

Abstract
Circadian disruption has been identified as a risk factor for health disorders such as obesity, cardiovascular disease, and cancer. Although epidemiological studies suggest an increased risk of various cancers associated with circadian misalignment due to night shift work, the underlying mechanisms have yet to be elucidated. We sought to investigate the potential mechanistic role that circadian disruption of cancer hallmark pathway genes may play in the increased cancer risk in shift workers. In a controlled laboratory study, we investigated the circadian transcriptome of cancer hallmark pathway genes and associated biological pathways in circulating leukocytes obtained from healthy young adults during a 24-hour constant routine protocol following three days of simulated day shift or night shift. The simulated night shift schedule significantly altered the normal circadian rhythmicity of genes involved in cancer hallmark pathways. A DNA repair pathway showed significant enrichment of rhythmic genes following the simulated day shift schedule, but not following the simulated night shift schedule. In functional assessments, we demonstrated that there was an increased sensitivity to both endogenous and exogenous sources of DNA damage after exposure to simulated night shift. Our results suggest that circadian dysregulation of DNA repair may increase DNA damage and potentiate elevated cancer risk in night shift workers.
Keywords: Circadian misalignment, genomic stability, hallmarks of cancer, shift work
1. INTRODUCTION
Circadian disruption induced by chronic misalignment between behavioral rhythms and external light/dark cycles is common in today’s 24/7 society. Misaligned circadian rhythms are particularly prevalent in night shift workers1,2 and are associated with elevated risk for a wide range of chronic health problems3, including various types of cancer4. Significant disturbances in the circadian rhythms of biomolecular pathways have been reported in humans during and after exposure to a night shift schedule5–8. Nonetheless, while night shift work is increasingly recognized as carcinogenic9, the molecular mechanisms linking night shift work to cancer in humans have yet to be elucidated.
The circadian clock exerts a strong influence on cellular and biochemical processes central to the initiation, promotion, and progression of cancer10–14. Targeted studies with genetic mouse models have provided inconsistent results regarding the role of canonical clock genes in cancer prognosis12,15–20. Even so, there is mounting evidence that the circadian clock affects multiple pathways that regulate cancer outcomes, including cell cycle checkpoints, apoptosis, cell proliferation, DNA repair, and inflammation21–29. Such wide-ranging impact suggests that a systems approach should be considered to understand the role of the circadian clock in carcinogenesis.
Cancers are generally associated with mutations caused by replication errors or DNA damage induced either endogenously or exogenously by sources such as ultraviolet or ionizing radiation (IR)30–33. As there is a close link between the circadian pacemaker and DNA damage-related protective processes such as DNA repair18,34–36, it is plausible that biomolecular disruption caused by night shift work can increase cancer risk. Rodent studies corroborate this notion, showing that altered circadian rhythmicity disrupts the cell cycle, alters metabolism promotes tumorigenesis, and induces tumor cell growth and tumor-associated immune cell remodeling. under simulated shift work and chronic jetlag conditions20,37–39.
We hypothesized that circadian misalignment caused by a night shift schedule alters the circadian expression of canonical cancer hallmark pathway genes known to influence the risk of cancer development in humans. In a controlled in-laboratory study (Figure 1), we assessed circadian rhythmicity in the transcriptome of leukocytes collected at 3-hour intervals during a 24-hour constant routine protocol immediately after exposure to three days of simulated day shift work (normal schedule; n=7) or night shift work (circadian misalignment; n=7). Our study design allowed us to characterize circadian disturbances in the gene expression profiles of cancer hallmark pathways and canonical clock genes that can be linked directly to being on a night shift schedule, in the absence of environmental and behavioral confounders from light exposure, sleep, food intake, and physical activity7. We observed widespread disturbances in the circadian rhythmicity of gene expression in cancer hallmark pathways. The panel-defined DNA repair pathway exhibited prominent rhythmicity following the simulated day shift schedule, but not following the simulated night shift schedule.
FIGURE 1.
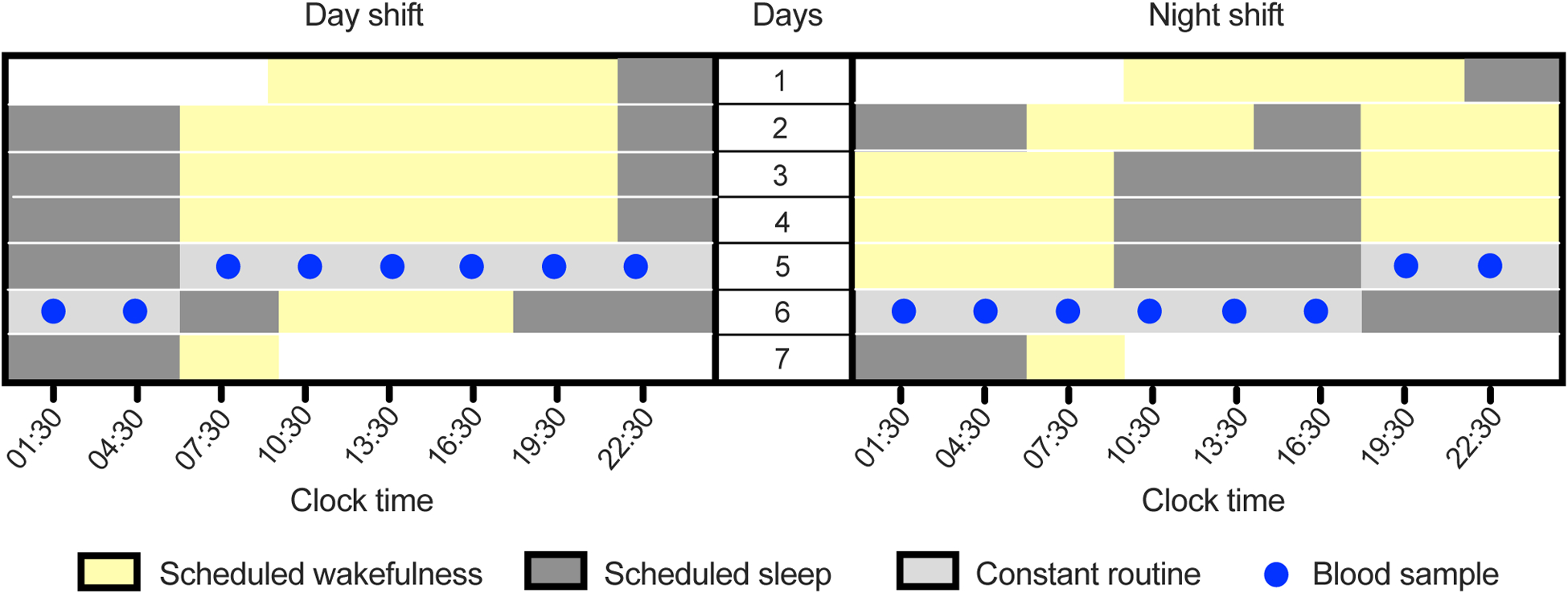
Design of the in-laboratory, simulated shift work study in humans. Left and right panels represent the simulated day and night shift conditions, respectively. After three days on the simulated day or night shift schedule, subjects underwent a 24-hour constant routine protocol during which blood samples were collected at 3-hour intervals.
We then hypothesized that loss of circadian rhythmicity in the expression of DNA repair genes could lead to defects in DNA damage repair following the simulated night shift schedule. We employed an alkaline comet assay to measure endogenous DNA damage in leukocytes and used immunofluorescence microscopy to monitor common biomarkers of the DNA damage response. Additionally, we measured sensitivity to exogenous DNA damage by repeating the assessments of the DNA damage response after exposure of the leukocytes to ionizing radiation. The results of these experiments suggest that circadian misalignment interferes with properly timed DNA damage repair in humans.
2. MATERIALS AND METHODS
2.1. Sample collection and processing
Fourteen healthy human adults (aged 22–34 years) with normal nighttime habitual sleep schedules participated in an in-laboratory study in the Sleep and Performance Research Center at Washington State University Health Sciences Spokane. Seven subjects were assigned to a simulated day shift schedule involving three days of daytime wakefulness (06:00–22:00); the other seven subjects were assigned to a simulated night shift schedule involving three days of nighttime wakefulness (18:00–10:00). Following the three days on the simulated shift work schedule, subjects began a 24-hour constant routine protocol during which they were kept awake under constant laboratory conditions, dim light (<50 lux), fixed semi-recumbent posture, and evenly distributed food intake (hourly isocaloric snacks). During the constant routine, blood samples were collected through intravenous catheter at 3-hour intervals (Figure 1). These samples were used for leukocyte transcriptome analysis and DNA damage assessment. Further details of the study participants, laboratory study, and sample collection protocol were reported previously7.
2.2. Gene expression analysis and normalization
After red blood cells (RBCs) were lysed from the blood samples using RBC lysis buffer (Zymo Research), leukocytes were collected by centrifugation at 12,000 g for 60 seconds at 4 °C followed by washing with 1x phosphate buffered saline (PBS). Cells were resuspended with TRI reagent (Zymo Research), snap frozen, and stored at −80 °C until RNA extraction (Whole-Blood RNA MiniPrep, Zymo Research). Purity and yield of RNA was verified by measuring the ratio of light absorbance at 260 and 280 nm (NanoDrop 2000 Spectrophotometer). Total leukocyte RNA (100 ng per sample) from each blood sample was used for measuring gene expression40. We selected 770 mRNA targets, including 40 internal controls, of the NanoString nCounter PanCancer Pathways panel (NanoString Technologies), and manually assigned 10 additional genes associated with cancer hallmark pathways to the panel (EGR1, KI67, MITF, P73, PARP1, RPA1, SIRT1, TCTP, TREM, and XPC) for a total of 780 genes. In addition, 17 canonical clock genes (Data file S1) were used as positive controls for circadian analysis. Transcript counts of each gene were normalized using nSolver Analysis Software 4.0 (NanoString Technologies).
Background from raw counts was subtracted using the geometric mean of 8 NanoString negative control probes followed by analysis of overall assay efficiency by geometric mean of 6 NanoString positive control probes. Data were further normalized with the geometric mean of ACTB (actin) and 17 other internal control genes (ACAD9, C10orf76, DHX16, EIF2B4, FTSJ2, GPATCH3, MRPS5, PIK3R4, PRPF38A, SLC4A1AP, TTC31, USP39, VPS33B, ZKSCAN5, ZNF143, ZNF346, ZNF384) selected from 40 internal controls of the NanoString nCounter PanCancer Pathways panel, based on evidence of arrhythmic gene expression in rodent tissues26,28,41–47 as documented in the Circadian Expression Profiles Data Base (CircaDB)*. Normalization factors for positive and internal controls were used with default nSolver 4.0 settings. For 14 of the 780 cancer hallmark genes considered (COL4A5, FGF6, FN1, FOSL1, GDF6, GHR, ID1, IL24, LAMB3, LAMC2, LIFR, PTPRR, RPS6KA6, SPRY1), the grand mean was not significantly different from the detection floor (p>0.01). These could not be analyzed reliably for rhythmicity and were therefore omitted. After removal of the 40 internal controls, 726 genes were left in the data set (Data file S2). Data from time points 01:30 and 13:30 for two subjects in the simulated night shift condition were discarded due to abnormal signal intensity. Data analyses were not predicated on having complete data sets.
2.3. Circadian rhythm analysis
Circadian rhythms were analyzed for the expression profiles of each of the selected genes during the 24-hour constant routine protocol that followed the 3-day simulated day or night shift schedule. Condition-specific estimates of circadian rhythm amplitude, acrophase (peak timing), and mesor were assessed by means of cosinor analysis using mixed-effects regression7,48. The analysis was repeated with a first-order autoregressive covariance matrix to account for any serial correlation across time points, and with a heteroskedasticity-consistent covariance matrix to account for any non-homogeneity of variances across time points. There were no material differences in results; therefore, the results of the more parsimonious original cosinor analysis were retained. The presence of circadian rhythmicity was evaluated by t-test (p<0.05) of the circadian amplitude49. Here we were most concerned with type II error (i.e., falsely claiming the absence of circadian rhythmicity) and therefore did not correct the type I error threshold for multiple comparisons50. Differences in circadian parameters between the simulated day and night shift conditions were evaluated using planned contrasts (t-tests, p<0.05). Heat maps were generated by plotting condition-specific z-transformed expression values, using ggplot2 version 3.4.451. Genes were rank-ordered by acrophase in the day shift condition – or the night shift condition for the subset of genes without significant circadian rhythmicity in the day shift condition.
2.4. Functional enrichment analysis of rhythmic genes
The genes in the NanoString nCounter PanCancer Pathways panel considered here were subdivided into four sets based on the significance of circadian rhythmicity in their transcripts: 1) rhythmic after simulated day shift only; 2) rhythmic after simulated night shift only; 3) rhythmic in both shift conditions; and 4) rhythmic in both conditions, with a significant difference in acrophase between the simulated day and night shift conditions. The number of genes meeting the criteria of each set was assessed for each of the 13 cancer-related pathways defined in the NanoString Pancancer Pathway panel. Enrichment in terms of gene counts in each of the four sets, relative to a priori expectation based on the observed frequency of rhythmicity among all of the genes measured, was computed using Fisher’s exact test (p<0.05). Additionally, the degree of enrichment in each of the 13 cancer-related pathways following simulated night shift was compared to that following simulated day shift, using counts of rhythmic genes expressed relative to expectation based on all genes as observed following simulated day shift, as quantified by means of log likelihood ratio. It should be noted that while these assessments characterize the prevalence of rhythmicity among genes linked with specific pathways, they do not indicate to what extent the observed rhythms may or may not be phase locked.
2.5. Alkaline comet assay
An alkaline comet assay was performed to measure genomic DNA damage52 in leukocytes, using a high-throughput comet electrophoresis system (COMPAC-50, Cleaver Scientific). Whole blood samples used for the comet assay were preserved at −80 °C after adding an equal volume of freezing mix (80% RPMI, 20% DMSO). Leukocytes were extracted by lysing RBCs using ice-cold ammonium chloride lysis buffer (0.14 M ammonium chloride, 0.01 M sodium bicarbonate, 0.001 M EDTA) for 5 minutes. After lysis, leukocytes were centrifuged at 1,500 g for 3 minutes at 4 °C, then washed two times using 1x PBS. Leukocytes were resuspended in 0.6% low-melt agarose below 40 °C and pipetted onto 1% agarose precoated slides. After placing a coverslip on top of the cells, slides were incubated at 4 °C for 30 minutes.
Leukocytes were further lysed in alkaline lysis buffer (0.1 M EDTA, 2.5 M NaCl, 0.01 M Tris, 1% Triton X-100) with a pH of 10 by incubation at 4 °C for 18 hours. The slides were washed with ddH2O and transferred into alkaline electrophoresis buffer (0.3 M NaOH, 0.001 M EDTA) with a pH of 13 to unwind the DNA for comet electrophoresis. Electrophoresis was performed at 21 V for 50 minutes. Slides were transferred to neutralization buffer (0.4 M Tris, pH of 7.5) for 20 minutes followed by a wash in ice cold ddH2O for 20 minutes, then dried at 47 °C overnight. For imaging, slides were rehydrated in ice cold ddH2O for 30 minutes and stained with propidium iodide for 20 minutes, then imaged with red filter (excitation and emission of 587 and 612 nm, respectively) at 20x magnification using a Leica DMi8 microscope. Images were analyzed using CometScore software (TriTek) and the average percent tail DNA was calculated for a minimum of 49 cells per sample.
2.6. Immunofluorescence
Immunofluorescence methods were used to measure DNA damage response with antibodies of γH2AX and BRCA1. Leukocytes were isolated from whole blood by lysing RBCs using RBC lysis buffer (Zymo Research). After adding lysis buffer, cells were kept at room temperature for 5 minutes followed by centrifugation at 12,000 g for 1 minute. Lysis was repeated and the resulting leukocyte pellet was washed with 1 ml of 1x PBS. Subsequently, cells were fixed and permeabilized with 3.7% paraformaldehyde and 0.1% Tween-20 in 1x PBS at room temperature for 10 minutes. Cells were washed with 1x PBS and stored in 0.02% NaCN in 1x PBS at 4 °C until staining.
On the day of staining, cells were centrifuged at 3,000 g for 5 minutes at room temperature and the pellet was washed with 1x PBS and blocked with 3% BSA and 2% FBS in 1x PBS for 30 minutes. After blocking, cells were probed with 1:100 dilution of BRCA1 antibody (Cell Signaling, catalog number 9010) or 1:100 dilution of γH2AX antibody (Millipore, catalog number 05–636) for 2 hours followed by incubation with 1:100 dilution of Alexa Fluor 488 (Invitrogen, catalog numbers A-21202 for anti-mouse and A-21206 for anti-rabbit) for 1 hour. Nuclei in leukocytes were counter-stained with 1:10,000 dilution of 10 μg/ml DAPI stock solution (Sigma, catalog number D9542) for 5 minutes at room temperature. Leukocytes were washed twice with 1x PBS. Cells were then mounted onto slides and sealed with coverslips using transparent nail polish. Images were taken using a fluorescence microscope (Leica) at 100x magnification. Analysis was done by calculating number of cells with and without foci.
2.7. Irradiation of blood samples
Blood samples collected from human subjects at two different times of the day (07:30 and 19:30) were used for examination of DNA damage upon ionizing radiation (IR) exposure. Samples were treated with 2.5 Gy of IR at a rate of 0.5 Gy per minute using an X-Rad 320 (Precision X-Ray). Following the IR exposure, samples were mixed with an equal volume of RPMI 1640 media supplemented with 10% FBS and incubated in a mammalian cell culture incubator at 37 °C until leukocyte collection. The leukocytes were then isolated (as described above) and measured for DNA damage response using immunoblotting at time points 1, 4, and 24 h post IR.
2.8. Immunoblotting
Immunoblotting was performed to measure the DNA damage response of double-strand breaks after exposure to IR. Leukocytes were extracted from whole blood as described above for immunofluorescence. After isolation of leukocytes, proteins were extracted by adding 200 μl of 1x RIPA lysis buffer (0.02 M Tris-HCl pH 7.5, 0.15 M NaCl, 0.001 M EDTA, 0.001 M EGTA, 1% Nonidet P-40, and 1% sodium deoxycholate with 1x Protease inhibitors and Phosphatase inhibitors). Protein estimation was performed using Bradford reagent (Bio-Rad). Equal amount of total protein was resolved by SDS-PAGE and then transferred to nitrocellulose membrane at 20 V for 30 minutes (Bio-Rad).
After transfer, membrane was washed with ddH2O and stained with 0.2% Ponceau S for 1 minute to confirm protein transfer. Membrane was further washed with Tris Buffered Saline containing 0.05% Tween 20 (TBST) for 5 minutes, then blocked with blocking solution (5% non-fat dried milk powder in TBST), followed by three washes with TBST for 5 minutes. Membrane was subsequently probed using pATM, pCHK2, C-CASPASE 3, PCNA (Cell Signaling, catalog numbers 4526, 2661, 9664, and 2586) and p53 (Santa Cruz Biotechnology, catalog number sc-126) primary antibodies (1:1000) at 4 °C overnight and appropriate secondaries (1:5000) were added and incubated at room temperature for 60 minutes. In between probing with primary and secondary antibodies, membrane was washed three times with TBST for 5 minutes. After probing, protein bands were detected with an enhanced chemiluminescence detection kit (GE Healthcare). Actin (Sigma Aldrich) was used as a loading control.
3. RESULTS
3.1. Simulated night shift alters the rhythmicity of cancer hallmark genes and pathways
To evaluate the level of circadian dysregulation induced by the simulated shift work protocol in the laboratory, we first analyzed the circadian rhythmicity of the expression of 17 canonical clock genes (Data file S1) in leukocytes from blood samples collected during a 24-hour constant routine protocol immediately after exposure to three days of either a day shift or a night shift schedule (Figure 1). The circadian expression of canonical clock genes was substantially altered by the simulated night shift schedule as compared to the simulated day shift schedule. Four genes, CRY1, CRY2, PER2 and NR1D2, lost their normal day shift rhythmicity after the night shift schedule. NPAS2 expression was not rhythmic in the simulated day shift condition but exhibited circadian rhythmicity in the simulated night shift condition. NR1D1, PER3, and DBP were significantly rhythmic in both shift conditions (Figure S1).
To investigate the effects of a simulated night shift schedule on circadian rhythmicity in cancer hallmark genes, we analyzed the transcriptome of 726 genes using the NanoString nCounter PanCancer Pathways panel. The panel consists of a preselected set of genes representing 13 canonical cancer-related pathways, to which we added 10 additional cancer hallmark genes for this study (Data file S2). Cosinor analysis7,48 showed that 257 (35.4%) of the transcripts were rhythmic after at least one of the two simulated shift work conditions (Data file S2). Among them, 113 (15.6%) were rhythmic in the day shift condition only (Figure 2A, top panel); 96 (13.2%) were rhythmic in the night shift condition only (Figure 2A, top middle panel); and 48 (6.6%) were rhythmic in both the day and night shift conditions (Figure 2A, bottom two panels). Among genes in this last category, a subset of 10 (1.4%) exhibited a significant phase advance (3.7 to 8.3 hours) or phase delay (2.8 to 7.0 hours) in the night shift condition relative to the day shift condition (Figure 2A, bottom panel). Thus, simulated night shift work caused significant disturbances in the rhythmicity of gene expression in cancer hallmark pathways.
FIGURE 2.
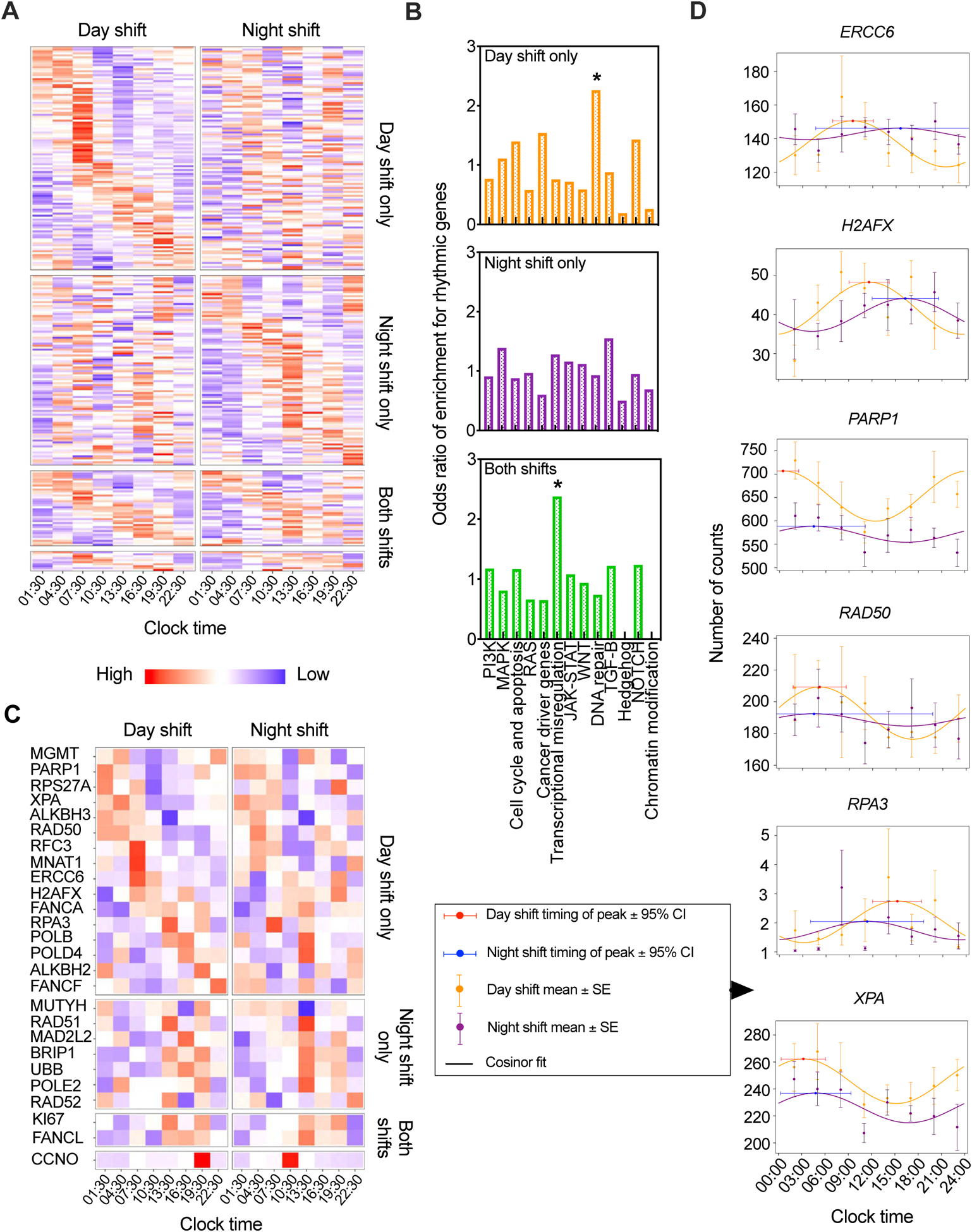
Circadian analysis of cancer hallmark gene expression as observed under 24-hour constant routine after exposure to simulated day or night shift schedule. (A) Heatmap of significantly (p<0.05, cosinor analysis t-test) rhythmic expression profiles of cancer hallmark genes during 24-hour constant routine following the three-day simulated day shift (left) and night shift (right) conditions. Genes are ordered in the heatmap by time of peak expression (acrophase) for genes significantly rhythmic after day shift only, after night shift only, and after both – with genes displaying a significant timing difference between conditions set apart in the bottom rows. (B) Enrichment for rhythmic genes in cancer hallmark pathways after day shift only, after night shift only, or after both (*p<0.05, Fisher’s exact test). (C) Heatmap of significantly rhythmic (p<0.05, cosinor analysis t-test) transcripts in the DNA repair pathway during 24-hour constant routine following the simulated day shift (left) and night shift (right) conditions. Genes are ordered in the heatmap as in panel (A). (D) Circadian rhythms in the expression of selected genetic markers of DNA repair during 24-hour constant routine after simulated day versus night shift.
Comparing enrichment of rhythmic gene transcripts in the 13 cancer-related pathways between the day and night shift conditions revealed enrichment in the transcriptional misregulation and DNA repair pathways. The transcriptional misregulation pathway, which consists of a group of genes where errors in their transcription such as over- and under-expression are known to be linked to tumorigenesis, showed significant enrichment of rhythmic genes in both the simulated day and night shift conditions. This enrichment of rhythmic genes, while not disrupted in the simulated night shift condition, may represent an area of vulnerability to tumorigenesis that may be targeted under other circumstances. However, the DNA repair pathway showed significant enrichment of rhythmic genes after the simulated day shift schedule, but corresponding enrichment was not observed after the simulated night shift schedule (Figure 2B, Table 1). Out of 57 DNA repair pathway genes, 26 were rhythmic in at least one of the two conditions (Data file S2). For more than 60% of the DNA repair pathway genes that were rhythmic in the day shift condition, the rhythm was statistically significantly different in the night shift condition (Figure 2C). This included the key DNA repair genes, ERCC6, H2AFX, PARP1, RAD50, RPA3 and XPA35,53 (Figure 2D). Though we found that these genes did not exhibit the same acrophase, the difference between their rhythmicity in the simulated day shift condition relative to the simulated night shift condition underscores the impact of circadian misalignment on this important process.
TABLE 1.
Enrichment of pathways in the NanoString nCounter PanCancer Pathways panel for genes exhibiting significant circadian rhythmicity in their expression during the 24-hour constant routine protocol after three days on the day shift or night shift schedule. The table shows likelihood ratios of genes found to be rhythmic after day shift only, after night shift only, or after both day and night shift without or with significant phase difference. The last column shows an odds ratio comparison between the night and day shift conditions.
| Pathway | Rhythmic after day shift only | Rhythmic after night shift only | Rhythmic after both day and night shift | Rhythmic after both day and night shift with phase difference | Odds ratio between night and day shifts |
|---|---|---|---|---|---|
| PI3K | 0.78 | 0.88 | 1.24 | 0.67 | 0.95 |
| MAPK | 1.13 | 1.36 | 0.85 | 1.60 | 0.97 |
| Cell cycle and apoptosis | 1.42 | 0.86 | 1.23 | 1.02 | 0.72 |
| RAS | 0.59 | 0.95 | 0.69 | 0.46 | 1.14 |
| Cancer driver genes | 1.56 | 0.59 | 0.68 | 0.53 | 0.53 |
| Transcriptional misregulation | 0.67 | 1.23 | *2.69 | 2.61 | 1.15 |
| JAK-STAT | 0.73 | 1.14 | 1.13 | 1.98 | 1.13 |
| WNT | 0.60 | 1.10 | 0.98 | 0.93 | 1.23 |
| DNA repair | *2.30 | 0.91 | 0.77 | 1.31 | 0.53 |
| TGF-β | 0.89 | 1.52 | 1.27 | 3.55 | 1.18 |
| Hedgehog | 0.19 | 0.49 | 0.00 | 0.00 | 2.00 |
| NOTCH | 1.44 | 0.93 | 1.29 | 0.00 | 0.71 |
| Chromatin modification | 0.53 | 0.65 | 0.00 | 0.00 | 1.00 |
p<0.05, Fisher’s exact test
3.2. Simulated night shift increases endogenous and exogenous DNA damage
Effective DNA damage repair protects the genome from the accumulation of DNA damage and prevents mutational events that drive carcinogenesis. Based on the loss of significant enrichment for rhythmic genes in the DNA repair pathway (Figure 2B) and the disruption of circadian rhythmicity in key DNA repair genes (Figure 2D) after simulated night shift, we hypothesized that the DNA repair machinery may be acting in a reduced capacity, leading to an accumulation of unrepaired DNA damage following a night shift schedule.
We measured endogenous DNA damage by means of the alkaline “comet” assay52 (Figure 3A). Across the 24 hours of the constant routine protocol (Figure 1), endogenous DNA damage was generally higher after the night shift schedule compared to the day shift schedule, with statistically significant increases (asterisks) in damage detected at 01:30, 07:30, 16:30, and 19:30 (Figure 3B). Further, we investigated DNA damage biomarkers BRCA1 and γH2AX by immunofluorescence microscopy in order to assess evidence of damage response to double-strand DNA breaks. Across the 24-hour constant routine, the percentage of cells with BRCA1 and γH2AX foci was significantly higher in the night shift condition compared to the day shift condition (Figure 3C and Figure S2A). There was no significant difference between the two conditions in the circadian rhythm of BRCA1 gene expression (Figure S2B), indicating that the increased formation of BRCA1 foci was due to DNA damage signaling and not due to shift condition-dependent changes in mRNA. Collectively, these findings support our hypothesis that compromised DNA repair due to circadian misalignment in the night shift condition leads to greater unrepaired endogenous DNA damage.
FIGURE 3.
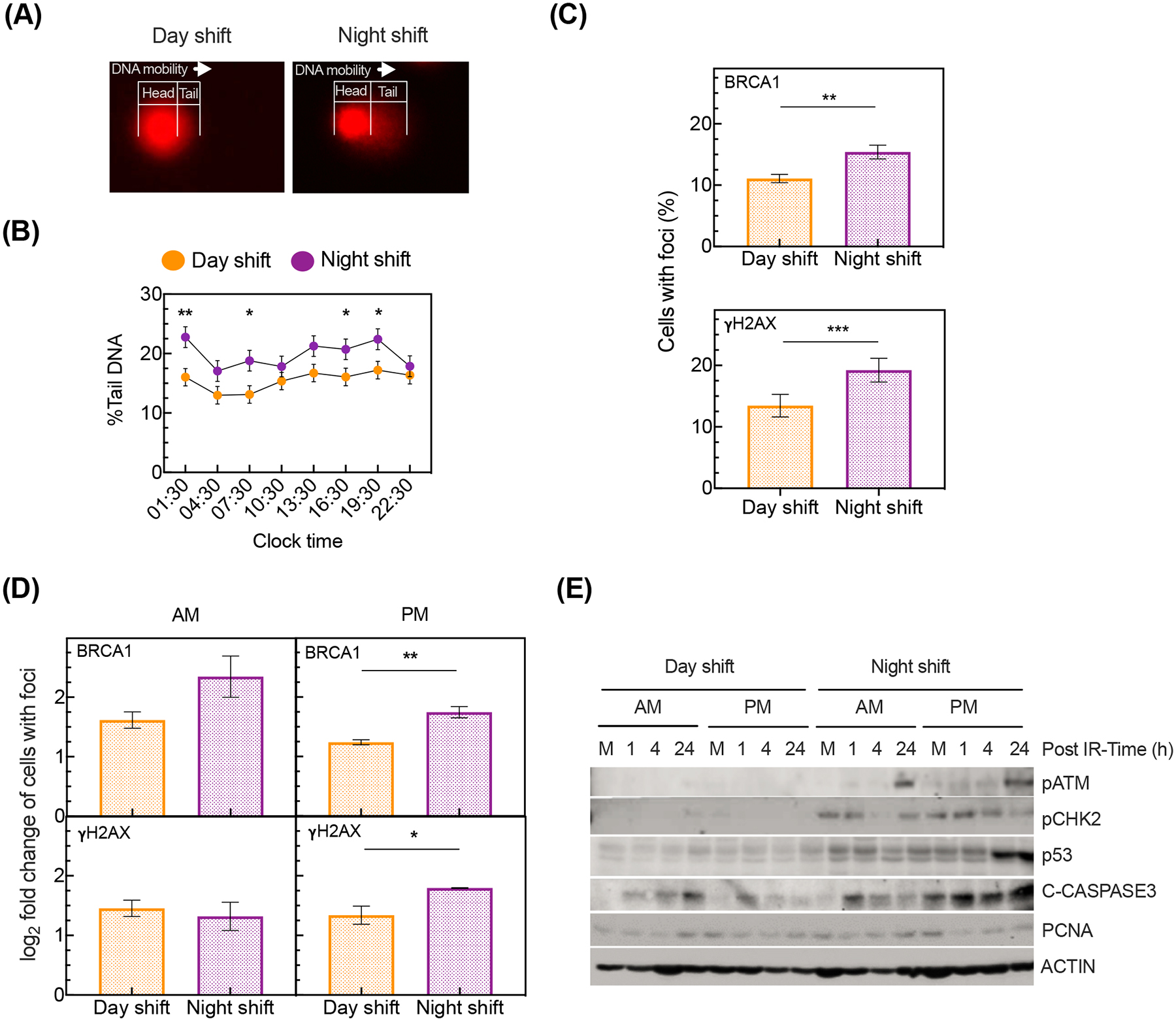
DNA damage under 24-hour constant routine after exposure to simulated day or night shift schedule. (A) Representative DNA migration (head to tail) in leukocytes after simulated day shift (left) versus night shift (right) as assessed with alkaline comet assay. (B) Percentage (mean ± SE) of tail genomic DNA across the 24-hour constant routine, after simulated day shift or night shift (F7,70=4.55, p<0.001, mixed-effects ANOVA condition by time interaction; *p<0.05, **p<0.01, pairwise contrasts). (C) Immunofluorescence quantification of leukocytes with BRCA1 (top) and γH2AX (bottom) foci averaged over the 24-hour constant routine after simulated day or night shift (**p<0.01, ***p<0.001, two-sample t-test). The y-axis indicates the average percentage ± SE. (D) Immunofluorescence quantification of BRCA1 (top) and γH2AX (bottom) foci in leukocytes upon IR treatment of samples collected at 07:30 (AM) or 19:30 (PM) during the 24-hour constant routine after simulated day or night shift (*p<0.05, **p<0.01, two-sample t-test). The y-axis indicates log2 fold change ± SE of the number of foci in IR treated cells versus untreated controls. (E) Representative immunoblot of the DNA damage response upon IR exposure of samples collected at 07:30 (AM) or 19:30 (PM) during the 24-hour constant routine after a simulated day or night shift. M denotes “mock” (i.e., sample not exposed to IR).
Shift workers in a variety of around-the-clock operations, such as health care, aviation, security, and the military, additionally face increased probability of exposure to IR, a known risk factor for DNA damage54 and carcinogenesis55,56. Therefore, we also investigated exogenously induced DNA damage from ionizing radiation (IR). Based on evidence that the accumulation of DNA damage from IR exposure varies by time of day57,58, we used the blood samples collected at both 07:30 and 19:30 for this experiment. Leukocytes harvested from the blood samples were immediately irradiated with a radiation dose of 2.5 Gy at a rate of 0.5 Gy per minute, which is the average IR exposure associated with various occupational accidents documented for night shift workers in radiation exposure environments59. Biomarkers of DNA double-strand breaks were measured over the 24-h period immediately after IR treatment. Although there was no difference in the percentage of cells with foci between the two shift conditions for the 07:30 sample, the percentage of cells with BRCA1 and γH2AX foci following IR exposure was significantly higher for the night shift condition in the 19:30 sample (Figure 3D). Immunoblotting revealed changes in biomarkers of DNA damage response signaling, including DNA damage sensors (pATM, pCHK2, p53), apoptosis (cleaved C-CASPASE3), and proliferation (PCNA), where the DNA damage response was high after simulated night shift compared to simulated day shift, especially in the 19:30 sample (Figure 3E). These results indicate that simulated night shift potentiated leukocyte sensitivity to IR-mediated DNA damage, consistent with our hypothesis of a reduced DNA repair following a night shift schedule.
4. DISCUSSION
Circadian disruption has been robustly documented to increase cancer risk and promote tumorigenesis in rodents12,20,38,60. This correlation translates to humans, where circadian misalignment in humans induced by night shift work is associated with the increased risk of cancers of the breast, prostate, colon, rectum, and blood61–63. We studied underlying mechanisms in a strictly controlled laboratory study, in which human volunteers were assigned to three days of either a simulated day shift schedule or a simulated night shift schedule that induces circadian misalignment. This was followed by a 24-hour constant routine protocol – identical for both groups – during which blood was collected at 3-hour intervals. The constant routine protocol allowed for the investigation of biological rhythms free of exogenous influences64,65, and the juxtaposition of constant routine measurements following the day shift schedule versus the night shift schedule thus revealed the impact of the prior night shift schedule on endogenous biological processes7.
Our analysis of circadian rhythmicity in the human leukocyte transcriptome revealed circadian dysregulation of canonical clock genes, confirming circadian misalignment in our protocol, as has been observed previously6. Circadian misalignment induced by the night shift schedule also caused circadian dysregulation of genes involved in key DNA repair pathways. Moreover, the effectiveness of the processes to repair DNA damage arising from endogenous sources as well as DNA damage induced by exogenous IR were compromised in leukocytes obtained in the night shift condition.
These findings align with results of studies in rodent models showing that the circadian clock regulates DNA repair, cell cycle, and apoptotic processes, which mediate DNA damage outcomes23. In particular, a well-studied circadian-regulated gene involved in nucleotide excision repair, Xpa35, as well as genes related to the cell cycle and apoptosis including Wee1, Tp53, Myc, Atr, Cdk4 and Cdkn1c, have been found to be rhythmic under conditions equivalent to day shift25. Disruption of circadian regulation of these genes may lead to excess DNA damage from single- and double-strand breaks – base lesions that may cause mutagenesis and genomic instability, which is a primary hallmark of cancer34,66.
In this study, we extended this line of research to humans, reporting for the first time that circadian expression of regulators of cell cycle and DNA repair were dysregulated in human leukocytes following a night shift schedule (Figure 2D and Figure S3). These genes are critical for recognizing DNA damage through cell cycle checkpoint activation (ATR, CDK4, CDKN1C, TP53, WEE1). They also play important roles in fixing DNA damage resulting from bulky DNA lesions and single- and double-strand breaks through nucleotide excision repair (ERCC6, TP53, RPA3, XPA) and double-strand break repair (ERCC6, H2AFX, PARP1, RAD50). In light of our observations of increased DNA damage from endogenous and exogenous sources in leukocytes from subjects studied under simulated night shift conditions, the circadian disruption of the expression of these critical genes points to a direct connection between cellular circadian clock mechanisms and the elevated risk of carcinogenesis in humans working night shifts.
Temporal coordination between DNA replication, DNA repair, and cell cycle checkpoints is essential for maintaining genomic integrity12,23,35,37,67. Cancer primarily results from mutagenesis through genomic instability as a result of DNA damage caused by endogenous agents such as free radicals generated as byproducts of normal metabolism or replicational errors arising during DNA replication, and exogenous agents such as ultraviolet radiation and IR from the environment30–33. Rodent models and in vitro experiments have shown that DNA damage varies with the time of day of IR exposure57,58, but there are no studies in humans associated with the conditions of the modern working world. As expected68, IR exposure increased DNA damage induction and/or dysregulated double-strand break repair (represented by γH2AX and BRCA1 foci) in human leukocytes following both simulated shift conditions. However, evening IR exposure resulted in more DNA damage in leukocytes from the simulated night shift group than from the simulated day shift group.
The composition of leukocytes – a heterogeneous collection of cell types including lymphocytes, granulocytes, and monocytes – may vary over the circadian cycle,69 and any such circadian variation may have been differently affected by the simulated night shift condition as compared to the simulated day shift condition. Indeed, this might be a source of the differential effect we observed for endogenous DNA damage in the night shift condition as shown in Figs. 3A–C (although this would not be a likely explanation for the exogenous DNA damage results shown in Figs. 3D–E). Further, any condition-based differences in circadian variation of the composition of leukocytes may have impacted our gene expression data. However, whether such an effect could have pathway-specific impacts is unclear, and the observed timing mismatch between DNA damage and DNA repair mechanisms in the night shift condition remains important nonetheless.
Limitations of our research include the focus on a specific set of genes predefined in the NanoString Pancancer Pathway panel and the selection of circulating leukocytes as the tissue of interest. While leukocytes may offer a convenient target for future clinical diagnostics, we do not know to what extent our results extend to other tissues or may be relevant with regard to tissue-specific cancers likely to be heterogeneous both cellularly and molecularly70. Although these limitations do not undermine our findings with respect to circadian disruption of DNA damage and repair mechanisms, which may underlie the elevated cancer risk in night shift work, further research is needed to examine the translational generalizability of our findings.
In summary, our results indicate that disruption of the circadian rhythmicity of gene expression after exposure to a night shift schedule dysregulates temporal coordination between the cellular circadian clock and DNA repair genes, and that this amplifies sensitivity to endogenous and exogenous sources of DNA damage while rendering the signaling and execution of DNA repair less efficient (Figure 4). Together, these phenomena may contribute to the elevated cancer risk in night shift workers4,62. As such, our findings suggest that a principal mechanism underlying the elevated cancer risk associated with night shift work in humans involves increased DNA damage and dysregulation of circadian rhythmicity in DNA repair gene expression. Our findings may also inform the development of circadian biomarkers for cancer diagnostics and therapeutics.
FIGURE 4.
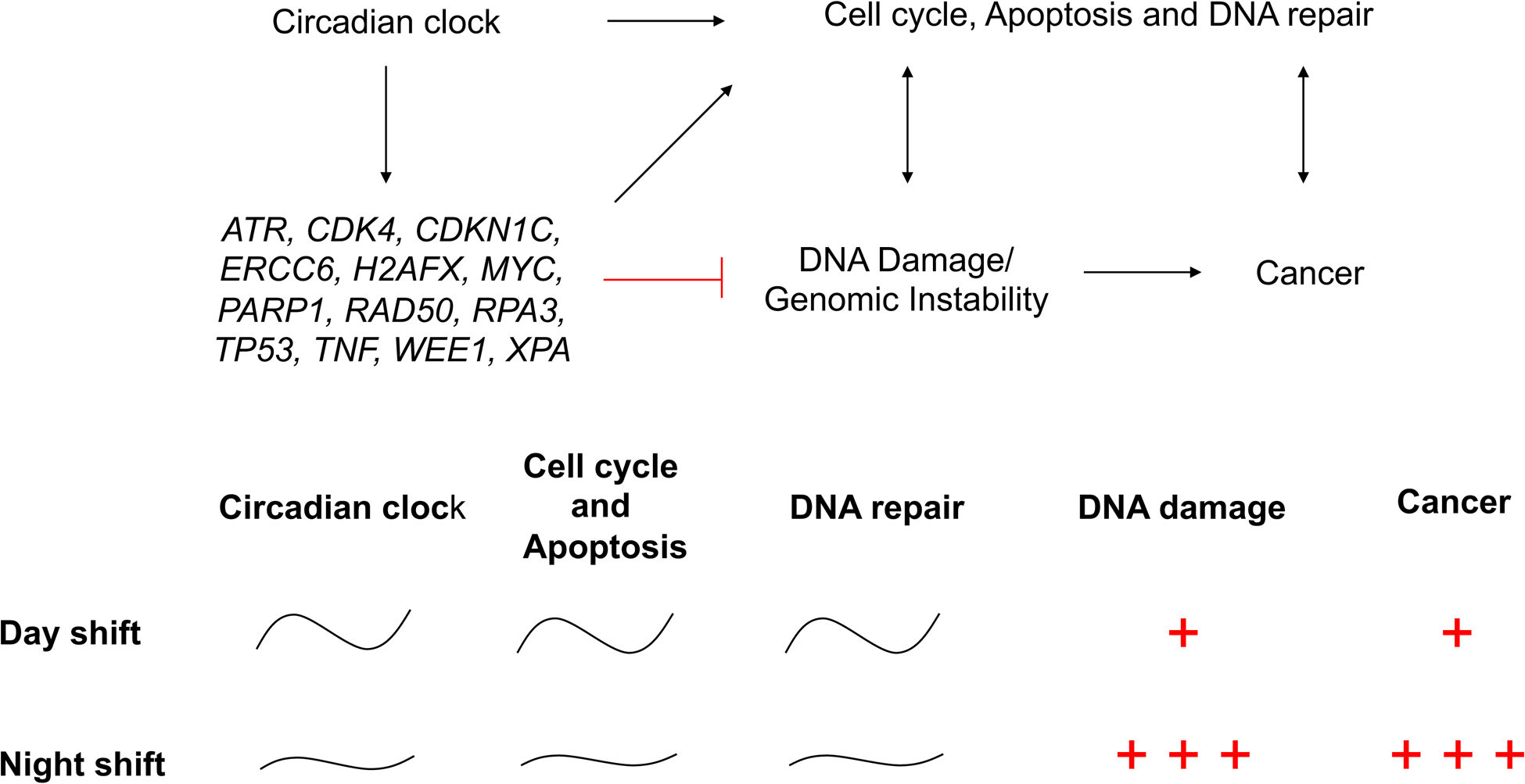
Schematic drawing showing how a night shift schedule may elevate cancer risk through circadian dysregulation of cell cycle, apoptosis and DNA repair mechanisms leading to increased DNA damage.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff of the human sleep laboratory in the Sleep and Performance Research Center at Washington State University Health Sciences Spokane for their help conducting the clinical study, Dr. Matt Layton for serving as physician of record for the clinical study, and Drs. Donna Goodenow and Michael G. Kemp for critical manuscript reading and useful comments. This work was supported by start-up funds from Washington State University and CHHE grant P30ES025128 from North Carolina State University (S.G.) - and in part by United States Army Medical Research and Development Command award W81XWH-18-1-0100 (H.P.A.V.D.); National Institutes of Health grants R01ES030113 and R21CA227381 and CDMRP Peer Reviewed Cancer Research Program award CA171123 (S.G.); and the BRAVE investment, a component of the Laboratory Directed Research and Development Program at Pacific Northwest National Laboratory, a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy under Contract DE-AC05-76RL01830 (J.T. and J.E.M.).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest. This manuscript reflects the views of the authors and does not necessarily reflect those of the U.S. Food and Drug Administration.
To ascertain that our selection of control genes did not predetermine our results, we re-ran analyses using only the two most temporally stable and abundant normalization genes, TTC31 (day condition 24-hour variation mean ± SE = 2.2% ± 4.2%, p=0.31; night condition 24-hour variation mean ± SE = 1.9% ± 3.7%, p=0.31) and ZNF384 (day condition 24-hour variation mean ± SE = 3.3% ± 3.4%, p=0.19; night condition 24-hour variation mean ± SE = 3.6% ± 3.4%, p=0.16). Categorization for functional enrichment analysis of rhythmic genes was nearly identical in this approach (contingency table assessment, χ216=1565.3, p<0.0001), and observed phase differences between the simulated night and day shift conditions were affected only slightly relative to the 24-hour cycle period (mean ± SD over genes: −0.3 ± 0.6 h or −1.4% ± 2.4%; range −1.4 to 1.2 h or −5.6% to 5.1%).
REFERENCES
- 1.Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol. 2016;26(10):R432–443. [DOI] [PubMed] [Google Scholar]
- 2.Waterhouse JM, DeCoursey PJ. Human circadian organization. In: Dunlap JC, Loros JJ, DeCoursey PJ, eds. Chronobiology: Biological Timekeeping. Sunderland: Sinauer Associates; 2003:291–323. [Google Scholar]
- 3.James SM, Honn KA, Gaddameedhi S, Van Dongen HPA. Shift work: disrupted circadian rhythms and sleep-implications for health and well-being. Curr Sleep Med Rep. 2017;3(2):104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haus EL, Smolensky MH. Shift work and cancer risk: potential mechanistic roles of circadian disruption, light at night, and sleep deprivation. Sleep Med Rev. 2013;17(4):273–284. [DOI] [PubMed] [Google Scholar]
- 5.Depner CM, Melanson EL, McHill AW, Wright KP Jr., Mistimed food intake and sleep alters 24-hour time-of-day patterns of the human plasma proteome. Proc Natl Acad Sci U S A. 2018;115(23):E5390–E5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kervezee L, Cuesta M, Cermakian N, Boivin DB. Simulated night shift work induces circadian misalignment of the human peripheral blood mononuclear cell transcriptome. Proc Natl Acad Sci U S A. 2018;115(21):5540–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skene DJ, Skornyakov E, Chowdhury NR, et al. Separation of circadian- and behavior-driven metabolite rhythms in humans provides a window on peripheral oscillators and metabolism. Proc Natl Acad Sci U S A. 2018;115(30):7825–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Resuehr D, Wu G, Johnson RL Jr., Young ME, Hogenesch JB, Gamble KL. Shift work disrupts circadian regulation of the transcriptome in hospital nurses. J Biol Rhythms. 2019;34(2):167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.IARC. Night Shift Work. IARC Monographs on the identification of carcinogenic hazards to humans. 2020;124:1–371. [Google Scholar]
- 10.Shafi AA, Knudsen KE. Cancer and the circadian clock. Cancer Res. 2019;79(15):3806–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sulli G, Lam MTY, Panda S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer. 2019;5(8):475–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111(1):41–50. [DOI] [PubMed] [Google Scholar]
- 13.Lee S, Donehower LA, Herron AJ, Moore DD, Fu L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS One. 2010;5(6):e10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puram RV, Kowalczyk MS, de Boer CG, et al. Core Circadian Clock Genes Regulate Leukemia Stem Cells in AML. Cell. 2016;165(2):303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gauger MA, Sancar A. Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res. 2005;65(15):6828–6834. [DOI] [PubMed] [Google Scholar]
- 16.Korkmaz T, Aygenli F, Emisoglu H, et al. Opposite Carcinogenic Effects of Circadian Clock Gene BMAL1. Sci Rep. 2018;8(1):16023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ozturk N, Lee JH, Gaddameedhi S, Sancar A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci U S A. 2009;106(8):2841–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sancar A, Van Gelder RN. Clocks, cancer, and chronochemotherapy. Science. 2021;371(6524). [DOI] [PubMed] [Google Scholar]
- 19.Antoch MP, Toshkov I, Kuropatwinski KK, Jackson M. Deficiency in PER proteins has no effect on the rate of spontaneous and radiation-induced carcinogenesis. Cell Cycle. 2013;12(23):3673–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papagiannakopoulos T, Bauer MR, Davidson SM, et al. Circadian rhythm disruption promotes lung tumorigenesis. Cell Metab. 2016;24(2):324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savvidis C, Koutsilieris M. Circadian rhythm disruption in cancer biology. Mol Med. 2012;18:1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahar S, Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nat Rev Cancer. 2009;9(12):886–896. [DOI] [PubMed] [Google Scholar]
- 23.Sancar A, Lindsey-Boltz LA, Gaddameedhi S, et al. Circadian clock, cancer, and chemotherapy. Biochemistry. 2015;54(2):110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, Liu Z, Selby CP, Sancar A. Long-term, genome-wide kinetic analysis of the effect of the circadian clock and transcription on the repair of cisplatin-DNA adducts in the mouse liver. J Biol Chem. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaucher J, Montellier E, Sassone-Corsi P. Molecular Cogs: Interplay between Circadian Clock and Cell Cycle. Trends Cell Biol. 2018;28(5):368–379. [DOI] [PubMed] [Google Scholar]
- 26.Miller BH, McDearmon EL, Panda S, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci U S A. 2007;104(9):3342–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheiermann C, Kunisaki Y, Frenette PS. Circadian control of the immune system. Nat Rev Immunol. 2013;13(3):190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller M, Mazuch J, Abraham U, et al. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci U S A. 2009;106(50):21407–21412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masri S, Cervantes M, Sassone-Corsi P. The circadian clock and cell cycle: interconnected biological circuits. Curr Opin Cell Biol. 2013;25(6):730–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cadet J, Douki T. Formation of UV-induced DNA damage contributing to skin cancer development. Photochem Photobiol Sci. 2018;17(12):1816–1841. [DOI] [PubMed] [Google Scholar]
- 31.Borrego-Soto G, Ortiz-Lopez R, Rojas-Martinez A. Ionizing radiation-induced DNA injury and damage detection in patients with breast cancer. Genet Mol Biol. 2015;38(4):420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477(1–2):7–21. [DOI] [PubMed] [Google Scholar]
- 33.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–228. [DOI] [PubMed] [Google Scholar]
- 34.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. [DOI] [PubMed] [Google Scholar]
- 35.Kang TH, Reardon JT, Kemp M, Sancar A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci U S A. 2009;106(8):2864–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J, Zhou XA, Zhang N, Wang J. Evolving insights: how DNA repair pathways impact cancer evolution. Cancer Biol Med. 2020;17(4):805–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee Y, Lahens NF, Zhang S, Bedont J, Field JM, Sehgal A. G1/S cell cycle regulators mediate effects of circadian dysregulation on tumor growth and provide targets for timed anticancer treatment. PLoS Biol. 2019;17(4):e3000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Dycke KC, Rodenburg W, van Oostrom CT, et al. Chronically alternating light cycles increase breast cancer risk in mice. Curr Biol. 2015;25(14):1932–1937. [DOI] [PubMed] [Google Scholar]
- 39.Aiello I, Fedele MLM, Roman F, et al. Circadian disruption promotes tumor-immune microenvironment remodeling favoring tumor cell proliferation. Sci Adv. 2020;6(42). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26(3):317–325. [DOI] [PubMed] [Google Scholar]
- 41.Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A. 2014;111(45):16219–16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hughes ME, DiTacchio L, Hayes KR, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5(4):e1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andrews JL, Zhang X, McCarthy JJ, et al. CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proc Natl Acad Sci U S A. 2010;107(44):19090–19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Panda S, Antoch MP, Miller BH, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109(3):307–320. [DOI] [PubMed] [Google Scholar]
- 45.Rudic RD, McNamara P, Curtis AM, et al. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2004;2(11):e377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoogerwerf WA, Sinha M, Conesa A, et al. Transcriptional profiling of mRNA expression in the mouse distal colon. Gastroenterology. 2008;135(6):2019–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pizarro A, Hayer K, Lahens NF, Hogenesch JB. CircaDB: a database of mammalian circadian gene expression profiles. Nucleic Acids Res. 2013;41(Database issue):D1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mikulich SK, Zerbe GO, Jones RH, Crowley TJ. Comparing linear and nonlinear mixed model approaches to cosinor analysis. Stat Med. 2003;22(20):3195–3211. [DOI] [PubMed] [Google Scholar]
- 49.Skornyakov E, Gaddameedhi S, Paech GM, et al. Cardiac autonomic activity during simulated shift work. Ind Health. 2019;57(1):118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDONALD J. Handbook of biological statistics. 3 ed. Baltimore, Maryland, U.S.A: SPARKY HOUSE PUBLISHING; 2014. [Google Scholar]
- 51.Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2 ed. New York: Springer-Verlag; 2016. [Google Scholar]
- 52.Ding W, Bishop ME, Lyn-Cook LE, Davis KJ, Manjanatha MG. In vivo alkaline comet assay and enzyme-modified alkaline comet assay for measuring DNA strand breaks and oxidative DNA damage in rat liver. J Vis Exp. 2016(111). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shostak A. Circadian clock, cell division, and cancer: from molecules to organism. Int J Mol Sci. 2017;18(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ravanat JL, Breton J, Douki T, et al. Radiation-mediated formation of complex damage to DNA: a chemical aspect overview. Br J Radiol. 2014;87(1035):20130715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leuraud K, Richardson DB, Cardis E, et al. Ionising radiation and risk of death from leukaemia and lymphoma in radiation-monitored workers (INWORKS): an international cohort study. Lancet Haematol. 2015;2(7):e276–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hadj-Hamou NS, Ugolin N, Ory C, et al. A transcriptome signature distinguished sporadic from postradiotherapy radiation-induced sarcomas. Carcinogenesis. 2011;32(6):929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corra S, Salvadori R, Bee L, Barbieri V, Mognato M. Analysis of DNA-damage response to ionizing radiation in serum-shock synchronized human fibroblasts. Cell Biol Toxicol. 2017;33(4):373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Plikus MV, Vollmers C, de la Cruz D, et al. Local circadian clock gates cell cycle progression of transient amplifying cells during regenerative hair cycling. Proc Natl Acad Sci U S A. 2013;110(23):E2106–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dainiak N. Hematologic consequences of exposure to ionizing radiation. Exp Hematol. 2002;30(6):513–528. [DOI] [PubMed] [Google Scholar]
- 60.Huber AL, Papp SJ, Chan AB, et al. CRY2 and FBXL3 cooperatively degrade c-MYC. Mol Cell. 2016;64(4):774–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davis S, Mirick DK, Stevens RG. Night shift work, light at night, and risk of breast cancer. J Natl Cancer Inst. 2001;93(20):1557–1562. [DOI] [PubMed] [Google Scholar]
- 62.group IMV. Carcinogenicity of night shift work. Lancet Oncol. 2019;20(8):1058–1059. [DOI] [PubMed] [Google Scholar]
- 63.Parent ME, El-Zein M, Rousseau MC, Pintos J, Siemiatycki J. Night work and the risk of cancer among men. Am J Epidemiol. 2012;176(9):751–759. [DOI] [PubMed] [Google Scholar]
- 64.Duffy JF, Dijk DJ. Getting through to circadian oscillators: why use constant routines? J Biol Rhythms. 2002;17(1):4–13. [DOI] [PubMed] [Google Scholar]
- 65.Wittenbrink N, Ananthasubramaniam B, Munch M, et al. High-accuracy determination of internal circadian time from a single blood sample. J Clin Invest. 2018;128(9):3826–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tubbs A, Nussenzweig A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell. 2017;168(4):644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ashok Kumar PV, Dakup PP, Sarkar S, Modasia JB, Motzner MS, Gaddameedhi S. It’s about time: advances in understanding the circadian regulation of DNA damage and repair in carcinogenesis and cancer treatment outcomes. Yale J Biol Med. 2019;92(2):305–316. [PMC free article] [PubMed] [Google Scholar]
- 68.Behjati S, Gundem G, Wedge DC, et al. Mutational signatures of ionizing radiation in second malignancies. Nat Commun. 2016;7:12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scheiermann C, Gibbs J, Ince L, Loudon A. Clocking in to immunity. Nat Rev Immunol. 2018;18(7):423–437. [DOI] [PubMed] [Google Scholar]
- 70.Almendro V, Marusyk A, Polyak K. Cellular heterogeneity and molecular evolution in cancer. Annu Rev Pathol. 2013;8:277–302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.