

Abstract
Mitochondrial dysfunction has been recognized as an essential contributor to many human diseases including neurodegenerative disorders. However, the exact pathological role of mitochondrial dysfunction, especially for mitochondrial reactive oxygen species associated oxidative stress, remains elusive, partially due to the lack of chemical probes with well-defined mechanisms of action. Herein, we describe the characterization and discovery of the rationally designed small molecule ZCM-I-1 as a selective modulator of the production of reactive oxygen species from mitochondrial complex I that does not alter mitochondria membrane potential and bioenergetics. Chemical biology studies employing photoaffinity probes derived from ZCM-I-1 demonstrated its novel mechanism of action of modulating complex I via interactions with the flavin mononucleotide site, proximal in the reaction pathway within complex I.
INTRODUCTION
Mitochondria are dynamic organelles contributing vital roles in cellular functions that control energy production, redox signaling, and cell fate under stress.1, 2 Mitochondrial dysfunction and oxidative stress have been linked to the development of many pathological conditions such as cancer, cardiovascular disease, and neurodegenerative disorders including Alzheimer’s disease (AD).3–12 The bioenergetic function of mitochondria is coordinated by oxidative phosphorylation (OXPHOS) driven by the electron transport chain (ETC). This is also where the majority of reactive oxygen species (ROS) are produced through complexes I and III of the ETC.13–15 Despite the fact that the pathological roles of mitochondrial dysfunction and oxidative stress in these diseases have become increasingly clear, it is still under debate whether they are the cause or merely a consequence of other pathological injuries. For example, in AD, a mitochondria cascade hypothesis suggests a causal role in AD development.16–19 However, there are also studies arguing against mitochondrial dysfunction being the early driver of AD pathogenesis.20–29 Therefore, it would be important and valuable if mitochondria-targeted small molecule probes with specificity and well-defined mechanisms of action (MOA) were available to complement ongoing molecular and genetic studies to elucidate the exact pathological roles of mitochondrial dysfunction in disease development and progression. As many of these diseases are not well served by currently available treatments, isolating the role for mitochondria through such probes may lead to paths towards novel, effective and highly specific therapeutics.
Recently, our laboratory has successfully developed a new chemical scaffold by incorporating some of the essential structural features of known natural products that show protective activities in models of neurodegenerative disorders.30, 31 Our pilot medicinal chemistry studies to optimize the chemical scaffold and to remove the concerns of the promiscuous Pan-Assay Interference Compounds (PAINS)32 led to the identification of one lead compound, ZCM-I-1 (1, Fig. 1), with promising neuroprotective activities in AD models both in vitro and in vivo. Although the MOA for the protective activity offered by ZCM-I-1 was not clear at first, our studies confirmed that it is apparently different from curcumin, melatonin, or the combination of these two.30 Furthermore, early studies using the “rule-out principle” suggested mitochondria to be the likely target of this lead compound.30 Herein, we report the characterization of ZCM-I-1 using in vitro, ex vivo, and in vivo models combined with a photoaffinity labeling technique and molecular docking. The results revealed ZCM-I-1 is a selective mitochondrial complex I modulator via interactions with the flavin mononucleotide (FMN) site of mitochondrial complex I (IF), which represents a novel MOA.
Figure 1.
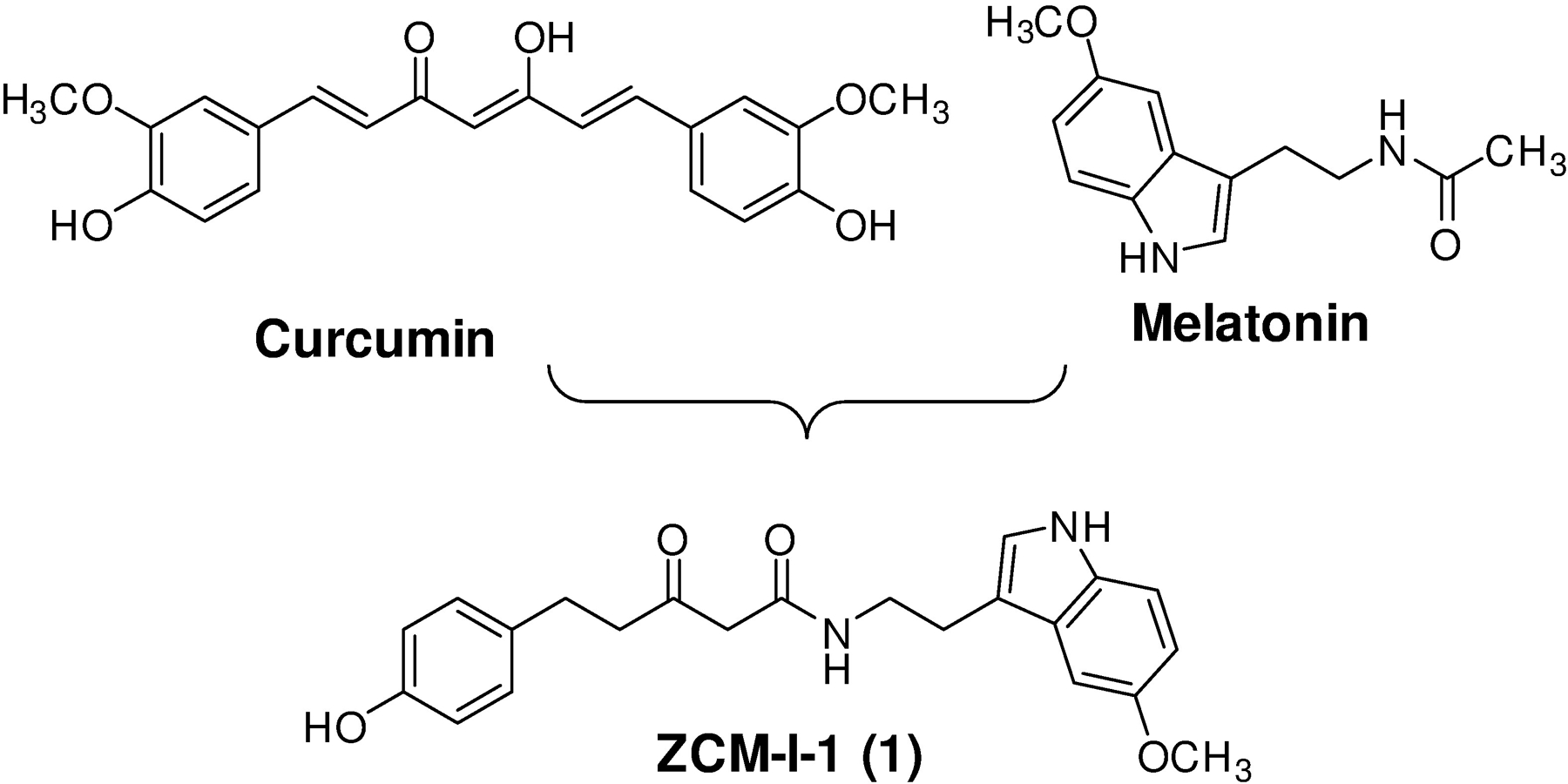
Identification of ZCM-I-1 as a lead neuroprotectant.
RESULTS
ZCM-I-1 selectively suppresses the production of ROS from complex I.
Since our early studies suggested mitochondria as the potential target organelle for ZCM-I-1 and this compound suppressed the production of total ROS in MC65 cells,30 we examined the effects of ZCM-I-1 on the production of mitochondrial ROS (mitoROS) in MC65 cells. As shown in Fig. 2A, ZCM-I-1 dose-dependently suppressed mitoROS production as measured by mitoSOX fluorescence, comparable to its protective activity (Supplemental Fig. 1A). Since most of the ROS produced by mitochondria are from the activity of complexes I and III of the ETC,13–15 we next evaluated how ZCM-I-1 would affect the mitoROS production induced by corresponding known inhibitors. As shown in Fig. 2B, without rotenone,, a complex I inhibitor that binds to the ubiquinone site (IQ), no mitoROS was produced in MC65 cells, while addition of rotenone induced the production of mitoROS with time. Notably, treatment with ZCM-I-1 suppressed the production of mitoROS induced by rotenone (Fig. 2B, ROS release rate: 8.3 vs 5.8 RFU/min for vehicle treated compared to ZCM-I-1 treated). A similar suppression was also observed in cultured primary mouse cortical neurons (Fig. 2C, ROS release rate: 3.3 vs 2.0 RFU/min for vehicle treated compared to ZCM-I-1 treated). No effect was observed on mitoROS induced by antimycin A, a complex III inhibitor (Supplemental Fig. 1B). Furthermore, ZCM-I-1 dose dependently reversed mitochondrial depolarization induced by MPP+, a known complex I inhibitor,33 in both primary mouse cortical neurons (Fig. 2E) and SHSY5Y cells (Fig. 2D). However, known antioxidants NAC and trolox did not show any rescue effects at 10 μM (Fig. 2D). This suggests that the rescue effects on mitochondrial membrane potential by ZCM-I-1 might be due to itsspecific targeting of complex I of mitochondria, not due to the general antioxidant activity. In addition, we confirmed that ZCM-I-1 has no effects on mitochondrial membrane potential and the level of ATP (Supplemental Fig. 1C and Fig. 1D) in MC65 cells, suggesting no effects on the coupling and bioenergetics of mitochondria. We then tested this compound using detergent-solubilized mouse brain mitochondria to confirm this. As shown in Fig. 2F, ZCM-I-1 suppressed the production of mitoROS when NADH was used as the complex I substrate. Taken together, the results strongly assert that mitochondrial complex I is the target of ZCM-I-1.
Figure 2. Effects of ZCM-I-1 on complex I ROS production.
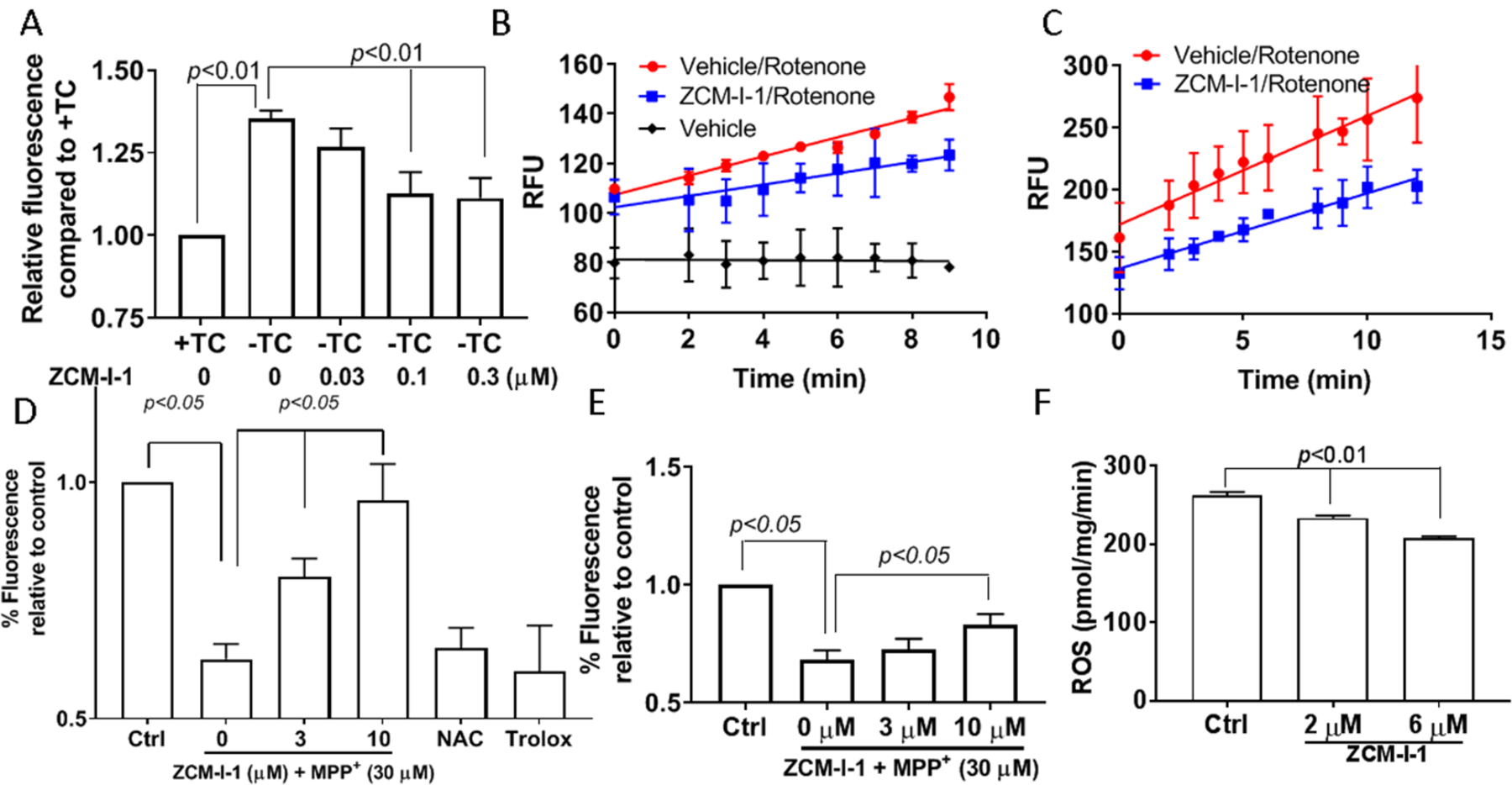
A) MC65 cells were treated with ZCM-I-1 in the presence of tetracycline (+TC) or absence of tetracycline (−TC) conditions for 48 h. After incubation with mitoSOX (2.5 μM), cells were analyzed by flow cytometry; MC65 cells (B) or mouse cortical neurons (DIV14) (C) were treated with vehicle or ZCM-I-1 (1 μM) for 12 h, then the cells were collected, washed, and suspended in HBSS. Upon addition of mitoSOX (2.5 μM) and rotenone (500 nM), the samples were analyzed by flow cytometry; SHSY5Y cells (D) or mouse cortical neurons (DIV14) (E) were treated with ZCM-I-1 at indicated concentrations or NAC (10 μm) or trolox (10 μM) and MPP+ (30 μM) for 24 h. After incubation with TMRM (100 nM), the samples were analyzed by flow cytometry; F) ROS production in permeabilized mouse brain mitochondria (N=6) pretreated with ZCM-I-1 was measured upon addition of NADH (10 μM) using Amplex red and HRP. Data presented as mean ± SEM. Statistical analysis by student t-test.
ZCM-I-1 selectively targets mitochondrial complex I via interactions with the IF site.
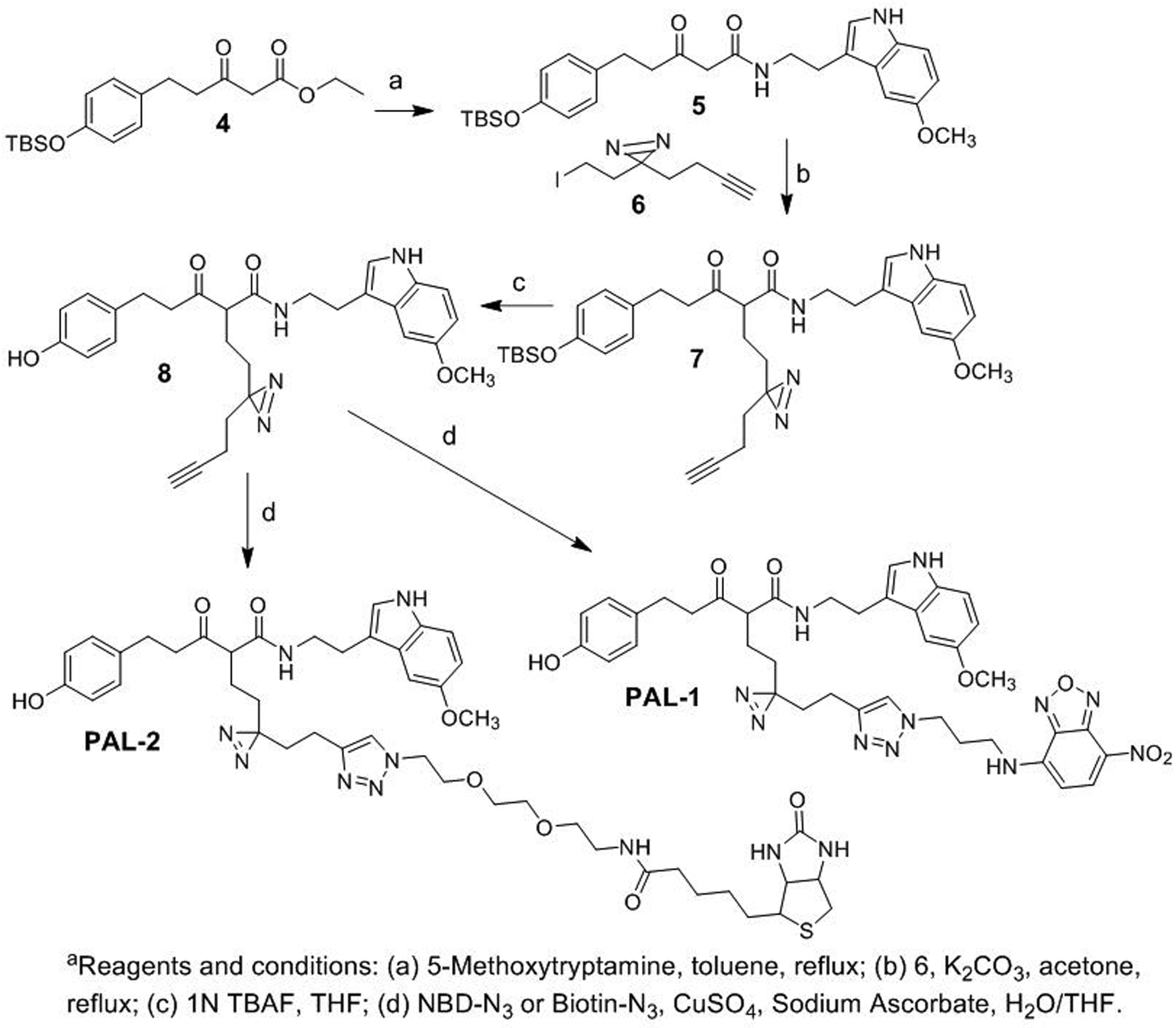
To elucidate the mode of action of ZCM-I-1 on its specific targeting of complex I, we designed a photoaffinity chemical probe PAL-1 (2, Fig. 3) to investigate its subcellular destination and interactions with complex I. The pilot structure-activity relationship (SAR) studies of ZCM-I-1 revealed that structural modifications on the methylene moiety between the carbonyl moieties would not decrease the activity. Therefore, in the design of PAL-1, the linker was attached to the methylene position of ZCM-I-1 and a diazirine was incorporated as the photoaffinity labeling moiety to covalently link interacting proteins. On the other end of the linker, a 7-nitrobenzo-2-oxa-1,3-diazole (NBD) moiety was incorporated as the fluorescent tag via click chemistry to allow the following visualization and detection. We also designed a second photoaffinity probe PAL-2 (3, Fig. 3) with a biotin tag instead of the NBD fluorescent tag to further confirm the interaction of ZCM-I-1 with its binding proteins. Upon incubation with PAL-2 and photo-activation, the interacting proteins from complex I with the probe should be identifiable by pull down assay using streptavidin resins. The chemical syntheses of the designed PAL-1 and PAL-2 were achieved by the conditions shown in Scheme 1. Briefly, ester-amide exchange reaction of 4 with 5-methoxyrtryptamine yielded compound 5. Alkylation of 5 with 6, synthesized following reported procedure, afforded the alkyne intermediate 7. Removal of the protecting TBS group of 7 followed by click reaction with corresponding NBD-N3 or Biotin-N3 gave the designed probes PAL-1 and PAL-2.
Figure 3.
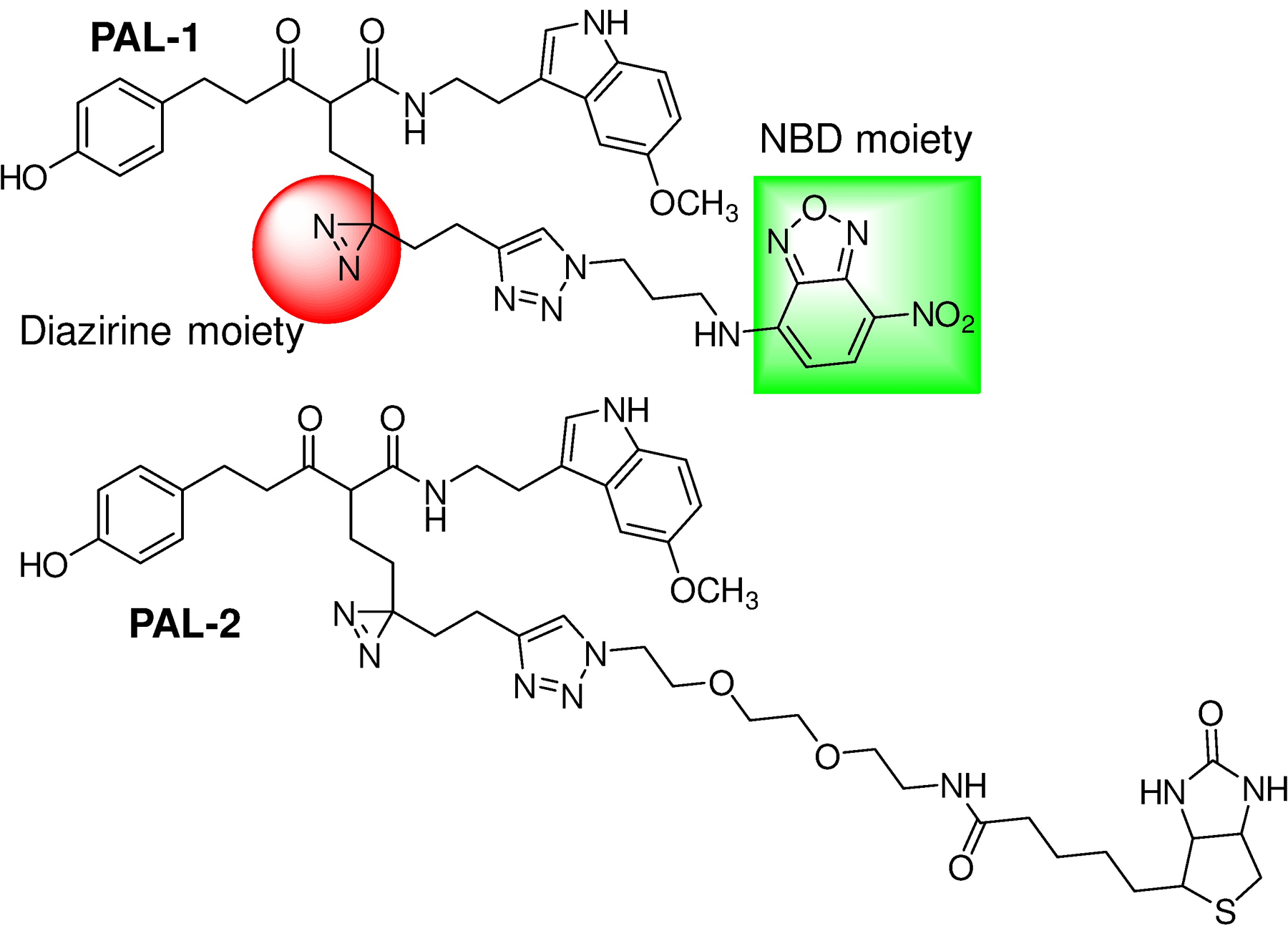
Design of photoaffinity probes PAL-1 (2) and PAL-2 (3).
Scheme 1a.
After chemical synthesis, rescue studies of MC65 cells confirmed PAL-1’s protective potency being 96.8 ± 2.9 nM, a ~ 3-fold decrease in potency compared to that of ZCM-I-1. However, PAL-2 lost its potency (> 3 μM) and this is probably due to cell penetration issues as we observed either significant decrease or loss of activities in compounds when a biotin moiety was attached (unpublished data). PAL-2 (1 μM) also shows no effects to suppress the mitoROS production in MC65 cells upon TC removal (Supplemental Fig. 1E). We expect that this probe should nonetheless recognize the interacting proteins from purified complex I since no membrane penetration is required. Immunocytochemistry (ICC) studies indicated that PAL-1 co-localizes with the mitochondria of MC65 cells and mouse primary cortical neurons (DIV 10–12) (Fig. 4A, 4B). Photoaffinity labeling studies using mitochondria isolated from the cerebral cortex of C57BL/6 mice with PAL-1 and colorless native gel isolation clearly demonstrated dose-dependent labeling of complex I by this probe (Fig. 4C). Next, complex I was purified from mouse brain mitochondria to study how PAL-1 interacts with the subunits of this complex. The results should shed light on the mode of action for ZCM-I-1. As shown in Fig. 5A, the probe PAL-1 could label subunits of 75 kDa, 51 kDa, 49 kDa, 24 kDa and 13 kDa in a dose-dependent manner. These subunits correspond to the human complex I subunit NDUFS1, NDUFV1, NDUFS2, NDUFV2, and NDUFS6 orthologues, respectively. Importantly, the labeled subunits, except for that of 49 kDa, compose the IF site of complex I,34–36 indicating that ZCM-I-1 binds to the IF site to exert the observed activities on its selective modulation of mitochondrial respiration and mitoROS production. The fact that PAL-1 labeling of these subunits was outcompeted by ZCM-I-1, NADH, and FMN, but not by rotenone, a known IQ site inhibitor,37 also supports this notion (Fig. 5A). To further confirm the interaction of ZCM-I-1 with the IF site, PAL-2 was incubated with purified complex I and the samples were photo-activated to induce covalent interactions. As shown in Fig. 5B, among the recruited proteins from purified mouse complex I by PAL-2 are subunits 75 kDa, 49 kDa and 24 kDa. In addition, the interactions are blocked by ZCM-I-1, consistent with the photoaffinity labeling results of PAL-1. Taken together, the results from the photoaffinity labeling and pull down studies using designed chemical probes established that ZCM-I-1 binds to the IF site of mitochondria.
Figure 4. PAL-1 localizes to the mitochondria and labels complex I.
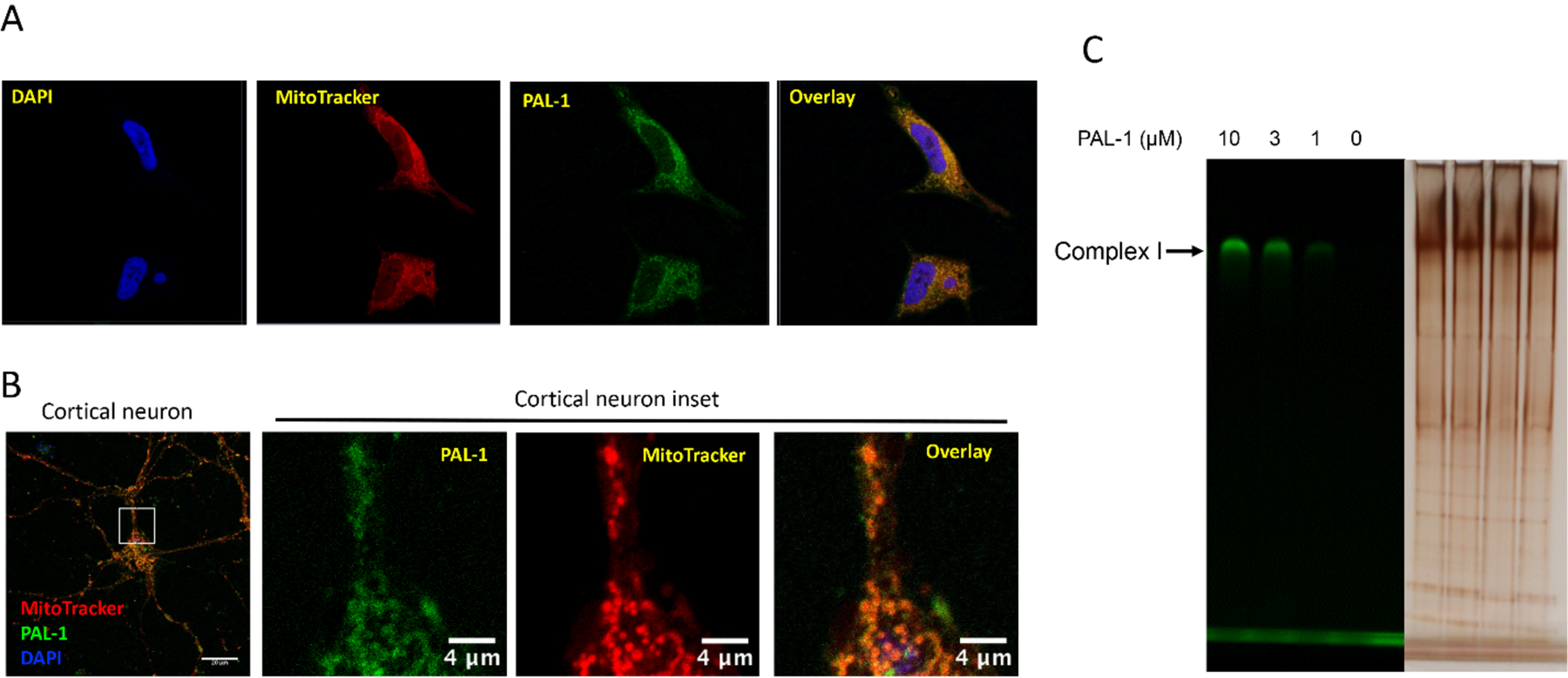
A) MC65 cells were treated with PAL-1 (3 μM) for 3.5 h. After incubation with MitoTracker Red (300 nM) and DAPI (2 μg/mL), samples were fixed and imaged with confocal microscopy. Image adjustments are as follows: DAPI unmodified, MitoTracker Red gamma adjusted to 1.15, PAL-1 NBD channel white level reduced to 13000, black increased to 350; B) Cultured neurons (DIV 10 to 12) were incubated with PAL-1 (25 μM) for 4 h, then incubated with Mitotracker Red CMXRos (25 nM) for 30 min. Neurons were washed × 3 by ½ media exchanges with neuronal culture media without phenol red. Neurons were imaged live on a Zeiss LSM710 confocal laser scanning microscope using a 63x/1.4 NA oil-immersion objective with pin hole of 1 Airy disc unit and Nyquist sampling; C) Mouse brain mitochondria (0.1 mg) were incubated with DMSO or PAL-1 at indicated concentrations for 1 h at 4 °C. The samples were UV irradiated at 365 nm for 10 min. After lysis, the samples were resolved by colorless Native PAGE and the gel was imaged with a BioRad imager system with excitation of 488 nm;
Figure 5. PAL-1 interacts with the IF site.
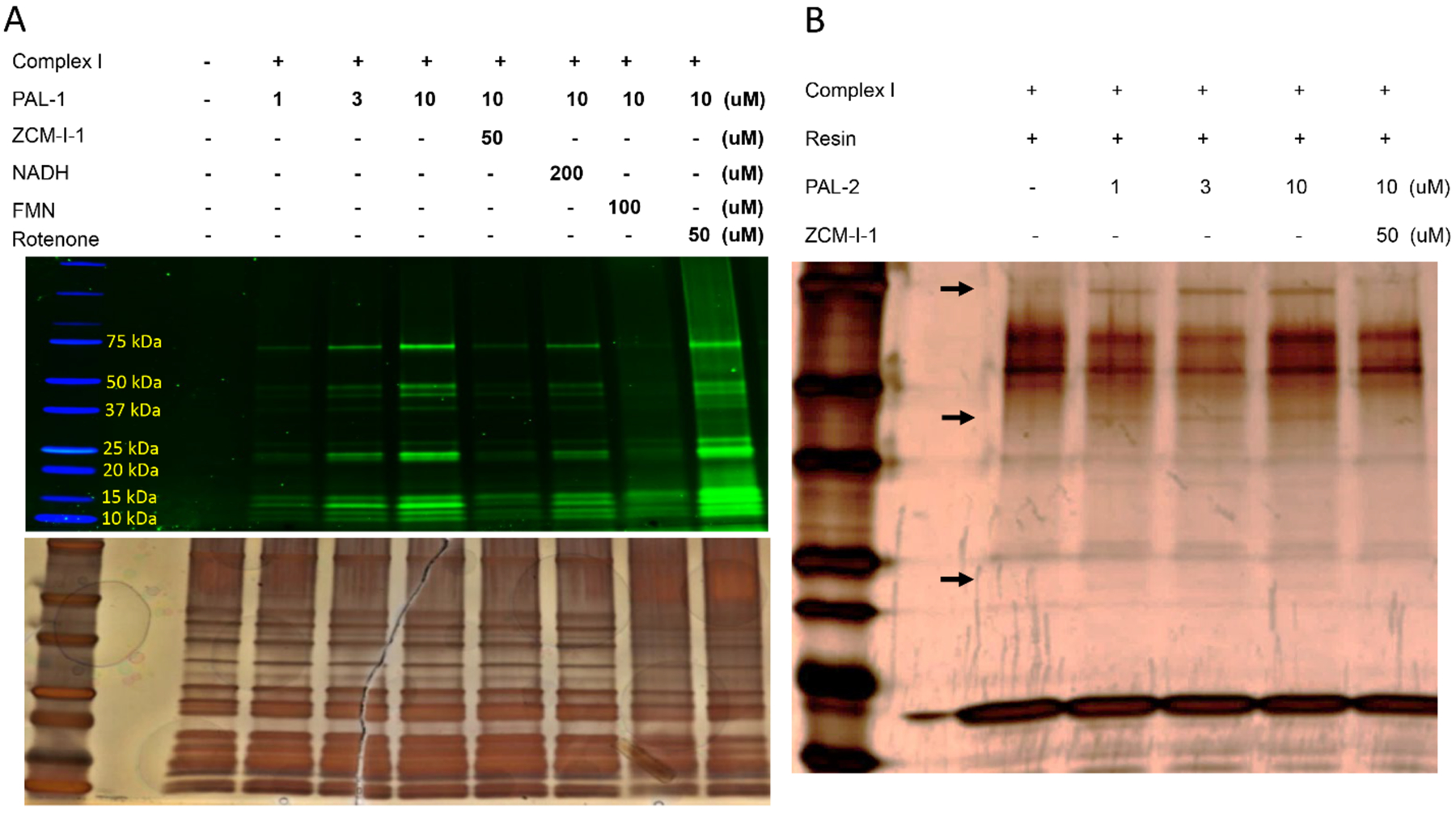
A) Complex I was purified from mouse brain mitochondria using an immuno-capture kit (Abcam, ab109711) and was incubated with PAL-1 under indicated conditions for 30 min (1.3 μg proteins/sample). Samples were UV irradiated at 365 nm for 10 min. After SDS-PAGE separation, the gel was imaged with a BioRad imager system with excitation of 488 nm. Image represents one of three independent experiments. Silver staining of the same gel to illustrate the presence of proteins; B) Complex I (2.4 μg/sample) was incubated with PAL-2 under indicated conditions for 30 min, then UV irradiated at 365 nm for 10 min. Samples were incubated with streptavidin resin (100 μL, Thermo Scientific#20349) for 1 h. After washing and adding loading buffer, samples were resolved by SDS-PAGE and visualized by silver staining. Arrows indicate the subunits 75 kDa, 49 kDa and 24 kDa.
Next, we conducted computational docking studies to illuminate how ZCM-I-1 interacts with the IF site, given that X-ray or cryo-EM structures of complex I of different species have been reported.36, 38, 39 To this end, we exploited as the template for homology modeling the T. thermophiles complex I X-ray structure (pdbid: 3IAM)40 within which the IF site shares 76.7% identity with that of human complex I and explored the potential interactions of ZCM-I-1 with key residues within the IF site. Poses were generated using GOLD5.641 and rescored with HINT,42 a program developed by us. The results (Fig. 6A) supported the interactions of ZCM-I-1 with the IF site with a HINT score of 410, comparable to NADH’s HINT score of 412. Specifically, as shown in Fig. 6B, the model suggests that residues E185, A68 and Y180 can form H-bond interactions with the indole nitrogen, the amide, and the phenol of ZCM-I-1, respectively. A potential hydrophobic pocket may be formed with the 5-CH3O-indole ring of ZCM-I-1 by residues of F70, F78, and F205. Notably, each of these residues is involved in the binding of NADH to subunit nqo1,40 an orthologue of the NDUFV1 subunit (51 kDa), consistent with the photoaffinity labeling results.
Figure 6. Molecular docking studies of ZCM-I-1.
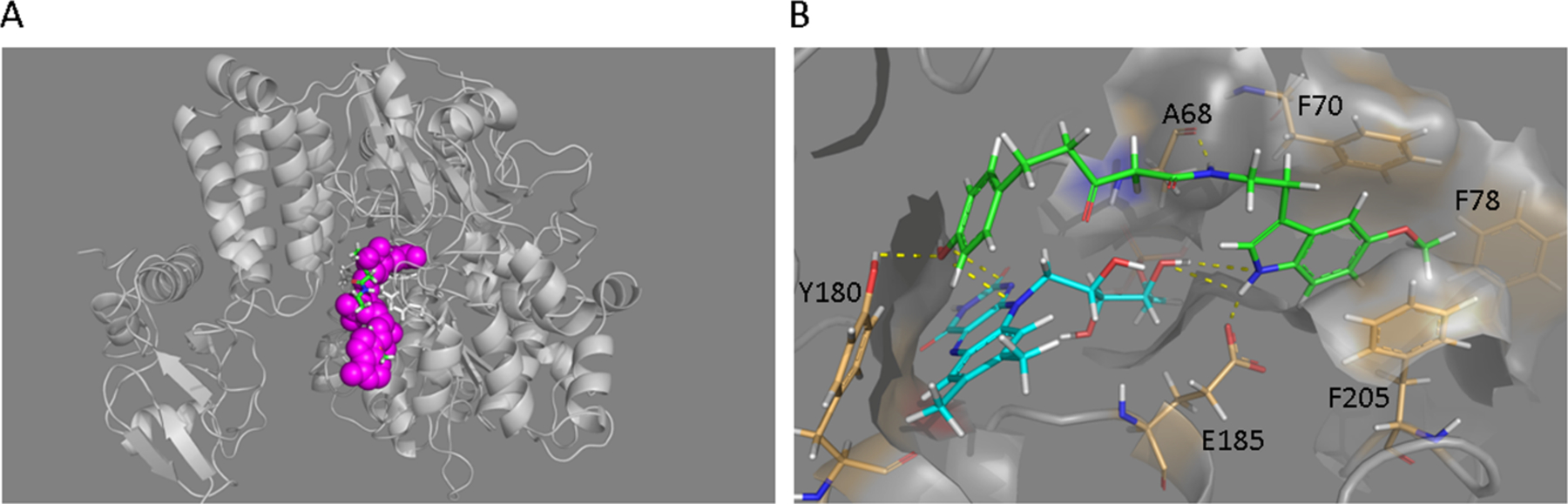
A) Docking orientations of ZCM-I-1 in the IF site of T. thermophiles complex I (pdbid: 3IAM). Upper image illustrates the overlap of ZCM-I-1 (green) with NADH (magenta) within the IF site of nqo1 subunit; B) Image illustrates the interactions of ZCM-I-1 (green) with residues of the IF site and FMN (cyan).
ZCM-I-1 selectively modulates the respiration of mitochondrial complex I both ex vivo and in vivo.
With the established MOA, we next tested how this compound influences the respiration of mitochondria and whether its action remains specific to complex I. First, we tested ZCM-I-1 in isolated mitochondria from the cerebral cortex of C57BL/6 mice to determine the effect on OXPHOS. As shown in Fig. 7A, ZCM-I-1 modulated the maximal rate of ADP stimulated respiration of pyruvate as the complex I substrate, but not with succinate as the complex II substrate. The respiratory control ratio (RCR, state 3/state 4) and the ADP/O ratio were not changed by the treatment with ZCM-I-1 (4.7 and 3.1, respectively) compared to vehicle treatment (5.3 and 2.9, respectively). The results indicated that despite the suppression of oxidation of pyruvate, the coupling and bioenergetic ability of mitochondria are preserved following treatment with ZCM-I-1, consistent with results from studies in MC65 cells. ZCM-I-1 also only moderately inhibited the complex I activity (~20%) when the ETC enzymatic activities were measured (Fig. 7B).
Figure 7. Effects of ZCM-I-1 on mouse brain mitochondria OXPHOS.
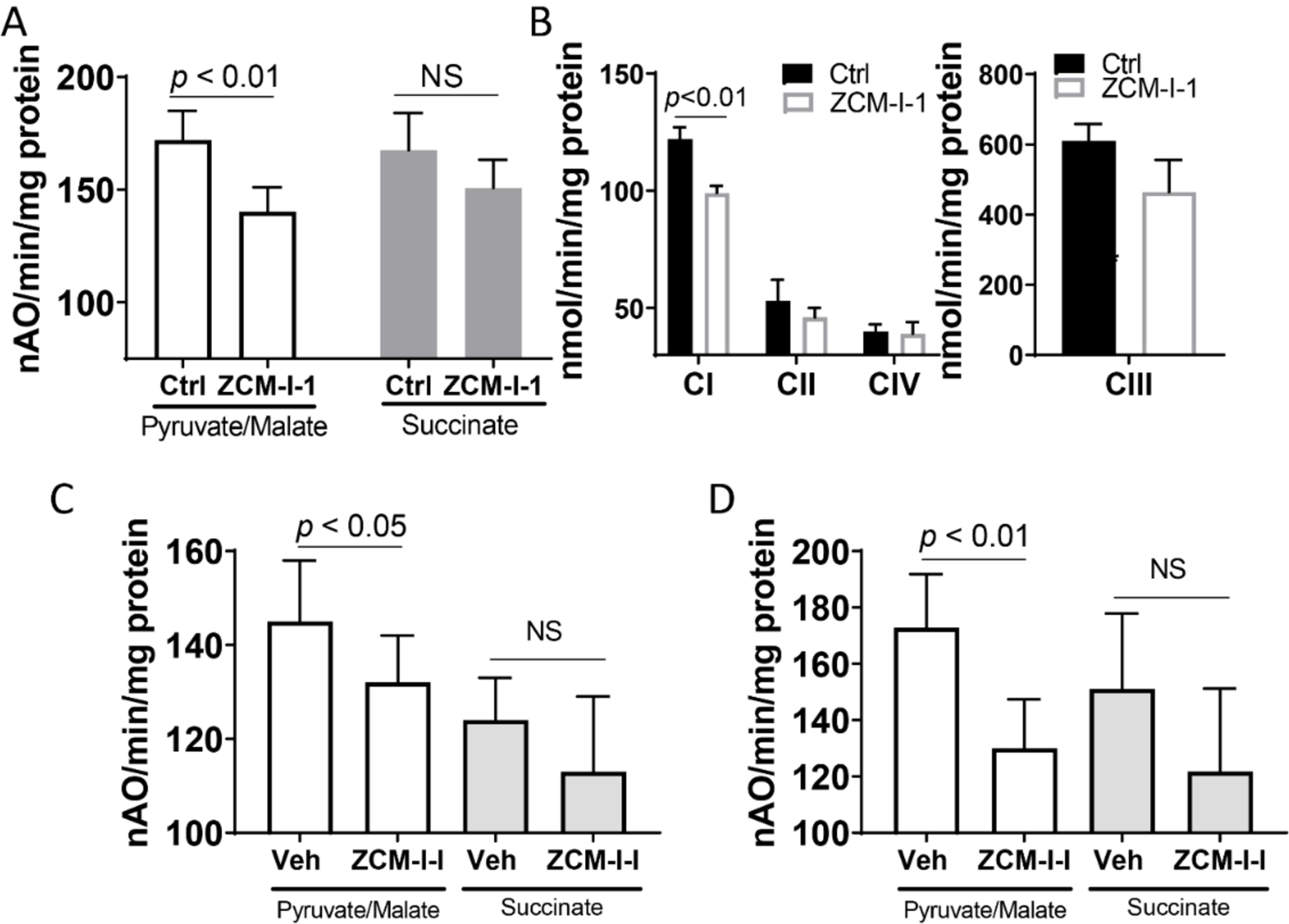
A) Oxygen consumption in isolated mouse brain mitochondria (N=6) was measured using a Clark-type oxygen electrode at 30 °C using pyruvate/malate (20/5 mM) or succinate (20 mM) in the presence of vehicle or ZCM-I-1 (2 μM); B) Detergent solubilized mouse brain mitochondria (N=6) were treated with ZCM-I-1 (2 μM) and the ETC activities were determined; C) 3xTg AD mice (5 months old) were treated with vehicle or ZCM-I-1 (50 mg/kg, oral gavage, 5 times/week, N=8 per group) for 2 weeks. Cortex/hippocampal mitochondria were isolated and oxygen consumption was measured as described above; D) 3xTg AD mice (10 months old) were treated with vehicle or ZCM-I-1 (50 mg/kg by oral gavage, once daily for 8 weeks, N=8 per group). Cortex/hippocampal mitochondria were isolated and measured for oxygen consumption. Data as mean ± SD, statistical analysis by student t-test.
We then decided to examine whether such selective modulation on mitochondria OXPHOS would be maintained by ZCM-I-1 in vivo using transgenic AD mice since mitochondrial dysfunction has been indicated as an important contributor to AD development.10, 16 To this end, we employed 3xTg AD mice, a triple transgenic AD mouse model that exhibits both plaques and tangles as well as early mitochondrial dysfunction.43–45 As shown in Fig. 7C, compared to the vehicle group, ZCM-I-1 treatment (50 mg/kg, oral gavage, 5 times/week) only led to moderate but statistically significant suppression (9%) of the maximal rate of ADP stimulated respiration of pyruvate, the complex I substrate, in 5 months old 3xTg AD mice (N=8 per group, half males and half females), an age by which intracellular Aβ and mitochondrial dysfunction start to appear.43, 44 This is consistent with the results from the ex vivo studies. In addition, moderate but significant suppression on the state 3 respiration of pyruvate was observed (Supplemental Fig. 2A), and the RCR and ADP/O ratio were not changed by the treatment of ZCM-I-1 (Table 1). In another study cohort, 3xTg AD mice at 10 months old, an age that AD pathologies become apparent,43, 44 were treated with ZCM-I-1 (50 mg/kg, once daily dosing by oral gavage, N=8 per group, half males and half females) for 8 weeks to mimic a clinically relevant therapeutic treatment. A similar modulation pattern upon treatment with ZCM-I-1 on the maximal ADP stimulated respiration (Fig. 7D) and state 3 respiration (Supplementary Fig. 2B) of pyruvate was observed as that from studies in the younger 3xTg AD mice, though the suppression is more significant in the 3xTg AD mice of 10 months old (25% suppression vs. 9% suppression).
Table 1.
RCR and ADP/O of mitochondrial OXPHOS from 3xTg AD mice of 5 months old treated with vehicle or ZCM-I-1.
| Pyruvate + Malate | Succinate | |||
|---|---|---|---|---|
| RCR | ADP/O | RCR | ADP/O | |
| Mice treated with vehicle | 4.1±0.7 | 2.6±0.2 | 2.5±0.2 | 1.4±0.1 |
| Mice treated with ZCM-I-1 | 4.2±0.8 | 2.6±0.2 | 2.7±0.3 | 1.3±0.1 |
3xTg AD mice (5 months old) were treated with vehicle or ZCM-I-1 (50 mg/kg, oral gavage, 5 times/week, N=8 per group) for 2 weeks. Cortex/hippocampal mitochondria were isolated and oxygen consumption was measured using a Clark-type oxygen electrode at 30 °C using pyruvate/malate (20/5 mM) or succinate (20 mM).
DISCUSSION
Mitochondria are the organelle that produce ATP, but they also generate the majority of ROS that have both physiological functions as signaling molecules and pathological roles associated with oxidative stress.13–15 Mitochondrial dysfunction and oxidative stress have been shown to have roles in a variety of human diseases including neurodegenerative disorders.4, 5, 8, 12 However, the contributions of mitochondria produced ROS are not clear, especially whether their pathological roles are causal versus merely a consequence of other disease-mediated injuries. This lack of understanding is due to, at least partially, the lack of effective and specific in vivo chemical probes with well-defined MOA.
The production of mitoROS is mostly from the reduction of molecular oxygen by electrons leaked from complexes during the OXPHOS.46, 47 Multiple sites on the mitochondrial ETC, mainly located on complex I, complex II, and complex III, have been shown to be responsible for the production of mitoROS.13, 48, 49 Numerous compounds, such as mitochondria-targeted antioxidants and inhibitors of the ETC complexes,50, 51 have been identified to target mitochondria. However, many drawbacks associated with these compounds have limited their further use as chemical probes. For example, most of the mitochondria-targeted antioxidants are generic ROS scavengers and cannot differentiate the site-specific roles of mitoROS. The majority of the known complex I inhibitors are also mitochondria toxins and typically interfere with the overall function of mitochondria including its bioenergetics. In addition, although the IQ site has been indicated as the binding site, the binding sites for most complex I inhibitors remain unknown. Recently, small molecule inhibitors that selectively target the ubiquinone site of complex I and complex III have been identified,46, 52, 53 and studies employing these inhibitors as chemical probes have provided valuable information on the pathological roles of site-specific mitoROS on different pathological disorders. The discovery of such inhibitors attests to the interest in developing novel small molecule compounds that specifically target the ETC of mitochondria and possess well-defined MOA as chemical probes and potential therapeutics.
Recently, we successfully developed a novel chemical scaffold and identified a lead compound ZCM-I-1 with promising neuroprotective activities in animal models of neurodegenerative disorders including AD.30, 31 In this study, we conducted experiments using in vitro, ex vivo, in vivo, and used chemical biology approaches to elucidate the MOA of this compound. Our studies demonstrated that ZCM-I-1 dose-dependently inhibited the production of mitoROS in MC65 cells, a cellular AD model,54, 55 and in isolated mouse brain mitochondria when NADH was used as the complex I substrate. Further studies using cellular models established that ZCM-I-1 suppresses mitoROS production and reverses the mitochondrial membrane potential change induced by known complex I inhibitors. More importantly, this compound does not interfere with the mitochondria membrane potential and ATP production in MC65 cells. The results suggested that ZCM-I-1 specifically targets mitochondrial complex I, while not interfering with the coupling and bioenergetics roles of mitochondria. ICC studies using chemical probe derived from ZCM-I-1 in cell cultures confirmed that the probe co-localizes with the mitochondria. Photoaffinity labeling studies in isolated mouse brain mitochondria and purified mitochondrial complex I demonstrated that the probe selectively interacts with and labels subunits that comprise the IF site. Competition studies using the parent compound, endogenous ligands and the IQ site inhibitor also support this notion. In addition, molecular docking studies using the structure of complex I of T. thermophilies (pbdid: 3IAM) supported the interactions of ZCM-I-1 with the IF site via H-bond and hydrophobic interactions. Further analysis of its effects on the OXPHOS of mouse brain mitochondria revealed that ZCM-I-1 selectively modulates the respiration of pyruvate, a NADH-linked complex I substrate, but not succinate, a complex II substrate. Its effects on the RCR and ADP/O ratio are also consistent with the results from studies using cellular models in that ROS production is decreased yet coupling of respiration and mitochondria membrane potential to support OXPHOS is not altered. The selective modulation on the respiration of pyruvate was observed in vivo in 3xTg AD mice after treatment with ZCM-I-1 (50 mg/kg) from two cohorts of studies with animals at different ages. The in vivo study results provided evidence to support the compound’s selective engagement and modulatory effects on mitochondrial complex I.
CONCLUSIONS
The mechanistic studies from in vitro, ex vivo, in vivo and chemical biology models established that ZCM-I-1 is a selective modulator of the complex I without effects on mitochondria membrane potential and bioenergetics via interaction with the IF site, thus representing a first-in-class MOA. Screening of ZCM-I-1 against the major CNS receptors using services from the NIMH PDSP program confirmed that no significant inhibition was observed at 10 μM. Given the crucial roles of mitochondrial complex I in the regulation of ATP, selective and moderate modulation of complex I, rather than strong inhibition, may be the key for development of potential therapeutics, especially for AD, as supported by studies from heart mitochondria.56, 57 Thus, the current study strongly favors the use of ZCM-I-1 as a chemical tool for the further study of the roles of complex I mitoROS produced at the IF site in human health and diseases, and SAR studies based on ZCM-I-1 as a lead compound may lead to development of novel and effective AD therapeutics.
EXPERIMENTAL SECTION
Chemistry.
Reagents and solvents were obtained from commercial suppliers and used as received unless otherwise indicated. Flash column chromatography was performed on silica gel (200–300 mesh, Fisher Scientific) using solvents as indicated. 1H NMR and 13C NMR spectra were recorded on Bruker Ultrashield Plus-400MHz spectrometer. The NMR solvent used was CDCl3 or DMSO-d6 as indicated. Tetramethylsilane (TMS) was used as the internal standard. HRMS were recorded on PerkinElmer AxION 2 TOF mass spectrometer. The purity of target compounds was determined by HPLC using a Varian 100–5 C18 250×4.6 mm column with UV detection (280 nm and 360 nm) (50% H2O in acetonitrile and 0.1% TFA, and 30–50% H2O in methanol and 0.1% TFA, two solvent systems) to be ≥ 95%.
5-(4-((tert-Butyldimethylsilyl)oxy)phenyl)-N-(2-(5-methoxy-1H-indol-3-yl)ethyl)-3-oxopentanamide (5):
Compound 4 (3.2 mmol) and 2-(5-methoxy-1H-indol-3-yl)ethan-1-amine (3.8 mmol) were dissolved in toluene (30 mL). The mixture was refluxed overnight. After cooling to room temperature, the solvents were removed by rotavap under reduced pressure. The residue was purified by flash chromatography (CH2Cl2/methanol: 100/1) to obtain 2. H-NMR (400 MHz, DMSO-d6) 0.15 (s, 6H), 0.93 (s, 9H), 2.66–2.71 (m, 2H), 2.76–2.80 (m, 4H), 3.30–3.31 (m, 4H), 3.75 (s, 3H), 6.69–6.75 (m, 3H), 7.00–7.11 (m, 4H), 7.23 (d, J=8.72 Hz, 1H), 8.15 (t, J=5.60 Hz, 1H), 10.63 (s, 1H).
2-(2-(3-(But-3-yn-1-yl)-3H-diazirin-3-yl)ethyl)-5-(4-((tert-butyldimethylsilyl)oxy)-phenyl)-N-(2-(5-methoxy-1H-indol-3-yl)ethyl)-3-oxopentanamide (7):
Compound 5 (0.2 mmol) and K2CO3 (0.5 mmol) were added to acetone (4 mL) at 0 °C, then compound 6 (0.5 mmol), synthesized following the reported procedure,58 was added. The mixture was heated to 80 °C for 12 h. After cooling to room temperature, solvents were removed by rotavap under reduced pressure. The residue was dissolved in CH2Cl2 (20 mL) and dried over anhydrous Na2SO4 overnight. After filtration and removal of solvents, the crude product was purified by flash chromatography (CH2Cl2/methanol: 100/1) to obtain 7. 1H NMR (400 MHz, CDCl3) 0.17 (s, 6H), 0.97 (s, 9H), 1.48–1.55 (m, 4H), 1.91–1.95 (m, 3H), 2.63 (s, 1H), 2.73–2.75 (m, 3H), 2.91 (t, J=6.64 Hz, 2H), 3.16 (t, J=7.24 Hz, 1H), 3.54 (s, 4H), 3.87 (s, 3H), 6.02 (t, J=5.64 Hz, 1H), 6.74 (d, J=8.40 Hz, 2H), 6.88 (dd, J1=2.44 Hz, J2=8.80 Hz, 1H), 6.93 (d, J=2.32 Hz, 1H), 6.98 (d, J=8.40 Hz, 2H), 7.01 (d, J=2.44 Hz, 1H), 7.24–7.25 (m, 1H), 7.92 (s, 1H).
2-(2-(3-(But-3-yn-1-yl)-3H-diazirin-3-yl)ethyl)-5-(4-hydroxyphenyl)-N-(2-(5-methoxy-1H-indol-3-yl)ethyl)-3-oxopentanamide (8):
Compound 7 (0.5 mmol) was dissolved in THF (5 mL), then 1N TBAF (1 mmol) was added. The mixture was stirred at room temperature. After adding EtOAc (30 mL), the organic layer was washed with water for 3 times, dried over anhydrous Na2SO4. After filtration and removal of solvents, the crude product was purified by flash chromatography (CH2Cl2/methanol: 50/1) to obtain 8. H-NMR (400 MHz, DMSO-d6) 1.21–1.28 (m, 2H), 1.38–1.46 (m, 2H), 1.57 (t, J=7.28 Hz, 2H), 1.97–2.01 (m, 2H), 2.64–2.72 (m, 4H), 2.80–2.85 (m, 3H), 3.29–3.31 (m, 1H), 3.37–3.41 (m, 2H), 3.79 (s, 3H), 6.67 (d, J=8.48 Hz, 2H), 6.76 (dd, J1=2.40 Hz, J2=8.68 Hz, 1H), 6.95 (d, J=8.48 Hz, 2H), 7.04 (d, J=2.44 Hz, 1H), 7.10 (d, J=2.36 Hz, 1H), 7.27 (d, J=8.56 Hz, 1H), 8.36 (t, J=5.64 Hz, 1H), 9.15 (s, 1H), 10.68 (s, 1H).
5-(4-Hydroxyphenyl)-N-(2-(5-methoxy-1H-indol-3-yl)ethyl)-2-(2-(3-(2-(1-(3-((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino)propyl)-1H-1,2,3-triazol-4-yl)ethyl)-3H-diazirin-3-yl)ethyl)-3-oxopentanamide (PAL-1):
Compound 8 (0.05 mmol) and NBD-N3 (0.05 mmol) were dissolved in THF/H2O (2/1, 2 mL), then sodium ascorbate (0.025 mmol) and CuSO4 (0.025 mmol) were added. The mixture was stirred for 1 h at room temperature. CH2Cl2 (15 mL) was added and the mixture was washed with water, and dried over anhydrous Na2SO4. After filtration and removal of solvents, the crude product was purified by flash chromatography (CH2Cl2/methanol: 50/3) to obtain PAL-1. H-NMR (400 MHz, DMSO-d6) 1.16–1.24 (m, 2H), 1.35–1.42 (m, 2H), 1.70 (t, J=8.08 Hz, 2H), 2.20–2.27 (m, 2H), 2.39 (t, J=7.6 Hz, 2H), 2.55–2.66 (m, 4H), 2.79 (t, J=7.40 Hz, 2H), 3.28–3.30 (m, 1H), 3.35–3.38 (m, 2H), 3.75 (s, 3H), 4.12 (q, J=5.28 Hz, 2H), 4.47 (t, J=6.92 Hz, 2H), 6.36 (d, J=9.00 Hz, 1H), 6.63 (d, J=8.44 Hz, 2H), 6.72 (dd, J1=2.40 Hz, J2=8.72 Hz, 1H), 6.91 (d, J=8.52 Hz, 2H), 6.99 (d, J=2.44 Hz, 1H), 7.06 (d, J=2.32 Hz, 1H), 7.22 (d, J=8.76 Hz, 1H), 7.87 (s, 1H), 8.33 (t, J=5.60 Hz, 1H), 8.50 (d, J=8.92 Hz, 1H), 9.11 (s, 1H), 9.49 (s, 1H), 10.64 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 204.87, 167.74, 155.35, 152.92, 145.30, 137.79, 131.35, 130.88, 128.92, 127.40, 123.25, 122.14, 116.07, 114.97, 111.95, 111.23, 110.96, 100.08, 99.48, 99.33, 68.35, 58.60, 55.31, 52.91, 46.92, 42.33, 40.67, 31.69, 29.65, 28.39, 28.07, 25.00, 21.89, 19.58.
HRMS (AP-ESI) m/z calcd for C38H41N11O7 [M + Na]+ 786.3088, found 786.3105.
PAL-2 was prepared similarly using 8 and Biotin-N3. H-NMR (400 MHz, DMSO-d6) 1.22–1.64 (m, 10H), 1.71 (t, J=7.96 Hz, 2H), 2.08 (t, J=7.44 Hz, 2H), 2.41 (t, J=7.88 Hz, 2H), 2.56–2.68 (m, 5H), 2.76–2.83 (m, 3H), 3.06–3.11 (m, 1H), 3.15–3.21 (m, 2H), 3.27–3.29 (m, 1H), 3.33–3.39 (m, 4H), 3.45–3.52 (m, 4H), 3.76 (s, 3H), 3.79 (t, J=5.28 Hz, 2H), 4.11–4.14 (m, 1H), 4.32 (t, J=6.92 Hz, 1H), 4.47 (t, J=5.24 Hz, 2H), 6.40 (d, J=21.76 Hz, 2H), 6.64 (d, J=8.44 Hz, 2H), 6.73 (dd, J1=2.40 Hz, J2=8.76 Hz, 1H), 6.92 (d, J=8.44 Hz, 2H), 7.01 (d, J=2.44 Hz, 1H), 7.07 (d, J=2.28 Hz, 1H), 7.23 (d, J=8.72 Hz, 1H), 7.78–7.81 (m, 2H), 8.33 (t, J=5.68 Hz, 1H), 9.12 (s, 1H), 10.64 (s, 1H). 13C-NMR (101 MHz, DMSO-d6) δ 204.88, 172.10, 167.75, 162.66, 155.36, 152.93, 145.13, 131.36, 130.89, 128.93, 127.42, 123.26, 122.34, 114.98, 111.96, 111.24, 110.98, 100.08, 69.48, 69.36, 69.09, 68.74, 61.01, 59.17, 58.60, 55.37, 55.32, 49.24, 42.35, 38.38, 35.06, 31.77, 29.65, 28.39, 28.15, 28.08, 28.00, 25.22, 25.00, 21.90, 19.58.
HRMS (AP-ESI) m/z calcd for C45H60N10O8S [M + Na]+ 923.4214, found 923.4227.
Biological assays
Animals:
All animal experiments were conducted under the guidelines of the “Guide for the care and use of laboratory animals” published by National Institutes of Health (revised 2011) and approved by the Institutional Animal Care and Use Committees (IACUC) of the McGuire VA Medical Center and Virginia Commonwealth University. C57BL/6 mice were purchased from the National Cancer Institute (Bethesda, MD). 3xTg AD mice carrying the APP695 gene with Swedish mutations (KM670/671NL/M596L), along with the PSEN1 mutation (M146V) and the MAPT mutation (P301L) on the C57B/6J background were purchased from Jackson Laboratory (Bar Harbor, ME).
For in vivo studies, mixed male and female 3xTg AD mice at the age of 5 mo or 10 mo were used. Animals were divided into two groups blinded receiving either ZCM-I-1 (50 mg/kg) or vehicle (2% DMSO + 10% cremophore) via oral gavage (N=8/age/group, half males and half females). For 5 mo animals, treatment was given 5 times/week for 2 weeks, whereas the 10 mo animals received single daily dosing for 8 weeks to mimic clinically relevant therapeutic strategies. After completing the treatment, animals were sacrificed, and fresh cerebral cortex and hippocampus were dissected and processed for mitochondria isolation and measurement as previously reported.59
Cells:
MC65 cells were kindly provided by Dr. George M. Martin at the University of Washington, Seattle and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies, Inc., Grand Island, NY) supplemented with 10% of heat-inactivated fetal bovine serum (FBS) (Hyclone, Logan, UT), 1% penicillin/streptomycin (P/S) (Sigma-Aldrich), tetracycline (TC) (1 μg/mL, Sigma-Aldrich) and 0.2 mg/mL G418 (Invitrogen). SHSY5Y cells (ATCC, Manassas, VA) were cultured in DMEM supplemented with 10% FBS and 1% P/S. All cells were maintained at 37 °C in a fully humidified atmosphere containing 5% CO2.
Cortex from E15.5 embryos of C57BL/6J mice were dissected then enzymatically dissociated in neurobasal medium containing trypsin (2.5 mg/mL, Sigma-Aldrich) and DNase I (15 μg/mL, Sigma-Aldrich) for 30 min at 37 °C, washed in neurobasal medium, then triturated using fire-polished Pasteur pipettes. Cell suspensions were passed through a 70 μm strainer to remove debris. Cells were plated on poly-D-lysine (Sigma Aldrich) coated plates or glass-bottomed dish and cultured in neuronal culture media (neurobasal medium supplemented with B27, GlutaMax (0.5 mM) and penicillin/streptomycin/amphotericin B). Neuronal culture media were changed by one-half volume exchange every 3 days. Cytarabine (1 μM final, Sigma-Aldrich) was added from day in vitro (DIV) 3 to 6 to inhibit glial proliferation.
MitoROS Assay
MC65 cells suspended in Opti-MEM (Invitrogen) were seeded in 6-well plates (1.6×106 cells/well), and incubated with ZCM-I-1 at 37 °C under +TC or −TC condition for 48 h. Cells were collected and suspended in HBSS. MitoSOX (2.5μM) was added at 37 °C for 20 min. After centrifugation and washing twice with HBSS, cells were suspended in HBSS and the mean fluorescent intensity was recorded by flow cytometry.
Mouse primary cortical neurons (DIV14) were seeded in 24-well plates (1×105 cells/well). MC65 cells were seeded in 6-well plates (1.6×106 cells/well) under +TC conditions. After incubation with ZCM-I-1 at 37 °C for 1 h, cells were collected and suspended in HBSS. MitoSOX (2.5μM) was added for 20 min. After centrifugation and washing twice with HBSS, cells were suspended in HBSS and added rotenone (500 nM), then the mean fluorescent intensity was recorded at different time point by flow cytometry.
Mitochondrial Membrane Potential Assay
SH-SY5Y cells (4×105 cells) plated in 12 well plates or mouse primary cortical neurons (DIV 14) plated in 96 well plates (5×104 cells/well) were treated with ZCM-I-1 and MPP+ for 24 h. TMRM was then added to a final concentration of 100 nM (SHSY5Y cells) or 1 μM (cortical neurons) and the cells were further incubated for 30 min. For SHSY5Y cells, cells were detached by trypsinization. After centrifugation, the cell pellet was suspended in PBS and fluorescence was analyzed by flow cytometry. For cortical neurons, cells were washed twice with PBS and then solubilized with 50% DMSO and 0.5% Triton in distilled water to disperse TMRM from the cells. The fluorescence intensity was measured using a FlexStation 3 plate reader at Ex/Em: 548/574 nm.
Mitochondria ROS assay
The rate of H2O2 production in mitochondria was determined using the oxidation of the fluorogenic indicator amplex red in the presence of HRP as we reported before.47 The concentrations of HRP and amplex red in the incubation were 0.1 unit/ml and 50 μM, respectively. Fluorescence was recorded in a microplate reader (1420 Victor2, PerkinElmer Life Sciences) with 530 nm excitation and 590 nm emission wavelengths. Standard curves obtained by adding known amounts of H2O2 to assay medium in the presence of the reactants (amplex red and HRP) were linear up to 2 μM. Isolated mitochondria were incubated at 0.1 mg of protein/ml at 30 °C with compound for 1 h. H2O2 production was initiated in mitochondria using glutamate (10 mM) + malate (2.5 mM), or succinate (5 mM) as substrates. Rotenone (2.4 μM) was added into incubation medium to inhibit the activities of complex I when succinate was used as complex II substrate. The rate of H2O2 production was linear with respect to mg of mitochondrial protein.
Confocal Microscopy Images
MC65 cells were plated for 24 h on coverslips coated with poly-D-lysine. Cells were treated with PAL-1 (3 μM) for 3.5 h, after which Mitotracker Red (300 nM) was added for 30 min. After washing with PBS, the cells were fixed with 4% formaldehyde for 15 min and washed with PBS once. DAPI (2.5 μg/mL) was then added and incubated for 5 min. Cells were washed with PBS three times, mounted, and solidified overnight. The samples were analyzed on a Zeiss LSM 700 on an Axio Imager 2 stand using the Plan-Apochromat 63x/1.40 oil DIC M27 objective lens. Images were analyzed using both the Zen Blue (version 2.6) and Zen Black software suites for colocalization coefficients and visualization.
Photoaffinity Labeling and Pull-down assays with Purified Complex I
Mitochondria were isolated from the brain cortex of C57BL/6 mice as described previously.59 Isolation of complex I was performed using a complex I immunocapture kit (Abacam, ab109711) according to the instructions provided by the manufacturer. Purified complex I was added to a solution of no BSA buffer (215 mM mannitol, 75 mM sucrose, 1 mM EGTA, 20 mM HEPES in purified water, pH 7.4). Samples were incubated with DMSO or ZCM-I-1 at the indicated concentration for 45 min at 4 °C. DMSO or PAL-1 at the indicated concentration was then added and incubated for 30 min at 4 °C. Samples were irradiated at 365 nm for 10 min on ice. After adding the Laemmli with β-ME buffer, samples were denatured and resolved by SDS-PAGE. After washing with water, gels were fixed overnight with 50% methanol and 10% acetic acid in purified water. After washing with purified water, gels were visualized on a Bio-Rad ChemiDoc MP imaging system with excitation of 488 nm.
Purified complex I (2.4 μg) in no BSA buffer (215 mM mannitol, 75 mM sucrose, 1 mM EGTA, 20 mM HEPS in purified water, pH 7.4) was incubated with DMSO or ZCM-I-1 at the indicated concentration for 45 min at 4 °C. DMSO or PAL-2 at the indicated concentrations was then added and the samples were incubated for 30 min at 4 °C. After irradiation at 365 nm for 10 min on ice, streptavidin Agarose Resins (100 μL, Number20349, Thermo Scientific, #20349, Pre-washed with PBS three times) was added. The samples were then transferred to the tube with PBS (final volume: 500 μL), and incubated overnight at 4 °C. The resins were spun down at 2000 × g and 4 °C and washed with 1% NP-40 in PBS (pH 7.4) three times, 0.1% SDS in PBS (pH 7.4) three times, and PBS three times. After adding loading buffer (1x, 50 μL), the samples were centrifuged for 3 min at 2000x g at 4 °C. Supernatants (30 μL per lane) were resolved by SDS-PAGE and stained following the silver staining protocol.
Photoaffinity Labeling with Mitochondria Lysate
Mitochondria (0.1 mg) were incubated with DMSO or ZCM-I-1 for 1 h at 4 °C. DMSO or PAL-1 were added and the mixture was incubated for an additional 1 h at 4 °C. The samples were irradiated at 365 nm for 10 min on ice. Samples were incubated with NativePAGE™ sample buffer, purified H2O, and 1% digitonin (final concentration) on ice for 30 min. After centrifugation at 16000g for 20 min at 4 °C, to the supernatant was added 2 μL Ponceau S solution (10 mg Ponceau S, 5 mL 50% v/v glycerol). The samples were separated with a 3–12% colorless NativePAGE Bis-Tris gel. Gels were then visualized with excitation of 488 nm on a Bio-Rad ChemiDoc MP imaging system.
Computational modeling
Models were prepared using the crystal structure of the hydrophilic portion of complex I (PDB ID: 3IAM) using Sybyl-X 2.1.1 software. All subunits except nqo1 were deleted and hydrogens were added to the remaining protein model. Protonation states of E185, E184 and K202 were evaluated and appropriate atom type modifications were made. NADH was extracted and the protein was prepared for docking in the GOLD program. Using the extracted NADH to define the binding site, ZCM-I-1 was docked into nqo1 already containing FMN to produce 100 unique binding poses. Each pose was merged with the unbound protein structure and minimized in Sybyl. Minimization parameters are as follows: 100000 steps for minimizing biopolymer hydrogens, sidechains, biopolymer without C-alpha, ligands, and finally all atoms using Gastiger:Huckel charges and Tripos force fields over 100000 iterations with a 0.02 kcal/(mol*A) cutoff energy. The resulting structures were then scored using HINT to identify important residues for binding. As a control, NADH was also minimized and scored using HINT. Important residues for binding included: E185 (1.9 Å) contributing H-bond and ionic interactions with the indole nitrogen of ZCM-I-1 via the sidechain carboxylic acid, A68 (2.0 Å) contributing H-bond interaction with the amide of ZCM-I-1 via the backbone carbonyl moiety, Y180 (2.5 Å) contributing H-bond interactions with between the phenol groups. Additionally, hydrophobic interactions were formed between residues of F70, F78, F205 and the tryptamine substructure of ZCM-I-1.
Supplementary Material
ACKNOLEDGEMENTS
The work was supported in part by the NIA of the NIH under award number R01AG058673 (SZ), Alzheimer’s Drug Discovery Foundation 20150601 (SZ), Office of Research and Development, Medical Research Service Merit Review Award (2IO1BX001355-01A2) (QC, EJL), the National Institute of Aging (NIA) R21AG054975-01, and the Pauley Heart Center, Virginia Commonwealth University (QC, JT, EJL). Microscopy was performed at the VCU Microscopy Facility, supported, in part, by funding from NIH-NCI Cancer Center Support Grant P30 CA016059. The screening of ZCM-I-1 for receptors/transporters was conducted by the NIMH PDSP at University of North Carolina at Chapel Hill. We thank Mr. Jeremy Thompson for his technical assistance in the mitochondria OXPHOS studies.
ABBREVIATIONS
- AD
Alzheimer’s disease
- ADP/O
adenosine diphosphate/oxygen ratio
- AP-ESI
atmospheric pressure-electrospray interface
- ATP
adenosine triphosphate
- BBB
blood-brian barrier
- CNS
Central nervous system
- DAPI
4′,6-diamidino-2-phenylindole
- DCM
Dichloromethane
- DMEM
Dulbecco’s Modified Eagle Medium
- DMF
N,N-Dimethylformamide
- DMSO
dimethyl sulfoxide
- EDCI
N-(3-(dimethylamino)propyl)-N-ethyl-carbodiimide hydrochloride
- EGTA
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- ETC
electron transport chain
- FBS
fetal bovine serum
- FMN
flavin mononucleotide
- HBSS
Hank’s Balanced Salt Solution
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HINT
hydropathic interactions
- HOBt
hydroxybenzotriazole
- HRMS
high resolution mass spectra
- ICC
immunocytochemistry
- IF
complex I flavin nucleotide binding site
- IP
intraperitoneal
- IQ
complex I quinone binding site
- IV
intravenous injection
- LC-MS/MS
Liquid chromatography tandem mass spectrometry
- LSM
laser scanning microscope
- MOA
mechanisms of action
- mitoROS
mitochondrial reactive oxygen species
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NADH
nicotinamide adenine dinucleotide
- NBD
nitrobenzofurazan
- NIA
National Institute of Aging
- NMR
nuclear magnetic resonance
- OXPHOS
oxidative phosphorylation
- PAL
photoaffinity label
- PBS
phosphate-buffered saline
- PDB ID
protein data bank identifier
- PK
pharmacokinetic
- PO
per os
- RCR
respiratory control ratio
- ROS
reactive oxygen species
- SD
standard deviation
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SEM
standard error of the mean
- TBAF
tert-butylammonium fluoride
- TC
tetracycline
- TEA
triethylamine
- 3xTG
triple transgenic
- THF
tetrahydrofuran
- TMRM
tetramethylrhodamine, methyl ester
Footnotes
SUPPORTING INFORMATION
Molecular formula strings *CSV) and supplemental Figures 1 and 2.
REFERENCES
- 1.Nicholls DF,S, Bioenergetics. 4th ed.; Elsevier, London, UK, 2013. [Google Scholar]
- 2.Scheffler I Mitochondria. 2d ed.; Wiley, New Jersey, USA, 2007. [Google Scholar]
- 3.Kudryavtseva AV; Krasnov GS; Dmitriev AA; Alekseev BY; Kardymon OL; Sadritdinova AF; Fedorova MS; Pokrovsky AV; Melnikova NV; Kaprin AD; Moskalev AA; Snezhkina AV Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peoples JN; Saraf A; Ghazal N; Pham TT; Kwong JQ Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med 2019, 51, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atamna H; Frey WH, 2nd. Mechanisms of mitochondrial dysfunction and energy deficiency in alzheimer’s disease. Mitochondrion 2007, 7, 297–310. [DOI] [PubMed] [Google Scholar]
- 6.Balaban RS; Nemoto S; Finkel T Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [DOI] [PubMed] [Google Scholar]
- 7.Gadaleta MN; Cormio A; Pesce V; Lezza AM; Cantatore P Aging and mitochondria. Biochimie 1998, 80, 863–870. [DOI] [PubMed] [Google Scholar]
- 8.Hirai K; Aliev G; Nunomura A; Fujioka H; Russell RL; Atwood CS; Johnson AB; Kress Y; Vinters HV; Tabaton M; Shimohama S; Cash AD; Siedlak SL; Harris PL; Jones PK; Petersen RB; Perry G; Smith MA Mitochondrial abnormalities in alzheimer’s disease. J Neurosci 2001, 21, 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenaz G; D’Aurelio M; Merlo Pich M; Genova ML; Ventura B; Bovina C; Formiggini G; Parenti Castelli G Mitochondrial bioenergetics in aging. Biochim Biophys Acta 2000, 1459, 397–404. [DOI] [PubMed] [Google Scholar]
- 10.Su B; Wang X; Bonda D; Perry G; Smith M; Zhu X Abnormal mitochondrial dynamics--a novel therapeutic target for alzheimer’s disease? Mol Neurobiol 2010, 41, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace DC Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science 1992, 256, 628–632. [DOI] [PubMed] [Google Scholar]
- 12.Young-Collier KJ; McArdle M; Bennett JP The dying of the light: Mitochondrial failure in alzheimer’s disease. J Alzheimers Dis 2012, 28, 771–781. [DOI] [PubMed] [Google Scholar]
- 13.Brand MD Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 2016, 100, 14–31. [DOI] [PubMed] [Google Scholar]
- 14.Goncalves RL; Bunik VI; Brand MD Production of superoxide/hydrogen peroxide by the mitochondrial 2-oxoadipate dehydrogenase complex. Free Radic Biol Med 2016, 91, 247–255. [DOI] [PubMed] [Google Scholar]
- 15.Quinlan CL; Perevoschikova IV; Goncalves RL; Hey-Mogensen M; Brand MD The determination and analysis of site-specific rates of mitochondrial reactive oxygen species production. Methods Enzymol 2013, 526, 189–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swerdlow RH Mitochondria and cell bioenergetics: Increasingly recognized components and a possible etiologic cause of alzheimer’s disease. Antioxid Redox Signal 2012, 16, 1434–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swerdlow RH; Burns JM; Khan SM The alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim Biophys Acta 2014, 1842, 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swerdlow RH; Khan SMA “mitochondrial cascade hypothesis” for sporadic alzheimer’s disease. Med Hypotheses 2004, 63, 8–20. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B; Gaiteri C; Bodea L-G; Wang Z; McElwee J; Podtelezhnikov Alexei A.; Zhang C; Xie T; Tran L; Dobrin R; Fluder E; Clurman B; Melquist S; Narayanan M; Suver C; Shah H; Mahajan M; Gillis T; Mysore J; MacDonald Marcy E.; Lamb John R.; Bennett David A.; Molony C; Stone David J.; Gudnason V; Myers Amanda J.; Schadt Eric E.; Neumann H; Zhu J; Emilsson V Integrated systems approach identifies genetic nodes and networks in late-onset alzheimer’s disease. Cell 2013, 153, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown MD; Shoffner JM; Kim YL; Jun AS; Graham BH; Cabell MF; Gurley DS; Wallace DC Mitochondrial DNA sequence analysis of four alzheimer’s and parkinson’s disease patients. Am J Med Genet 1996, 61, 283–289. [DOI] [PubMed] [Google Scholar]
- 21.Ehrenkrantz D; Silverman JM; Smith CJ; Birstein S; Marin D; Mohs RC; Davis KL Genetic epidemiological study of maternal and paternal transmission of alzheimer’s disease. Am J Med Genet 1999, 88, 378–382. [DOI] [PubMed] [Google Scholar]
- 22.Hudson G; Sims R; Harold D; Chapman J; Hollingworth P; Gerrish A; Russo G; Hamshere M; Moskvina V; Jones N; Thomas C; Stretton A; Holmans PA; O’Donovan MC; Owen MJ; Williams J; Chinnery PF; Consortium G No consistent evidence for association between mtdna variants and alzheimer disease. Neurology 2012, 78, 1038–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khalil B; El Fissi N; Aouane A; Cabirol-Pol MJ; Rival T; Lievens JC Pink1-induced mitophagy promotes neuroprotection in huntington’s disease. Cell Death Dis 2015, 6, e1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magrane J; Cortez C; Gan WB; Manfredi G Abnormal mitochondrial transport and morphology are common pathological denominators in sod1 and tdp43 als mouse models. Hum Mol Genet 2014, 23, 1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Payami H; Hoffbuhr K Lack of evidence for maternal effect in familial alzheimer’s disease. Genet Epidemiol 1993, 10, 461–464. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez-Santiago B; Casademont J; Nunes V Is mitochondrial DNA depletion involved in alzheimer’s disease? Eur J Hum Genet 2001, 9, 279–285. [DOI] [PubMed] [Google Scholar]
- 27.Wang W; Li L; Lin WL; Dickson DW; Petrucelli L; Zhang T; Wang X The als disease-associated mutant tdp-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet 2013, 22, 4706–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waterham HR; Koster J; van Roermund CW; Mooyer PA; Wanders RJ; Leonard JV A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 2007, 356, 1736–1741. [DOI] [PubMed] [Google Scholar]
- 29.Zsurka G; Kalman J; Csaszar A; Rasko I; Janka Z; Venetianer P No mitochondrial haplotype was found to increase risk for alzheimer’s disease. Biol Psychiatry 1998, 44, 371–373. [DOI] [PubMed] [Google Scholar]
- 30.Chojnacki JE; Liu K; Yan X; Toldo S; Selden T; Estrada M; Rodriguez-Franco MI; Halquist MS; Ye D; Zhang S Discovery of 5-(4-hydroxyphenyl)-3-oxo-pentanoic acid [2-(5-methoxy-1h-indol-3-yl)-ethyl]-amide as a neuroprotectant for alzheimer’s disease by hybridization of curcumin and melatonin. ACS Chem Neurosci 2014, 5, 690–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerenu G; Liu K; Chojnacki JE; Saathoff JM; Martinez-Martin P; Perry G; Zhu X; Lee HG; Zhang S Curcumin/melatonin hybrid 5-(4-hydroxy-phenyl)-3-oxo-pentanoic acid [2-(5-methoxy-1h-indol-3-yl)-ethyl]-amide ameliorates ad-like pathology in the app/ps1 mouse model. ACS Chem Neurosci 2015, 6, 1393–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson KM; Dahlin JL; Bisson J; Graham J; Pauli GF; Walters MA The essential medicinal chemistry of curcumin. J Med Chem 2017, 60, 1620–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dauer W; Przedborski S Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [DOI] [PubMed] [Google Scholar]
- 34.Sanchez-Caballero L; Guerrero-Castillo S; Nijtmans L Unraveling the complexity of mitochondrial complex i assembly: A dynamic process. Biochim Biophys Acta 2016, 1857, 980–990. [DOI] [PubMed] [Google Scholar]
- 35.Vinothkumar KR; Zhu J; Hirst J Architecture of mammalian respiratory complex i. Nature 2014, 515, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zickermann V; Wirth C; Nasiri H; Siegmund K; Schwalbe H; Hunte C; Brandt U Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex i. Science 2015, 347, 44–49. [DOI] [PubMed] [Google Scholar]
- 37.Friedrich T; Ohnishi T; Forche E; Kunze B; Jansen R; Trowitzsch W; Hofle G; Reichenbach H; Weiss H Two binding sites for naturally occurring inhibitors in mitochondrial and bacterial nadh:Ubiquinone oxidoreductase (complex i). Biochem Soc Trans 1994, 22, 226–230. [DOI] [PubMed] [Google Scholar]
- 38.Baradaran R; Berrisford JM; Minhas GS; Sazanov LA Crystal structure of the entire respiratory complex i. Nature 2013, 494, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parey K; Brandt U; Xie H; Mills DJ; Siegmund K; Vonck J; Kuhlbrandt W; Zickermann V Cryo-em structure of respiratory complex i at work. Elife 2018, 7, e39213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berrisford JM; Sazanov LA Structural basis for the mechanism of respiratory complex i. J Biol Chem 2009, 284, 29773–29783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verdonk ML; Cole JC; Hartshorn MJ; Murray CW; Taylor RD Improved protein-ligand docking using gold. Proteins 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]
- 42.Amadasi A; Surface JA; Spyrakis F; Cozzini P; Mozzarelli A; Kellogg GE Robust classification of “relevant” water molecules in putative protein binding sites. J Med Chem 2008, 51, 1063–1067. [DOI] [PubMed] [Google Scholar]
- 43.Oddo S; Caccamo A; Kitazawa M; Tseng BP; LaFerla FM Amyloid deposition precedes tangle formation in a triple transgenic model of alzheimer’s disease. Neurobiol Aging 2003, 24, 1063–1070. [DOI] [PubMed] [Google Scholar]
- 44.Oddo S; Caccamo A; Shepherd JD; Murphy MP; Golde TE; Kayed R; Metherate R; Mattson MP; Akbari Y; LaFerla FM Triple-transgenic model of alzheimer’s disease with plaques and tangles. Neuron 2003, 39, 409–421. [DOI] [PubMed] [Google Scholar]
- 45.Yao J; Irwin RW; Zhao L; Nilsen J; Hamilton RT; Brinton RD Mitochondrial bioenergetic deficit precedes alzheimer’s pathology in female mouse model of alzheimer’s disease. Proc Natl Acad Sci U S A 2009, 106, 14670–14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brand MD; Goncalves RL; Orr AL; Vargas L; Gerencser AA; Borch Jensen M; Wang YT; Melov S; Turk CN; Matzen JT; Dardov VJ; Petrassi HM; Meeusen SL; Perevoshchikova IV; Jasper H; Brookes PS; Ainscow EK Suppressors of superoxide-h2o2 production at site iq of mitochondrial complex i protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab 2016, 24, 582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Q; Vazquez EJ; Moghaddas S; Hoppel CL; Lesnefsky EJ Production of reactive oxygen species by mitochondria: Central role of complex iii. J Biol Chem 2003, 278, 36027–36031. [DOI] [PubMed] [Google Scholar]
- 48.Zorov DB; Juhaszova M; Sollott SJ Mitochondrial reactive oxygen species (ros) and ros-induced ros release. Physiol Rev 2014, 94, 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy MP How mitochondria produce reactive oxygen species. Biochem J 2009, 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zielonka J; Joseph J; Sikora A; Hardy M; Ouari O; Vasquez-Vivar J; Cheng G; Lopez M; Kalyanaraman B Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev 2017, 117, 10043–10120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teixeira J; Deus CM; Borges F; Oliveira PJ Mitochondria: Targeting mitochondrial reactive oxygen species with mitochondriotropic polyphenolic-based antioxidants. Int J Biochem Cell Biol 2018, 97, 98–103. [DOI] [PubMed] [Google Scholar]
- 52.Watson MA; Wong HS; Brand MD Use of s1qels and s3qels to link mitochondrial sites of superoxide and hydrogen peroxide generation to physiological and pathological outcomes. Biochem Soc Trans 2019, 47, 1461–1469. [DOI] [PubMed] [Google Scholar]
- 53.Orr AL; Vargas L; Turk CN; Baaten JE; Matzen JT; Dardov VJ; Attle SJ; Li J; Quackenbush DC; Goncalves RL; Perevoshchikova IV; Petrassi HM; Meeusen SL; Ainscow EK; Brand MD Suppressors of superoxide production from mitochondrial complex iii. Nat Chem Biol 2015, 11, 834–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hong HS; Maezawa I; Yao N; Xu B; Diaz-Avalos R; Rana S; Hua DH; Cheng RH; Lam KS; Jin LW Combining the rapid mtt formazan exocytosis assay and the mc65 protection assay led to the discovery of carbazole analogs as small molecule inhibitors of abeta oligomer-induced cytotoxicity. Brain Res 2007, 1130, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sopher BL; Fukuchi K; Kavanagh TJ; Furlong CE; Martin GM Neurodegenerative mechanisms in alzheimer disease. A role for oxidative damage in amyloid beta protein precursor-mediated cell death. Mol Chem Neuropathol 1996, 29, 153–168. [DOI] [PubMed] [Google Scholar]
- 56.Szczepanek K; Chen Q; Larner AC; Lesnefsky EJ Cytoprotection by the modulation of mitochondrial electron transport chain: The emerging role of mitochondrial stat3. Mitochondrion 2012, 12, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szczepanek K; Chen Q; Derecka M; Salloum FN; Zhang Q; Szelag M; Cichy J; Kukreja RC; Dulak J; Lesnefsky EJ; Larner AC Mitochondrial-targeted signal transducer and activator of transcription 3 (stat3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem 2011, 286, 29610–29620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z; Hao P; Li L; Tan CY; Cheng X; Chen GY; Sze SK; Shen HM; Yao SQ Design and synthesis of minimalist terminal alkyne-containing diazirine photo-crosslinkers and their incorporation into kinase inhibitors for cell- and tissue-based proteome profiling. Angew Chem Int Ed Engl 2013, 52, 8551–8556. [DOI] [PubMed] [Google Scholar]
- 59.He L; Jiang Y; Liu K; Gomez-Murcia V; Ma X; Torrecillas A; Chen Q; Zhu X; Lesnefsky E; Gomez-Fernandez JC; Xu B; Zhang S Insights into the impact of a membrane-anchoring moiety on the biological activities of bivalent compounds as potential neuroprotectants for alzheimer’s disease. J Med Chem 2018, 61, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.