.38.24008. [DOI] [PubMed] [Google Scholar]
- Poisson L., Lopez-Charcas O., Chadet S., Bon E., Lemoine R., Brisson L., Ouaissi M., Baron C., Besson P., Roger S., Moussata D. Rock inhibition promotes NaV1.5 sodium channel-dependent SW620 colon cancer cell invasiveness. Sci. Rep. 2020;10:13350. doi: 10.1038/s41598-020-70378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato P.E., Payen V.L., Baselet B., Sonveaux P. Metabolic changes associated with tumor metastasis, part 2: mitochondria, lipid and amino acid metabolism. Cell. Mol. Life Sci. 2016;73:1349–1363. doi: 10.1007/s00018-015-2100-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., Mcnamee C. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- Rahgozar K., Wright E., Carrithers L.M., Carrithers M.D. Mediation of protection and recovery from experimental autoimmune encephalomyelitis by macrophages expressing the human voltage-gated sodium channel NaV1.5. J. Neuropathol. Exp. Neurol. 2013;72:489–504. doi: 10.1097/NEN.0b013e318293eb08. [DOI] [PubMed] [Google Scholar]
- Roger S., Besson P., Le Guennec J.Y. Involvement of a novel fast inward sodium current in the invasion capacity of a breast cancer cell line. Biochim. Biophys. Acta. 2003;1616:107–111. doi: 10.1016/j.bbamem.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Roger S., Gillet L., Le Guennec J.Y., Besson P. Voltage-gated sodium channels and cancer: is excitability their primary role? Front. Pharmacol. 2015;6:152. doi: 10.3389/fphar.2015.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger S., Rollin J., Barascu A., Besson P., Raynal P.I., Iochmann S., Lei M., Bougnoux P., Gruel Y., Le Guennec J.Y. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int. J. Biochem. Cell Biol. 2007;39:774–786. doi: 10.1016/j.biocel.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Rose D.P., Connolly J.M., Coleman M. Effect of omega-3 fatty acids on the progression of metastases after the surgical excision of human breast cancer cell solid tumors growing in nude mice. Clin. Cancer Res. 1996;2:1751–1756. [PubMed] [Google Scholar]
- Rose D.P., Connolly J.M., Liu X.H. Fatty acid regulation of breast cancer cell growth and invasion. Adv. Exp. Med. Biol. 1997;422:47–55. doi: 10.1007/978-1-4757-2670-1_4. [DOI] [PubMed] [Google Scholar]
- Rose D.P., Rayburn J., Hatala M.A., Connolly J.M. Effects of dietary fish oil on fatty acids and eicosanoids in metastasizing human breast cancer cells. Nutr. Cancer. 1994;22:131–141. doi: 10.1080/01635589409514338. [DOI] [PubMed] [Google Scholar]
- Sanchez-Sandoval A.L., Gomora J.C. Contribution of voltage-gated sodium channel beta-subunits to cervical cancer cells metastatic behavior. Cancer Cell Int. 2019;19:35. doi: 10.1186/s12935-019-0757-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M., Huibregtse J.M., Vierstra R.D., Howley P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- Sessler D.I., Ben-Eliyahu S., Mascha E.J., Parat M.O., Buggy D.J. Can regional analgesia reduce the risk of recurrence after breast cancer? Methodology of a multicenter randomized trial. Contemp. Clin. Trials. 2008;29:517–526. doi: 10.1016/j.cct.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Sessler D.I., Pei L., Huang Y., Fleischmann E., Marhofer P., Kurz A., Mayers D.B., Meyer-Treschan T.A., Grady M., Tan E.Y. Recurrence of breast cancer after regional or general anaesthesia: a randomised controlled trial. Lancet. 2019;394:1807–1815. doi: 10.1016/S0140-6736(19)32313-X. [DOI] [PubMed] [Google Scholar]
- Shaheen U., Akka J., Hinore J.S., Girdhar A., Bandaru S., Sumithnath T.G., Nayarisseri A., Munshi A. Computer aided identification of sodium channel blockers in the clinical treatment of epilepsy using molecular docking tools. Bioinformation. 2015;11:131–137. doi: 10.6026/97320630011131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H., Li Z., Jiang Y., Pan X., Wu J., Cristofori-Armstrong B., Smith J.J., Chin Y.K.Y., Lei J., Zhou Q. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science. 2018;362:eaau2596. doi: 10.1126/science.aau2596. [DOI] [PubMed] [Google Scholar]
- Shen H., Liu D., Wu K., Lei J., Yan N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science. 2019;363:1303–1308. doi: 10.1126/science.aaw2493. [DOI] [PubMed] [Google Scholar]
- Shen H., Zhou Q., Pan X., Li Z., Wu J., Yan N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science. 2017;355:eaal4326. doi: 10.1126/science.aal4326. [DOI] [PubMed] [Google Scholar]
- Sjoblom T., Jones S., Wood L.D., Parsons D.W., Lin J., Barber T.D., Mandelker D., Leary R.J., Ptak J., Silliman N. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Stollings L.M., Jia L.J., Tang P., Dou H., Lu B., Xu Y. Immune modulation by volatile anesthetics. Anesthesiology. 2016;125:399–411. doi: 10.1097/ALN.0000000000001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sula A., Booker J., Ng L.C., Naylor C.E., Decaen P.G., Wallace B.A. The complete structure of an activated open sodium channel. Nat. Commun. 2017;8:14205. doi: 10.1038/ncomms14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillman T.S., Cascio M. Effects of membrane lipids on ion channel structure and function. Cell Biochem. Biophys. 2003;38:161–190. doi: 10.1385/CBB:38:2:161. [DOI] [PubMed] [Google Scholar]
- Tomasic T., Hartzoulakis B., Zidar N., Chan F., Kirby R.W., Madge D.J., Peigneur S., Tytgat J., Kikelj D. Ligand- and structure-based virtual screening for clathrodin-derived human voltage-gated sodium channel modulators. J. Chem. Inf. Model. 2013;53:3223–3232. doi: 10.1021/ci400505e. [DOI] [PubMed] [Google Scholar]
- Wang J., Lu Z., Wu C., Li Y., Kong Y., Zhou R., Shi K., Guo J., Li N., Liu J. Evaluation of the anticancer and anti-metastasis effects of novel synthetic sodium channel blockers in prostate cancer cells in vitro and in vivo. Prostate. 2019;79:62–72. doi: 10.1002/pros.23711. [DOI] [PubMed] [Google Scholar]
- Wang J., Ou S.W., Bai Y.F., Wang Y.J., Xu Z.D., Luan G.M. Downregulation of adult and neonatal Nav1.5 in the dorsal root ganglia and axon of peripheral sensory neurons of rats with spared nerve injury. Int. J. Mol. Med. 2018;41:2225–2232. doi: 10.3892/ijmm.2018.3446. [DOI] [PubMed] [Google Scholar]
- Wang J., Ou S.W., Wang Y.J., Kameyama M., Kameyama A., Zong Z.H. Analysis of four novel variants of Nav1.5/SCN5A cloned from the brain. Neurosci. Res. 2009;64:339–347. doi: 10.1016/j.neures.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Wang J., Ou S.W., Wang Y.J., Zong Z.H., Lin L., Kameyama M., Kameyama A. New variants of Nav1.5/SCN5A encode Na+ channels in the brain. J. Neurogenet. 2008;22:57–75. doi: 10.1080/01677060701672077. [DOI] [PubMed] [Google Scholar]
- Wang J., Ou S.W., Zhang Z.Y., Qiu B., Wang Y.J. Molecular expression of multiple Nav1.5 splice variants in the frontal lobe of the human brain. Int. J. Mol. Med. 2018;41:915–923. doi: 10.3892/ijmm.2017.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wannous R., Bon E., Gillet L., Chamouton J., Weber G., Brisson L., Gore J., Bougnoux P., Besson P., Roger S., Chevalier S. Suppression of PPARbeta, and DHA treatment, inhibit NaV1.5 and NHE-1 pro-invasive activities. Pflugers Arch. 2015;467:1249–1259. doi: 10.1007/s00424-014-1573-4. [DOI] [PubMed] [Google Scholar]
- Wannous R., Bon E., Maheo K., Goupille C., Chamouton J., Bougnoux P., Roger S., Besson P., Chevalier S. PPARbeta mRNA expression, reduced by n-3 PUFA diet in mammary tumor, controls breast cancer cell growth. Biochim. Biophys. Acta. 2013;1831:1618–1625. doi: 10.1016/j.bbalip.2013.07.010. [DOI] [PubMed] [Google Scholar]
- Webb B.A., Chimenti M., Jacobson M.P., Barber D.L. Dysregulated pH: a perfect storm for cancer progression. Nat. Rev. Cancer. 2011;11:671–677. doi: 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- Wisedchaisri G., Tonggu L., Mccord E., Gamal El-Din T.M., Wang L., Zheng N., Catterall W.A. Resting-state structure and gating mechanism of a voltage-gated sodium channel. Cell. 2019;178:993–1003 e12. doi: 10.1016/j.cell.2019.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J., Huang N., Huang H., Sun L., Dong S., Su J., Zhang J., Wang L., Lin L., Shi M. Voltage-gated sodium channel Nav 1.7 promotes gastric cancer progression through MACC1-mediated upregulation of NHE1. Int. J. Cancer. 2016;139:2553–2569. doi: 10.1002/ijc.30381. [DOI] [PubMed] [Google Scholar]
- Xiao Y.F., Ke Q., Wang S.Y., Auktor K., Yang Y., Wang G.K., Morgan J.P., Leaf A. Single point mutations affect fatty acid block of human myocardial sodium channel alpha subunit Na+ channels. Proc. Natl. Acad. Sci. U S A. 2001;98:3606–3611. doi: 10.1073/pnas.061003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.F., Wright S.N., Wang G.K., Morgan J.P., Leaf A. Fatty acids suppress voltage-gated Na+ currents in HEK293t cells transfected with the alpha-subunit of the human cardiac Na+ channel. Proc. Natl. Acad. Sci. U S A. 1998;95:2680–2685. doi: 10.1073/pnas.95.5.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Brackenbury W.J. Membrane potential and cancer progression. Front. Physiol. 2013;4:185. doi: 10.3389/fphys.2013.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., James A.D., Suman R., Kasprowicz R., Nelson M., O'toole P.J., Brackenbury W.J. Voltage-dependent activation of Rac1 by Nav 1.5 channels promotes cell migration. J. Cell. Physiol. 2020;235:3950–3972. doi: 10.1002/jcp.29290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Kozminski D.J., Wold L.A., Modak R., Calhoun J.D., Isom L.L., Brackenbury W.J. Therapeutic potential for phenytoin: targeting Na(v)1.5 sodium channels to reduce migration and invasion in metastatic breast cancer. Breast Cancer Res. Treat. 2012;134:603–615. doi: 10.1007/s10549-012-2102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yapa K., Deuis J., Peters A.A., Kenny P.A., Roberts-Thomson S.J., Vetter I., Monteith G.R. Assessment of the TRPM8 inhibitor AMTB in breast cancer cells and its identification as an inhibitor of voltage gated sodium channels. Life Sci. 2018;198:128–135. doi: 10.1016/j.lfs.2018.02.030. [DOI] [PubMed] [Google Scholar]
- Yaqoob P., Shaikh S.R. The nutritional and clinical significance of lipid rafts. Curr. Opin. Clin. Nutr. Metab. Care. 2010;13:156–166. doi: 10.1097/MCO.0b013e328335725b. [DOI] [PubMed] [Google Scholar]
- Yildirim S., Altun S., Gumushan H., Patel A., Djamgoz M.B. Voltage-gated sodium channel activity promotes prostate cancer metastasis in vivo. Cancer Lett. 2012;323:58–61. doi: 10.1016/j.canlet.2012.03.036. [DOI] [PubMed] [Google Scholar]
- Yin X., Yu X.W., Zhu P., Zhang Y.M., Zhang X.H., Wang F., Zhang J.J., Yan W., Xi Y., Wan J.B. Endogenously synthesized n-3 fatty acids in fat-1 transgenic mice prevent melanoma progression by increasing E-cadherin expression and inhibiting beta-catenin signaling. Mol. Med. Rep. 2016;14:3476–3484. doi: 10.3892/mmr.2016.5639. [DOI] [PubMed] [Google Scholar]
- Yu J.J., Xiao W., Dong S.L., Liang H.F., Zhang Z.W., Zhang B.X., Huang Z.Y., Chen Y.F., Zhang W.G., Luo H.P. Effect of surgical liver resection on circulating tumor cells in patients with hepatocellular carcinoma. BMC Cancer. 2018;18:835. doi: 10.1186/s12885-018-4744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaric O., Pinker K., Zbyn S., Strasser B., Robinson S., Minarikova L., Gruber S., Farr A., Singer C., Helbich T.H. Quantitative sodium MR imaging at 7 T: initial results and comparison with diffusion-weighted imaging in patients with breast tumors. Radiology. 2016;280:39–48. doi: 10.1148/radiol.2016151304. [DOI] [PubMed] [Google Scholar]