

Abstract
The palladium/norbornene (Pd/NBE) cooperative catalysis has received enormous attention and found numerous synthetic applications in the past two decades. Considering the critical roles that NBE plays in the catalytic cycle, the use of structurally modified NBEs (smNBEs), starting from 2015, has become an important approach to address limitations and modulate reaction selectivity in the Pd/NBE catalysis. This perspective highlights the development of three types of smNBEs: C1-substituted, C2-substituted, and C5-substituted or C5, C6-disubstituted NBEs, as well as their synthetic applications towards site-selective C–H functionalization. A focus is given to the structure-activity relationship of smNBEs in these reactions, and rationales of using smNBEs in many cases have also been provided.
Graphical Abtract

1. Introduction
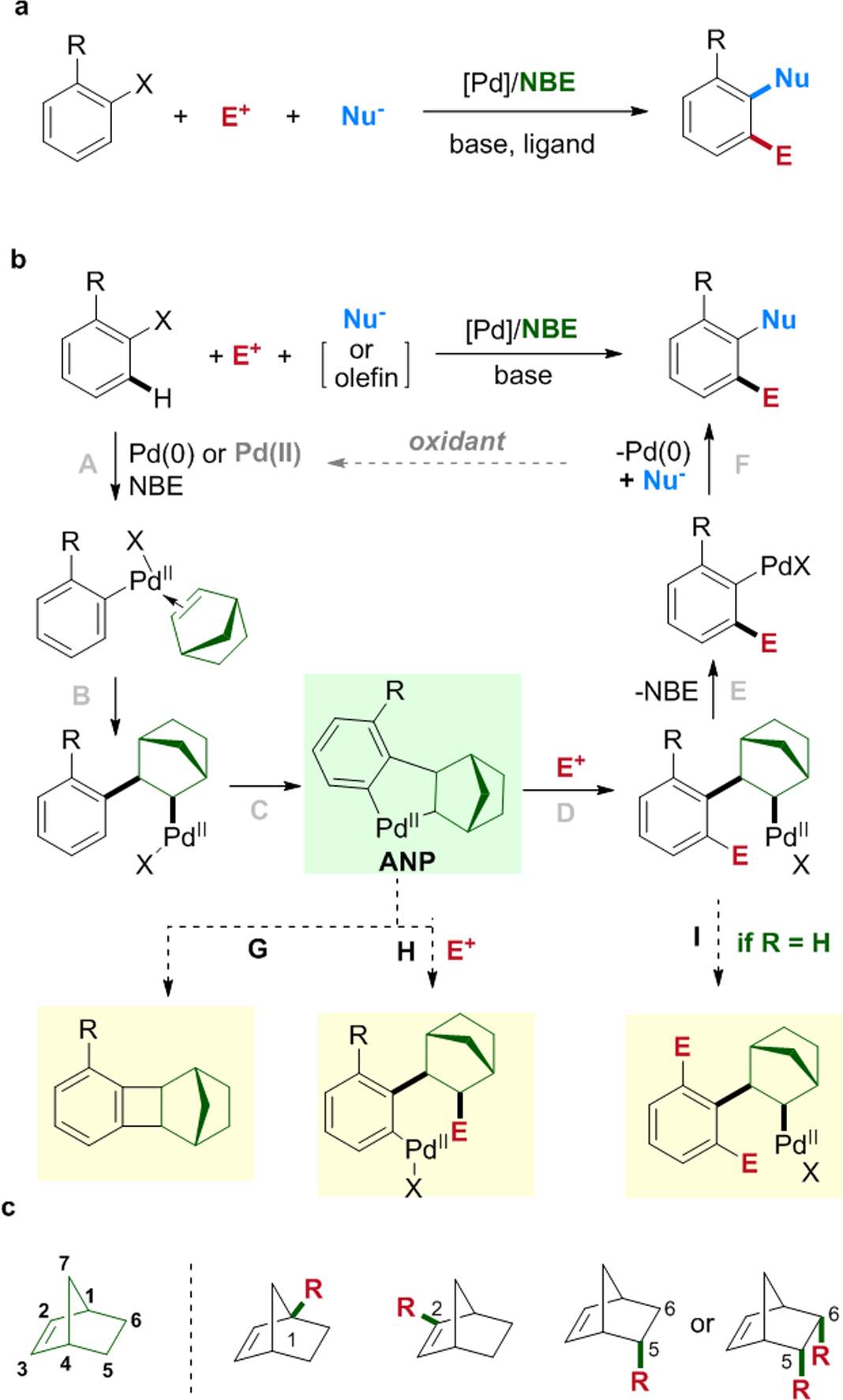
Since the seminal discovery by Catellani in 1997, the palladium/norbornene (Pd/NBE) co-catalyzed arene functionalization reactions have received increasing amounts of attention and led to many synthetic applications in the past two decades.1 Compared to the widely adopted cross-coupling and nucleophilic aromatic substitution (SNAr) reactions where the number and position of newly installed functional groups (FGs) are dictated by those of the leaving groups in the substrates, the Catellani-type reactions can introduce multiple different FGs simultaneously in a site-selective manner (Scheme 1a). Considering that polysubstituted aromatics are ubiquitously found in pharmaceuticals and agrochemicals, the Pd/NBE catalysis could become an incredible useful tool for functionalization and streamlined syntheses of complex arenes and heteroarenes. Despite the great promise, some intrinsic constraints due to the use of the NBE co-factor had nevertheless become roadblocks to practical applications of these reactions.
Scheme 1.
Pd/NBE Cooperative Catalysis
As illustrated in Scheme 1b, a general mechanism of the Catellani reaction involves first forming an aryl-Pd(II) species from either oxidative addition of Pd(0) with aryl halides or transmetallation or direct C–H palladation from a Pd(II) catalyst. The subsequent NBE migratory insertion and palladation at the ortho position give a unique aryl-norbornyl-palladacycle (ANP) intermediate that can react with an electrophile to introduce an ortho FG (relative to the original reaction site).2 After β-carbon elimination to exclude NBE, the resulting aryl-Pd(II) species can either react with a nucleophile (including olefins) to install an ipso FG and regenerate the Pd(0) catalyst, or be quenched by a proton to regenerate the Pd(II) catalyst. Due to the high complexity of the mechanism, many competing side reactions can take place. For example, the ANP can undergo direct C−C reductive elimination to form benzocyclobutenes (step G);3 the reaction with electrophiles can oxidize the C(sp3)−Pd bond in ANP instead of the desired C(sp2)−Pd bond (step H);4–5 the β-carbon elimination of NBE could be problematic in the absence of an existing sizable ortho substituent and a second C–H palladation may take place (known as “ortho constraint”, step I),6 etc. Clearly, the structure of NBE plays critical roles in almost every single step of the catalytic cycle, and most side-products contain the NBE moieties. Thus, in the past five years researchers in this field have been motivated to use structurally modified NBEs (smNBE) to solve these selectivity problems in the Catellani-type reactions.
The strained and rigid [2.2.1] bicyclic scaffold of NBE offers unique features for the success of the Catellani-type reactions: it enables rapid migratory insertion to outcompete direct ipso functionalization, avoids β-hydrogen elimination to promote the desired ortho C–H palladation, and allows β-carbon elimination to regenerate NBE. Hence, the current successful modifications on NBE usually maintain the original [2.2.1] skeleton, and to date effective variations are located at the C1, C2, C5 and C6 positions (Scheme 1c). In general, the size and position of the substituents are critical. For instance, increasing the steric hindrance of NBE would promote β-carbon elimination, but reduce its binding affinity to Pd, thereby causing formation of more undesired direct ipso functionalization side-products. The C1-substituted NBEs can inhibit the undesired second C–H metalation and promote β-carbon elimination; thus, they were developed for addressing the “ortho constraint” issue. The reactivity of the C5- and C6-substituted NBEs are close to (yet sometimes more reactive than) simple unsubstituted NBE. The C2-substituted NBEs play a more complex role: they can inhibit direct reductive elimination of ANP to form norbornyl benzocyclobutenes, prompt NBE insertion and C–H palladation (when the C2-substituent can form hydrogen bonding). More detailed discussions are provided in the following sections.
This perspective article was inspired by several excellent review articles published recently7–17 and will primarily focus on three types of smNBE cocatalysts: C1-substituted, C2-substituted and C5-substituted or C5, C6-disubstituted smNBEs.18
2. C1-Substituted smNBEs
Typically, the aryl-halide substrates used in the Pd/NBE catalysis contain an ortho-substituent. When ortho unsubstituted haloarenes were used, di-ortho-functionalization (with para-substituted substrates) or formation of a mixture of NBE-containing side-products (with meta-substituted substrates) would dominate, whereas the desired mono-ortho-functionalization generally cannot be achieved (Scheme 2a). This has been referred as “ortho constraint”.6 The origin of the problem is due to slow β-carbon elimination of NBE in the absence of the ortho substituent, which promotes a second ortho C–H palladation or pre-mature termination.
Scheme 2.

Overcoming the ortho constraint with C1-smNBEs
To address this challenge, the Dong group developed a class of C1-substituted smNBEs in 2018, which effectively allowed the use of ortho-unsubstituted aryl halides as substrates for mono-ortho-functionalization.6 Compared to regular NBE, the bridgehead substitution was anticipated to inhibit the undesired second C–H palladation by destabilizing its transition state (TS) through repulsive interaction with the newly installed ortho FG; meanwhile, it can also reduce the barrier for β-carbon elimination to extrude NBE through controlling the orientation of the arene (Scheme 2b). The C1 alkyl-substituted NBEs, e.g. N2–N5, were found particularly effective when the ortho amination was used as the model reaction (Scheme 2c).19 Stoichiometric smNBEs were used here to minimize undesired ipso functionalization. The typical procedure for synthesis of C1-substituted smNBEs is shown in Scheme 2d with N3 as an example.
While the n-heptyl-substituted N3 smNBE was optimal for most meta-substituted substrates, the cyclohexyl-substituted N4 was more effective when the meta-substituents are small, such as F, OMe and CO2Me (Scheme 3). Higher yield was observed when aryliodides with a bulkier meta substituent were used (4–6). Heck reaction (4–6), hydrogenation (7), Suzuki (8) and Sonogashira (9) reactions could all take place at the ipso position; besides ortho amination, ortho acylation (10) and arylation (11) also worked under slightly modified reaction conditions. Without such ortho constraint, vicinal difunctionalization or site-selective derivatization of complex bioactive molecules (12–15) could now be conveniently realized via a two-step sequence—arene iodination and this method. When ipso hydrogenation was employed, complementary site-selectivity to electrophilic aromatic substitution (EAS) was achieved for arene C–H functionalization because FGs can be installed at the positions that are not favorable for EAS reactions.
Scheme 3.

Scope and Late-stage Functionalization of Bioactive Compounds
Finally, selective mono ortho functionalization of the more challenging para-substituted aryl iodides could be realized with a doubly bridgehead-substituted NBE (N8). The density functional theory (DFT) calculation suggested that steric repulsion caused by the bridgehead substituents in N8 significantly increased the barrier of the second C–H metalation step (thus preventing di-ortho-functionalization), whereas the β-carbon elimination and migratory insertion steps exhibit reduced activation energy, which makes forming the mono-ortho-product kinetically favored.
The C1-substituted smNBEs provide a viable approach to address the longstanding ortho constraint in the Catellani-type transformations. Future work may focus on improving the efficiency of C1-smNBEs and enabling new synthetic applications.
3. C2-Substituted smNBEs
Compared to C1-substituted smNBEs, the C2-substituted ones exhibit different functions. First, the C2 substituents directly introduce steric hindrance on the carbon that binds to Pd, which greatly suppresses direct C−C reductive elimination of ANP (Scheme 4a).20 Second, the electron-withdrawing C2 substituent can promote migratory insertion of NBE from the electronic prospect to compromise the weakened olefin binding and steric disadvantage (Scheme 4b). Third, when a secondary amide moiety is introduced at the C2 position, the resulting hydrogen bonding with the concerted metalation deprotonation (CMD) ligand on Pd can reduce the overall activation barrier (vide infra, Scheme 13).21 While a number of C2-substituted smNBEs have been prepared, to date the most effective two types are: the ester-substituted one pioneered by Yu20 and the amide-substituted ones developed by Dong (Scheme 4c).22–23 The preparation of N9 was shown as an example (Scheme 4d).24
Scheme 4.

C2-Substituted smNBEs
Scheme 13.

Asymmetric ortho arylation of Aryl Iodides
3.1. Pd(II)-Catalyzed Meta C–H Functionalization Involving C2-Substituted smNBEs
While the directing group (DG)-promoted meta C–H arylation and alkylation of arenes via the Pd/NBE cooperative catalysis was firstly realized using simple NBE,25,26 these reactions suffered from relatively limited substrate scopes. For example, aryl iodides without an ortho DG typically gave less than 5% yield, and formation of norbornyl benzocyclobutene side-products outcompeted the desired meta-alkylation pathway, especially when using alkyl iodides bearing β-hydrogens. To address these drawbacks, Yu and co-workers utilized a C2 methyl carboxylate-substituted NBE (N9) to greatly expand the scope of aryl and alkyl electrophiles for meta C–H functionalization (Scheme 5).20 The new condition allowed the use of simple alkyl iodides with β-hydrogens and aryl iodides without an ortho DG; it also minimized benzocyclobutene side-products. The ester moiety was more effective than a more electron-withdrawing ketone or nitrile group as the corresponding N13 and N14 NBEs showed much lower reactivity.
Scheme 5.

Meta Alkylation and Arylation of Substituted Phenylacetamides Using C2-Substituted smNBEs
Besides aryl and alkyl iodides, other electrophiles such as o-benzoyl hydroxylamines and alkynyl bromides were successfully utilized in the C–H activation-initiated meta-functionalization reactions. Owing to the use of C2 methyl carboxylate-substituted smNBE (N9) as well as the 2-pyridone-type ligands, the Yu group realized the first meta amination and alkynylation (Scheme 6a) of aniline- and phenol-derivatives in 2016.27 For example, the yield of meta amination product was improved significantly by switching simple NBE N1 to N9. Heterocycles including indole, indoline and indazole were compatible in the meta amination and alkynylation reactions. In terms of the alkyne electrophiles, only bulky silyl-protected alkynyl bromides afforded the desired products in good yields at the current stage, while simple alkyl and aryl alkynyl bromides only led to trace amounts of meta products. In addition, smNBE N9 also enabled the first Pd(II)-initiated meta C–H chlorination of aniline and phenol derivatives also by Yu in 2017 (Scheme 6b).28 An aryl chlorosulfate reagent29 was used as a mild chlorinating reagent to exclusively oxidize Pd(II) to Pd(IV)30 without direct ortho C–H chlorination. The two new hydroxypyridine ligands L1 and L2 were also important for enhanced yields and scope. As an important application, the chloride group in the products could be conveniently converted to other FGs through various cross-coupling reactions.
Scheme 6.

Meta Amination, Alkynylation and Chlorination of Aniline and Phenol Derivatives
The unsymmetrical structure of C2-substituted smNBEs also offers opportunities for developing enantioselective transformations. In 2018, the Yu group reported the first example of enantioselective meta C–H arylation and alkylation using enantiopure N9* (Scheme 7).31 It was proposed that the chiral NBE structure could differentiate the enantiomeric ortho C–H palladation intermediates during the NBE migratory insertion step. Compared with other smNBEs, the C2 substitution was more important for the enantio-determining step. Addition of a catalytic amount of chiral or achiral phosphoric acids as the additive was beneficial for both yield and enantioselectivity, while control experiments indicated that the enantioenriched smNBE was mostly responsible for the chiral induction.
Scheme 7.

Enantioselective Meta C–H Arylation and Alkylation Enabled by a Chiral smNBE
Besides diarylbenzylamines, homobenzylamines can also be used as the substrates, despite forming a more distal stereocenter (Scheme 7b). The use of nosyl-protected amines as DGs proved to be efficient. Besides desymmetrization, kinetic resolution of unsymmetrical aryl substrates was also achieved. Note that a contemporary discovery of an enantioenriched C2-smNBE-promoted Pd(0)-catalyzed asymmetric annulation with aryl iodides was reported by Dong (vide infra, Scheme 12).22
Scheme 12.

Asymmetric Annulation between Aryl Iodides and Epoxides
The meta C–H functionalization could also be realized for substrates without covalently bound auxiliary DGs, in which the C2-substiutted smNBEs were again found to be important. In 2019, the Yu group reported the meta C–H arylation of aryl ethers, which offered complementary site-selectivity to EAS reactions (Scheme 8a).32 Interestingly, a dual-ligand system was applied: an electron-deficient quinoxaline-6-carbonitrile L3 along with a 3-pyridinesulfonic acid L4 were used to generate more electrophilic cationic Pd(II) species, thus facilitating the first C–H palladation. A series of aryl ethers can be used as substrates; besides anisoles, 2,3-dihydrobenzofuran and chromane moieties (36 and 37) were also suitable, affording exclusively meta selectivity. Heterocycles, such as pyridine (34), were tolerated. Various complex moieties such as estrone (35) and tasimelteon (36) were also comparable. Based on the DFT calculation, the first C–H palladation was proved to be the rate-determining step.33 Very recently, this method has been extended to electron-deficient heterocycles34 and fluorobenzenes35 by the Yu group (Scheme 8b).
Scheme 8.

Direct Meta Arylation of Electron-Rich Arenes, Electron-Deficient Heteroarenes and Fluorobenzenes.
Very recently, a remote site-selective C–H arylation of quinolines and other heterocycles was achieved by Yu and Houk using the N9 smNBE (Scheme 9a).36 This work merged their previously developed template-directed approach37 and the Pd/NBE catalysis to realize complementary remote site-selectivity. The DFT calculation indicated that the nitrile group on the side arm of the Pd template directed the initial C–H palladation with the second Pd center to reach the C5 position of the quinoline substrate, and an N-acetylglycine ligand promoted the CMD step. This approach was applicable to a variety of heterocycles, including quinolines (40), benzoxazoles (41) and benzothiazoles (42). A late-stage modification of an antileukemic and antitumor alkaloid, camptothecin (43), was demonstrated. Besides the nitrile template, this strategy also worked with their previously reported U-shape templates (Scheme 9b). Tetrahydroisoquinolines (44) was able to undergo C–H arylation at the C7 position, which is one bond further away from the “normal” C8 selectivity offered by the U-shape DG alone. Finally, para arylation of phenylpropanoic acid derivatives (45) was realized in a similar manner, while the two ortho positions adjacent to the DG had to be blocked likely due to the ortho constraint of the Catellani-type reactions (vide supra, Scheme 2).
Scheme 9.

Remote C–H Arylation of Benzoazines, Tetrahydroisoquinolines and Phenylpropanoic Acids
3.2. Pd(II)-Initiated Vicinal Difunctionalization Involving C2-Substituted smNBEs
While the meta C–H functionalization via the Pd/NBE cooperative catalysis has been extensively developed, the corresponding vicinal difunctionalization through sequential double C–H activation was not reported until 2019. From the synthetic efficiency viewpoint, it could be attractive to simultaneously convert two adjacent C–H bonds into different FGs in a site-selective manner. In 2019, the Dong group reported a direct vicinal difunctionalization of thiophenes and furans without aid of DGs, where a C2 amide-substituted smNBE (N12) was found superior to other NBEs (Scheme 10a).38 N12 has been known to prompt both the migratory insertion and CMD processes (vide infra, Scheme 14). The reaction was initiated through a reversible C–H palladation at thiophene C2 (or C5) positions. The use of a weakly coordinative π-acidic arsine ligand was important to prevent chelation of the sulfur moiety on the palladium without compromising the ANP formation.26 The scope of the ortho arylation/ipso Heck reaction was relatively broad (Scheme 10b). A series of mono- and disubstituted thiophenes were difunctionalized site- and regio-selectively at the C4 and C5 positions in good yields. Various FGs including bromide (51) and internal alkynes (50) were tolerated. Preliminary success was obtained with a furan substrate (49). In addition, this method was applied to vicinal difunctionalization of complex bioactive compounds (52–55). Besides, an open-flask gram-scale preparation was demonstrated using commercial ethyl acetate without further purification (Scheme 10c). Finally, a kinetic study implied a simultaneous difunctionalization pathway instead of a stepwise functionalization pathway. The high reactivity and selectivity shown by the C2-amide-substituted smNBE (N12) could inspire vicinal di-C–H-functionalization of other arenes and heteroarenes.
Scheme 10.

Vicinal Difunctionalization of Thiophenes
Scheme 14.

NBE Effect for the Meta-Substituted Aryl Iodides
3.3. Pd(0)-Initiated Difunctionalization of Aryl Halides Involving C2-Substituted smNBEs
In most of Pd(0)-initiated Catellani-type reactions, simple NBE is sufficient to achieve good yields and selectivity. However, in some special cases, particularly those with less reactive electrophiles or substrates, side reactions, e.g. multi-NBE insertion or direct reductive elimination from ANP,3, 39 can dominate; the C2-substiutted smNBEs have been found to be effective to enhance selectivity of such reactions. For example, when epoxides were employed as the electrophile in an annulation with aryl iodides for synthesis of 2,3-dihydrobenzofurans (DHBFs), the use of simple NBE (N1) generated a significant amount of multi-NBE insertion products.40 This was likely due to the relatively low electrophilicity of epoxides, which makes them less reactive to couple with ANP compared with N1. The Dong group in 2017 disclosed that the multi-NBE insertion pathway can be effectively suppressed using C2-substituted smNBEs as the steric hindrance introduced in the ANP intermediate can prevent insertion of another bulky smNBE. While the C2 isopropyl ester-substituted NBE (N10) proved to be optimal, further increasing the steric hindrance, e.g. use of tert-butyl ester-substituted N23, significantly diminished the yield. Note that, while most prior Pd/NBE-catalyzed reactions require a high loading of or excess NBE due to the formation of undesired NBE-attached side-product (vide supra, Scheme 1), only 20 mol% N10 was found to be sufficient in this reaction. In addition, a bulky phosphine-derived Buchwald’s precatalyst, Ruphos-Pd-G4,41 was employed to inhibit β−H elimination of the Pd alkoxide intermediate and promote the final C−O reductive elimination step.42–43
This reaction tolerates various primary epoxides and aryl iodides with diverse FGs and heterocycles. When an enantiopure epoxide was used as the coupling partner, the annulation reaction proceeded with complete stereo-retention (61). Finally, this method was used to realize a concise synthesis of fufenozide. Note that almost concurrently, Zhou reported an elegant transformation using epoxides as electrophiles in an ortho alkylation/ipso Heck reaction, whereas a C5-substituted NBE was found more important (vide infra, Scheme 18).44–45
Scheme 18.

Pd/NBE-Catalyzed Ortho Alkylation Reaction with Epoxides
While enantioenriched epoxides can be obtained from kinetic resolution or asymmetric synthesis, it would still be attractive to utilize more available racemic epoxides to realize an asymmetric synthesis of DHBFs. In 2018, the Dong group reported their initial study on a Pd-catalyzed asymmetric annulation between aryl iodides and epoxides enabled by an enantioenriched C2-substituted smNBE cocatalyst.22 This work represents the first chiral NBE scaffold-promoted asymmetric reaction via Pd(0)-initiated Catellani-type reactions. The chirality of the enantiopure NBE was anticipated to create a chiral pocket around the metal center in ANP, consequently promoting one enantiomer of the epoxide to react faster than the other (Scheme 12a). A reliable route was first developed to prepare these enantiopure smNBEs using 2,10-camphorsultam as a chiral auxiliary (Scheme 12b). The pyrrolidine amide-substituted (+)-N11 gave the highest enantioselectivity (45% ee) but diminished reactivity; the enantiopure isopropyl ester-substituted NBE (−)-N10 gave slightly lower enantioselectivity but significantly better yield (Scheme 12c). The kinetic monitoring of the reaction suggested that the stereochemistry of (−)-N10 matches the (S)-epoxide, which nevertheless reacted significantly slower if using another enantiomer of NBE (+)-N10 (Scheme 12d).
The Pd(0)-initiated asymmetric Catellani-type reactions enabled by chiral smNBEs was further expanded to asymmetric ortho arylation reaction by Zhou group very recently (Scheme 13).46 The C2 ethyl ester-substituted N21* gave the highest enantioselectivity (96% ee), while the methylamide-substituted N12* afforded 86% ee and diminished reactivity. In contrast, the C5 ester-substituted N33* and C5 acid-substituted N34* gave significantly lower reactivity and enantioselectivity. A series of axially chiral biaryl compounds were obtained in decent yield and enantioselectivity and good FG tolerance was demonstrated as well. Note that besides Heck reaction, other ipso quenching, e.g. Sonogashira coupling (70), was also realized. When an ortho acetyl aryl bromide was used, a fluorenol product (71) was obtained in decent yield and enantioselectivity, which is consistent with Lautens’ prior observation.47 In terms of the stereo-induction model, it was proposed that the aryl moiety is located at the planar position of the Pd(IV) complex to minimize the steric repulsion; the coordination of the ester group would help fixing the orientation of the aryl moiety through a stable dihedral angle (Scheme 13b).
One long-standing limitation of the Catellani-type reactions is that sizeable substituents (e.g. methyl or isopropyl group) at the meta position of aryl halides inhibit ortho functionalization, which has been referred as “meta constraint”.48 The steric hindrance at the meta position makes the ANP formation and the following steps more difficult. Electron-withdrawing meta substituents were also detrimental to the reaction. These substrates often lead to NBE-attached side-products or direct ipso substitution. In 2020, the Dong group reported the use of C2 amide-derived smNBEs to realize Catellani-type reactions with aryl iodides bearing diverse meta substituents, which provided rapid access to 1,2,3,4-tetrasubstituted arenes (Scheme 14).21 Compared with other NBEs, the C2 smNBE that contains a secondary amide moiety (N12) proved to be most efficient. Based on the DFT calculations, N12 with a mild electron-withdrawing substituent exhibited the lowest barrier for the NBE-insertion step through balancing the opposite electronic requirements in the NBE binding and the migratory insertion steps. In addition, the existence of a hydrogen bonding interaction between the N−H bond of the amide moiety in N12 and the oxygen of the CMD promoter stabilizes the transition state of the ortho C–H palladation step (TS2), which is the turnover-limiting step. Moreover, the C2 substituent should also inhibit the direct reductive elimination of ANP to form norbornyl benzocyclobutene side-products. It is noteworthy that, while mechanistically only a catalytic amount of NBE is sufficient to promote this transformation, a higher NBE concentration can suppress the undesired direct ipso-Heck pathway.
Through addressing such meta constraint, the substrate scope of the Catellani-type reaction was substantially expanded (Scheme 14b). Meta substituents with different sizes and electronic properties (72-76) were tolerated, and diverse ipso/ortho functionalizations were realized (77-78). Consequently, streamlined syntheses of several bioactive compounds were demonstrated with this method.
3.4. Pd(0)-Initiated Difunctionalization of Alkenyl Halides Involving C2-Substituted smNBEs
Beyond arene substrates, alkene-based substrates were seldom used in the Pd/NBE cooperative catalysis. One major difficulty was caused by the more reactive olefin π bond toward the undesired cyclopropanation reactions (Scheme 15a). In addition, the lack of an ortho substituent in general alkenyl substrates also makes the NBE extrusion more challenging. In 2019, the Dong group found the same C2-methylamide smNBE (N12) was highly efficient for the vicinal difunctionalization of alkenyl halides and triflates, which offers a rapid approach to build all-carbon tetrasubstituted olefins (Scheme 15b).23 The undesired 3-exo-trig process (cyclopropane formation, 80) was significantly inhibited by the rigid amide moiety; meanwhile the β-carbon elimination was promoted due to the bulkiness of N12.6, 38 For comparison, regular NBE N1, the C2-ester smNBE N9 and those with substituents at other positions were not reactive and/or selective.
Scheme 15.

Ortho/Ipso Difunctionalization of Vinyl Triflates/Halides via Pd/NBE Catalysis
Both cyclic and linear vinyl triflates or bromides were competent substrates. Besides ipso Heck reaction, other ipso terminating reactions, such as hydrogenation (84) and Suzuki (85) coupling, could also take place. Both ortho alkylation and arylation were realized. As an interesting feature, the PhDavePhos ligand underwent in situ C–H/P−C bond activation to generate the corresponding phosphafluorene as the actual active ligand.
Overall, compared to the simple unsubstituted NBE N1 and other smNBEs, the C2-substituted smNBEs own some unique features due to the direct substitution on the reactive C=C double bond. Adjusting the steric and electronic properties of the C2 substituents can greatly influence the NBE-insertion and the downstream reactions with ANP, thereby capable of expanding the reaction capacity. They also provide promising directions for developing enantioselective transformations.
4. C5-Substituted or C5, C6-Disubstituted smNBEs
In general, C5-substituted and C5, C6-disubstituted smNBEs exhibit similar reactivity as simple unsubstituted NBE, while in some specific cases they have been found to be more efficient. To date, at least eleven types of C5-substituted or C5, C6-disubstituted NBEs have been developed and employed in Pd/NBE cooperative catalysis (Scheme 16). All of them contain EWGs, as the skeletons are typically prepared via the Diels−Alder reaction between cyclopentandiene and electron-deficiency alkenes.49 Unlike C1- or C2-substituted smNBEs, the steric and electronic environment of the NBE alkene in these smNBEs is almost identical to simple NBE, though the remote steric effect or coordinative properties of the C5/C6 substituents could play an important role in modulating reaction selectivity.
Scheme 16.

Reported C5-Substituted and C5, C6-Disubstituted NBEs
4.1. Pd(0)-Initiated Difunctionalization of Aryl Halides Involving C5-Substituted or C5, C6-Disubstituted smNBEs
The first use of a C5-substituted smNBE was reported by the Dong group in the ortho C–H acylation of haloarenes in 2015 (Scheme 17a),50 which also represents the first use of smNBEs in the catalytic Catellani reactions. In the ortho acylation/ipso hydrogenation reaction, a bifunctional isopropyl-carbonate anhydride was employed as both an acyl electrophile and a “masked” hydride source, i.e. isopropoxide, which prevented undesired esterification if homo-anhydrides and isopropanol were used. The C5 methylamide-substituted NBE (N19) not only provided consistently higher yields than unsubstituted NBE, but also eased isolation of pure products from NBE-containing side-products due to the polar amide moiety. This transformation tolerated a broad range of FGs, including various heterocycles. Besides ipso hydrogenation, Heck (92) and Suzuki (93) couplings could also be used for the ipso quench under similar conditions. Note that the ortho acylation of haloarenes was also concurrently reported by the Liang51 and Gu52 groups. This ortho acylation approach was further extended to a rapid construction of substituted indenones via direct annulation between aryl iodides and unsaturated carboxylic acid anhydrides by the same group in 2019.53 Significant yield improvement was found using the C5-amide NBE (N19) compared to simple NBE (N1) (Scheme 17b). Diverse aryl iodides and conjugated anhydrides were suitable substrates. Mechanistic studies indicated the ipso functionalization was likely realized through a Heck-type coupling. Utility of this method was demonstrated in concise syntheses of indenone-based nature products, pauciflorol F and acredinone A. Notably, the total synthesis of acredinone A features a strategy involving two Pd/N19-catalyzed ortho acylations to construct both penta-substituted arene cores, including the use of a new ortho acylation/ipso borylation method.54
Scheme 17.

Ortho Acylation of Haloarenes Enabled by C5 Methylamide-Substituted NBE
In 2018, the Zhou group reported the successful use of a unique potassium salt of 5-norbornene-2-carboxylic acid (N25) in the Pd/NBE-catalyzed ortho alkylation with epoxides as the electrophile (Scheme 18).44 The carboxylate moiety in N25 was proposed to serve as a CMD promoter to promote the ortho C–H activation, while comparable yield was obtained using the combination of simple NBE N1 and CsOAc. Unlike the direct annulation approach (vide supra, Scheme 11), olefins were utilized here as the termination reagent. With additional base, a one-pot oxa-Michael addition could be realized, providing a rapidly access to isochroman scaffolds (Scheme 18b). The substrate scope was broad for both aryl iodides and terminal epoxides. Stereo-retention was observed (101) using an enantiopure epoxide; pyridine (102) and styrene (103) were all tolerated. Note that an intramolecular ortho alkylation/ipso Heck coupling was achieved using an alkene-tethered epoxide, which afforded 14- and 13-membered macrocycles (104 and 105). Finally, a gram-scale preparation was demonstrated using only 1 mol% Pd(OAc)2.
Scheme 11.

Direct Annulation between Aryl Iodides and Epoxides
In 2018, the same group extended the use of in situ generated 5-norbornene-2-carboxylate in an ortho alkylation/redox-Heck relay reaction, which allowed for a rapid construction of tetrahydronaphthalene and indane scaffolds that contain quaternary centers (Scheme 19).55,56 Interestingly, the C5/C6-dicarboxylic acid-derived NBE was also examined but only giving a trace amount of product. Regarding the reaction scope, both allyl and homo-allyl alcohols could be employed as the reagent to yield the corresponding aldehydes and ketones as the products, and various aryl iodides (e.g. 107 and 108) were suitable substrates. An indane-type product (108) was obtained in moderate yield when using a shorter-linked reagent. Besides carbon linkers, substrates bearing an oxygen linker (109) were also suitable, affording a seven-membered product albeit in lower yield. The synthetic utility was nicely demonstrated through a concise synthesis of (±)-eptazocine (Scheme 19b). The asymmetric ortho alkylation/ipso redox-relay Heck cascade has also been investigated using a chiral phosphine ligand (L8) and an achiral C5, C6-disubstituted smNBE (N29). Promising enantioselectivity (78% ee) was obtained albeit in 27% yield (Scheme 19c).
Scheme 19.

Ortho Alkylation/Ipso Redox-Relay Heck Cascade
Analogous to epoxides, aziridines have also been used as electrophiles in ortho alkylation of aryl iodides. In 2018, Liang and coworkers reported a direct annulation between aryl iodides and Ts-aziridines to form indoline-type products using unsubstituted simple NBE (N1).57 Shortly after, the Zhou group discovered that the C5, C6-disubstituted imide-type NBE (N29) was more effective than other smNBEs or simple NBE for the aziridine alkylation when using olefins as the ipso terminating reagent (Scheme 20).58 By large, C5-substiutted and C5,C6-disubstiuted smNBEs were more efficient than the C2-substituted one in this reaction. Owing to a broad substrate scope and good FG tolerance, this method provides a streamlined assembly of substituted tetrahydroisoquinolines in a stereoselective manner from readily available starting materials.
Scheme 20.

Aziridines as Electrophiles in Catellani-type Reactions
Considering that sulfur-substituted aromatic compounds are widely found in pharmaceuticals, agrochemicals, and organic materials, the ortho-thiolation of aryl halides via the Pd/NBE catalysis has attracted significant attentions. In 2019, the Gu59 and Dong60 groups independently reported ortho thiolation reactions of aryl iodides with different thiolating reagents (Scheme 21). Thiosulfonates and sulfenamides were used as the electrophiles by the two groups, respectively. This also represents the first time when a heteroatom besides nitrogen was installed at the ortho position of aryl halides via the Pd/NBE catalysis. Gu’s work features the use of a C5 aldehyde-substituted NBE (N28), which gave a better result than simple NBE (N1). Both aryl and alkyl thiolate moieties can be installed using this method.
Scheme 21.

Ortho Thiolation of Aryl Iodide with Thiosulfonates
Methyl groups have played a profound role in medicinal chemistry, as the introduction of a methyl group could potentially modulate physical properties and even conformation of drug candidates.61 The coupling of a methyl group at the arene ortho positions via the Catellani reaction was first reported in 2007 by Lautens using methyl iodide as the electrophile with simple NBE.62 The use of MeONs and tetramethylammonium salts as electrophiles in the Pd/NBE catalysis were later reported in 2018 and 2019, respectively.21,63 In 2019, the Zhou group systematically studied the ortho methylation and trideuteriomethylation of haloarenes, and they nicely enhanced the efficiency and expanded the scope of the reaction (Scheme 22).64 Besides simple NBE, the C5 nitrile-substituted NBE (N27) was found to be an efficient and general co-catalyst. Two types of methyl electrophiles, MeOTs and trimethylphosphate, were utilized as electrophiles. The ipso position of substrates could be terminated by a range of olefins and nucleophiles, e.g. t-butyl acrylate (122 and 123), arylboronic acids (124), terminal alkynes (125), zinc cyanide (126), B2(pin)2 (127) and sodium formate as a hydride source (128), suggesting broad utilities in modification of bioactive compounds. For the ipso hydrogenation, a C5, C6-disubstituted NBE (N30) was found to be more effective. Finally, use of the corresponding CD3OTs or 13CH3OTs as the electrophiles instead led to isotope-labelled methylation (123 and 128).
Scheme 22.

Ortho Methylation of Aryl Halides
Given that C-linked glycosides have been widely used as carbohydrate mimetics owing to their relatively high chemical and metabolic stability compared to O-linked glycoside, developing a transition metal-catalyzed C–H glycosylation reaction has attracted significant attentions.65 In 2020, the Liang66 and Cheng67 groups independently realized the Pd/NBE-catalyzed ortho glycosylation reaction of aryl iodides using glycosyl chlorides as the electrophile (Scheme 23). Liang’s work features the use of a 5-norbornene-2-carbonitrile (N27), which gave a better result than other NBEs. The major side reaction was observed to be the direct ipso Heck termination, which was significantly inhibited using C5-substituted (N27, N36 and N38) and C5, C6-disubstituted (N31) NBEs. In Cheng’s work a C5 anilide-substituted NBE (N44) proved to be the optimal co-catalyst. Note that both reactions proceed in a stereoselective manner: α isomers of the resulting C−aryl glycoside products were obtained predominantly in most examples.
Scheme 23.

Ortho Glycosylation of Aryl Iodides
4.2. Pd(II)-Initiated Difunctionalization of Aryl Halides Involving C5-Substituted or C5, C6-Disubstituted smNBEs
Besides through oxidative addition of Pd(0) with aryl halides, as illustrated in Scheme 1b, the initial aryl-Pd(II) species can also be formed through transmetallation of arylboronic acids to Pd(II). After the Catellani process, the Pd(II) catalyst can be regenerated via oxidation of the Pd(0) after ipso termination. In 2018, the Zhang68 and Zhou69 groups independently reported a Pd(II)-initiated Catellani-type reaction with arylboron substrates. While simple NBE was efficient for vicinal difunctionalization of arylboronic acids in Zhang’s method, Zhou found 5-norbornene-2-carbonitrile (N27) was more efficient than other NBEs when using arylboronic acid pinacol esters as substrates (Scheme 24a). Besides ortho alkylation, ortho arylation of arylboronic acid pinacol esters was also realized by the Zhou group in 2019 using aryl bromides as the electrophiles, in which a C5, C6 diester-substituted NBE (N20) proved to be optimal (Scheme 24b).70 Air was employed as the terminal oxidant in Zhou’s reactions, which is beneficial compared to the use of stoichiometric metallic oxidants. In this context, a related redox-neutral arylboron-based Pd(II)-initiated ortho functionalization was reported by Dong71 and later Zhou72, in which the Pd(II) was regenerated through a protonation process, therefore avoiding stoichiometric oxidants and bases. A key merit of the Pd(II)-initiated processes is the tolerance of aryl iodides and bromides that are otherwise reactive under the Pd(0) conditions. Such orthogonal reactivity could be beneficial for strategic planning.
Scheme 24.

Pd(II)-Initiated Ortho Alkylation and Arylation of Arylboronates
4.3. Pd(II)-Catalyzed Distal C–H Functionalization with C5, C6-Disubstituted smNBEs
In 2018, the Ding group reported a meta alkylation reaction of nosyl-protected phenethylamines, which was enabled by a C5, C6-disubstituted NBE (N32) and a simple pyridine ligand (Scheme 25).73 Compared to Yu’s C2 ester smNBE (N9) or simple NBE, the 5,6-di-isopropyl ester-substituted NBE (N32) afforded much enhanced yields. While high efficiency was generally observed for using alkyl electrophiles without β-hydrogens, moderate yield was nevertheless obtained for the ortho ethylation reaction.
Scheme 25.

Pd(II)-Catalyzed Meta Alkylation of Nosyl-Protected Phenylalanines
The concept of the distal C–H functionalization via the Pd/NBE catalysis was recently extended to alkene substrates by the Dong group, which provides a regio- and stereo-selective preparation of trisubstituted alkenes from 1,2-disubstituted ones (Scheme 26).74 Given that the π bond in olefin substrates is more reactive than arene substrates and easily undergoes various π-breaking side-reactions,23 the key of this reaction was to use an appropriate combination of a DG and a NBE cocatalyst to realize a fast and reversible proximal C–H palladation. The N-Ph-imide-based NBE (N31) was found to be most efficient; an oxime ether-based DG proved to be superb as it can be easily installed and removed. Both alcohol and amine-based substrates could be used. Besides distal C–H arylation, alkylation with α-halogenated esters was realized in good yield under slightly modified conditions (143).
Scheme 26.
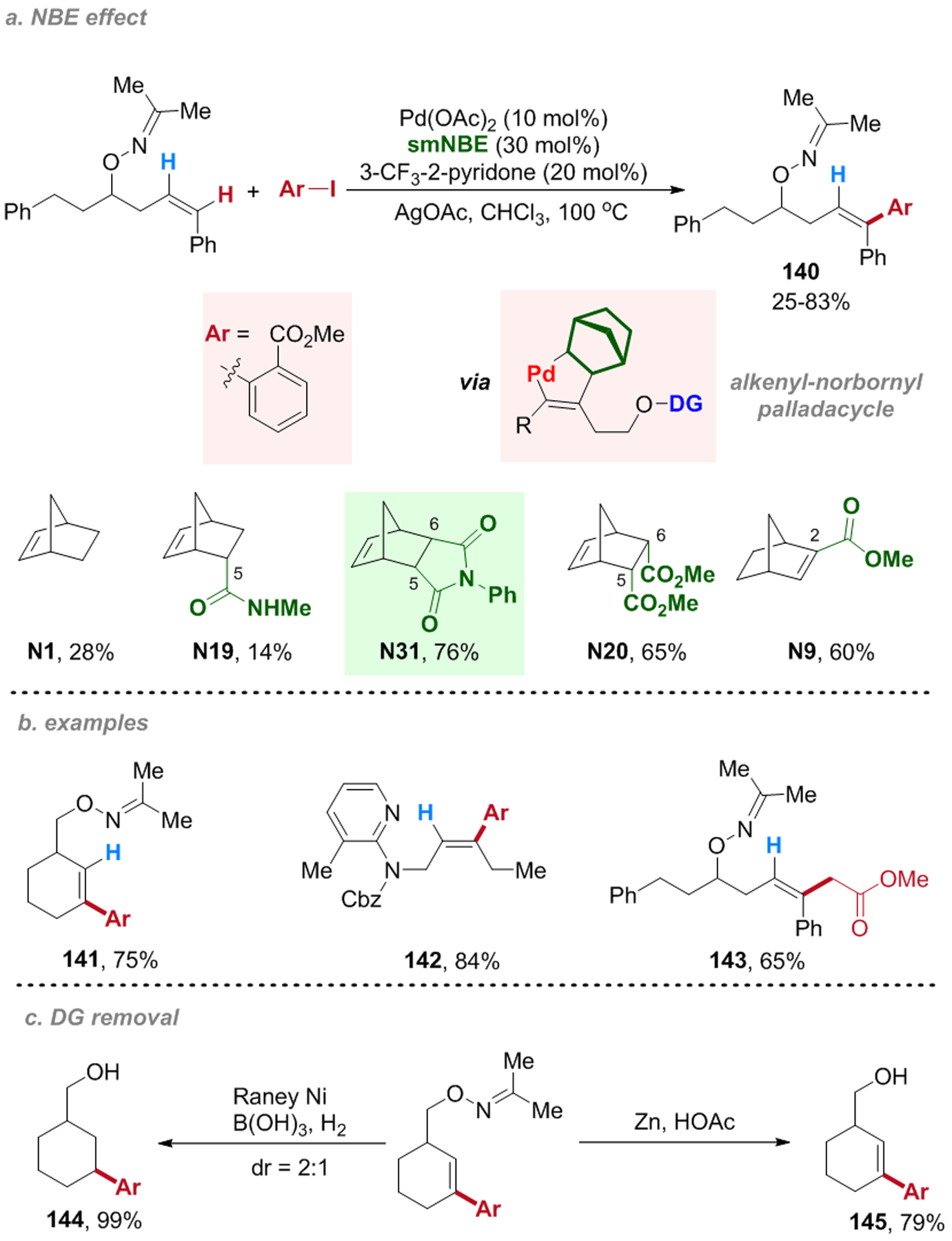
Pd(II)-Catalyzed Distal Alkenyl C–H Functionalization
While C5-substituted and C5, C6-disubstituted smNBEs generally exhibit similar reactivity/selectivity to simple unsubstituted NBE, remarkably improved efficiency can nevertheless be observed in many cases shown above. Compared to C1- and C2-substituted smNBEs, the effect of the C5/C6 substituents that are distal to the reactive site remains to be well understood, which could be an important topic to be explored in the future.
5. Conclusion and Outlook
In summary, the Pd/NBE cooperative catalysis has emerged as a powerful tool for regio- and site-selective functionalization of arenes, heteroarenes and alkenes, and smNBEs have significantly contributed to the growth of this field. Many breakthroughs, including broader scope, better reactivity, higher selectivity, and new applications, have been realized, and some intrinsic limitations have been overcome owing to these structural modifications on the NBE scaffold. For example, the ortho constraint in the Catellani-type reactions was addressed using the C1-substituted smNBE; the meta constraint and the problem of forming norbornyl benzocyclobutene side-products were solved by the C2-substituted smNBEs; enhanced reaction efficiency can be achieved through modifying the NBE C5 and C6 positions. In addition, the Pd/NBE-catalyzed alkene C–H functionalization and enantioselectivity transformations have also been enabled by smNBEs.
Despite the enormous achievements made to date, future advancement in the field of Pd/NBE catalysis would benefit from developing more versatile smNBEs. First, the catalyst efficiency in most reactions still requires further improvement. Due to the complex nature of the Catellani-type reactions, the amounts of NBE or smNBEs used are typically 50% or higher, and the loading of Pd pre-catalysts is often 10 mol%. It is anticipated that deep and systematic mechanistic understanding of these reactions could guide development of more efficient catalyst systems. In addition, it would be attractive to realize more synthetically useful enantioselective transformations. Given that most of the smNBEs contain a chiral skeleton, there is sufficient room for developing new asymmetric Pd/NBE catalysis methods. Moreover, the applicability of the Pd/NBE catalysis would be aided by a broadened reaction scope. For example, discovery of new and compatible electrophiles for ortho functionalization has been an ongoing challenge. These “tailor-made” smNBEs with fine-tuned steric and electronic properties are expected to become a key factor for accommodating unusual electrophiles that are currently unknown for the Catellani-type reactions. It is our expectation that, through addressing these challenges, the Pd/NBE catalysis could ultimately become one of the “go-to” methods for preparing polysubstituted arenes and alkenes in the future.
ACKNOWLEDGMENT
Financial supports from the University of Chicago and NIGMS (1R01GM124414-01A1) are acknowledged. We thank Dr. Jianchun Wang for helpful discussions.
ABBREVIATIONS
- smNBE
structurally modified norbornene
- NBE
norbornene
- E
electrophile
- Nu
nucleophile
- FG
functional group
- SNAr
nucleophilic aromatic substitution
- ANP
aryl-norbornyl-palladacycle
- L
ligand
- Cy
cyclohexyl
- EAS
electrophilic aromatic substitution
- RuPhos
2-dicyclohexylphosphino-2’,6’-diisopropoxybiphenyl
- SPhos
2-dicyclohexylphosphino-2’,6’-dimethoxybiphenyl
- XPhos
2-dicyclohexylphosphino-2’,4’,6’-triisopropylbiphenyl
- DFT
density functional theory
- CMD
concerted metalation deprotonation
- R.E.
reductive elimination
- EWG
electron withdrawing group
- DG
directing group
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- BNDHP
1,1’-binaphthyl-2,2’-diyl hydrogenphosphate
- TBME
tert-butyl methyl ether
- HFIP
1,1,1,3,3,3-hexafluoro-2-propanol
- Ac-Gly-OH
N-acetylglycine
- DMAP
4-dimethylaminopyridine
- BQ
1,4-benzoquinone
- DHBF
2,3-dihydrobenzofuran
- DMF
dimethylformamide
- PhDavePhos
2’-(diphenylphosphino)-N,N’-dimethyl-(1,1’-biphenyl)-2-amine
- TFP
tri(2-furyl)phosphine
- DTBPF
1,1’-bis(di-tert-butylphosphino)ferrocene
- THF
tetrahydrofuran
- NMP
1-methyl-2-pyrrolidinone
- dba
dibenzylideneacetone
- DMA
N,N-dimethylacetamide
- DME
1,2-dimethoxyethane
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Catellani M; Frignani F; Rangoni A, A Complex Catalytic Cycle Leading to a Regioselective Synthesis of o,o′-Disubstituted Vinylarenes. Angew. Chem. Int. Ed 1997, 36, 119–122. [Google Scholar]
- (2).An Z.-w.; Catellani M; Chiusoli GP, Palladium-catalyzed synthesis of coumarin. J. Organomet. Chem 1989, 371, C51–C52. [Google Scholar]
- (3).Bocelli G; Catellani M; Chiusoli GP, Palladium-catalyzed C–C bond formation involving aromatic C–H activation: III. Aspects of aromatic substitution and structure of 1-bromo-3-[3-(2-methylenecyclohex-5-en-1-yl)bicyclo[2.2.1]hept-2-yl]-4-bicyclo[2.2.1]hept-2-ylbenzene. J. Organomet. Chem 1985, 279, 225–232. [Google Scholar]
- (4).Catellani M; Chiusoli GP, Palladium-catalyzed synthesis of 1,2,3,4,4a,12b-hexahydro-1,4-methanotriphenylenes. J. Organomet. Chem 1985, 286, c13–c16. [Google Scholar]
- (5).Maestri G; Motti E; Della Ca’ N; Malacria M; Derat E; Catellani M, Of the Ortho Effect in Palladium/Norbornene-Catalyzed Reactions: A Theoretical Investigation. J. Am. Chem. Soc 2011, 133, 8574–8585. [DOI] [PubMed] [Google Scholar]
- (6).Wang J; Li R; Dong Z; Liu P; Dong G, Complementary site-selectivity in arene functionalization enabled by overcoming the ortho constraint in palladium/norbornene catalysis. Nat. Chem 2018, 10, 866–872. [DOI] [PubMed] [Google Scholar]
- (7).Catellani M, Catalytic Multistep Reactions via Palladacycles. Synlett 2003, 2003, 0298–0313. [Google Scholar]
- (8).Catellani M, Novel Methods of Aromatic Functionalization Using Palladium and Norbornene as a Unique Catalytic System. Top. Organomet. Chem 2005, 14, 21–53. [Google Scholar]
- (9).Catellani M; Motti E; Della Ca’ N, Catalytic Sequential Reactions Involving Palladacycle-Directed Aryl Coupling Steps. Acc. Chem. Res 2008, 41, 1512–1522. [DOI] [PubMed] [Google Scholar]
- (10).Martins A; Mariampillai B; Lautens M, Synthesis in the Key of Catellani: Norbornene-Mediated ortho C–H Functionalization. Top. Curr. Chem 2009, 292, 1–33. [DOI] [PubMed] [Google Scholar]
- (11).Ye J; Lautens M, Palladium-catalysed norbornene-mediated C–H functionalization of arenes. Nat. Chem 2015, 7, 863. [DOI] [PubMed] [Google Scholar]
- (12).Della Ca’ N; Fontana M; Motti E; Catellani M, Pd/Norbornene: A Winning Combination for Selective Aromatic Functionalization via C–H Bond Activation. Acc. Chem. Res 2016, 49, 1389–1400. [DOI] [PubMed] [Google Scholar]
- (13).Wegmann M; Henkel M; Bach T, C–H alkylation reactions of indoles mediated by Pd(ii) and norbornene: applications and recent developments. Org. Biomol. Chem 2018, 16, 5376–5385. [DOI] [PubMed] [Google Scholar]
- (14).Liu Z-S; Gao Q; Cheng H-G; Zhou Q, Alkylating Reagents Employed in Catellani-Type Reactions. Chem. Eur. J 2018, 24, 15461–15476. [DOI] [PubMed] [Google Scholar]
- (15).Zhao K; Ding L; Gu Z, Development of New Electrophiles in Palladium/Norbornene-Catalyzed ortho-Functionalization of Aryl Halides. Synlett 2019, 30, 129–140. [Google Scholar]
- (16).Cheng H-G; Chen S; Chen R; Zhou Q, Palladium(II)-Initiated Catellani-Type Reactions. Angew. Chem. Int. Ed 2019, 58, 5832–5844. [DOI] [PubMed] [Google Scholar]
- (17).Wang J; Dong G, Palladium/Norbornene Cooperative Catalysis. Chem. Rev 2019, 119, 7478–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).The use of norbornadiene and 7-oxa-[2.2.1]bicyclic alkenes as stoichiometric reagents will not be covered.
- (19).Dong Z; Dong G, Ortho vs Ipso: Site-Selective Pd and Norbornene-Catalyzed Arene C–H Amination Using Aryl Halides. J. Am. Chem. Soc 2013, 135, 18350–18353. [DOI] [PubMed] [Google Scholar]
- (20).Shen P-X; Wang X-C; Wang P; Zhu R-Y; Yu J-Q, Ligand-Enabled Meta-C–H Alkylation and Arylation Using a Modified Norbornene. J. Am. Chem. Soc 2015, 137, 11574–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang J; Zhou Y; Xu X; Liu P; Dong G, Entry to 1,2,3,4-Tetrasubstituted Arenes through Addressing the “Meta Constraint” in the Palladium/Norbornene Catalysis. J. Am. Chem. Soc 2020, 142, 3050–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li R; Liu F; Dong G, Palladium-catalyzed asymmetric annulation between aryl iodides and racemic epoxides using a chiral norbornene cocatalyst. Org. Chem. Front 2018, 5, 3108–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang J; Dong Z; Yang C; Dong G, Modular and regioselective synthesis of all-carbon tetrasubstituted olefins enabled by an alkenyl Catellani reaction. Nat. Chem 2019, 11, 1106–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Verkruijsse HD; Brandsma L, Preparative metallation of norbornene and norbornadiene. Recl. Trav. Chim. Pays-Bas 1986, 105, 66–68. [Google Scholar]
- (25).Wang X-C; Gong W; Fang L-Z; Zhu R-Y; Li S; Engle KM; Yu J-Q, Ligand-enabled meta-C–H activation using a transient mediator. Nature 2015, 519, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dong Z; Wang J; Dong G, Simple Amine-Directed Meta-Selective C–H Arylation via Pd/Norbornene Catalysis. J. Am. Chem. Soc 2015, 137, 5887–5890. [DOI] [PubMed] [Google Scholar]
- (27).Wang P; Li G-C; Jain P; Farmer ME; He J; Shen P-X; Yu J-Q, Ligand-Promoted meta-C–H Amination and Alkynylation. J. Am. Chem. Soc 2016, 138, 14092–14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shi H; Wang P; Suzuki S; Farmer ME; Yu J-Q, Ligand Promoted meta-C–H Chlorination of Anilines and Phenols. J. Am. Chem. Soc 2016, 138, 14876–14879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).DeBergh JR; Niljianskul N; Buchwald SL, Synthesis of Aryl Sulfonamides via Palladium-Catalyzed Chlorosulfonylation of Arylboronic Acids. J. Am. Chem. Soc 2013, 135, 10638–10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhao X; Dimitrijević E; Dong VM, Palladium-Catalyzed C–H Bond Functionalization with Arylsulfonyl Chlorides. J. Am. Chem. Soc 2009, 131, 3466–3467. [DOI] [PubMed] [Google Scholar]
- (31).Shi H; Herron AN; Shao Y; Shao Q; Yu J-Q, Enantioselective remote meta-C–H arylation and alkylation via a chiral transient mediator. Nature 2018, 558, 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liu L-Y; Qiao JX; Yeung K-S; Ewing WR; Yu J-Q, meta C–H Arylation of Electron-Rich Arenes: Reversing the Conventional Site Selectivity. J. Am. Chem. Soc 2019, 141, 14870–14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ma X; Zhao X; Zhu R; Zhang D, Computational Study on Why and How of Nonconventional meta-C–H Arylation of Electron-Rich Arenes via Pd/Quinoxaline-Based Ligand/Norbornene Cooperative Catalysis. J. Org. Chem 2020, 85, 5995–6007. [DOI] [PubMed] [Google Scholar]
- (34).Liu L-Y; Qiao JX; Ewing WR; Yeung K-S; Yu J-Q, β-Selective C–H Arylation of Electron-Deficient Thiophenes, Pyrroles, and Furans. Isr. J. Chem 2020, 60, 416–418. [Google Scholar]
- (35).Liu L-Y; Qiao JX; Yeung K-S; Ewing WR; Yu J-Q, meta-Selective C–H Arylation of Fluoroarenes and Simple Arenes. Angew. Chem. Int. Ed 2020, 59, 13831–13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Shi H; Lu Y; Weng J; Bay KL; Chen X; Tanaka K; Verma P; Houk KN; Yu J-Q, Differentiation and functionalization of remote C–H bonds in adjacent positions. Nat. Chem 2020, 12, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhang Z; Tanaka K; Yu J-Q, Remote site-selective C–H activation directed by a catalytic bifunctional template. Nature 2017, 543, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Li R; Zhou Y; Xu X; Dong G, Direct Vicinal Difunctionalization of Thiophenes Enabled by the Palladium/Norbornene Cooperative Catalysis. J. Am. Chem. Soc 2019, 141, 18958–18963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Catellani M; Cugini F, A Catalytic Process Based on Sequential Ortho-alkylation and Vinylation of Ortho-alkylaryl Iodides via Palladacycles. Tetrahedron 1999, 55, 6595–6602. [Google Scholar]
- (40).Li R; Dong G, Direct Annulation between Aryl Iodides and Epoxides through Palladium/Norbornene Cooperative Catalysis. Angew. Chem. Int. Ed 2018, 57, 1697–1701. [DOI] [PubMed] [Google Scholar]
- (41).Bruno NC; Niljianskul N; Buchwald SL, N-Substituted 2-Aminobiphenylpalladium Methanesulfonate Precatalysts and Their Use in C–C and C–N Cross-Couplings. J. Org. Chem 2014, 79, 4161–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Torraca KE; Huang X; Parrish CA; Buchwald SL, An Efficient Intermolecular Palladium-Catalyzed Synthesis of Aryl Ethers. J. Am. Chem. Soc 2001, 123, 10770–10771. [DOI] [PubMed] [Google Scholar]
- (43).Kuwabe S.-i.; Torraca KE; Buchwald SL, Palladium-Catalyzed Intramolecular C−O Bond Formation. J. Am. Chem. Soc 2001, 123, 12202–12206. [DOI] [PubMed] [Google Scholar]
- (44).Cheng H-G; Wu C; Chen H; Chen R; Qian G; Geng Z; Wei Q; Xia Y; Zhang J; Zhang Y; Zhou Q, Epoxides as Alkylating Reagents for the Catellani Reaction. Angew. Chem. Int. Ed 2018, 57, 3444–3448. [DOI] [PubMed] [Google Scholar]
- (45).Wu C; Cheng H-G; Chen R; Chen H; Liu Z-S; Zhang J; Zhang Y; Zhu Y; Geng Z; Zhou Q, Convergent syntheses of 2,3-dihydrobenzofurans via a Catellani strategy. Org. Chem. Front 2018, 5, 2533–2536. [Google Scholar]
- (46).Liu Z-S; Hua Y; Gao Q; Ma Y; Tang H; Shang Y; Cheng H-G; Zhou Q, Construction of axial chirality via palladium/chiral norbornene cooperative catalysis. Nat. Catal 2020, 3, 727–733. [Google Scholar]
- (47).Zhao Y-B; Mariampillai B; Candito DA; Laleu B; Li M; Lautens M, Exploiting the Divergent Reactivity of Aryl–Palladium Intermediates for the Rapid Assembly of Fluorene and Phenanthrene Derivatives. Angew. Chem. Int. Ed 2009, 48, 1849–1852. [DOI] [PubMed] [Google Scholar]
- (48).Lautens M; Paquin J-F; Piguel S; Dahlmann M, Palladium-Catalyzed Sequential Alkylation−Alkenylation Reactions and Their Application to the Synthesis of Fused Aromatic Rings. J. Org. Chem 2001, 66, 8127–8134. [DOI] [PubMed] [Google Scholar]
- (49).Davies DI; Gomez PM; Hallett P, Synthesis of 2,5- and 2,6-norbornane derivatives with prostaglandin-like side chains. J. Chem. Soc., Perkin Trans 1 1984, 843–848. [Google Scholar]
- (50).Dong Z; Wang J; Ren Z; Dong G, Ortho C–H Acylation of Aryl Iodides by Palladium/Norbornene Catalysis. Angew. Chem. Int. Ed 2015, 54, 12664–12668. [DOI] [PubMed] [Google Scholar]
- (51).Zhou P-X; Ye Y-Y; Liu C; Zhao L-B; Hou J-Y; Chen D-Q; Tang Q; Wang A-Q; Zhang J-Y; Huang Q-X; Xu P-F; Liang Y-M, Palladium-Catalyzed Acylation/Alkenylation of Aryl Iodide: A Domino Approach Based on the Catellani–Lautens Reaction. ACS Catal 2015, 5, 4927–4931. [Google Scholar]
- (52).Huang Y; Zhu R; Zhao K; Gu Z, Palladium-Catalyzed Catellani ortho-Acylation Reaction: An Efficient and Regiospecific Synthesis of Diaryl Ketones. Angew. Chem. Int. Ed 2015, 54, 12669–12672. [DOI] [PubMed] [Google Scholar]
- (53).Liu F; Dong Z; Wang J; Dong G, Palladium/Norbornene-Catalyzed Indenone Synthesis from Simple Aryl Iodides: Concise Syntheses of Pauciflorol F and Acredinone A. Angew. Chem. Int. Ed 2019, 58, 2144–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Shi H; Babinski DJ; Ritter T, Modular C–H Functionalization Cascade of Aryl Iodides. J. Am. Chem. Soc 2015, 137, 3775–3778. [DOI] [PubMed] [Google Scholar]
- (55).Liu Z-S; Qian G; Gao Q; Wang P; Cheng H-G; Wei Q; Liu Q; Zhou Q, Palladium/Norbornene Cooperative Catalysis To Access Tetrahydronaphthalenes and Indanes with a Quaternary Center. ACS Catal 2018, 8, 4783–4788. [Google Scholar]
- (56).Liu Z-S; Qian G; Gao Q; Wang P; Cheng H-G; Hua Y; Zhou Q, 5-Norbornene-2-carboxylic acid: Another catalytic mediator for Catellani-type reactions. Tetrahedron 2019, 75, 1774–1780. [Google Scholar]
- (57).Liu C; Liang Y; Zheng N; Zhang B-S; Feng Y; Bi S; Liang Y-M, Synthesis of indolines via a palladium/norbornene-catalyzed reaction of aziridines with aryl iodides. Chem. Commun 2018, 54, 3407–3410. [DOI] [PubMed] [Google Scholar]
- (58).Qian G; Bai M; Gao S; Chen H; Zhou S; Cheng H-G; Yan W; Zhou Q, Modular One-Step Three-Component Synthesis of Tetrahydroisoquinolines Using a Catellani Strategy. Angew. Chem. Int. Ed 2018, 57, 10980–10984. [DOI] [PubMed] [Google Scholar]
- (59).Cai W; Gu Z, Selective Ortho Thiolation Enabled by Tuning the Ancillary Ligand in Palladium/Norbornene Catalysis. Org. Lett 2019, 21, 3204–3209. [DOI] [PubMed] [Google Scholar]
- (60).Li R; Zhou Y; Yoon K-Y; Dong Z; Dong G, Sulfenamide-enabled ortho thiolation of aryl iodides via palladium/norbornene cooperative catalysis. Nat. Commun 2019, 10, 3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Schönherr H; Cernak T, Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem. Int. Ed 2013, 52, 12256–12267. [DOI] [PubMed] [Google Scholar]
- (62).Mariampillai B; Alliot J; Li M; Lautens M, A Convergent Synthesis of Polysubstituted Aromatic Nitriles via Palladium-Catalyzed C–H Functionalization. J. Am. Chem. Soc 2007, 129, 15372–15379. [DOI] [PubMed] [Google Scholar]
- (63).Dong Z; Lu G; Wang J; Liu P; Dong G, Modular ipso/ortho Difunctionalization of Aryl Bromides via Palladium/Norbornene Cooperative Catalysis. J. Am. Chem. Soc 2018, 140, 8551–8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Gao Q; Shang Y; Song F; Ye J; Liu Z-S; Li L; Cheng H-G; Zhou Q, Modular Dual-Tasked C–H Methylation via the Catellani Strategy. J. Am. Chem. Soc 2019, 141, 15986–15993. [DOI] [PubMed] [Google Scholar]
- (65).Wang Q; An S; Deng Z; Zhu W; Huang Z; He G; Chen G, Palladium-catalysed C–H glycosylation for synthesis of C-aryl glycosides. Nat. Catal 2019, 2, 793–800. [Google Scholar]
- (66).Ding Y-N; Shi W-Y; Liu C; Zheng N; Li M; An Y; Zhang Z; Wang C-T; Zhang B-S; Liang Y-M, Palladium-Catalyzed ortho-C–H Glycosylation/ipso-Alkenylation of Aryl Iodides. J. Org. Chem 2020, 85, 11280–11296. [DOI] [PubMed] [Google Scholar]
- (67).Lv W; Chen Y; Wen S; Ba D; Cheng G, Modular and Stereoselective Synthesis of C-Aryl Glycosides via Catellani Reaction. J. Am. Chem. Soc 2020, 142, 14864–14870. [DOI] [PubMed] [Google Scholar]
- (68).Shi G; Shao C; Ma X; Gu Y; Zhang Y, Pd(II)-Catalyzed Catellani-Type Domino Reaction Utilizing Arylboronic Acids as Substrates. ACS Catal 2018, 8, 3775–3779. [Google Scholar]
- (69).Chen S; Liu Z-S; Yang T; Hua Y; Zhou Z; Cheng H-G; Zhou Q, The Discovery of a Palladium(II)-Initiated Borono-Catellani Reaction. Angew. Chem. Int. Ed 2018, 57, 7161–7165. [DOI] [PubMed] [Google Scholar]
- (70).Wang P; Chen S; Zhou Z; Cheng H-G; Zhou Q, Chemoselective Borono-Catellani Arylation for Unsymmetrical Biaryls Synthesis. Org. Lett 2019, 21, 3323–3327. [DOI] [PubMed] [Google Scholar]
- (71).Li R; Liu F; Dong G, Redox-Neutral ortho Functionalization of Aryl Boroxines via Palladium/Norbornene Cooperative Catalysis. Chem 2019, 5, 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Chen S; Wang P; Cheng H-G; Yang C; Zhou Q, Redox-neutral ortho-C–H amination of pinacol arylborates via palladium(ii)/norbornene catalysis for aniline synthesis. Chem. Sci 2019, 10, 8384–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Liu J; Ding Q; Fang W; Wu W; Zhang Y; Peng Y, Pd(II)/Norbornene-Catalyzed Meta-C–H Alkylation of Nosyl-Protected Phenylalanines. J. Org. Chem 2018, 83, 13211–13216. [DOI] [PubMed] [Google Scholar]
- (74).Wu Z; Fatuzzo N; Dong G, Distal Alkenyl C–H Functionalization via the Palladium/Norbornene Cooperative Catalysis. J. Am. Chem. Soc 2020, 142, 2715–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]