

Summary
It remains unclear whether plasma phospholipid transfer protein (PLTP) is involved in hypercoagulation or hypocoagulation. This study investigated the direct effect of PLTP on platelet aggregation and the underlying mechanism. Washed platelets from humans or mice and mouse platelet–rich plasma and human recombinant PLTP were isolated. PLTP is present in human platelets. We assessed ADP-, collagen-, and thrombin-induced platelet aggregation, phosphatidylserine externalization, and photothrombosis-induced cerebral infarction in mice. PLTP overexpression increased platelet aggregation, while PLTP deficiency had the opposing reaction. Human recombinant PLTP increased both mouse and human platelet aggregation in a dose-dependent manner. Phosphatidylserine externalization provides a water/lipid surface for the interaction of coagulation factors, which accelerates thrombosis. Compared with wild type controls, platelets from PLTP transgenic mice had significantly more phosphatidylserine on the exterior surface of the plasma membrane, whereas platelets from PLTP-deficient mice had significantly less on the surface, thus PLTP influences fibrinogen binding on the plasma membrane. Moreover, recombinant PLTP together with ADP significantly increased phosphatidylserine exposure on the plasma membrane of PLTP-deficient platelets, thereby increasing fibrinogen binding. PLTP overexpression significantly accelerated the incidence of photothrombosis-induced infarction in mice, whereas PLTP deficiency significantly reduced the frequency of infarction. We concluded that PLTP promotes phosphatidylserine externalization at the plasma membrane of platelets and accelerates ADP- or collagen-induced platelet aggregation. This effect plays an important role in the initiation of thrombin generation and platelet aggregation under sheer stress conditions. Thus, PLTP is involved in hypercoagulation. Therefore, PLTP inhibition could be a novel approach for countering thrombosis.
Keywords: Phospholipid transfer protein, platelet aggregation, phosphatidylserine externalization
Introduction
The expression of phospholipid transfer protein (PLTP) is increased in different pathologies associated with cardiovascular diseases (CVD), including obesity (1, 2), insulin resistance (3), and type II diabetes (4). Fifteen years ago, we reported that serum PLTP activity is increased in CVD patients (5). Despite many unresolved questions, we have since suggested that PLTP might be a therapeutic target for CVD (5). Within the past decade, the majority of human studies have shown a positive association between plasma PLTP activity and CVD (6–12). By contrast, PLTP abundance was lower in a small group of CVD patients compared with controls (13), although plasma PLTP concentration is not the preferred marker for PLTP-associated disease risk factors (14, 15). In addition, the reported effects of PLTP concentration on peripheral artery disease are both limited and inconsistent (16, 17). In mouse models, systemic PLTP deficiency reduces atherosclerosis (18), whereas its overexpression has the opposite effect (19). PLTP deficiency also is associated with a reduced frequency of developing abdominal aortic aneurysms (20). In rabbits fed a high-fat diet, PLTP overexpression increases the number of atherosclerotic lesions (21).
A few mechanisms have been suggested for the relationship between PLTP and CVD. Plasma PLTP activity is positively associated with production of atherogenic apolipoprotein B–containing lipoprotein (18, 22, 23). PLTP deficiency protects circulating lipoprotein from oxidation owing to the enhanced accumulation of vitamin E (24), whereas PLTP overexpression has the opposite effect (25). However, these mechanisms need further investigation and clarification.
PLTP knockout (KO) mice exhibit a longer clotting time following tail bleeding compared with WT mice. This may be related to a relative decrease in PS externalization due to reduced vitamin E level in erythrocytes (26). Consistent with this result, Desrumaux et al. reported that plasma PLTP deficiency is associated with reduced thrombotic responses to acute intravascular oxidative stress (27). Thus, PLTP seems to be involved in hypercoagulation. However, other research suggests that plasma PLTP has an anticoagulation effect (28, 29). Although we cannot explain the discrepancy between these studies, the previous studies have not directly evaluated the relationship between plasma PLTP levels and platelet aggregation. We thus utilized PLTP transgenic (Tg) and KO mice, as well as human recombinant PLTP (rPLTP), to systemically evaluate the effect of circulating PLTP level on mouse and human platelets.
Methods
Mice and Diet
Human PLTP transgenic (Tg) mice were a gift from Dr. de Crom R (Department of Cell Biology & Genetics, Erasmus Medical Center, The Netherlands). The mouse expresses whole human PLTP gene (15 kb 5′ to the first exon and ≈3.5 kb 3′ to the last exon) (30). PLTP KO homozygous, PLTP Tg, and wild-type (WT) mice, each in a C57BL/6 background and 10–13 weeks old, were fed a standard chow diet and had free access to water. Mice were housed in a controlled environment (22 ± 2°C, humidity 40–60%) with a 12 h light/12 h dark cycle. The Taishan Medical University Animal Care and Use Committee approved all animal procedures.
Reagents
Fluorescein isothiocyanate (FITC)-labeled mouse fibrinogen was purchased from Fisher Scientific (Silver Spring, MD, USA), and apyrase, prostaglandin E1, human fibrinogen, ADP, bovine serum albumin (BSA), and HEPES were purchased from Sigma-Aldrich (St. Louis, MO, USA). Annexin V–conjugated FITC was obtained from BD Pharmingen (San Jose, CA, USA). Collagen was from Chrono-Log (Havertown, PA, USA). TRIZOL reagent was from Invitrogen (Carlsbad, CA, USA). MuLV reverse transcriptase was from Applied Biosystems (Foster City, CA, USA). SYBR green PCR master mix kit was from TianGen Biotech (Beijing, China).
Reverse Transcription-PCR
Total RNA from washed platelets was isolated with TRIZOL Reagent. cDNA was synthesized using MuLV reverse transcriptase. The primers used for reverse transcription (RT)-PCR were as follows: PLTP Forward, CATGCGGGATTCCTCACC; PLTP Reverse, GAGGGGGCA CTACAGGCTAT; Flt-1 Forward, GGCCCGGGATATTTATAGAAC; Flt-1 Reverse, CCATCCATTTTAGGGGAAGTC; Flk-1 Forward, CAGTGGTACTGGCAGCTAGAAG; Flk-1 Reverse, ACAAGCATACGGGCTTGTTT; GAPDH Forward, ACCACAGTCCATGCCATCAC; GAPDH Reverse, TCCACCACCCTG TTGCTGTA. Amplification of cDNA with primers for GAPDH served as an internal control. PCR was performed in an iCycler (Bio-Rad, Hercules, CA, USA). The expected PCR product was confirmed by agarose gel electrophoresis with ethidium bromide staining.
Recombinant PLTP (rPLTP) preparation
A cDNA encoding His-tagged human rPLTP was used to transfect Chinese hamster ovary cells. After subjecting the cells to selection with methotrexate, serum-free medium was collected, and rPLTP was isolated by chromatography through a Ni2+-nitrilotriacetic acid resin column. The isolated rPLTP fractions were assayed for PLTP activity and evaluated for purity by SDS-PAGE.
Blood Collection
Mice were anesthetized by subcutaneous injection of a mixture of xylazine (5 mg/mL) and ketamine (40 mg/L), and blood was subsequently collected by cardiac puncture. For human blood sampling, venipuncture was performed from a free-flowing vein using a sterile 21G butterfly needle (Fisher Scientific).
Preparation of Washed Platelet-rich plasma
Platelet-rich plasma (PRP) was collected and then washed three times with freshly Tyrode’s buffer. Finally, platelets were re-suspended in the buffer. Platelets were allowed to equilibrate for 30 min before use in experiments.
PLTP Secretion from Platelets
Washed platelets (2~4×108/ml) were adjusted to the final volume of 500μl to incubate with collagen (final conc. 4μg/ml) and fibrinogen (final conc. 1μg/ml) for aggregation at 37°C for 15 min. The incubated mixture was centrifuged at 750 × g for 10 min to harvest supernatant and PLTP was detected by Western Blotting.
Mouse Tail Bleeding Time Measurement
Anaesthetized mice were placed on a heating pad warmed at 37°C. The mouse tail was amputated 5mm from the tip and immediately submersed in normal saline at 37°C. The bleeding times was defined as the time from the start of transection to complete cessation of bleeding.
Platelet Aggregation Assay
Washed platelets and platelets in PRP were counted using a PE-6800 VET Fully Auto-hematology Analyzer (Procan Electronics, Shenzhen, China) and adjusted to 250 × 109 platelets/L with Tyrode’s buffer or physiological saline, respectively. Platelet-poor plasma was diluted at the same proportion using normal saline. Platelet-poor plasma or Tyrode’s buffer was used to set 100% light transmission. A total of 250 μL PRP was added to a cuvette, and aggregation was initiated by adding ADP or collagen. The maximum aggregation rate of platelets was recorded with an aggregometer (Model 700–2s/N, Chrono-Log).
PS Externalization Measurement
Washed PLTP KO mouse platelets (3 × 108/mL) and thrombin (2 U/mL) were incubated with BSA (20 μg/mL) or rPLTP (20 μg/mL) at 37°C for 30 min. Annexin V binding buffer Supplier was then mixed with pre-treated platelets. The volume of buffer, FITC-annexin V, and platelets were at a ratio of 50:10:1. Samples were incubated at room temperature for 15 min and then subjected to flow cytometry with a FACSCalibur (BD Pharmingen). The occurrence of PS on the platelet surface was reported as the percentage of platelets that bound to annexin V or the geometric mean fluorescence intensity.
Platelet Fibrinogen Binding Assay
Washed platelets (2 × 107/mL) of PLTP KO mice were incubated with BSA (20 μg/mL) or rPLTP (20 μg/mL) for 15 min before stimulation (30 μM ADP) in the presence of 100 μg/mL FITC-fibrinogen for 10 min. Samples were fixed with 1% paraformaldehyde for 30 min and analyzed by flow cytometry (FACSCalibur). Platelet fibrinogen binding was presented as the percentage of fibrinogen-bound cells in the platelet population.
Shear-induced platelet aggregation (SIPA)
Mouse PRP, suspended to a final concentration of 3 × 107/mL in normal saline with 2 mg/mL fibrinogen and 5 μg/mL VWF, was sheared at 450/second for 10 minutes at 37°C in a computer-controlled 0.5° Cone-and-Plate Rheometer (Pulisheng, Beijing, China) after incubating with PE-CD61 at room temperature for 15 minutes. Samples were fixed with 1% paraformaldehyde for 30 min and platelet aggregation was measured by flow cytometry (FACSCalibur).
Thrombin-induced platelet activation
PRP of mouse was re-suspended to a final concentration of 3 × 107/mL in normal saline. Platelets were then incubated with PE-CD61 (BD Bioscience, to define platelets ) for 15 min before stimulation (0 and 0.5 U/mL thrombin) in the presence of 25 μg/mL FITC-CD62p or 10 μg/mL APC-CD63 (Ebioscience Inc. San Diego, CA, USA) for 15 min at room temperature. Samples were fixed with 1% paraformaldehyde for 30 min and analyzed by flow cytometry (FACSCalibur). CD62p and CD63 stained platelets were counted and the percentage of these platelets in whole population were presented.
Thrombin Generation in Mouse PRP
Mouse PRP and the fluorescent thrombin substrate Z-GGR-AMC (833μmol/L) was added into the thrombin generation assay system. The final concentrations therefore were: platelets, 2.53×108 platelets/mL; substrate, 833 μM; recombinant tissue factor, 0.17 pM; CaCl2, 16.7 mM. Fluorescence was measured in each well at 30 s intervals during 70 min. Thrombin production was calculated from a calibration curve constructed with known amounts of calibrated thrombin.
Photothrombosis and Assessment of Cerebral Infarct Size
Anesthetized mice were injected intravenously with the photosensitizer dye Rose Bengal (50 mg/kg), and a laser beam was focused via an optic fiber through the skull onto the right hemisphere. The skull was exposed to the laser for 3 min beginning 5 min after dye injection. Mice were euthanized 24 h later. Each brain was then harvested, sliced into 2-mm-thick coronal sections, stained with 1% 2,3,5-triphenyltetrazolium chloride, and fixed in 4% paraformaldehyde. The caudal face of each section was scanned with a flatbed color scanner. The percentage of infarct area was calculated by dividing the infarct area (white) by the total area (white + red) using Image-Pro1 Plus Version 6.0.
Statistical Analysis
Results are expressed as mean ± standard deviation (SD). The statistical significance of the difference between two data means was determined with the unpaired two-tailed Student’s t-test, and differences among multiple groups were assessed by analysis of variance followed by the Student-Newman-Keuls test. A difference for which P was <0.05 was considered statistically significant.
Results
PLTP Deficiency and Overexpression Have Opposite Effects on Platelet Aggregation and Fibrinogen Binding
We simultaneously measured plasma PLTP activity in PLTP KO and human PLTP Tg mice. As expected, negligible and strongly increased phospholipid transfer activities have been observed in PLTP KO and PLTPTg mice, respectively (Supplement Table 1). Both PLTP KO and PLTP Tg mice have significantly lower plasma total phospholipids, cholesterol, HDL-cholesterol (Supplement Table 1), as reported before (30, 31). We next measured the bleeding time of PLTP KO as well as control mice and found PLTP KO mice exhibit a longer clotting time (Fig. 1A). This phenomenon was observed before (26). However, the mechanism is still unknown. We next utilized platelet rich plasma (PRP) from PLTP-deficient mice to assess platelet aggregation. Collagen treatment significantly induced platelet aggregation in WT mouse PRP, however, such an induction was greatly diminished in PLTP KO mouse PRP (50%, P < 0.01; Fig. 1B). Compared with collagen, ADP treatment had a more pronounced effect [WT vs. PLTP homozygous KO (−/−), 79%, P < 0.01] in a gene dose–dependent manner [WT vs. PLTP heterozygous KO (+/−), 42%, P < 0.05; PLTP heterozygous KO (+/−) vs. PLTP homozygous KO (−/−), 63%, P < 0.05] (Fig. 1C).
Figure 1. Mouse PLTP expression influences clotting time and platelet aggregation.
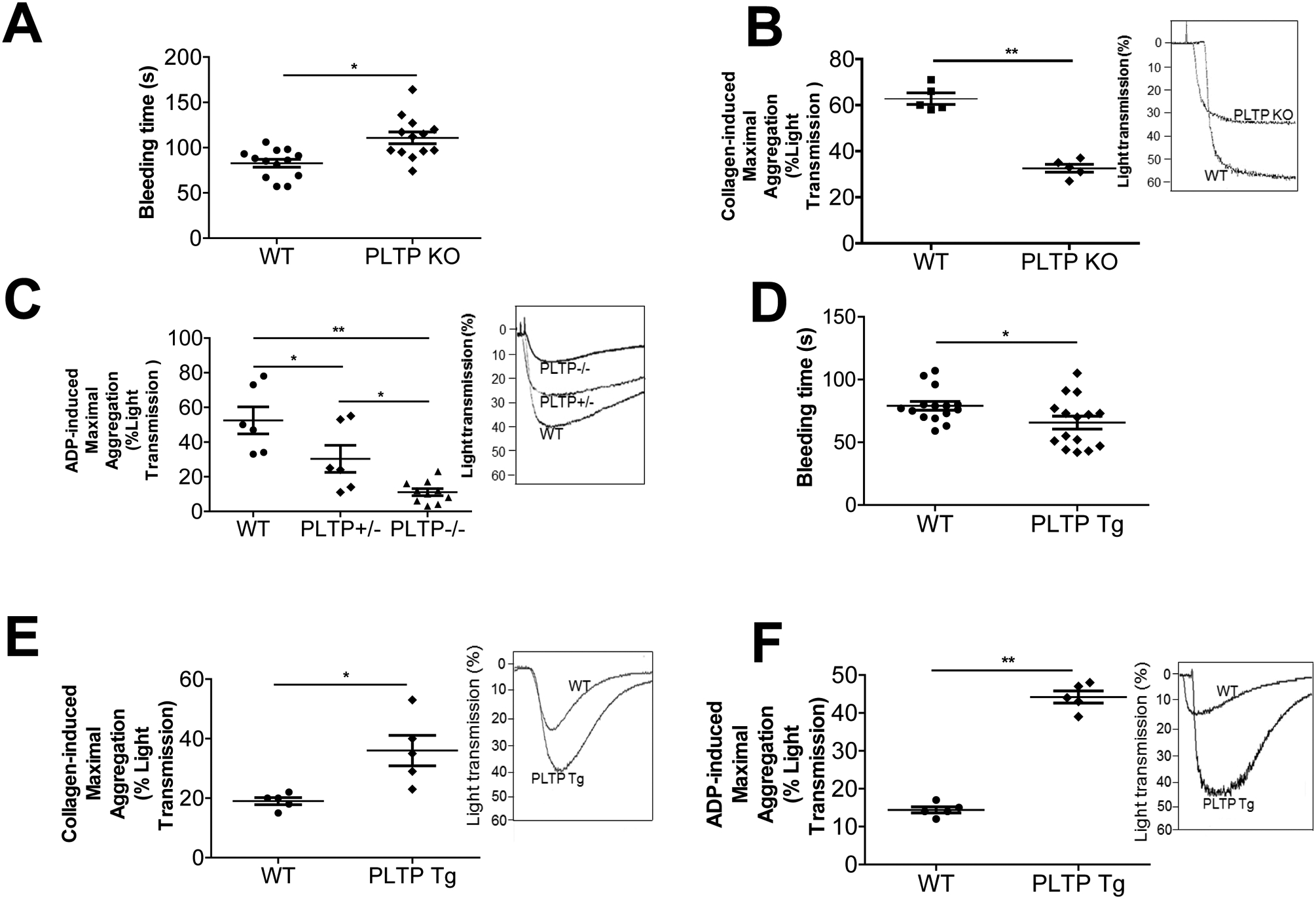
(A) PLTP KO mouse bleeding time measurement. (B) Collagen (4 μg/mL) was used to induce platelet aggregation. (C) ADP (30 μM) was used to induce platelet aggregation. (D) PLTP Tg mouse bleeding time measurement. (E) Collagen (4 μg/mL) was used to induce platelet aggregation. (F) ADP (30 μM) was used to induce platelet aggregation. PLTP+/−, heterozygous PLTP KO mice; PLTP−/−, homozygous PLTP KO mice; PLTP Tg, PLTP transgenic mice. Values represent the mean ± SD, n = 5, *P < 0.05, **P < 0.01.
We next sought to measure the bleeding time of PLTP Tg as well as control mice and found PLTP Tg mice exhibit a shorter clotting time (Fig. 1D). We also utilized PLTP Tg mice, to perform the same experiment as PLTP KO mice. Collagen treatment significantly induced platelet aggregation in PRP (1.9-fold, P < 0.05; Figure 1E), whereas ADP treatment, again, had a more pronounced effect (3.1-fold, P < 0.01; Fig. 1F).
We also measured aspirin effect of ADP-mediated platelet aggregation using PLTP Tg and control mouse PRP. Again, we found that PLTP overexpression promotes platelet aggregation, however, aspirin treatment can mask the difference between PLTP Tg and control (Supplement Fig. 1A and B).
We next utilized flow cytometry analysis to measure fibrinogen binding, a prerequisite of platelet aggregation. We found that fibrinogen binding to PLTP-overexpressing platelets was significantly increased (Fig. 2A), whereas binding to PLTP-deficient platelets was significantly reduced, compared with controls (Fig. 2B). CD63 and CD62P have been recognized as platelet activation markers (32). We also performed CD62 and CD63 measurement on platelet surface after thrombin treatment and found PLTP overexpression induced both on the surface of platelet (Fig. 2C and D).
Figure 2. The effect of PLTP on platelet fibrinogen binding and CD63/CD62 levels.
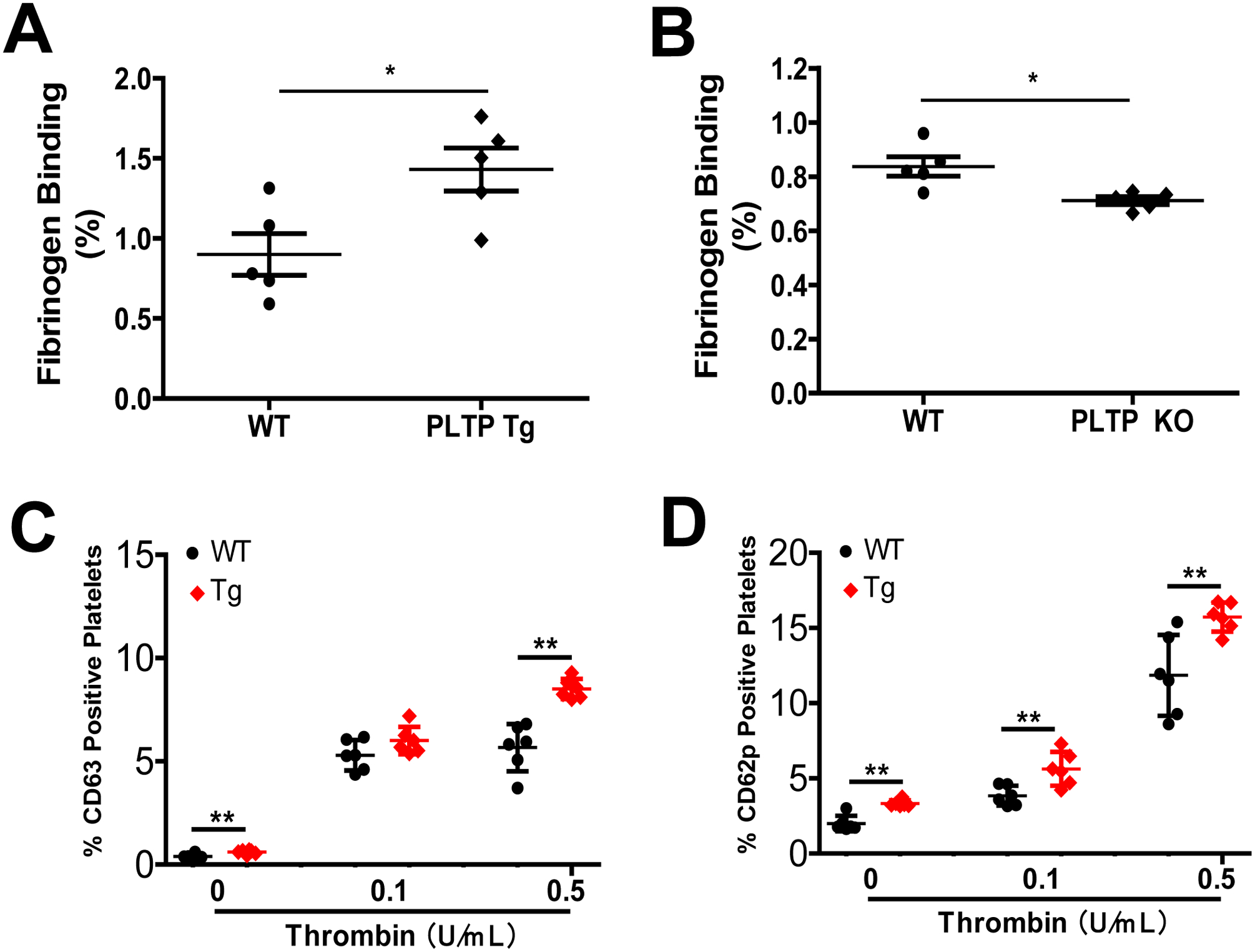
(A) Effect of PLTP overexpression on platelet fibrinogen binding. (B) Effect of PLTP deficiency on platelet fibrinogen binding. (C) and (D) Flow cytometry analysis of the percentage CD63 and CD62p positive platelets after stimulated with thrombin (0, 0.1 and 0.5 U/mL). Values represent the mean ± SD, n = 5, *P < 0.05, **P < 0.01.
Effect of rPLTP on Fibrinogen Binding and Aggregation of Human or Mouse Platelets
We prepared His-tagged human rPLTP of high purity (Fig. 3A) that had high phospholipid transfer activity (Fig. 3B). We next utilized flow cytometry analysis to investigate fibrinogen binding on human platelet after rPLTP and ADP treatment. We found that rPLTP treatment significantly increased fibrinogen binding (Fig. 3C, Supplement Fig. 2). To determine the effect of rPLTP on platelet aggregation, we treated PLTP-deficient mouse PRP with rPLTP (20 μg/mL) or BSA (20 μg/mL, control). We found that rPLTP dramatically increased ADP-mediated platelet aggregation (Fig. 3D and E). Moreover, we found that rPLTP stimulated both ADP-induced and collagen-induced human platelet aggregation in a dose-dependent manner (Fig. 3F and G). Thus, in these studies, exogenous rPLTP had a consistent effect on the aggregation of both mouse and human platelets.
Figure 3. The effect of rPLTP on platelet fibrinogen and aggregation.
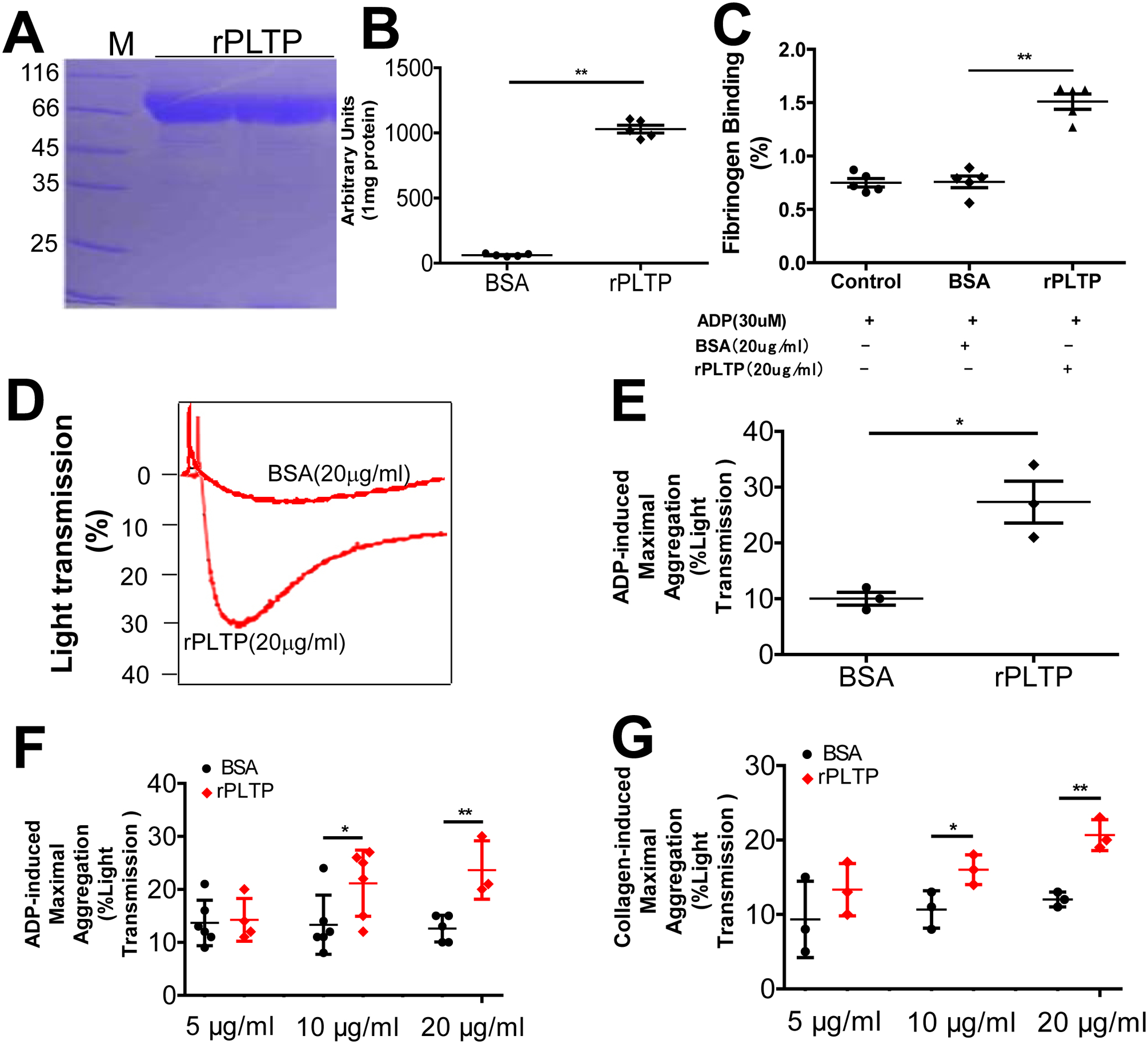
(A) SDS PAGE of His-tagged human rPLTP stained with Coomassie brilliant blue. (B) PLTP activity of rPLTP. (C) Flow cytometry analysis of fibrinogen binding on human platelet after rPLTP and ADP treatment. (D) and (E) the effect of rPLTP on ADP-induced mouse platelet aggregation. PLTP-deficient mouse PRP was treated with rPLTP (20 μg/mL) or BSA (20 μg/mL, control) and the aggregation was examined. (F) Dose effect of rPLTP on ADP-induced human platelet aggregation. (G) Dose effect of rPLTP on collagen-induced human platelet aggregation. Values represent the mean ± SD, n = 3–5, *P < 0.05, **P < 0.01.
PLTP Promotes PS Externalization
We next explored the mechanism underlying PLTP-mediated platelet aggregation. It has been reported that erythrocytes from PLTP-deficient mice can inhibit coagulation owing to a reduction in PS incorporation in the outer leaflet of the erythrocyte plasma membrane (26); however, whether this holds for platelets is unknown. PS externalization was assessed with annexin V–conjugated FITC labeling followed by flow cytometry. Platelets from PLTP Tg mice had significantly more PS on the exterior surface of the platelet plasma membrane compared with WT platelets (Fig. 4A and B), whereas platelets from PLTP-deficient mice had significantly less PS on the surface (Fig. 4C). Moreover, for PLTP-deficient washed platelets treated with ADP, rPLTP significantly increased PS externalization on the plasma membrane compared with control treatment (Fig. 4D).
Figure 4. Effect of PLTP on PS externalization.
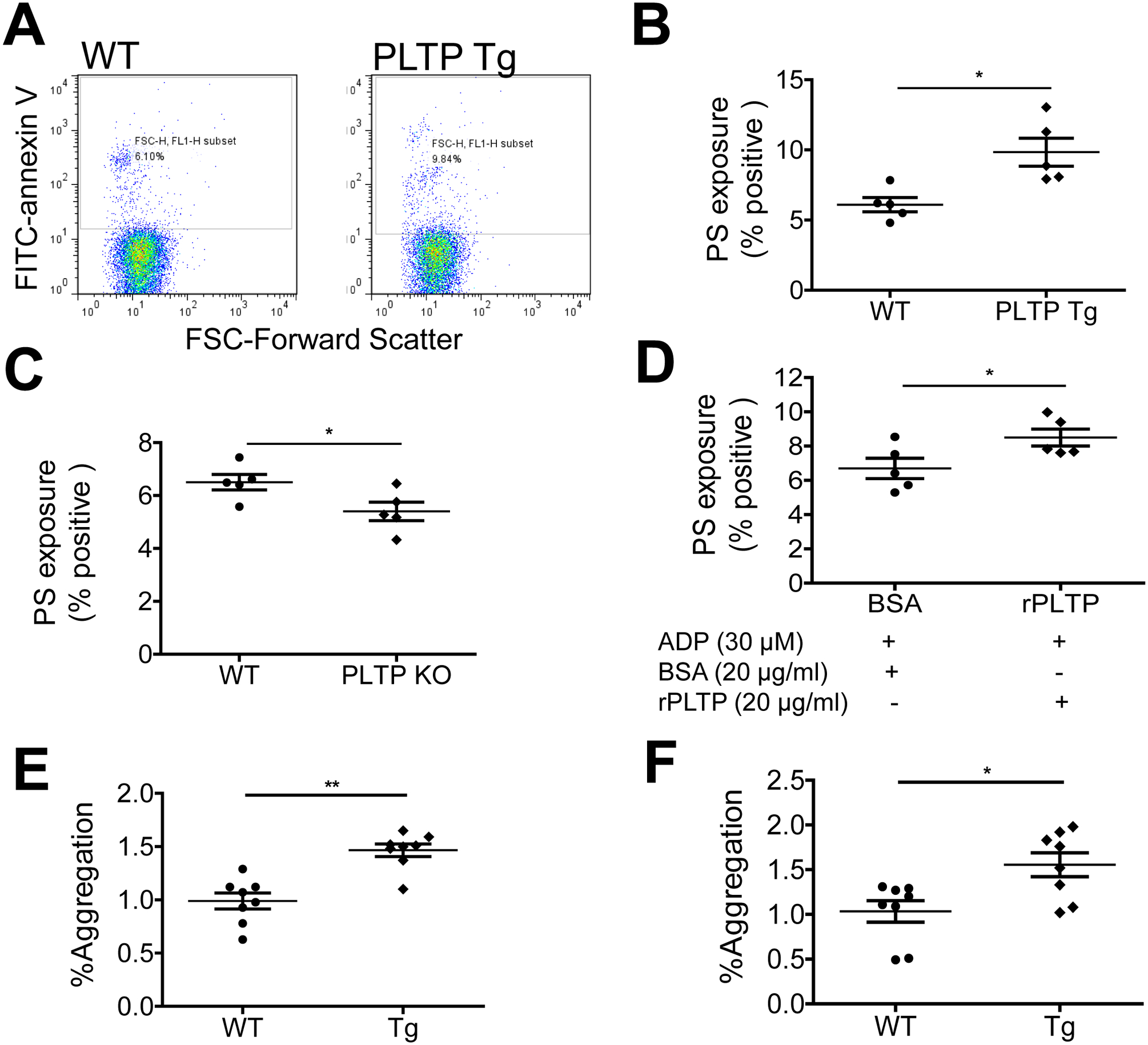
PS externalization was assessed by annexin V–FITC labeling followed by flow cytometry. (A) and (B) flow cytometry analysis and quantification of the effect of PLTP overexpression (Tg) on PS externalization on platelets. (C) Effect of PLTP deficiency on PS externalization. (D) The effect of rPLTP on PLTP deficient platelet PS externalization. (E) Effect of PLTP overexpression on shear-induced (SIPA) induced platelet aggregation. Flow cytometric plots for the measurement of platelet aggregation. SIPA was estimated as increase in platelet population outside the single platelet gate compared to static control mouse platelet rich plasma. (F) Flow cytometric plots for the measurement of platelet aggregation after gated with PE-CD61 for platelet population. Values represent the mean ± SD, n = 5–8, *P < 0.05, **P < 0.01.
Mobilization of PS plays an important role in platelet aggregation under sheer stress conditions (33). We did shear-induced platelet aggregation (SIPA) experiment. We found that PLTP overexpression significantly increase SIPA (Fig. 4E and F). We also did thrombin substrate Z-GGR-AMC stimulated thrombin generation. However, no significant difference was found between control and PLTP overexpressed platelet-rich plasma (Supplement Fig. 3). We also evaluated the effect of rPLTP on the secretion of platelet α-granules and dense granules. Washed platelets were treated with rPLTP for 15 min followed by activation with thrombin. We found that platelet factor 4 (PF4) (released from α-granules) and ATP (released from dense granules) were significantly increased compared with control (Supplement Fig. 4A and B).
PLTP binds to platelet surface
We assumed that PLTP binding on platelet surface could promotes platelet aggregation. Thus, we evaluated whether exogenous active rPLTP binds to the surface of human platelets. We incubated human washed platelet with active and inactive rPLTP-Histag (60°C incubation for 3 hours) and then anti-Histag antibody (Alexa Fluor-488 labeled, R&D System). Finally, we utilized FACS to evaluate rPLTP binding. Indeed, active rPLTP (10 μg) has a significant stronger ability to promote the fluorescent curve rightward shift, compared with inactive one, suggesting that rPLTP can bind on the surface of human platelet and such a binding requires PLTP activity (Fig. 5A and B).
Figure 5. Active rPLTP binds on human platelet.
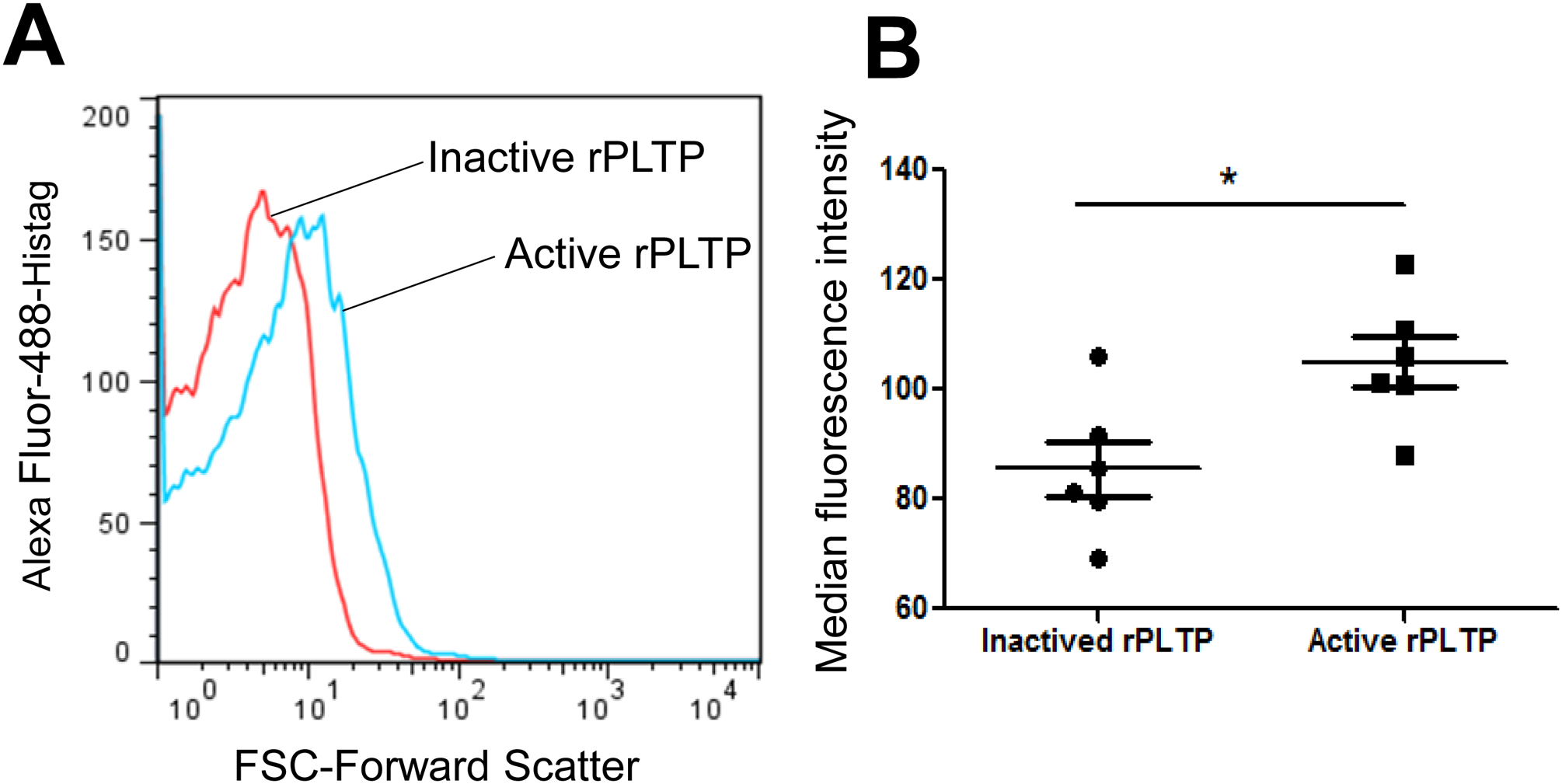
rPLTP inactivation was performed at 60°C for 3 hours and PLTP activity was completely blocked. A total 250μL Human platelet (1×108/ml) was incubated with active rPLTP or inactive rPLTP (10 μg) at room temperature for 10 min, then, incubated with His-Tab Alexa Fluor 488-conjugated antibody at room temperature for 15 min, and then subjected to flow cytometry with a FACSCalibur (BD Pharmingen). (A) flow cytometry analysis. (B) quantification. Values represent the mean ± SD, n = 6, *P < 0.05.
PLTP Deficiency Countervails and PLTP Overexpression promotes Cerebral Thrombosis
Next, we compared focal cerebral photothrombosis among the mice. At 24 h post-induction of photothrombotic stroke, the mice were euthanized, and live versus dead cells were localized in triphenyltetrazolium chloride–stained sections. Sections from PLTP KO mice had significantly smaller infarct lesion volumes compared with WT mice (P < 0.05; Fig. 6A and B). This result confirmed what has been reported (27). Importantly, PLTP Tg mice showed significantly larger lesion volumes compared with WT mice (P < 0.05; Fig. 6C and D). We also found that mRNA levels of Flt-1and Flk-1, two well-known markers of anoxia/hypoxia (34, 35), were induced in mouse brains after photothrombotic stroke induction. Importantly, PLTP Tg mouse brain has much higher levels of both mRNAs (Supplement Fig. 5A and B).
Figure 6. Photothrombosis and assessment of cerebral infarct size.
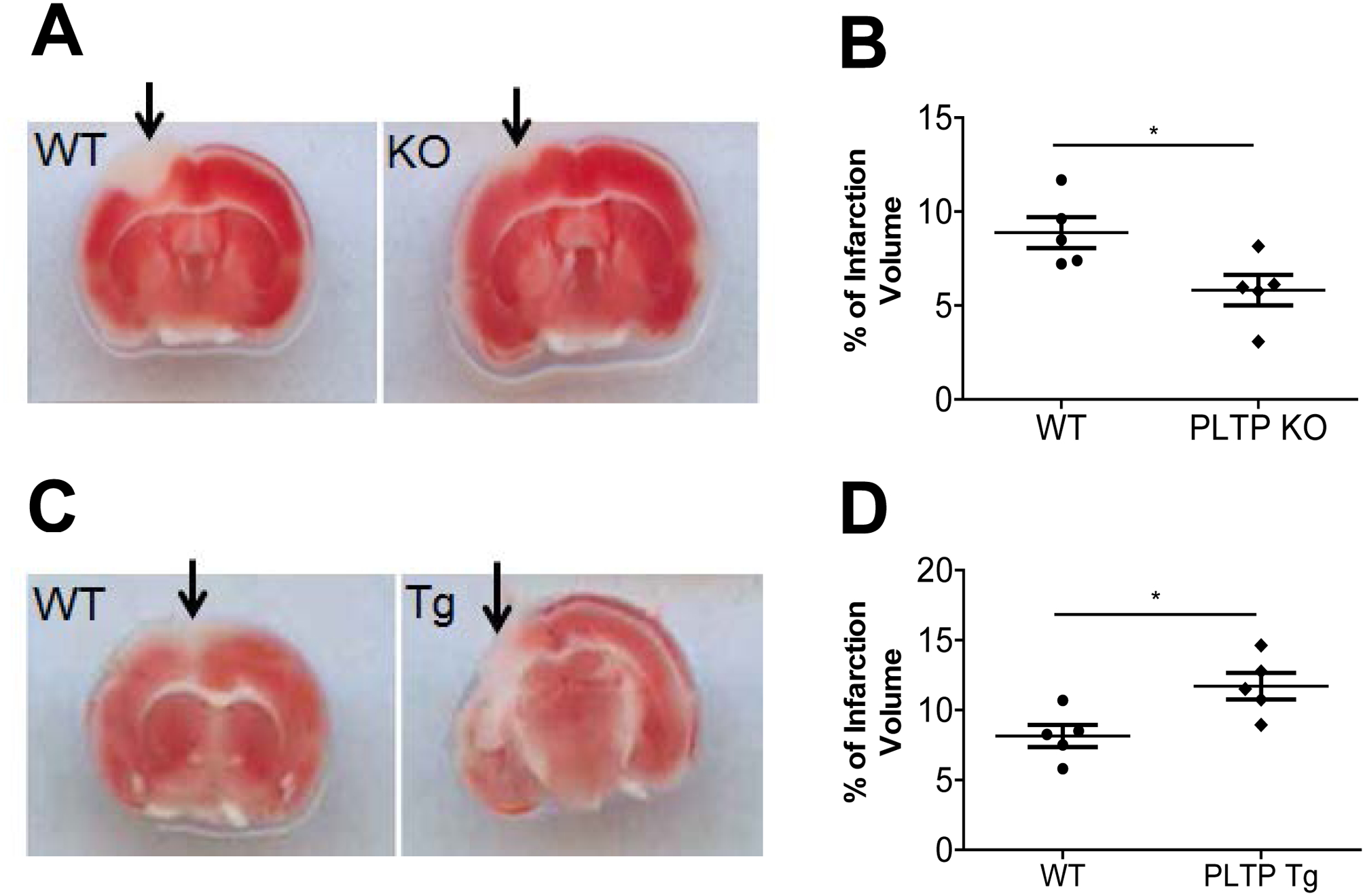
Photothrombosis was performed as described in “Materials and Methods”. At 24 h post-induction of photothrombotic stroke, the mice were euthanized, and living versus dead cells were quantified in triphenyl tetrazolium chloride–stained sections. (A) and (B) Images of intravascular thrombosis (A) and quantification (B) in WT and PLTP KO mouse brain. (C) and (D) Images of intravascular thrombosis (C) and quantification (D) in WT and PLTP Tg mouse brain. Arrows indicate the infarct site. Values represent the mean ± SD, n = 5, *P < 0.05.
Human platelet produces PLTP
We prepared homogenates from washed human platelets and performed western blotting for PLTP, which confirmed that the platelets indeed produce PLTP (Fig. 7A and B). We also evaluate whether human platelet can secrete PLTP under activation and found that collagen and fibrinogen treated platelets (2 × 108) secreted PLTP (Fig. 7C and D).
Figure 7. Human platelets produce PLTP, and mouse PLTP deficiency reduces platelet aggregation.
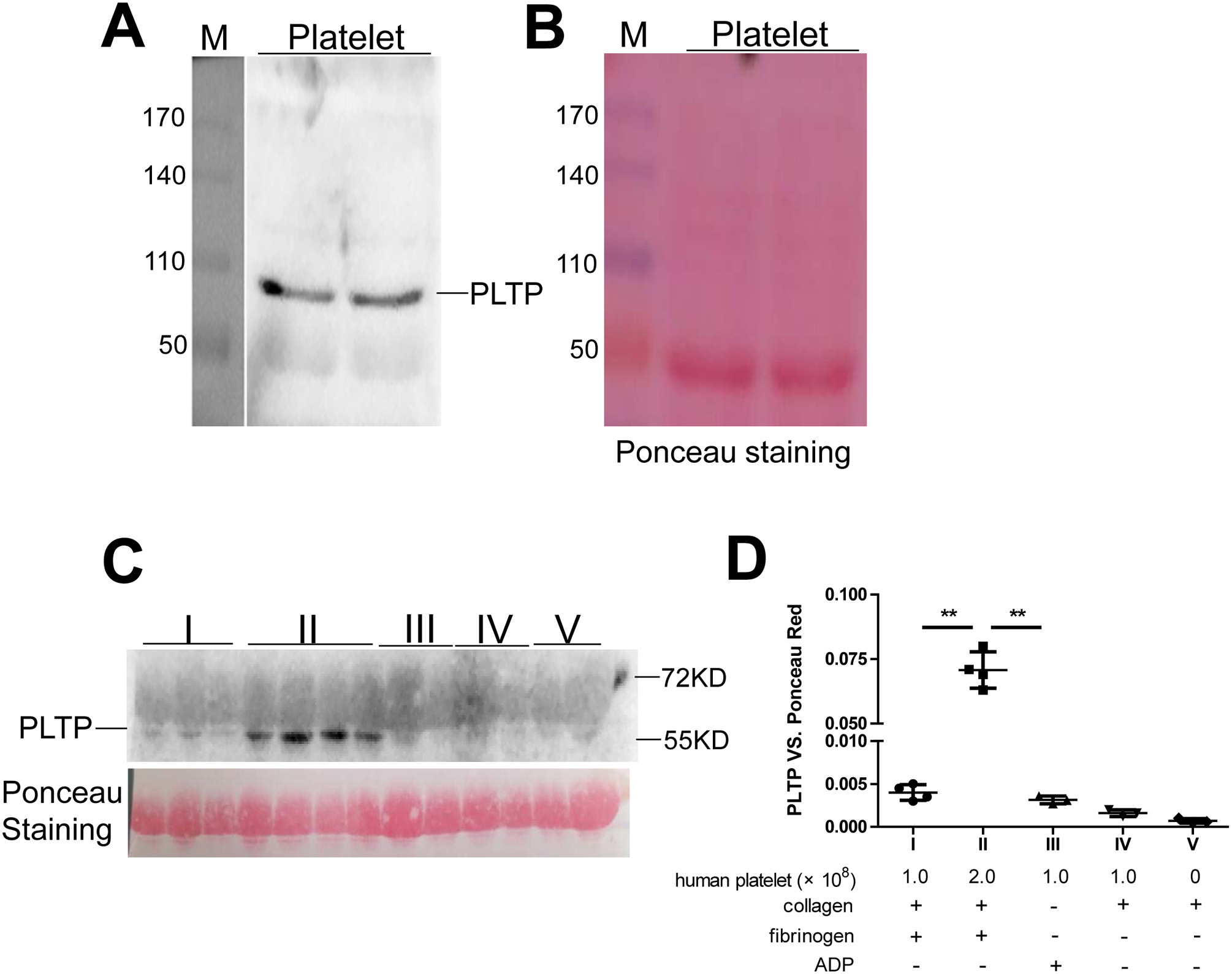
(A) PLTP in human platelet homogenate (arrow) measured by western blotting. (B) Loading control (Ponceau S staining). M, molecular size markers (kDa). (C) PLTP secreted from aggregated platelets was measured by western blotting. The Ponceau S Staining was used as a loading control. (D) quantification of the western blotting; the components layout of fraction I-V was listed. Values represent the mean ± SD, n = 3–4, **P < 0.01.
Discussion
Besides the ability of PLTP to promote the formation of atherosclerotic lesions (5, 18, 21), our results show for the first time that human platelets contain PLTP and that PLTP promotes platelet aggregation. We found that: 1) PLTP overexpression increases ADP- or collagen- or thromobin-induced aggregation of mouse platelets, while PLTP deficiency had the opposing reaction; 2) human rPLTP accelerates ADP- or collagen-induced aggregation of mouse or human platelets in a dose-dependent manner; 3) PLTP overexpression promotes whereas PLTP deficiency attenuates PS externalization to the exterior surface of platelets; 4) PLTP-mediated mobilization of PS plays an important role in the initiation of thrombin generation and platelet aggregation under sheer stress conditions; 5) treatment of platelets with human rPLTP increases PS externalization and, hence, fibrinogen binding; and 6) PLTP overexpression significantly increases the volume of photothrombosis-induced cerebral infarction, whereas PLTP deficiency significantly decreases the infarction volume.
It remains controversial whether PLTP is involved in hypocoagulation or hypercoagulation. On one hand, PLTP has the ability (in vitro) to favor the scavenging of anionic phospholipids (mainly PS), which has the effect of attenuating their procoagulation effect (28). A recent paper also reported that, when added to plasma in increasing amounts, rPLTP reduced the total amount of thrombin generated and prolonged the lag time for thrombin generation in plasma activated by sulfatides (29). Based on their results, the authors even suggested a negative association between venous thrombosis risk and PLTP activity in humans (29). On the other hand, PLTP has been shown to have hypercoagulant properties. For example, PLTP-deficient mice displayed fewer externalized PS molecules in erythrocytes compared with WT mice, thereby modulating in vivo PS-dependent procoagulant properties (26). It was also shown that PLTP deficiency in mice was associated with an extended tail bleeding time, a slowdown of the coagulation process, and a significant reduction in ischemic tissue damage (27). A potential explanation for the prolonged bleeding times seen in PLTP KO mice has been attributed to reduction in PS exposure on the erythrocytes (26), since erythrocyte PS can activate the contact pathway and support thrombin generation in vitro (36). However, none of the previous studies directly evaluated the relationship between PLTP and platelet aggregation. Therefore, we utilized PLTP-deficient and PLTP-overexpressing mice (in vivo) as well as rPLTP (in vitro) to assess any direct relationship between PLTP and platelet aggregation. Our results clearly indicate that PLTP promotes platelet aggregation and is critical for hypercoagulation, although our findings cannot fully resolve the discrepancy among the above-mentioned studies. In fact, mobilization of PS is irrelevant to platelet aggregation under static condition and platelet activation is intact in patients with Scott syndrome, who show impaired PS externalization (37). However, mobilization of PS plays an important role in platelet aggregation under sheer stress conditions (33) and we confirmed this (Figs. 4E and F).
Hypercoagulation is always associated with loss of lipid symmetry in membrane bilayers, including sickle-shaped erythrocytes (from sickle-cell anemia patients), apoptotic cells, glucose-treated erythrocytes (38), and shedded membrane microparticles such as those derived from atherosclerotic plaques (39). A major finding of our present work is that PS externalization at the plasma membrane was significantly decreased in PLTP-deficient platelets, whereas rPLTP treatment could increase PS externalization. Interestingly, PS exposure under pathological conditions has been referred to as an “apoptotic-like” event (40).
Does plasma PLTP have activities other than phospholipid transfer activity in the circulation? PLTP deficiency is related to PS externalization through its ability to produce vitamin E–poor erythrocytes (26). This could also be true for platelets, although there are other possibilities. Under resting conditions, the transverse asymmetry of platelet plasma membrane phospholipids could be maintained through the action of a flippase and floppase at the expense of ATP (41). Moreover, scramblase is a calcium-dependent enzyme that catalyzes the bidirectional transfer of phospholipids, and PS externalization is one of the consequences (42). PLTP belongs to a family of lipid transfer/lipopolysaccharide-binding proteins, including cholesteryl ester transfer protein, lipopolysaccharide-binding protein, and bactericidal/permeability-increasing protein (43). Besides transferring phosphatidylcholine, PLTP also efficiently transfers sphingolipids, cholesterol, diacylglycerol, α-tocopherol, cerebroside, lipopolysaccharide (31, 44), and perhaps PS and phosphatidylethanolamine in a “lipoprotein-lipoprotein” or “lipoprotein-cell” manner (45). We previously reported that PLTP facilitates sphingosine-1-phosphate transport from erythrocytes to lipoproteins (46). PLTP may use its amphipathic helical region to associate with plasma platelets and mediate phospholipid exchange between the inner and outer leaflets, an action similar to scramblase (42) but calcium independent. PLTP may also transfer PS from lipoproteins in the circulation to the membrane of platelets. Thus, the actual physiological consequences of PLTP-mediated PS externalization in platelets deserve further investigation.
Because platelets contain PLTP (Fig. 7), our results may be related to endogenous PLTP (originated by platelets) or exogenous PLTP (from the circulation). However, the results of Figure 3D and 3E, in which PLTP-deficient mouse PRP was treated with rPLTP, indicated that both ADP- and collagen-mediated platelet aggregations were significantly induced in a dose-dependent manner, suggesting that exogenous PLTP or secreted PLTP exerts its effect on platelets. Another uncertainty is that we could not exclude a potential effect of rPLTP on platelet activation which is not related to its phospholipid transfer activity. However, we showed that active rPLTP has a significant stronger ability to bind on the surface of human platelet than that of inactive one (Fig. 5), suggesting that both PS externalization and PS transfer activity, from platelet membrane to the circulation, may require PLTP activity. This aspect deserves further investigation.
The brain is a highly vascularized organ, and under certain stimulated conditions the volume of infarcted tissue is a reliable measure of the extent of thrombosis in the vascular network (47). The photothrombotic stroke mouse model is a well-established means of inducing ischemic damage within a given cortical area via photo-activation of a previously injected light-sensitive dye (48). We compared focal cerebral photothrombosis between WT and PLTP KO mice and between WT and PLTP Tg mice. The extent of the in situ infarcted tissue was found to be significantly reduced in PLTP KO mice compared with WT mice (Fig. 6A and B), and this result confirms the results of a previous study (27). Importantly, we found that PLTP Tg mice had a significantly larger infarction volume on average compared with WT mice (Fig. 6C and D), suggesting that, indeed, PLTP is involved in hypercoagulation.
PLTP activity was correlated with the inflammatory marker CRP in acute phase response (49) and in patients with cardiovascular diseases (1) and type 2 diabetes (50). It has been suggested that increased PLTP activity may enable more transfer of PL to cells to maintain their membrane integrity under conditions of inflammatory stress (51). Present study is particularly compelling when contemplated in the context of a recent publication with regard to the interaction between the different CRP conformations and lipid microparticles (52).
Although the presently known risk factors have some predictive value for CVD, a major part of the variability in predicting CVD remains unexplained. Our finding that PLTP plays a role in platelet aggregation considerably increases the prospect that inhibition of PLTP activity could be a novel therapeutic approach toward mitigating thrombosis. Still, more studies are needed to elucidate all aspects of PLTP function related to CVD.
Supplementary Material
What is known on this topic?
PLTP is a risk factor for atherosclerosis.
Phospholipid transfer protein (PLTP) knockout mice exhibit a longer clotting time.
PLTP is involved in coagulation.
What this paper adds?
PLTP transgenic mice exhibit a shorter clotting time.
PLTP promotes platelet aggregation through inducing membrane phosphatidylserine externalization.
PLTP is involved in hypercoagulation.
Inhibition of PLTP activity could be a novel therapeutic approach toward mitigating thrombosis.
Grants:
This work was supported by China Natural Science Funds (31371190, 91539114, 81173061), Taishan Scholar Foundation of Shandong Province of China (ts201511057), and Natural Science Foundation of Shandong Province of China (ZR2014HQ007, ZR2017MH048), Veterans Affairs Merit award 000900-01, and National Institutes of Health grant R56HL121409.
Abbreviations:
- PLTP
phospholipid transfer protein
- PLTP KO
PLTP gene knockout
- PLTP Tg
PLTP transgenic
- PS
phosphatidylserine
- PRP
platelet-rich plasma
- rPLTP
recombinant PLTP
- WT
wild type
Footnotes
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Kaser S, Sandhofer A, Foger B, et al. Influence of obesity and insulin sensitivity on phospholipid transfer protein activity. Diabetologia 2001; 44(9): 1111–7. [DOI] [PubMed] [Google Scholar]
- 2.Tzotzas T, Dumont L, Triantos A, et al. Early decreases in plasma lipid transfer proteins during weight reduction. Obesity (Silver Spring) 2006; 14(6): 1038–45. [DOI] [PubMed] [Google Scholar]
- 3.Borggreve SE, De Vries R, Dullaart RP. Alterations in high-density lipoprotein metabolism and reverse cholesterol transport in insulin resistance and type 2 diabetes mellitus: role of lipolytic enzymes, lecithin:cholesterol acyltransferase and lipid transfer proteins. Eur J Clin Invest 2003; 33(12): 1051–69. [DOI] [PubMed] [Google Scholar]
- 4.van Tol A Phospholipid transfer protein. Current opinion in lipidology 2002; 13(2): 135–9. [DOI] [PubMed] [Google Scholar]
- 5.Schlitt A, Bickel C, Thumma P, et al. High plasma phospholipid transfer protein levels as a risk factor for coronary artery disease. Arteriosclerosis, thrombosis, and vascular biology 2003; 23(10): 1857–62. [DOI] [PubMed] [Google Scholar]
- 6.de Vries R, Dallinga-Thie GM, Smit AJ, et al. Elevated plasma phospholipid transfer protein activity is a determinant of carotid intima-media thickness in type 2 diabetes mellitus. Diabetologia 2006; 49(2): 398–404. [DOI] [PubMed] [Google Scholar]
- 7.Dullaart RP, van Tol A, Dallinga-Thie GM. Phospholipid transfer protein, an emerging cardiometabolic risk marker: is it time to intervene? Atherosclerosis 2013; 228(1): 38–41. [DOI] [PubMed] [Google Scholar]
- 8.Colhoun HM, Scheek LM, Rubens MB, et al. Lipid transfer protein activities in type 1 diabetic patients without renal failure and nondiabetic control subjects and their association with coronary artery calcification. Diabetes 2001; 50(3): 652–9. [DOI] [PubMed] [Google Scholar]
- 9.Schlitt A, Blankenberg S, Bickel C, et al. PLTP activity is a risk factor for subsequent cardiovascular events in CAD patients under statin therapy: the AtheroGene study. Journal of lipid research 2009; 50(4): 723–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vergeer M, Boekholdt SM, Sandhu MS, et al. Genetic variation at the phospholipid transfer protein locus affects its activity and high-density lipoprotein size and is a novel marker of cardiovascular disease susceptibility. Circulation 2010; 122(5): 470–7. [DOI] [PubMed] [Google Scholar]
- 11.Robins SJ, Lyass A, Brocia RW, et al. Plasma lipid transfer proteins and cardiovascular disease. The Framingham Heart Study. Atherosclerosis 2013; 228(1): 230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavusoglu E, Marmur JD, Chhabra S, et al. Elevated baseline plasma phospholipid protein (PLTP) levels are an independent predictor of long-term all-cause mortality in patients with diabetes mellitus and known or suspected coronary artery disease. Atherosclerosis 2015; 239(2): 503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yatsuya H, Tamakoshi K, Hattori H, et al. Serum phospholipid transfer protein mass as a possible protective factor for coronary heart diseases. Circ J 2004; 68(1): 11–6. [DOI] [PubMed] [Google Scholar]
- 14.Huuskonen J, Ekstrom M, Tahvanainen E, et al. Quantification of human plasma phospholipid transfer protein (PLTP): relationship between PLTP mass and phospholipid transfer activity. Atherosclerosis 2000; 151(2): 451–61. [DOI] [PubMed] [Google Scholar]
- 15.Dullaart RP, De Vries R, Scheek L, et al. Type 2 diabetes mellitus is associated with differential effects on plasma cholesteryl ester transfer protein and phospholipid transfer protein activities and concentrations. Scand J Clin Lab Invest 2004; 64(3): 205–15. [DOI] [PubMed] [Google Scholar]
- 16.Ruhling K, Lang A, Richard F, et al. Net mass transfer of plasma cholesteryl esters and lipid transfer proteins in normolipidemic patients with peripheral vascular disease. Metabolism 1999; 48(11): 1361–6. [DOI] [PubMed] [Google Scholar]
- 17.Schgoer W, Mueller T, Jauhiainen M, et al. Low phospholipid transfer protein (PLTP) is a risk factor for peripheral atherosclerosis. Atherosclerosis 2008; 196(1): 219–26. [DOI] [PubMed] [Google Scholar]
- 18.Jiang XC, Qin S, Qiao C, et al. Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat Med 2001; 7(7): 847–52. [DOI] [PubMed] [Google Scholar]
- 19.van Haperen R, van Tol A, van Gent T, et al. Increased risk of atherosclerosis by elevated plasma levels of phospholipid transfer protein. The Journal of biological chemistry 2002; 277(50): 48938–43. [DOI] [PubMed] [Google Scholar]
- 20.Deckert V, Kretz B, Habbout A, et al. Development of Abdominal Aortic Aneurysm Is Decreased in Mice with Plasma Phospholipid Transfer Protein Deficiency. Am J Pathol 2013. [DOI] [PubMed] [Google Scholar]
- 21.Masson D, Deckert V, Gautier T, et al. Worsening of Diet-Induced Atherosclerosis in a New Model of Transgenic Rabbit Expressing the Human Plasma Phospholipid Transfer Protein. Arterioscler Thromb Vasc Biol. 2011;31:766–74. [DOI] [PubMed] [Google Scholar]
- 22.Manchekar M, Liu Y, Sun Z, et al. Phospholipid transfer protein plays a major role in the initiation of apolipoprotein B-containing lipoprotein assembly in mouse primary hepatocytes. The Journal of biological chemistry 2015;290(13): 8196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yazdanyar A, Jiang XC. Liver phospholipid transfer protein (PLTP) expression with a PLTP-null background promotes very low-density lipoprotein production in mice. Hepatology 2012; 56(2): 576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang XC, Tall AR, Qin S, et al. Phospholipid transfer protein deficiency protects circulating lipoproteins from oxidation due to the enhanced accumulation of vitamin E. The Journal of biological chemistry 2002; 277(35): 31850–6. [DOI] [PubMed] [Google Scholar]
- 25.Yang XP, Yan D, Qiao C, et al. Increased atherosclerotic lesions in apoE mice with plasma phospholipid transfer protein overexpression. Arteriosclerosis, thrombosis, and vascular biology 2003; 23(9): 1601–7. [DOI] [PubMed] [Google Scholar]
- 26.Klein A, Deckert V, Schneider M, et al. Alpha-tocopherol modulates phosphatidylserine externalization in erythrocytes: relevance in phospholipid transfer protein-deficient mice. Arteriosclerosis, thrombosis, and vascular biology 2006; 26(9): 2160–7. [DOI] [PubMed] [Google Scholar]
- 27.Desrumaux C, Deckert V, Lemaire-Ewing S, et al. Plasma phospholipid transfer protein deficiency in mice is associated with a reduced thrombotic response to acute intravascular oxidative stress. Arteriosclerosis, thrombosis, and vascular biology 2010; 30(12): 2452–7. [DOI] [PubMed] [Google Scholar]
- 28.Oslakovic C, Krisinger MJ, Andersson A, et al. Anionic phospholipids lose their procoagulant properties when incorporated into high density lipoproteins. The Journal of biological chemistry 2009; 284(9): 5896–904. [DOI] [PubMed] [Google Scholar]
- 29.Deguchi H, Wolfbauer G, Cheung MC, et al. Inhibition of thrombin generation in human plasma by phospholipid transfer protein. Thrombosis journal 2015; 13: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Haperen R, van Tol A, Vermeulen P, et al. Human plasma phospholipid transfer protein increases the antiatherogenic potential of high density lipoproteins in transgenic mice. Arteriosclerosis, thrombosis, and vascular biology 2000; 20(4): 1082–8. [DOI] [PubMed] [Google Scholar]
- 31.Jiang XC, Bruce C, Mar J, et al. Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. The Journal of clinical investigation 1999; 103(6): 907–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukunaga K, Shimoyama T, Yamaji K, et al. In vitro comparison study of CD63 and CD62P expression after contacting leukocyte filters. Artificial organs 1999; 23(1): 108–13. [DOI] [PubMed] [Google Scholar]
- 33.Briede JJ, Wielders SJ, Heemskerk JW, et al. von Willebrand factor stimulates thrombin-induced exposure of procoagulant phospholipids on the surface of fibrin-adherent platelets. Journal of thrombosis and haemostasis : JTH 2003; 1(3): 559–65. [DOI] [PubMed] [Google Scholar]
- 34.Das B, Yeger H, Tsuchida R, et al. A hypoxia-driven vascular endothelial growth factor/Flt1 autocrine loop interacts with hypoxia-inducible factor-1alpha through mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 pathway in neuroblastoma. Cancer research 2005; 65(16): 7267–75. [DOI] [PubMed] [Google Scholar]
- 35.Bhatt AJ, Amin SB, Chess PR, et al. Expression of vascular endothelial growth factor and Flk-1 in developing and glucocorticoid-treated mouse lung. Pediatric research 2000; 47(5): 606–13. [DOI] [PubMed] [Google Scholar]
- 36.Whelihan MF, Zachary V, Orfeo T, et al. Prothrombin activation in blood coagulation: the erythrocyte contribution to thrombin generation. Blood 2012; 120(18): 3837–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toti F, Satta N, Fressinaud E, et al. Scott syndrome, characterized by impaired transmembrane migration of procoagulant phosphatidylserine and hemorrhagic complications, is an inherited disorder. Blood 1996; 87(4): 1409–15. [PubMed] [Google Scholar]
- 38.Wilson MJ, Richter-Lowney K, Daleke DL. Hyperglycemia induces a loss of phospholipid asymmetry in human erythrocytes. Biochemistry 1993; 32(42): 11302–10. [DOI] [PubMed] [Google Scholar]
- 39.Mallat Z, Hugel B, Ohan J, et al. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation 1999; 99(3): 348–53. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki J, Denning DP, Imanishi E, et al. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 2013; 341(6144): 403–6. [DOI] [PubMed] [Google Scholar]
- 41.Seigneuret M, Devaux PF. ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. Proceedings of the National Academy of Sciences of the United States of America 1984; 81(12): 3751–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lhermusier T, Chap H, Payrastre B. Platelet membrane phospholipid asymmetry: from the characterization of a scramblase activity to the identification of an essential protein mutated in Scott syndrome. Journal of thrombosis and haemostasis : JTH 2011; 9(10): 1883–91. [DOI] [PubMed] [Google Scholar]
- 43.Bruce C, Beamer LJ, Tall AR. The implications of the structure of the bactericidal/permeability-increasing protein on the lipid-transfer function of the cholesteryl ester transfer protein. Curr Opin Struct Biol 1998; 8(4): 426–34. [DOI] [PubMed] [Google Scholar]
- 44.Massey JB, Hickson D, She HS, et al. Measurement and prediction of the rates of spontaneous transfer of phospholipids between plasma lipoproteins. Biochim Biophys Acta 1984; 794(2): 274–80. [DOI] [PubMed] [Google Scholar]
- 45.Oram JF, Wolfbauer G, Tang C, et al. An amphipathic helical region of the N-terminal barrel of phospholipid transfer protein is critical for ABCA1-dependent cholesterol efflux. The Journal of biological chemistry 2008; 283(17): 11541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu Y, Guo S, Feng Y, et al. Phospholipid transfer protein deficiency decreases the content of S1P in HDL via the loss of its transfer capability. Lipids 2014; 49(2): 183–90. [DOI] [PubMed] [Google Scholar]
- 47.Van Reempts J, Borgers M. Histopathological characterization of photochemical damage in nervous tissue. Histology and histopathology 1994; 9(1): 185–95. [PubMed] [Google Scholar]
- 48.Labat-gest V, Tomasi S. Photothrombotic ischemia: a minimally invasive and reproducible photochemical cortical lesion model for mouse stroke studies. Journal of visualized experiments : JoVE 2013(76). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levels JH, Pajkrt D, Schultz M, et al. Alterations in lipoprotein homeostasis during human experimental endotoxemia and clinical sepsis. Biochimica et biophysica acta 2007; 1771(12): 1429–38. [DOI] [PubMed] [Google Scholar]
- 50.Tan KC, Shiu SW, Wong Y, et al. Plasma phospholipid transfer protein activity and subclinical inflammation in type 2 diabetes mellitus. Atherosclerosis 2005; 178(2): 365–70. [DOI] [PubMed] [Google Scholar]
- 51.Barlage S, Frohlich D, Bottcher A, et al. ApoE-containing high density lipoproteins and phospholipid transfer protein activity increase in patients with a systemic inflammatory response. Journal of lipid research 2001; 42(2): 281–90. [PubMed] [Google Scholar]
- 52.Braig D, Nero TL, Koch HG, et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nature communications 2017; 8: 14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.