

Visual Abstract

Keywords: polycystic kidney disease, genetic kidney disease, ADPKD, human genetics, mutation
Abstract
Background and objectives
Progression of autosomal dominant polycystic kidney disease (ADPKD) is highly variable. On average, protein-truncating PKD1 mutations are associated with the most severe kidney disease among all mutation classes. Here, we report that patients with protein-truncating PKD1 mutations may also have mild kidney disease, a finding not previously well recognized.
Design, setting, participants, & measurements
From the extended Toronto Genetic Epidemiologic Study of Polycystic Kidney Disease, 487 patients had PKD1 and PKD2 sequencing and typical ADPKD imaging patterns by magnetic resonance imaging or computed tomography. Mayo Clinic Imaging Classification on the basis of age- and height-adjusted total kidney volume was used to assess their cystic disease severity; classes 1A or 1B were used as a proxy to define mild disease. Multivariable linear regression was performed to test the effects of age, sex, and mutation classes on log-transformed height-adjusted total kidney volume and eGFR.
Results
Among 174 study patients with typical imaging patterns and protein-truncating PKD1 mutations, 32 (18%) were found to have mild disease on the basis of imaging results (i.e., Mayo Clinic Imaging class 1A–1B), with their mutations spanning the entire gene. By multivariable analyses of age, sex, and mutation class, they displayed mild disease similar to patients with PKD2 mutations and Mayo Clinic Imaging class 1A–1B. Most of these mildly affected patients with protein-truncating PKD1 mutations reported a positive family history of ADPKD in preceding generations and displayed significant intrafamilial disease variability.
Conclusions
Despite having the most severe mutation class, 18% of patients with protein-truncating PKD1 mutations had mild disease on the basis of clinical and imaging assessment.
Podcast
This article contains a podcast at https://www.asn-online.org/media/podcast/CJASN/2021_02_18_CJN11100720_final.mp3
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease worldwide, and accounts for 5%–10% of patients requiring KRT (1,2). It is typically characterized by age-dependent development of innumerable cysts with bilateral kidney enlargement, which eventually leads to advanced kidney failure in a majority of patients (2). Mutations of two genes, PKD1 and PKD2, account for 60%–78% and 15%–26% of patients where a pathogenic mutation is identified, respectively (3–5). In more recent clinical studies, mutations of additional cystic disease genes (GANAB and DNAJB11) were discovered in <1% of patients (6,7), while 10%–15% of patients had no mutation detected (3–5).
Progression of kidney disease in ADPKD is highly variable between families, in part due to a strong gene locus (PKD1 versus PKD2) effect (8). Patients with PKD1 mutations have more kidney cysts, larger kidney volume, and require KRT 16–20 years earlier than patients with PKD2 mutations (3–5,8,9). More recent studies have further delineated a strong genotype-phenotype relationship: on average, protein-truncating PKD1 mutations are associated with the most severe disease, nontruncating PKD1 mutations with intermediate disease, and PKD2 mutations with mild disease (3–5). Significant within-family kidney disease variability is well documented and suggests a deleterious modifier effect from genetic and/or environmental factors in ADPKD (10–12).
With the recent approval of tolvaptan as the first disease-modifier drug for ADPKD (13,14), identifying patients at high risk for progression who may benefit from this treatment is a clinical priority (15–19). The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease has shown that total kidney volume expands quasi-exponentially during adult life in ADPKD at approximately 5%/yr (17–19). Both the Food and Drug Administration and European Medicines Agency have accepted total kidney volume as a validated prognostic imaging biomarker for enrichment of high-risk patients for clinical trials. Using age- and height-adjusted total kidney volume, the Mayo Clinic Imaging Classification provides a useful clinical tool to predict the rate of eGFR decline (20,21). Since March 2016, we routinely performed both mutation- and imaging-based risk assessment in all patients seen at the Center for Innovative Management of Polycystic Kidney Disease (https://www.cimpkd.ca/) in Toronto. As we collated the results of their genetic and imaging results, we noticed that some patients with protein-truncating PKD1 mutations had very mild cystic disease, a finding not previously well recognized. We therefore performed and report here a systematic study to define the prevalence and clinical characteristics of patients with protein-truncating PKD1 mutations and mild disease.
Materials and Methods
Study Population
The extended Toronto Genetic Epidemiologic Study of Polycystic Kidney Disease cohort included probands with ADPKD (n=612) and their affected relatives (n=778) from a total of 612 unrelated families. Study patients were seen at the Toronto General Hospital, University Health Network (n=877) between December 1, 2006 and July 31, 2018, or St. Joseph’s Healthcare in Hamilton (n=21) between September 1, 2016 and July 31, 2018. Diagnosis of ADPKD was confirmed in all patients using ultrasound or magnetic resonance imaging–based criteria (22,23). The median follow-up of patients with kidney volume imaging was 2.1 years (Quartile 1 to Quartile 3 [Q1–Q3], 1.5 to 2.8 years). All participants provided informed consent according to a prespecified protocol approved by the Institutional Ethics Review Boards from both sites.
Exposure
All recruited probands and their available affected relatives underwent standardized clinical evaluation, which included a detailed family history of ADPKD, noting kidney disease severity, age at KRT (if applicable), and survival status, and were comprehensively screened for PKD1 and PKD2 mutations (Supplemental Material) (4,24,25). Since March 2016, 500 patients from 442 unrelated families also underwent kidney volume measurement. All patients had serum creatinine and eGFR (calculated using the CKD Epidemiology Collaboration equation) within 3 months of their kidney imaging (26).
Kidney volume was measured by magnetic resonance imaging in 489 (98%) patients and by computed tomography in 11 (2%) patients using the ellipsoid method read by a single radiologist (MP); Mayo Clinic Imaging Class was assigned using age- and height-adjusted total kidney volume (20); all patients with atypical imaging patterns (class 2) were excluded. Patients with Mayo Clinic Imaging Class 1A or 1B were classified as “low risk” and 1C, 1D, or 1E were classified as “high risk” for the need of KRT, respectively.
Outcomes
Using low-risk Mayo Clinic Imaging Class 1A or 1B as a proxy for mild disease, we defined the prevalence and clinical characteristics of mild disease in 489 patients with protein-truncating PKD1 mutations. For patients with protein-truncating PKD1 mutations who did not have kidney imaging available, we used age at KRT >65 years as a proxy for mild ADPKD (27). To identify affected relatives with concordant or discordant disease to define the prevalence of within-family disease variability, we further used Mayo Clinic Imaging Class 1C–1E or age at KRT <55 years as proxies for severe disease, as previously reported (27).
Statistical Analyses
Continuous variables were reported as mean and 95% confidence interval (95% CI) if normally distributed, and medians and Q1–Q3 if not. Discrete variables were reported as percentages. Groups were compared with a t test; Wilcoxon or chi-squared tests were used when appropriate. Multivariable linear regression was performed to test the effects of age, sex, and mutation classes on log-transformed height-adjusted total kidney volume and eGFR using R (version 3.3.3, R Foundation for Statistical Computing). The main relationships were first tested, followed by tests for interaction of age, sex, and mutation classes. The beta of log10 transformed height-adjusted total kidney volume from the regression model was back transformed by exponentiation to obtain the percent difference per change in predictor variable.
Results
Characteristics of Study Cohort
From the extended Toronto Genetic Epidemiologic Study of Polycystic Kidney Disease (n=1390), 500 (36%) patients had both genetic testing and kidney volume measurement. After reviewing their imaging patterns, 13 patients with atypical ADPKD imaging patterns were excluded. Of the remaining 487 patients with typical imaging patterns, 190 (38%) had Mayo Clinic Imaging Class 1A–1B and 297 (59%) had Class 1C–1E; none of them required KRT at their last follow-up. Of 890 patients without kidney volume measurement, 602 (68%) had died or required KRT at their last follow-up.
A total of 570 (41%) patients in the study cohort had protein-truncating PKD1 mutations; 174 had kidney volume measurement and 34 (20%) had Mayo Clinic Imaging Class 1A–1B (Figure 1). Among 396 patients with protein-truncating PKD1 mutations but no kidney volume measurement, 290 required KRT at their last follow-up. The age distribution of patients with protein-truncating PKD1 mutations requiring (n=290) and not requiring (n=280) KRT at their last follow-up is shown in Figure 2. Of interest, 26 out of 290 (9%) patients with protein-truncating PKD1 mutations did not require KRT until age >65 years, and 15 out of 280 (5%) had CKD stages 2–4 at age ≥60 years (Figure 2B); all of these patients had mild disease similar to patients with PKD2 mutations.
Figure 1.
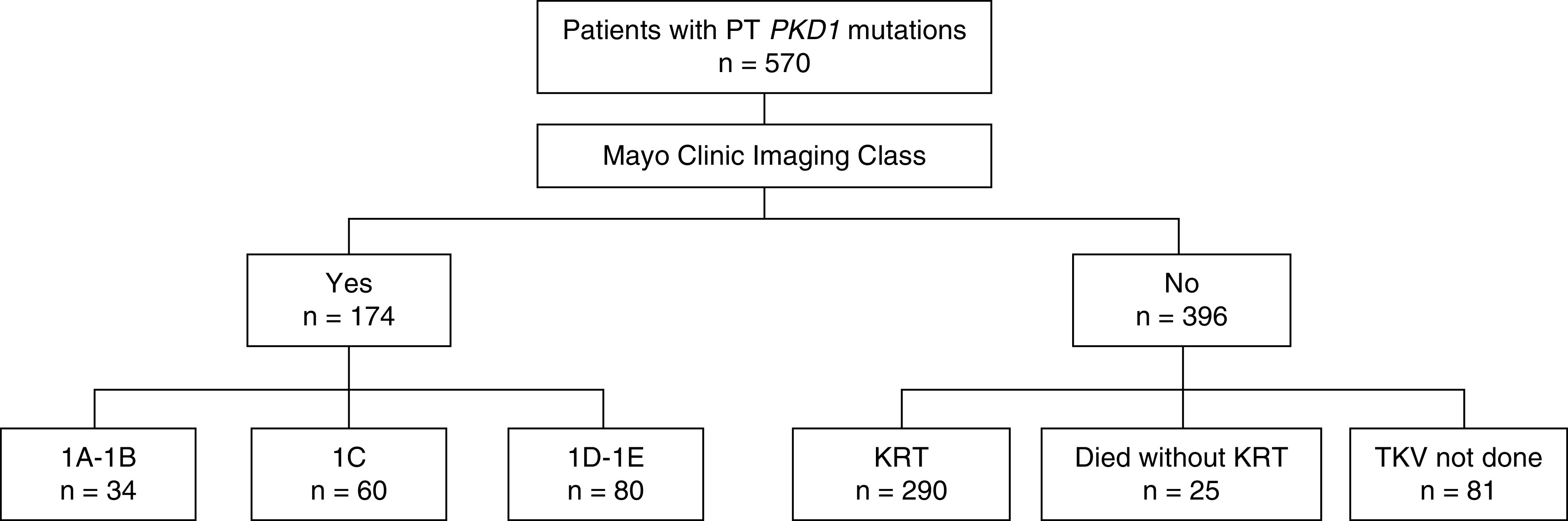
Study flow diagram. Patients with protein-truncating PKD1 mutations in the extended Toronto Genetic Epidemiology Study of Polycystic Kidney Disease with and without Mayo Clinic Imaging Class. PT, protein truncating; TKV, total kidney volume.
Figure 2.

Frequency plots of age distribution of patients with protein-truncating PKD1 mutations in the Toronto Genetic Epidemiology of Polycystic Kidney Disease Cohort (n=570). (A) Patients who required KRT (n=290). (B) Patients who did not require KRT at the time of death (n=25) or last follow-up (n=255).
Patients with Protein-Truncating PKD1 Mutations and Mild Disease
Of 34 patients from 29 unrelated families with protein-truncating PKD1 mutations and Mayo Clinic Imaging Class 1A–1B, two had advanced kidney failure (eGFR <45 ml/min per 1.73 m2) that was discordant from their mild cystic disease burden by imaging: one was a 59-year-old male with longstanding history of type 2 diabetes mellitus and the other, a 44-year-old female with a history of poorly controlled hypertension; both were excluded from further analysis. The clinical characteristics of the remaining 32 patients with confirmed mild disease are shown in Table 1; 23 (72%) were >30 years of age. Representative magnetic resonance images comparing age-matched cystic disease severity in patients with different mutation types and Mayo Clinic Imaging Class are shown in Supplemental Figure 1. The familial mutations of these patients include 16 frameshift insertions or deletions, 11 nonsense mutations, and two canonical splice site (c.11538–2A>C, p.R3846fs, reported in Mayo PKD database; c.1385+1G>C, p.R462fs10×, novel) mutations; they spanned the entire gene with no preferential location or accumulation in the 3′ exons (Supplemental Figure 2). The relative frequency of mutations in the duplicated versus nonduplicated regions of PKD1 was not different between patients with Mayo Clinic Imaging Class 1A–1B and 1C–1E (P>0.5).
Table 1.
Clinical characteristics of patients by mutation type and Mayo Clinic Imaging Class
| Patient Characteristics | Protein-Truncating PKD1 Mutation (Mild Imaging)a (n=32) | Protein-Truncating PKD1 Mutation (Severe Imaging)b (n=140) | PKD2 Mutation (Mild Imaging)c (n=66) |
|---|---|---|---|
| Number of families (%) | 29 | 132 | 61 |
| Age at time of MRI, yr, mean (95% CI) | 36 (32 to 41) | 38 (36 to 40) | 47 (43 to 50)d |
| Male, n (%) | 10 (31) | 57 (41) | 20 (30) |
| Body mass index, kg/m2, median (Q1–Q3) | 23.1 (21.5 to 24.9) | 25.4 (22.6 to 28.7)d | 24.5 (22.0 to 29.5) |
| Ethnicity (%) | |||
| European | 66 | 78 | 76 |
| Asian | 25 | 15 | 18 |
| Black | 6 | 2 | 4.5 |
| Other | 3 | 5 | 1.5 |
| Ht-TKV, ml/m, mean (95% CI) | 343 (273 to 405) | 923 (662 to 1331)d | 342 (249 to 556) |
| eGFR, ml/min per 1.73 m2, mean (95% CI) | 95 (85 to 110) | 75 (48 to 103)d | 90 (68 to 105) |
| CKD stage (%) | |||
| 1–2 | 94 | 65 | 79 |
| 3 | 6 | 25 | 18 |
| 4–5 | 0 | 10 | 3 |
MRI, magnetic resonance imaging; 95% CI, 95% confidence interval; Ht-TKV, height-adjusted total kidney volume.
Protein-truncating PKD1 mutations with Mayo Clinic Imaging Class 1A–1B (reference group).
Protein-truncating PKD1 mutations with Mayo Clinic Imaging Class 1C–1E.
PKD2 mutations with Mayo Clinic Imaging Class 1A–1B.
P<0.05 for pairwise comparisons to the reference.
Comparison of Disease Severity by Mutation Types and Imaging Class
Comparing patients with mild disease by imaging (Mayo Clinic Imaging Class 1A–1B), we did not find any difference in eGFR (P=0.18) between those with protein-truncating PKD1 versus PKD2 mutations; however, patients with PKD2 mutations were older than those with protein-truncating PKD1 mutations (47; 95% CI, 43 to 50 years versus 36; 95% CI, 32 to 41 years, P=0.002) (Table 1). The slopes of log-transformed height-adjusted total kidney volume (βPKD1&MCIC1A-B=1.0% larger kidney per year; 95% CI, 0.8 to 1.3 versus βPKD2&MCIC1A-B=1.3% larger per year; 95% CI, 1.1 to 1.5, P=0.3) and eGFR (βPKD1&MCIC1A-B =−1.3 ml/min per 1.73 m2 per year; 95% CI, −1.7 to −0.9 versus βPKD2&MCIC1A-B =−1.5 ml/min per 1.73 m2 per year; 95% CI, −1.8 to −1.2, P=0.6) overlap in patients with Mayo Clinic Imaging Class 1A–1B regardless of whether they had protein-truncating PKD1 or PKD2 mutations (Figure 3). As expected, patients with both protein-truncating PKD1 mutations and Mayo Clinic Imaging Class 1C–1E had distinctly more severe disease compared with either of the former groups.
Figure 3.

Disease severity is similar in patients with Mayo Clinic Imaging Class (MCIC) 1A–1B regardless of whether they had protein-truncating PKD1 (n=32) or PKD2 (n=66) mutations. Plots of log-transformed height-adjusted total kidney volume (A) and eGFR (B) versus age. Patients with protein-truncating PKD1 and MCIC 1C–1E (n=140) had more severe disease than either of the former groups.
By multivariable analysis, age, male sex, and Mayo Clinic Imaging Class 1C–1E were independently associated with larger kidney volume and reduced eGFR. Comparing patients with Mayo Clinic Imaging Class 1A–1B after adjusting for age and sex, patients with PKD2 mutations had marginally smaller kidney volumes (β=−7.7%; 95% CI: −13.9% to −0.01%; P=0.02) and their eGFR was not significantly different (β=9.0 ml/min per 1.73 m2; 95% CI: −0.4 to 18.3 ml/min per 1.73 m2, P=0.06) compared with those with protein-truncating PKD1 mutations (Table 2). There was no evidence of interaction between age and sex, age and mutation class, or sex and mutation class for either kidney size or eGFR (P>0.1, data not shown).
Table 2.
Predictors of kidney size and eGFR from multivariable regression analysis
| Interpretation of Results | |||||
|---|---|---|---|---|---|
| Predictors | B | 95% Confidence Interval | P | Percentage Difference | ml/min/1.73m2 |
| log10-transformed height-adjusted total kidney volume | |||||
| Age (per yr) | 0.013 | 0.011 to 0.015 | <0.001 | ||
| Back transformeda | 1.013 | 1.011 to 1.015 | 1.3 larger | ||
| Female (versus male) | –0.08 | −0.12 to−0.04 | <0.001 | ||
| Back transformed | 0.923 | 0.886 to 0.961 | 7.7 smaller | ||
| PT PKD1 mutations (severe) versus (mild) | 0.41 | 0.35 to 0.47 | <0.001 | ||
| Back transformed | 1.49 | 1.419 to 1.600 | 49 larger | ||
| PKD21A-1B versus PT PKD1 mutations (mild) | −0.08 | −0.15 to−0.01 | 0.02 | ||
| Back transformed | 0.923 | 0.861 to 0.990 | 7.7 smaller | ||
| eGFR | |||||
| Age (per yr) | −1.7 | −1.9 to −1.5 | <0.001 | 1.7 lower | |
| Female (versus male) | 6.4 | 0.7 to 12.1 | 0.02 | 6.4 higher | |
| PT PKD1 mutations (severe) versus (mild) | −15.9 | −24.1 to −7.6 | <0.001 | 15.9 lower | |
| PKD2 1A-1B versus PT PKD1 mutations (mild) | 9.0 | −0.4 to 18.3 | <0.06 | 9.0 higher | |
PT PKD1 mutations (mild): protein-truncating PKD1 mutations with Mayo Clinic Imaging Class 1A–1B.
PT PKD1 mutations (severe): protein-truncating PKD1 mutations with Mayo Clinic Imaging Class 1C–1E.
Percent difference obtained by exponentiation of the beta of log10 height-adjusted total kidney volume.
Within-Family Disease Variability in Patients with Protein-Truncating PKD1 Mutations and Mild Disease
A positive family history of ADPKD was confirmed in 23 of 29 (79%) probands with protein-truncating PKD1 mutations and mild disease by imaging. Of the remaining probands, three had no apparent family history and three had unknown or nonconfirmable family history. Within-family disease variability (on the basis of Mayo Clinic Imaging Class and/or age at KRT) of the affected relatives in these 23 families is shown in Figure 4 and Table 3. Of six families with Mayo Clinic Imaging Class available in at least two affected relatives, four (TOR094, TOR139, TOR173, and TOR198) demonstrated concordant disease, whereas two displayed discordant disease spanning at least two classes (TOR17 and TOR308). Patient TOR308.2 was found to have somatic mosaicism (PKD1: c.2605delC; p.R869FS28×; mutant allele fraction of approximately 10% in blood sample) and germline disease transmission to her daughter, as previously reported (24). Except for TOR308, all families shown in Table 3 had an older affected relative, which excludes younger generations as potential genetic mosaics.
Figure 4.

Within-family disease variability in families with PKD1-truncating mutations and mild disease by imaging. Each vertical line represents one family, and each dot represents an affected relative. Mild disease defined by Mayo Clinic Imaging Class 1A–1B are denoted by light blue open circles; severe disease defined by Mayo Clinic Imaging Class 1C–1E are denoted by dark blue open circles. Individuals requiring KRT with mild (i.e., >65 years) or severe disease (i.e., <55 years) are denoted by a light or dark blue solid circle, respectively. Individuals whose kidney imaging (black open circles) or age at KRT (solid black circle) did not allow assignment of mild or severe disease are deemed uninformative. A total of 16 families had at least one discordant (D) relative pair, and six families had at least one concordant (C) relative pair. The assessment of disease variability was indeterminate (I) in six families.
Table 3.
Within-family disease variability in affected relatives of patients with protein-truncating PKD1 mutations and mild kidney disease by imaging
| Patient ID | Age (yr) | htTKV (ml/m) | Mayo Clinic Imaging Class | eGFR (ml/min per 1.73 m2) | No. of Affected Relatives Requiring KRT | |
|---|---|---|---|---|---|---|
| Before Age 55 | After Age 65 | |||||
| Disease discordance by imaging in one or more relative pair | ||||||
| TOR308.2 | 57 | 927 | 1B | 65 | 0 | 0 |
| TOR308.1 | 25 | 637 | 1E | 97 | ||
| TOR17.1 | 51 | 623 | 1B | 48 | 4 | 0 |
| TOR17.13 | 28 | 655 | 1D | 102 | ||
| TOR17.12 | 23 | 712 | 1E | 133 | ||
| Disease concordance by imaging in one or more relative pair | ||||||
| TOR094.1 | 22 | 264 | 1B | 93 | 2 | 0 |
| TOR094.2 | 19 | 211 | 1B | 91 | ||
| TOR139.1 | 20 | 183 | 1A | 127 | 3 | 0 |
| TOR139.2 | 22 | 175 | 1A | 125 | ||
| TOR198.3 | 39 | 291 | 1B | 105 | 1 | 0 |
| TOR198.4 | 35 | 316 | 1B | 93 | ||
| TOR173.1 | 54 | 587 | 1B | 86 | 0 | 2 |
| TOR173.2 | 28 | 493 | 1C | 115 | ||
| Disease discordance by imaging and age at KRT in one or more relative pair | ||||||
| TOR094.1 | 22 | 264 | 1B | 93 | 2 | 0 |
| TOR139.1 | 22 | 183 | 1B | 127 | 3 | 0 |
| TOR198.3 | 39 | 291 | 1B | 105 | 1 | 0 |
| TOR420.1 | 39 | 275 | 1B | 109 | 1 | 0 |
| TOR431.1 | 25 | 254 | 1B | 102 | 2 | 0 |
| TOR438.1 | 25 | 296 | 1B | 122 | 2 | 0 |
| TOR48.9 | 35 | 352 | 1B | 96 | 3 | 0 |
| TOR595.1 | 32 | 344 | 1B | 85 | 1 | 0 |
| TOR663.1 | 39 | 324 | 1B | 85 | 1 | 0 |
| TOR833.1 | 36 | 221 | 1A | 90 | 1 | 0 |
| TOR841.1 | 18 | 237 | 1B | 113 | 1 | 0 |
| TOR845.1 | 34 | 378 | 1B | 83 | 1 | 0 |
| TOR968.1 | 59 | 371 | 1B | 64 | 1 | 0 |
| Disease concordance by imaging and age at KRT in one or more relative pair | ||||||
| TOR619.2 | 43 | 342 | 1B | 79 | 0 | 1 |
| Uninformative for assessment of within-family disease variability | ||||||
| TOR242.2 | 31 | 326 | 1B | 119 | 0 | 0 |
| TOR291.3 | 46 | 589 | 1B | 105 | 0 | 0 |
| TOR388.1 | 46 | 463 | 1B | 103 | 0 | 0 |
| TOR674.1 | 33 | 348 | 1B | 115 | 0 | 0 |
| TOR803.1 | 35 | 389 | 1B | 93 | 0 | 0 |
| TOR895.1 | 28 | 321 | 1B | 120 | 0 | 0 |
htTKV, height-adjusted total kidney volume.
In older patients with more advanced disease and without kidney imaging, we used age at KRT <55 years to define severe ADPKD (27). Using this criterion, disease discordance was noted in 16 of 18 (89%) families with a protein-truncating PKD1 mutation and mild disease by imaging in the proband. Similarly, using age at KRT >65 years or Mayo Clinic Imaging Class 1A–1B as a proxy for mild disease (27), six of 18 (33%) families displayed disease concordance in at least one affected relative pair (Figure 4).
Discussion
The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease has shown that kidney volume expands on average 5%/yr during adult life in ADPKD and is a sensitive imaging biomarker for predicting kidney-disease progression (17–19). Using age- and height-adjusted total kidney volume, the Mayo Clinic Imaging Classification provides a useful clinical tool for assessing disease severity in ADPKD (20,21). By integrating the results of both mutation- and imaging-based risk assessment, we defined the severity of our patients over a wide age range, including younger patients in whom eGFR poorly discriminates mild from severe disease. In a large and clinically well-characterized cohort, we found 18% (32/174) of patients with protein-truncating PKD1 mutations displayed mild disease by their kidney imaging pattern and eGFR; their mild disease was similar to patients with both PKD2 mutations and the same imaging classes. Among the older patients with protein-truncating PKD1 mutations without kidney volume measurements, we also found evidence for mild disease in 9% (26 out of 290) of patients who required KRT after age 65 years and in 5% (15 out of 280) of patients who did not require KRT at age ≥60 years. Taken together, these data indicate that mild disease described in the above patients is an extreme phenotype associated with protein-truncating PKD1 mutations.
Significant within-family kidney disease variability in ADPKD is well documented and implicates a modifier effect from genetic and environmental factors (10–12). Of the genetic modifiers identified to date, although uncommon, coinheritance of a second rare mutation in a cystic disease gene, such as PKD1, PKD2, or HNF1B, increased the severity of a PKD1 mutation (28–33), while coinheritance of a COL4A1 mutation increased the severity of a PKD2 mutation (34). Additionally, common genetic variants may increase the severity of ADPKD, as exemplified by the variant association observed with DKK3 (35). By comparison, concurrent medical conditions that are risk factors for CKD progression including diabetes, hypertension, and obesity; complications of ADPKD including kidney stone disease and cyst infections; and environmental and dietary factors including smoking and salt, carbohydrate, and protein intake may also increase the severity of ADPKD (36). Notably, all these modifiers are expected to be deleterious by increasing cystic disease severity.
In this study, we observed significant within-family disease variability in our patients with protein-truncating PKD1 mutations and mild disease. In this regard, our documentation of patient TOR308.2 as a somatic mosaic provides an explanation for her mild disease (24). Mosaicism refers to the occurrence of two genetically distinct cell populations within an individual resulting from a somatic mutation during embryogenesis (37). Due to dilution and variable involvement of the affected cells, a mosaic individual with ADPKD typically presents with de novo ADPKD with atypical imaging (i.e., focal, unilateral, or asymmetric) patterns. However, the presence of a positive parental family history of ADPKD in preceding generations in 84% of our patients and a typical ADPKD imaging pattern makes somatic mosaicism an unlikely explanation for the mild disease observed in most of our patients with protein-truncating PKD1 mutations. Overall, 72% of our patients with both protein-truncating PKD1 mutations and Mayo Clinic Imaging Class 1A–1B were older than 30 years of age when misclassification of mild disease is less likely; however, in the remaining younger patients, potential misclassification is a limitation of our study. Taken together, our findings strongly suggest the existence of a protective modifier effect in ADPKD from genetic and/or environmental factors.
Protective genetic factors have been identified in multiple pathologic conditions including HIV-1 infection, hypercholesterolemia, Alzheimer’s disease, nonalcoholic steatohepatitis, and various autoimmune disorders (38–42). For example, homozygous deletions (i.e., delta 32) of the HIV-1 coreceptor CCR5, present in approximately 1% of European population, protect against HIV infection (39); heterozygous loss of function (i.e., p.142×, p.679×) variants of proprotein convertase subtilisin/kexin type 9 (PCSK9), present in approximately 2% of Black individuals, lower LDL cholesterol and reduce the risk of coronary heart disease (40,41); and a heterozygous loss of function (i.e., rs72613567:TA) variant of the hepatic lipid droplet protein hydroxysteroid 17-beta dehydrogenase (HSD17B13), present in approximately 25% of European population, protects against nonalcoholic steatohepatitis (42). These studies provided important insight into disease pathobiology, advanced biomarker discovery, and identified novel targets for therapeutic development.
Mutations of multiple genes encoding for ciliary proteins cause cystic disease, suggesting a central role of the primary cilia in the pathogenesis of cystic kidney diseases (43). Of interest, a study of murine polycystic kidney disease models showed that ablation of primary cilia by inactivation of a ciliary gene (Kif3 or Ift20) resulted in milder disease, whereas inactivation of either Pkd1 or Pkd2 resulted in severe disease. Unexpectedly, ablation of the primary cilia together with inactivation of Pkd1 or Pkd2 in these models resulted in attenuated kidney disease compared with Pkd1 or Pkd2 inactivation, respectively (44). These data suggest that an unidentified cilia-based signaling pathway interacts with the polycystin complex to modulate cyst disease severity; loss of cilia function is protective in the context of ADPKD. Screening of our patients with protein-truncating PKD1 mutations and mild disease for loss-of-function ciliary gene mutations by next-generation sequencing may provide a promising approach to identify protective genetic modifiers.
Metabolic reprogramming and tissue inflammation are important pathogenic mechanisms that mediate the progression of experimental cystic kidney disease (45,46). By modulating the AMP-activated protein kinase–mammalian target of rapamycin signaling pathway, caloric restriction and intermittent fasting have been shown to attenuate the severity of experimental cystic kidney disease (47,48). Likewise, the gut microbiome has been shown to modulate the metabolism and tissue inflammation (49,50). The absence of systematic collection of metabolic comorbidities and dietary intake is a limitation of this study. Therefore, comparative studies of the dietary patterns and gut microbiomes in our discordant affected relative pairs with mild and severe cystic disease may provide another promising approach to unravel the potential environmental protective factors for ADPKD.
In conclusion, we found 18% of patients with protein-truncating PKD1 mutations displayed mild disease, on the basis of their kidney imaging pattern and eGFR. Similarly, among older patients with protein-truncating PKD1 mutations without kidney volume measurements, we also found mild disease in 9% who required KRT >65 years and 5% who did not require KRT at age ≥60 years. Taken together, these data indicate that mild kidney disease described here is not a rare phenotype associated with protein-truncating PKD1 mutation. On average, protein-truncating PKD1 mutations are associated with severe ADPKD; however, our data indicate that mutation class alone cannot be used to predict disease severity in individual patients with complete certainty. The presence of significant within-family disease variability in a majority of these mildly affected patients with protein-truncating PKD1 mutations strongly suggests a protective modifier effect from unidentified genetic and/or environmental factors.
Disclosures
A. Paterson reports employment with the Hospital for Sick Children. E. Guiard reports employment with Toronto General Hospital University Health Network. I.-A. Iliuta reports employment with Toronto General Hospital, University Health Network. M. Lanktree reports employment with St. Joseph Healthcare Hamilton and McMaster University; is a young investigator in the KRESCENT Program, a national kidney research training partnership of the Kidney Foundation of Canada, the Canadian Society of Nephrology, and the Canadian Institutes of Health Research; reports receiving grant funding from Canadian Institutes of Health Research, Hamilton Health Sciences, and Hamilton Academic Health Sciences Organization, and received compensation for participation as a speaker and an advisory board member with Otsuka Pharmaceuticals. S. Ahmed reports employment with Toronto General Hospital. Y. Pei reports employment with University Health Network and University of Toronto; consultancy agreements with Otsuka, Sanofi, and Vertex; receiving honoraria from Otsuka, Sanofi, and Vertex; receiving compensation for participation in advisory boards for Otsuka, Reata Pharmaceuticals, and Sanofi-Genzyme; and serving on the steering committee for a randomized controlled trial in ADPKD (Sanofi/Genzyme). All remaining authors have nothing to disclose.
Funding
This work was in part supported by Canadian Institutes of Health Research (CIHR) project grant PJT-376307 and CIHR Strategy for Patient Oriented Research program grant in CKD (CAN-Solve-CKD SCA-145103; to Y. Pei). M. Lanktree is a new investigator in the Kidney Research Scientist Core Education and National Training program funded by the CIHR Kidney Foundation of Canada and the Canadian Society of Nephrology.
Supplementary Material
Acknowledgments
The authors wish to thank all of the study patients and their families, and the research staff of the Center for Innovative Management of PKD at the Toronto General Hospital and the St. Joseph’s Healthcare Hamilton.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
Supplemental Material
This article contains the following supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.11100720/-/DCSupplemental.
Supplemental Material. Mutation screening protocol for study cohort.
Supplemental Figure 1. Representative axial (left) and coronal (right) magnetic resonance images comparing age-matched cystic disease severity by Mayo class in patients with different mutation types.
Supplemental Figure 2. Location of PKD1-truncating mutations and mild Mayo Imaging Class.
References
- 1.Spithoven EM, Kramer A, Meijer E, Orskov B, Wanner C, Abad JM, Aresté N, de la Torre RA, Caskey F, Couchoud C, Finne P, Heaf J, Hoitsma A, de Meester J, Pascual J, Postorino M, Ravani P, Zurriaga O, Jager KJ, Gansevoort RT; ERA-EDTA Registry; EuroCYST Consortium; WGIKD: Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: Prevalence and survival--An analysis of data from the ERA-EDTA Registry. Nephrol Dial Transplant 29[Suppl 4]: iv15–iv25, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ong AC, Devuyst O, Knebelmann B, Walz G; ERA-EDTA Working Group for Inherited Kidney Diseases: Autosomal dominant polycystic kidney disease: The changing face of clinical management [published correction appears in Lancet 385: 2576, 2015 10.1016/S0140-6736(15)61160-6]. Lancet 385: 1993–2002, 2015 [DOI] [PubMed] [Google Scholar]
- 3.Cornec-Le Gall E, Audrézet M-P, Chen JM, Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau E, Jousset P, Guillodo MP, Grall-Jezequel A, Saliou P, Férec C, Le Meur Y: Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 24: 1006–1013, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwang Y-H, Conklin J, Chan W, Roslin NM, Liu J, He N, Wang K, Sundsbak JL, Heyer CM, Haider M, Paterson AD, Harris PC, Pei Y: Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 1861–1868, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heyer CM, Sundsbak JL, Abebe KZ, Chapman AB, Torres VE, Grantham JJ, Bae KT, Schrier RW, Perrone RD, Braun WE, Steinman TI, Mrug M, Yu AS, Brosnahan G, Hopp K, Irazabal MV, Bennett WM, Flessner MF, Moore CG, Landsittel D, Harris PC; HALT PKD and CRISP Investigators: Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 2872–2884, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, Edwards ME, Madsen CD, Mauritz SR, Banks CJ, Baheti S, Reddy B, Herrero JI, Bañales JM, Hogan MC, Tasic V, Watnick TJ, Chapman AB, Vigneau C, Lavainne F, Audrézet MP, Ferec C, Le Meur Y, Torres VE, Harris PC; Genkyst Study Group, HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease: Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet 98: 1193–1207, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, Audrézet MP, Hopp K, Porath B, Shi B, Baheti S, Senum SR, Arroyo J, Madsen CD, Férec C, Joly D, Jouret F, Fikri-Benbrahim O, Charasse C, Coulibaly JM, Yu AS, Khalili K, Pei Y, Somlo S, Le Meur Y, Torres VE, Harris PC; Genkyst Study Group; HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease: Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet 102: 832–844, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D: Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet 353: 103–107, 1999 [DOI] [PubMed] [Google Scholar]
- 9.Harris PC, Bae KT, Rossetti S, Torres VE, Grantham JJ, Chapman AB, Guay-Woodford LM, King BF, Wetzel LH, Baumgarten DA, Kenney PJ, Consugar M, Klahr S, Bennett WM, Meyers CM, Zhang QJ, Thompson PA, Zhu F, Miller JP: Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 17: 3013–3019, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Paterson AD, Magistroni R, He N, Wang K, Johnson A, Fain PR, Dicks E, Parfrey P, St George-Hyslop P, Pei Y: Progressive loss of renal function is an age-dependent heritable trait in type 1 autosomal dominant polycystic kidney disease. J Am Soc Nephrol 16: 755–762, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Peters DJ, Breuning MH: Autosomal dominant polycystic kidney disease: Modification of disease progression. Lancet 358: 1439–1444, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Lanktree M, Guiard E, Li W, Akbari P, Haghighi A, Iliuta I-A, Shi B, Chen C, He N, Song X, Margetts PJ, Ingram AJ, Khalili K, Paterson AD, Pei Y: Intrafamilial variability of ADPKD. Kidney Int Rep 4: 995–1003, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS; TEMPO 3:4 Trial Investigators: Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367: 2407–2418, 2012. 23121377 [Google Scholar]
- 14.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Perrone RD, Koch G, Ouyang J, McQuade RD, Blais JD, Czerwiec FS, Sergeyeva O; REPRISE Trial Investigators: Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med 377: 1930–1942, 2017 [DOI] [PubMed] [Google Scholar]
- 15.Gansevoort RT, Arici M, Benzing T, Birn H, Capasso G, Covic A, Devuyst O, Drechsler C, Eckardt KU, Emma F, Knebelmann B, Le Meur Y, Massy ZA, Ong AC, Ortiz A, Schaefer F, Torra R, Vanholder R, Więcek A, Zoccali C, Van Biesen W: Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: A position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol Dial Transplant 31: 337–348, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soroka S, Alam A, Bevilacqua M, Girard L-P, Komenda P, Loertscher R, McFarlane P, Pandeya S, Tam P, Bichet DG: Updated Canadian expert consensus on assessing risk of disease progression and pharmacological management of autosomal dominant polycystic kidney disease. Can J Kidney Health Dis 5: 1–15, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF Jr, Wetzel LH, Baumgarten DA, Kenney PJ, Harris PC, Klahr S, Bennett WM, Hirschman GN, Meyers CM, Zhang X, Zhu F, Miller JP; CRISP Investigators: Volume progression in polycystic kidney disease. N Engl J Med 354: 2122–2130, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Yu ASL, Shen C, Landsittel DP, Harris PC, Torres VE, Mrug M, Bae KT, Grantham JJ, Rahbari-Oskoui FF, Flessner MF, Bennett WM, Chapman AB; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP): Baseline total kidney volume and the rate of kidney growth are associated with chronic kidney disease progression in autosomal dominant polycystic kidney disease. Kidney Int 93: 691–699, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perrone RD, Mouksassi MS, Romero K, Czerwiec FS, Chapman AB, Gitomer BY, Torres VE, Miskulin DC, Broadbent S, Marier JF: Total kidney volume is a prognostic biomarker of renal function decline and progression to end-stage renal disease in patients with autosomal dominant polycystic kidney disease. Kidney Int Rep 2: 442–450, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, Bae KT, Chapman AB, Grantham JJ, Mrug M, Hogan MC, El-Zoghby ZM, Harris PC, Erickson BJ, King BF, Torres VE; CRISP Investigators: Imaging classification of autosomal dominant polycystic kidney disease: A simple model for selecting patients for clinical trials. J Am Soc Nephrol 26: 160–172, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irazabal MV, Abebe KZ, Bae KT, Perrone RD, Chapman AB, Schrier RW, Yu AS, Braun WE, Steinman TI, Harris PC, Flessner MF, Torres VE; HALT Investigators: Prognostic enrichment design in clinical trials for autosomal dominant polycystic kidney disease: The HALT-PKD clinical trial. Nephrol Dial Transplant 32: 1857–1865, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, San Millan JL, Gibson R, Breuning M, Peters D, Ravine D: Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 20: 205–212, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pei Y, Hwang YH, Conklin J, Sundsbak JL, Heyer CM, Chan W, Wang K, He N, Rattansingh A, Atri M, Harris PC, Haider MA: Imaging-based diagnosis of autosomal dominant polycystic kidney disease. J Am Soc Nephrol 26: 746–753, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iliuta I-A, Kalatharan V, Wang K, Cornec-Le Gall E, Conklin J, Pourafkari M, Ting R, Chen C, Borgo AC, He N, Song X, Heyer CM, Senum SR, Hwang YH, Paterson AD, Harris PC, Khalili K, Pei Y: Polycystic kidney disease without an apparent family history. J Am Soc Nephrol 28: 2768–2776, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consugar MB, Wong WC, Lundquist PA, Rossetti S, Kubly VJ, Walker DL, Rangel LJ, Aspinwall R, Niaudet WP, Ozen S, David A, Velinov M, Bergstralh EJ, Bae KT, Chapman AB, Guay-Woodford LM, Grantham JJ, Torres VE, Sampson JR, Dawson BD, Harris PC; CRISP Consortium: Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int 74: 1468–1479, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration): A new equation to estimate glomerular filtration rate [published correction appears in Ann Intern Med 155: 408, 2011]. Ann Intern Med 150: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barua M, Cil O, Paterson AD, Wang K, He N, Dicks E, Parfrey P, Pei Y: Family history of renal disease severity predicts the mutated gene in ADPKD. J Am Soc Nephrol 20: 1833–1838, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pei Y, Paterson AD, Wang KR, He N, Hefferton D, Watnick T, Germino GG, Parfrey P, Somlo S, St George-Hyslop P: Bilineal disease and trans-heterozygotes in autosomal dominant polycystic kidney disease. Am J Hum Genet 68: 355–363, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pei Y, Lan Z, Wang K, Garcia-Gonzalez M, He N, Dicks E, Parfrey P, Germino G, Watnick T: A missense mutation in PKD1 attenuates the severity of renal disease. Kidney Int 81: 412–417, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, Chauveau D, Rees L, Barratt TM, van’t Hoff WG, Niaudet P, Torres VE, Harris PC: Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease [published correction appears in Kidney Int 75: 1359, 2009]. Kidney Int 75: 848–855, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, Hampel T, Pape L, Buske A, Jonsson J, Sarioglu N, Santos A, Ferreira JC, Becker JU, Cremer R, Hoefele J, Benz MR, Weber LT, Buettner R, Zerres K: Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol 22: 2047–2056, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Audrézet MP, Corbiere C, Lebbah S, Morinière V, Broux F, Louillet F, Fischbach M, Zaloszyc A, Cloarec S, Merieau E, Baudouin V, Deschênes G, Roussey G, Maestri S, Visconti C, Boyer O, Abel C, Lahoche A, Randrianaivo H, Bessenay L, Mekahli D, Ouertani I, Decramer S, Ryckenwaert A, Cornec-Le Gall E, Salomon R, Ferec C, Heidet L: Comprehensive PKD1 and PKD2 mutation analysis in prenatal autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 722–729, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cornec-Le Gall E, Torres VE, Harris PC: Genetic complexity of autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol 29: 13–23, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cornec-Le Gall E, Chebib FT, Madsen CD, Senum SR, Heyer CM, Lanpher BC, Patterson MC, Albright RC, Yu AS, Torres VE, Harris PC; HALT Progression of Polycystic Kidney Disease Group Investigators: The value of genetic testing in polycystic kidney diseases illustrated by a family with PKD2 and COL4A1 mutations. Am J Kidney Dis 72: 302–308, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu M, Shi S, Senthilnathan S, Yu J, Wu E, Bergmann C, Zerres K, Bogdanova N, Coto E, Deltas C, Pierides A, Demetriou K, Devuyst O, Gitomer B, Laakso M, Lumiaho A, Lamnissou K, Magistroni R, Parfrey P, Breuning M, Peters DJ, Torra R, Winearls CG, Torres VE, Harris PC, Paterson AD, Pei Y: Genetic variation of DKK3 may modify renal disease severity in ADPKD. J Am Soc Nephrol 21: 1510–1520, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowak KL, You Z, Gitomer B, Brosnahan G, Torres VE, Chapman AB, Perrone RD, Steinman TI, Abebe KZ, Rahbari-Oskoui FF, Yu ASL, Harris PC, Bae KT, Hogan M, Miskulin D, Chonchol M: Overweight and obesity are predictors of progression in early autosomal dominant polycystic kidney disease. J Am Soc Nephrol 29: 571–578, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell IM, Shaw CA, Stankiewicz P, Lupski JR: Somatic mosaicism: Implications for disease and transmission genetics. Trends Genet 31: 382–392, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harper AR, Nayee S, Topol EJ: Protective alleles and modifier variants in human health and disease. Nat Rev Genet 16: 689–701, 2015 [DOI] [PubMed] [Google Scholar]
- 39.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR: Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86: 367–377, 1996 [DOI] [PubMed] [Google Scholar]
- 40.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH: Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 37: 161–165, 2005 [DOI] [PubMed] [Google Scholar]
- 41.Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH: Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 354: 1264–1272, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, Liu Y, Kozlitina J, Stender S, Wood GC, Stepanchick AN, Still MD, McCarthy S, O’Dushlaine C, Packer JS, Balasubramanian S, Gosalia N, Esopi D, Kim SY, Mukherjee S, Lopez AE, Fuller ED, Penn J, Chu X, Luo JZ, Mirshahi UL, Carey DJ, Still CD, Feldman MD, Small A, Damrauer SM, Rader DJ, Zambrowicz B, Olson W, Murphy AJ, Borecki IB, Shuldiner AR, Reid JG, Overton JD, Yancopoulos GD, Hobbs HH, Cohen JC, Gottesman O, Teslovich TM, Baras A, Mirshahi T, Gromada J, Dewey FE: A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med 378: 1096–1106, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harris PC, Torres VE: Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 124: 2315–2324, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S: Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45: 1004–1012, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, Song XW, Xu H, Mari S, Qian F, Pei Y, Musco G, Boletta A: Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med 19: 488–493, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Zhou X, Fan LX, Yao Y, Swenson-Fields KI, Gadjeva M, Wallace DP, Peters DJ, Yu A, Grantham JJ, Li X: Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J Clin Invest 125: 2399–2412, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Warner G, Hein KZ, Nin V, Edwards M, Chini CC, Hopp K, Harris PC, Torres VE, Chini EN: Food restriction ameliorates the development of polycystic kidney disease. J Am Soc Nephrol 27: 1437–1447, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Torres JA, Kruger SL, Broderick C, Amarlkhagva T, Agrawal S, Dodam JR, Mrug M, Lyons LA, Weimbs T: Ketosis ameliorates renal cyst growth in polycystic kidney disease. Cell Metab 30: 1007–1023.e5, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lynch SV, Pedersen O: The human intestinal microbiome in health and disease. N Engl J Med 375: 2369–2379, 2016 [DOI] [PubMed] [Google Scholar]
- 50.Brown JM, Hazen SL: The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu Rev Med 66: 343–359, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.