

Abstract
Quiescent and contractile VSMC can switch to proliferative and migratory phenotype in response to growth factors and cytokines, an effect underscored by Nox family NADPH oxidases, particularly Nox1. We previously showed that quiescin/sulfhydryl oxidase 1 (QSOX1) has a role in neointima formation in balloon-injured rat carotid. Here, we investigated the intracellular redox mechanisms underlying these effects in primary VSMC. Our results show that exogenous incubation with wild type QSOX1b (wt QSOX), or with secreted QSOX1, but not with the inactive C452S QSOX 1b (C452S QSOX) or secreted inactive C455S QSOX1, induces VSMC migration and chemotaxis. PEG-catalase (PEG-CAT) prevented, while PEG-superoxide dismutase (PEG-SOD) increased migration induced by wt QSOX. Moreover, wt QSOX-induced migration was abrogated in NOX1-null VSMC. In contrast, both wt QSOX and C452S QSOX, and both secreted QSOX1 and C455S QSOX1, induce cell proliferation. Such effect was unaltered by PEG-CAT, while being inhibited by PEG-SOD. However, QSOX1-induced proliferation was not significantly affected in NOX1-null VSMC, compared with WT VSMC. These results indicate that hydrogen peroxide and superoxide mediate, respectively, migration and proliferation. However, Nox1 was required only for QSOX1-induced migration. In parallel, QSOX1-induced proliferation was independent of its redox activity, although mediated by intracellular superoxide.
Keywords: quiescin/sulfhydryl oxidase 1b, smooth muscle cells, Nox1, hydrogen peroxide, superoxide, cell migration
1. Introduction
Vascular smooth muscle cells (VSMC) migration and proliferation are key processes underlying vascular diseases such as atherosclerosis and restenosis. Quiescent and contractile VSMC can switch to a proliferative, migratory and synthetic phenotype in response to extracellular matrix components, mechanical force, mitogenic and chemotactic growth factors, cytokines and other small molecules [1]. These agents are often released from the injured artery and activate receptors located on the cell surface, which transduce the external signal to several pathways, leading to events that trigger cell migration, proliferation or both. Although molecular players that promote proliferation and migration are diverse [2,3], redox signaling has emerged as a fine tune regulator of both processes [4,5]. In the vessel wall, NADPH oxidases are major sources of the signaling redox species superoxide and hydrogen peroxide. NADPH oxidases are multisubunit enzymes in which the catalytic subunit consists of one of the Nox proteins [6]. Rodent VSMC from large arteries express only Nox1 and Nox4 [7]. Nox1 is the inducible isoform and is activated at multiple levels after growth factor and hormone stimulation, producing superoxide, whereas Nox4 has a constitutive expression and activity, producing hydrogen peroxide [8,9].
QSOX1 is a flavoprotein composed of two sequential thioredoxin-like domains, followed by a helix-rich region and a FAD-bound Erv/ALR (augmenter of liver regeneration) domain [10]. It contains three CxxC motifs, located at the first thioredoxin-like domain, at the Erv/ALR domain and immediately after the Erv/ALR domain. QSOX1 catalyzes disulfide formation, by reducing molecular oxygen to hydrogen peroxide. Heckler et al. demonstrated, in recombinant human QSOX1, that the first two CxxC motifs are essential to oxidize model substrates, such as dithiothreitol (DTT) or reduced RNAse, since single mutations in the cysteine residues of the CxxC motifs result in loss of activity, particularly from the second CxxC motif [11]. According to the proposed model, the reducing equivalents are transferred from the substrate thiols to the first CxxC disulfide, then to the second, to FAD and finally to molecular oxygen [11]. There are two QSOX1 isoforms: QSOX1b is the secreted short isoform, while QSOX1a is the Golgi-localized isoform [10]. QSOX1b has been found in chicken egg white [12], supernatant of fibroblasts [13,14] and smooth muscle progenitor cells [15], rat seminal fluid [16], normal serum [17,18], and in higher levels in serum from oncological [19] or from heart failure [20] patients. A QSOX1-knockout mouse was recently characterized [21]. These mice develop dilated cardiomyopathy associated with ER stress, although its extracellular function has not been assessed [21]. Interestingly, active extracellular QSOX1 mediates laminin trimer incorporation into extracellular matrix, promoting a proadhesive and promigratory surface [14], as well as invasion of cancer cells through matrigel [22]. Recently, using a loss-of-function approach we demonstrated the contribution of QSOX1 in neointima formation after balloon injury in rat carotid [23]. Moreover, overexpression of QSOX1b protein promoted both proliferation and migration of VSMC in vitro [23]. Thus, the aim of the current study was to dissect the redox-sensitive pathways involved in QSOX1b-induced proliferation and migration in primary VSMC.
2. Material and Methods
Constructs to eukaryotic expression of wt QSOX-HA and C455S QSOX-HA
Mouse seminal vesicle cDNA was synthesized from total RNA with Impron II™-reverse transcriptase (Promega) and oligo(dT) (Invitrogen) and used to clone the whole CDS of QSOX1b (NM_023268.2) into pCDNA3.1(−) vector (Invitrogen), as QSOX1b-HA (herein referred to as wt QSOX-HA). The hemagluttinin (HA) tag was inserted by adding its sequence into the reverse primer. Forward and reverse primers sequences were: 5’-CTCGAATTCACCATGAGGAGGTGCGGCCGC-3’ and 5’-CTCAAGCTTTCACGCATACGGCACATCATACGGATACAAGAGCAGCTCGGGGGTAG-3’. The amplicon was inserted into EcoRI and HindIII restriction sites. Competent Nova Blue™ cells (Novagen) were used to amplify the plasmids. To obtain the C455S (CxxS) mutant (herein referred to as C455S QSOX-HA), mutagenic primers were designed with the Primer X software. Mutagenic forward and reverse primers sequences were: 5’-CTTTGGCTGTCGTGACAGTGCGGACAATTTG-3’ and 5’-CAAAATGGTCCGCACTGTCACGACAGCCAAAG-3’). The wild type sequence was used as template, and amplification was carried out with 15 cycles by Pfu Ultra High Fidelity DNA polymerase AD (Agilent). After DpnI (Fermentas) digestion, competent Nova Blue cells (Novagen) were transformed. The sequences were confirmed by sequencing.
Cell cultures
Human embryonic kidney cell line HEK 293T was cultured in MEM containing 10% fetal bovine serum (FBS, Gibco) and 40 μg/mL gentamycin. Vascular smooth muscle cell (VSMC) was obtained by the explant method from aortas from male Wistar rats weighing 180-220 g (about 2 months of age). This protocol was approved by the Research Ethics Committee (CEUA) of the Biological Sciences Building, protocol number 882/2015. Confirmation of the smooth muscle cell identity was obtained by immunofluorescence with anti-smooth muscle actin (1:250; Sigma-Aldrich A5228). Aortic smooth muscle cells from male NOX1 null (Nox1−/y) and control wild-type (Nox1+/y) mice were mantained in DMEM containing 10% FBS and 40 μg/mL gentamycin. Cells were maintained in a humid atmosphere at 37 °C with 5% carbon dioxide. All experiments were performed after 16-24 h in DMEM (Gibco) containing 0.1% FBS and antibiotics. Low serum was used to induce cell quiescence, to induce PDGF receptor expression and to minimize the contribution of serum QSOX1 activity [17].
Conditioned medium (CM) from QSOX-HA overexpressing-cells
HEK293T cells (~1x106 cells/P100) were transfected with 12 μg plasmids encoding wt QSOX-HA or C455S QSOX-HA by calcium phosphate or Lipofectamine 2000 (Life Technologies). As a negative control, cells were transfected with pEGFP-C1 (Clontech). Five hours after the transfection, the transfection medium was changed to DMEM (Gibco) containing 10% FBS and incubated for 16 h. After this period, the medium was replaced by 4 mL of DMEM containing 0.1% FBS, which was mantained for additional 48 h to accumulate secreted QSOX1b.
Production of the recombinant mouse QSOX1b
Recombinant wild type short isoform QSOX1b (herein referred to as wt QSOX) and SxxC mutant inactive enzyme (herein referred to as C452S QSOX) were obtained as described [23]. Briefly, competent AD494 E. coli cells (Novagen) were transformed with pET32a-wt QSOX1b [24] or pET32a-C452S QSOX1b [23] by electroporation or heat shock and expression was induced by 0.1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) overnight at 20°C. Cells were lysed by French press in native buffer (50 mM phosphate buffer pH 8.0, 500 mM sodium chloride) containing 10 mM imidazole and 50 μM FAD, and the proteins were purified in nickel-agarose beads (Qiagen). Elution buffer consisted of 250 mM imidazole in native buffer. Recombinant proteins were dialyzed against PBS and stored in 50 mM phosphate buffer pH 7.5, containing 1 mM EDTA and 10% glycerol [11]. Proteins were stored at −20 °C or −80 °C.
QSOX activity
DTT oxidase activity was determined as previously described [25]. Briefly, the reaction mixture consisted of 0.2 μM QSOX (or 10 μL of CM), 1.4 μM HRP, 1 mM homovanillic acid in 50 mM phosphate buffer containing 0.3 mM EDTA pH 7.5, at 27°C. Reaction was started by addition of 150 μM dithiothreitol (DTT, Sigma). Fluorescence was monitored in a Tecan microplate reader (λem = 420 nm, λexc = 320 nm). Hydrogen peroxide concentration was determined by a calibration curve produced with (0-2.0 μM) hydrogen peroxide solutions. In some experiments with recombinant proteins, diphenylene iodonium (DPI, Sigma) was added at 20, 40 or 60 μM.
Scratch assay
The scratch migration assay was performed as previously described [26]. VSMC (25,000) were seeded in 24-well plates in complete medium. After cell attachment, medium was changed to DMEM with 0.1% FBS for 16 hours. A P-200 pipette tip was used to scratch the dish surface to generate a “wound” followed by two washes with PBS. The cells were then treated with 50 nM recombinant proteins, 20 ng/mL PDGF-BB (Proteintech; herein referred to as PDGF) in DMEM with 0.1% FBS or with vehicle (0.1% FBS). The wounds were photographed before (time 0) and 24 h after treatments. In some assays, cells were pre-incubated for 30 min with 200 U/mL polyethylene glycol-catalase (PEG-CAT, Sigma), 25 U/mL polyethylene glycol-superoxide dismutase (PEG-SOD, Sigma), 25 U/mL bovine liver SOD (Sigma), 20 μM DPI, 150 nM ML171 (a gift from Dr Lucia Lopes, University of São Paulo, Brazil), or 0.4 μg/mL mitomycin C (Roche). This assay was also performed with the conditioned media (CM) of HEK 293T cells overexpressing wt QSOX-HA, C455S QSOX-HA or GFP. The CM were added directly to wounded VSMC cultures. Migration distance was calculated as the difference between the edges of the scratch measured at times 0 and 24 h.
Transwell assay
We used a transwell device with a 8 μm-pores polycarbonate membrane (#140620 Nunc). The under side of the insert was coated with 10 μg/mL rat tail type I collagen (Gibco) in PBS for 30 min [27]. After ~15 min, the insert was seeded with 50,000 VSMC, previously cultivated in DMEM with 0.1% FBS for 16 hours. The lower part of the chamber was filled with 50 nM recombinant proteins or 20 ng/mL PDGF-BB in DMEM with 0.1% FBS or with vehicle. This assay was also performed with CM of HEK 293T cells overexpressing wt QSOX-HA, C455S QSOX-HA, or GFP. After 4 hours, cells from the upper side of the insert were removed with a cotton swab and the migrating cells were washed with PBS, permeabilized with methanol (5 min) and stained with 1 μg/mL DAPI for 5 min. Images were acquired on an inverted fluorescence microscope (Nikon Eclipse TS100) at 4x magnification and counting of migrating cells was performed in ImageJ in 4 fields.
Proliferation assay
VSMC (8,000) were seeded in 96-well plates in complete medium. After adhesion and 16-24 h starvation, they were incubated with 50, 75 and 100 nM recombinant proteins in DMEM with 0.1% FBS, 10% FBS or with vehicle for 48 h. Cells were then gently washed with PBS, fixed with 4% paraformaldehyde for 10 min, permeabilized with 2% methanol for 10 min and stained with 0.5% crystal violet (in 20 % methanol) for 10 min. The stain was removed with several washes of tap water and crystals were dissolved with 100 μL 0.1 M sodium citrate pH 4.2 in 50% methanol. Absorbance at 570 nm was determined in a microplate reader (BioRad). In some experiments, cells were preincubated with the same agents described for migration assay.
Catalase and superoxide dismutase activities
VSMC (5 x 105) were seeded in 60 mm-plates. On the next day, cells were treated or not with 200 U/mL PEG-CAT, 200 U/mL catalase or 25 U/mL PEG-SOD, for 30 minutes, in DMEM containing 10 % FBS. After this period, they were rinsed with PBS, pelleted, lysed (freeze-thawing) in PBS containing a protease inhibitor cocktail, and the supernatant (9,000 xg, 20 min at 4°C) was collected. Catalase activity was measured by decomposition of 10 mM hydrogen peroxide in PBS, by 5 μL of protein extract, monitored at 240 nm in a spectrophotometer (Hitachi U-2900) [28]. As a positive control of the reaction, the cell supernatant was replaced by 1 U/mL PEG-CAT. SOD activity in cell lysates (100 μg protein in 30 μL), or in SOD solution, was determined by the ability to inhibit nitroblue tetrazolium (NBT) reduction by the superoxide produced by autooxidation of hydroxylamine in alkaline solution [29]. NBT reduction was followed at 560 nm. The absorbances were measured at times 0, 30 and 60 min and normalization was performed by quantification of total proteins with Bradford reagent.
Western blotting
Proteins from the CM or wt QSOX were separated by SDS-PAGE 10% and subjected to Western blotting. Proteins were electrotransferred to nitrocellulose membrane, which was blocked with 5% nonfat milk, and incubated with anti-QSOX1 (1:500) [24]. Anti-rabbit IgG, coupled to HRP (1:4,000, A0545 Sigma) was used as secondary antibody. Reactions were developed with chemiluminescence kit (West or Femto Pico, Pierce), and detected with autoradiogram films (Carestream). The densitometry analysis was done with ImageJ (NIH).
Intracellular hydrogen peroxide
PEG-CAT-inhibitable fraction of dichlorofluorescein (DCF) intracellular fluorescence was calculated and considered as an estimate of hydrogen peroxide levels. VSMC (10,000) were seeded in 96-well black microplate with clear flat bottom (Corning) in complete medium. After attachment and starvation, cells were incubated with 20 μM 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) for 20 minutes, rinsed twice with PBS and then treated with 20 ng/mL PDGF-BB, 50 nM wt QSOX or 50 nM C452S QSOX in PBS, in the absence or in the presence of 200 U/mL PEG-CAT. In one experiment set, cells were treated only with 25 U/mL PEG-SOD, in the absence or in the presence of PEG-CAT. Fluorescence was measured at 0 (basal), 15 and 30 minutes after treatment in a microplate reader (Tecan, Infinite M200) with excitation at 488 nm and emission at 525 nm. The basal fluorescences were subtracted from the fluorescences at 15 or 30 minutes, and each condition was subtracted of its respective fluorescence obtained in the presence of PEG-CAT.
Statistical analysis
The results were reported as mean ± SD of at least 3 independent experiments. Differences between groups were analyzed using one-way ANOVA test with Tukey's post-test. The effect of hydrogen peroxide in DCFH-DA oxidation and the comparison of WT vs NOX1-null cells were analyzed with unpaired t-test. A value of P <0.05 was considered statistically significant.
3. Results
3.1. Sulfhydryl oxidase activity is required for QSOX1b-induced VSMC migration
We had previously shown that QSOX1b expression levels positively correlate with VSMC migratory capacity, and that recombinant mouse QSOX1b (wt QSOX), but not the inactive enzyme (C452S QSOX), induces VSMC migration when added into the culture medium [23]. Here, we performed a scratch assay to confirm that incubation of VSMC with 50 nM wt QSOX significantly enhanced cell migration by 2.5-fold that of the untreated control, while the inactive recombinant had no effect (Figure 1A). PDGF, a mitogen and chemoattractant for VSMC, at 20 ng/mL, significantly increased cell migration by 3.5-times (Figure 1A). We also assessed cell migration by using the transwell assay. The presence of 20 ng/mL PDGF in the lower chamber led to a significant 2.5-fold increase in migrating cell number in comparison with the untreated control (Figure 1B). When 50 nM wt QSOX was present in the lower chamber, a significant increase of ~2 fold was observed, compared with the control. But no effect was detected with 50 nM C452S QSOX (Figure 1B). To control for a possible contribution of cell proliferation, migration assay was also performed in the presence of mitomycin C, an inhibitor of DNA synthesis. The results obtained showed that VSMC migration was not significantly affected by pretreatment with mitomycin C (Suppl. Figure 1), indicating that proliferation was not interfering with the migration results.
Figure 1. Effect of wild type and inactive QSOX1b in VSMC migration and proliferation.
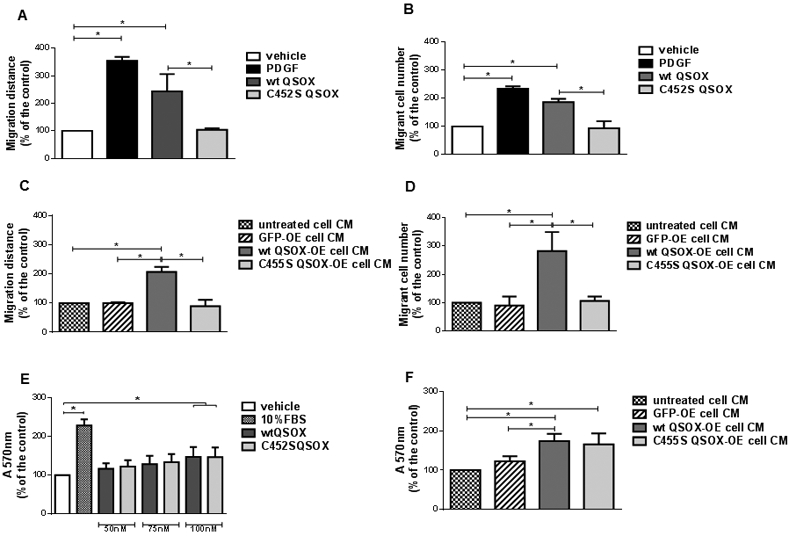
VSMC migration was determined by scratch (A, C) and by transwell (B, D) assays, while proliferation was determined by crystal violet staining (E, F). VSMC were exposed for 24 h (A) or for 4 h (through a 8 μm-membrane of a transwell device) (B) to vehicle (DMEM containing 0.1% FBS), 20 ng/mL PDGF-BB, 50 nM wt QSOX or 50 nM C452S QSOX, after which the migration distance or the number of migrant cells were measured, respectively. Alternatively, VSMC were exposed for 24 h (C) or for 4 h (through a 8 μm-membrane of a transwell device) (D) to conditioned media (CM) from untreated HEK293T cells, or from HEK293T transfected with a pEGFP, wt QSOX-HA or C455S QSOX constructs, after which the migration distance or the number of migrant cells were measured, respectively. For proliferation assay, VSMC were treated with vehicle, 10% FBS or 50, 75 or 100 nM wt QSOX or C452S QSOX for 48 h (E), or with 48 h-CM from non-transfected HEK293T, or from HEK293T transfected with pEGFP, wt QSOX-HA or C455S QSOX-HA constructs for additional 48 h (F). Data are mean ± SD of 3 independent experiments. *P<0.05
Given that recombinant proteins produced by E. coli may have aberrant biochemical properties, due to lack of glycosylation [16] for instance, we performed the same experiments with conditioned medium (CM) from HEK293T overexpressing (OE) either wt QSOX-HA or an inactive mutant C455S QSOX-HA. CM of non-transfected HEK293T and HEK293T transfected with pEGFP were used as negative controls. The secretion of the QSOX-HA into the medium was demonstrated by immunoblotting (Suppl. Figure 2). QSOX-HA concentration in CM was estimated by comparing the band density with bands of known amounts of wt QSOX detected under the same conditions, and it was found to be in the order of ~ 200 nM for both proteins (Suppl. Figure 2). In both scratch (Figure 1C) and transwell (Figure 1D) assays, wt QSOX-HA CM promoted significant enhancements of VSMC migration of ~2-2.5-fold that observed for the control, while C455S QSOX-HA did not. Recombinant wt QSOX and CM from cells overexpressing wt QSOX-HA were enzymatically active, while C452S QSOX and CM from cells overexpressing C455S QSOX-HA were inactive (Suppl. Figure 3). Overall, these data confirm that extracellular QSOX1b has a promigratory effect in VSMC, which requires QSOX1b enzymatic activity.
3.2. Both active and inactive QSOX1b contribute to VSMC proliferation
QSOX1b expression levels also positively correlated with VSMC proliferation as measured by crystal violet assay [23]. Furthermore, the addition of both wt and C452S QSOX in culture medium induced VSMC proliferation [23], as confirmed here (Figure 1E). Fetal bovine serum (10%), used as a mitogenic stimulus, promoted a significant 2.5-fold increase in cell number, when compared with the control condition. Recombinant wt and C452S QSOX proteins induced a concentration-dependent increase in cell number, with a significant increase of ~1.5-fold observed only at 100 nM of both recombinant QSOX, compared with the untreated cells (Figure 1E). Similar result was obtained by counting cells in a Neubauer chamber2. Conditioned media of non-transfected HEK293T or pEGPF-transfected cells had no effect in proliferation, while CM from cells overexpressing either wt or C455S QSOX-HA induced a significant increase in crystal violet staining, compared with the non-transfected or GFP CM (Figure 1F). Interestingly, the mitogenic action of QSOX1b does not depend on the sulfhydryl oxidase activity, and both active and inactive enzymes promote proliferation to the same extent. Overall, the results indicate that QSOX1b-induced proliferation is independent of its enzymatic activity.
3.3. Intracellular hydrogen peroxide is required for migration, but not for proliferation
Reactive oxygen species (ROS), particularly hydrogen peroxide and superoxide, have been shown to be involved in VSMC migration and proliferation, elicited by growth factors, cytokines and vasoactive hormones. Thus, we investigated whether ROS could be involved in QSOX1b signaling. We first investigated the effect of intracellular catalase activity, by pre-incubating PEG-CAT with cells. Conjugation of both CAT and SOD with PEG has been shown to enhance their cellular enzymatic activities and the cellular resistance to oxidative stress [30]. Migration induced by PDGF and by wt QSOX was significantly inhibited by PEG-CAT (Figure 2A). PEG-CAT inhibited proliferation induced by 10% FBS, but not that induced by wt or by C452S QSOX (Figure 2B). Catalase added to the culture medium did not affect migration (Suppl. Figure 4A) induced by PDGF or wt QSOX, or proliferation induced by FBS, wt QSOX or C452S QSOX (Suppl. Figure 4B), indicating that extracellular hydrogen peroxide is involved neither in migration nor in proliferation. To confirm that PEG-CAT was properly incorporated into the cells, the intracellular catalase activity was determined. An increased activity was measured in cell lysates from VSMC incubated with PEG-CAT, but not with catalase (Suppl. Figure 5), validating that intracellular hydrogen peroxide has a role in the migration induced by QSOX1b. The oxidase activity of extracellular QSOX1b could be the source of this peroxide, which could diffuse into the cell. However, the lack of effect of exogenous catalase weakens this hypothesis. Proliferation, in contrast, was not affected by intracellular or extracellular hydrogen peroxide.
Figure 2. Role of intracellular hydrogen peroxide in VSMC migration and proliferation induced by QSOX1b.
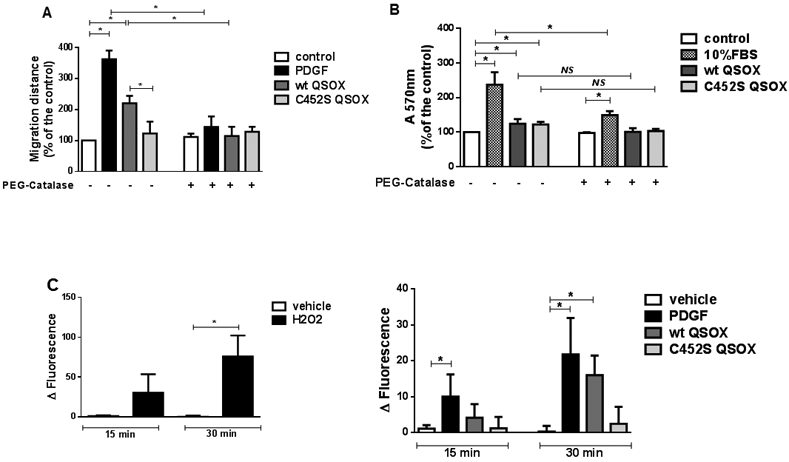
The effect of PEG-catalase (PEG-CAT) in VSMC migration (A) and proliferation (B) and the amount of DCF fluorescence inhibited by PEG-CAT (C) was assessed. PEG-CAT (200 U/mL) was pre-incubated for 30 min before the addition of vehicle (DMEM containing 0.1% FBS), 20 ng/ mL PDGF, 50 nM QSOX or 50 nM C452S QSOX (A) or before the addition of 10% FBS, 100 nM wt QSOX or 100 nM C452S QSOX (B). Migration was determined by the scratch assay and proliferation was determined by crystal violet staining, after 24 h- or 48 h-treatment, respectively. Amounts of intracellular H2O2 were estimated as PEG-CAT-inhibitable fraction (Δ fluorescence), which corresponds to the difference between the DCF fluorescence measured in the absence and in the presence of PEG-CAT. Intracellular H2O2 levels were measured in VSMC incubated for 15 and 30 min with 0.5 mM H2O2 (C, left panel), or with vehicle (DMEM containing 0.1% FBS), 20 ng/mL PDGF-BB, 50 nM wt QSOX or 50 nM C452S QSOX (C, right panel). Data are mean ± SD of 3 independent experiments. *P<0.05; NS: not significant.
Then, we used DCFH-DA to assess the levels of intracellular hydrogen peroxide in VSMC. Although this probe is not specific for this oxidant, we measured the PEG-CAT-inhibitable fraction of fluorescence as an estimate of intracellular hydrogen peroxide amounts. As a control, we incubated VSMC with or without 0.5 mM hydrogen peroxide (Figure 2C, left panel). A significant increase in DCF signal was measured after 30 min, compared with cells incubated only with vehicle and DCFH-DA. Levels of hydrogen peroxide were significantly increased in VSMC incubated with PDGF, but not with wt or C452S QSOX, after 15 min, compared with the control cells (Figure 2C, right panel). After 30 min, incubations with PDGF and wt QSOX led to significant increases in hydrogen peroxide levels compared with control cells (Figure 2C, right panel), confirming that production of this oxidant occurs during the first 30 min. These results indicate that PDGF and wt, but not C452S QSOX, induce intracellular hydrogen peroxide accumulation early after 30 minutes.
3.4. Superoxide-derived hydrogen peroxide from Nox1 is required for migration, but Nox1 is not the exclusive source of superoxide for proliferation
Hydrogen peroxide can be produced by several cellular sources, such as superoxide-producing enzymes, followed by superoxide dismutation. To assess the role of intracellular superoxide in migration and proliferation, we used PEG-SOD. Increased SOD activity in cells was demonstrated by the inhibition in NBT reduction rate by lysates of cells treated with PEG-SOD, in comparison with lysates of untreated cells (Suppl. Figure 6). PEG-SOD enhanced significantly migration of VSMC under all conditions, including the basal untreated condition (Figure 3A). This result agrees with the interpretation that migration is promoted by intracellular superoxide-derived hydrogen peroxide. Indeed, the ability of PEG-SOD-treated cells to increase the amount of intracellular hydrogen peroxide was shown, even under basal metabolism (Suppl. Figure 7), when superoxide production is usually due to mitochondrial electron transport chain. PEG-SOD, however, inhibited the proliferation induced by 10% FBS, wt QSOX and C452S QSOX (Figure 3B), pointing to a role of superoxide anion in proliferation. SOD added to the culture medium did not affect migration (Suppl. Figure 8A) or proliferation (Suppl. Figure 8B), in any condition, indicating that effector superoxide is intracellular. In addition, the fact that C452S QSOX positively regulates proliferation, but does not induce a detectable increase in hydrogen peroxide levels, may indicate that the superoxide production associated with proliferation takes place close to superoxide target, preventing its dismutation to hydrogen peroxide.
Figure 3. Role of superoxide (O2•−) in VSMC migration and proliferation induced by QSOX1b.

The effects of PEG-SOD (A, B), Nox inhibitor diphenylene iodonium (DPI) (C, D) and Nox1-specific inhibitor ML171 (E, F) in VSMC migration (A, C, E) and proliferation (B, D, F) were assessed. PEG-SOD (25 U/mL), DPI (20 μM) and ML171 (150 nM) were pre-incubated for 20 min before the addition of vehicle (DMEM with 0.1% FBS), 20 ng/ mL PDGF, 50 nM QSOX or 50 nM C452S QSOX (A, C, E) or the addition of vehicle, 10% FBS, 100 nM wt QSOX or 100 nM C452S QSOX (B, D, F). Migration was determined by the scratch assay and proliferation was determined by crystal violet staining, after 24 h- or 48 h-treatment, respectively. Data shown correspond to the mean ± SD of at least 3 experiments. *p< 0.05
Controlled ROS production in vascular tissue is prominently due to Nox proteins that compose the NADPH oxidases complexes. Rodent aortic VSMC express only Nox1 and Nox4 [6]. Nox1 is the inducible isoform [8], activated by vasoactive compounds [31], whereas Nox4 is constitutively active in these cells [6]. Thus, we used a general flavoprotein inhibitor diphenyleniodonium (DPI) and ML171, which is considered a more specific Nox1 inhibitor [32]. Migration induced by PDGF and by wt QSOX was suppressed by both inhibitors (Figure 3 C, E). Similarly, proliferation induced by wt and by C452S QSOX was also inhibited by both compounds (Fig 3D, 3F). However, while DPI inhibited proliferation induced by FBS, ML171 did not (Fig 3D, 3F), indicating that FBS induces a mitogenic pathway that does not depend on Nox1. Overall, these data suggest that Nox1 is a critical mediator of QSOX1b-mediated migration and proliferation of VSMC. However, to unequivocally confirm Nox1 contribution to migration and proliferation, we used NOX1-null cells. Our results indicated that Nox1 deficiency significantly inhibited migration induced by PDGF and by wt QSOX, compared with the respective WT cells (Figure 4A), supporting the critical role of Nox1 for the migration activated by PDGF and wt QSOX. Consistently, stimulation with PDGF and wt QSOX, but not with C452S QSOX, induced a significant production of hydrogen peroxide in WT cells, which was abrogated in NOX1-null cells (Figure 4C). Thus, hydrogen peroxide resulting from Nox1 activity parallels the migratory response observed for PDGF and wt QSOX treatments. NOX1-null cells proliferated less than the respective WT cells under all conditions, but those differences were not significant (Figure 4B), which suggests that Nox1 is not essential in QSOX1b-induced proliferation.
Figure 4. Role of Nox1 in VSMC migration, proliferation and hydrogen peroxided levels induced by QSOX1b.

NOX1-null (Nox1−/y) and wt (Nox1+/y) murine smooth muscle cells were treated with vehicle (DMEM containing 0.1% FBS), 20 ng/mL PDGF-BB, 50 nM wt QSOX or 50 nM C452S QSOX for 24 h, after which migration was determined by scratch assay (A). The same cell lines were treated with vehicle (DMEM containing 0.1% FBS), 10% FBS, 100 nM wt QSOX or 100 nM C452S QSOX for 48 h, and proliferation was analyzed by crystal violet assay (B). Amounts of intracellular H2O2 were estimated as PEG-CAT-inhibitable fraction (Δ fluorescence), which corresponds to the difference between the DCF fluorescence measured in the absence and in the presence of PEG-CAT. Intracellular H2O2 levels were measured in VSMC incubated for 30 min with vehicle (DMEM containing 0.1% FBS), 20 ng/mL PDGF-BB, 50 nM wt QSOX or 50 nM C452S QSOX (C). Data are mean ± SD of 4 (A,C) or 3 (B) independent experiments. *P<0.05.
4. Discussion
The results presented here confirmed that E. coli-produced QSOX1b induces VSMC migration and proliferation, by mechanisms dependent and independent of its sulfhydryl oxidase activity, respectively. Also, under our experimental conditions, VSMC migration was modified by QSOX1b to a greater extent than VSMC proliferation. We expanded these observations by showing that mammalian cell-produced QSOX1b promoted the same effects, demonstrating that they were not due to an aberrant QSOX1b folding or lack of glycosylation. In addition, the fact that the sulfhydryl oxidase activity is required for cell migration, but not for cell proliferation, indicates that QSOX1b signaling activates distinct cellular targets, as previously discussed [23]. In this paper, however, we aimed to investigate the intracellular redox signaling activated by extracellular QSOX1b that are associated with VSMC proliferation and migration. Using pharmacological inhibitors and genetically modified cells, we provided evidence that Nox1 is a common point of convergence of both migration and proliferation signaling induced by QSOX1b, although not essential for proliferation. Interestingly, Nox1 expression and activity can be regulated by protein disulfide isomerase (PDI) [33,34], a redox chaperone, from the same CxxC-containing protein superfamily, as QSOX1. Nox1 activation mediates physiopathological VSMC migration and proliferation stimulated by growth factors, cytokines and vasoactive agonists (reviewed by [31]), by producing superoxide anions, that further dismutate to hydrogen peroxide [9]. Nox1-derived ROS are produced either at the extracellular side of the plasma membrane or at the interior of intracellular vesicles after Nox1 endocytosis, depending on the activator [31]. Given that SOD and catalase effects were intracellular, QSOX1b appears to induce Nox1 endocytosis. The essential role of hydrogen peroxide during VSMC migration became clear as catalase inhibited migration induced by PDGF [35,36], thrombin [37] and bFGF [38]. Nox1 was shown to be relevant for migration induced by bFGF [38], PDGF-BB [39] and thrombin [40], indicating that Nox1-derived ROS are mediators of VSMC migration. Consistent with these data, QSOX1b induced an early production of hydrogen peroxide that was prevented in NOX1-null cells, confirming the role of Nox1. The contribution of hydrogen peroxide to VSMC migration was also demonstrated, since migration was prevented when hydrogen peroxide was depleted by PEG-CAT, and was increased when hydrogen peroxide levels were increased by pre-incubation of cells with PEG-SOD. Hydrogen peroxide may oxidize several proteins leading to enhanced motility [3]. For instance, exogenous or Nox1-derived hydrogen peroxide oxidizes 14-3-3 protein, disrupting its association with slingshot-1L phosphatase (SSH1L) [41]. Free SSH1L is activated by auto-dephosphorylation [42], which then triggers cofilin dephosphorylation and activation and finally cell migration. Cofilin itself can also be oxidized by hydrogen peroxide at cysteine residues, leading to a modified activity [43]. Furthermore, Nox1-derived hydrogen peroxide stimulates c-Jun N-terminal kinase (JNK) phosphorylation and activation during bFGF-induced VSMC migration [38], and activates p38 during thrombin-induced VSMC migration [37]. Alternatively, a recent proteomic study showed that hydrogen peroxide can upregulate the expression levels of proteins involved in migration, such as actin-related protein 2/3 complex subunit 2 (ARPC2), by a Nox1-dependent pathway [44].
While migration induced by QSOX1b displayed a clear and expected behavior, proliferation response was more complex. We found that both wild-type and mutant QSOX1b induced a discrete, but significant increase in VSMC proliferation, that was dependent on superoxide, but not on hydrogen peroxide. Several reports demonstrated that VSMC proliferation is redox-regulated. Catalase has been shown to inhibit proliferation induced by PDGF [35], thrombin [45] and EGF [46] indicating that hydrogen peroxide mediates proliferation. However, superoxide production has also been associated with a mitogenic condition, given the inhibitory effects of Cu/Zn-SOD in VSMC proliferation stimulated by xanthine /xanthine oxidase [47] and by EGF [46].
Nox1 has been considered a major source of the superoxide that mediates the signaling elicited by PDGF, angiotensin II (Ang II) [8] and urokinase plasminogen activator [48] in VSMC. NOX1-deficient smooth muscle cells or NOX1-deficient mice displayed an impaired proliferative response to arterial injury [39] and thrombin [40]. Additionally, the positive correlation between the proliferative rate of VSMC with Nox1 expression or with superoxide level supports the idea that Nox1-derived superoxide mediates proliferation [9, 49]. In agreement, we found that both wt and C452S QSOX induced VSMC proliferation, which was inhibited by PEG-SOD, DPI and ML171, thus indicating a dependency on Nox1-derived superoxide. However, proliferation of NOX1-deficient cells, although lesser, was not significantly different from the respective WT control cell, indicating that Nox1 may not be the unique source of superoxide to transduce mitogenic signals. Similarly, Ang II-stimulated VSMC proliferation in aorta of WT animals was not significantly decreased in NOX1-deficient mice [50], suggesting that Nox1 is not essential to Ang II-induced proliferation in those animals. Also, the inability of the C452S QSOX to increase the levels of hydrogen peroxide in WT VSMC (considering that the detected hydrogen peroxide results from Nox1-derived superoxide), with the concomitant ability to induce proliferation, corroborates the idea that superoxide may be produced by alternative sources, close to its target, to induce proliferation. Indeed, it was reported that downregulation of Nox1 with antisense technology led to increased levels of superoxide compared with the non-suppressed cells under basal conditions, suggesting that another source of superoxide may be upregulated to compensate for Nox1 deficiency [8]. Therefore, we can hypothesize that in our case NOX1-null cells might have upregulated this alternative superoxide source, which promoted a proliferation level similar to that of WT cells. In this context, mitochondria-generated superoxide may have a role. A decreased Mn-SOD activity, leading to increased mitochondrial superoxide, increased fibroblast proliferation [51]. Of note, a crosstalk between dysfunctional mitochondria and Nox1 has been reported [52], and a recent paper indicated the participation of superoxide from these two sources in VSMC proliferation [53].
In summary, in the present work we expanded our previous findings and demonstrated that extracellular QSOX1b transduces migratory and mitogenic responses in primary VSMC by distinct pathways. The migratory pathway is triggered by active QSOX1b and depends on hydrogen peroxide from Nox1-derived superoxide. The mitogenic pathway does not depend on QSOX1b oxidase activity, nor on hydrogen peroxide, but on superoxide, whose source is not exclusively Nox1.
Supplementary Material
Highlights.
QSOX1b promotes a Nox1-dependent migratory response in VSMC1.
Hydrogen peroxide mediates VSMC migration induced by active QSOX1b.
Nox1 contributes, but is not essential, for QSOX1-induced mitogenic response in VSMC.
Superoxide mediates VSMC proliferation induced by active and inactive QSOX1b.
5. Acknowledgements
Authors thank Prof. Dr. Francisco Laurindo for valuable discussion and Gisele T. Veiga for her help in proliferation experiments. This study was supported by CNPq (573530/2008-4, 401591/2014-0, 420782/2016-8), CAPES-Print (88881.311846/2018-01) and CAPES (Finance Code 001). Fellowships from CAPES (awarded to KCF, PAM, MLP, BEB, SML) and CNPq (awarded to LSN, SMZ) are also acknowledged.
Footnotes
Declarations of interest: none
Abbreviations:
AngII: angiotensin II, bFGF: basic fibroblast growth factor, ER stress: endoplasmic reticulum stress, FBS: fetal bovine serum, GFP: green fluorescent protein, PDGF: platelet-derived growth factor, PEG-CAT: polyethyleneglycol-catalase, PEG-SOD: polyethyleneglycol-superoxide dismutase, VSMC: vascular smooth muscle cells
Prado, ML (unpublished result)
6. References
- [1].Gerthoffer WT, Mechanisms of vascular smooth muscle cell migration, Circ. Res 100 (2007) 607–621. [DOI] [PubMed] [Google Scholar]
- [2].Berk BC, Vascular smooth muscle growth: autocrine growth mechanisms, Physiol. Rev 81 (2001) 999–1030. [DOI] [PubMed] [Google Scholar]
- [3].Louis SF, Zahradka P, Vascular smooth muscle cell motility: From migration to invasion, Exp. Clin. Cardiol 15 (2010) e75–85. [PMC free article] [PubMed] [Google Scholar]
- [4].San Martín A, Griendling KK, Redox control of vascular smooth muscle migration, Antioxid. Redox Signal 12 (2010) 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Byon CH, Heath JM, Chen Y, Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells, Redox Biol. 9 (2016) 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brandes RP, Schröder K, Composition and functions of vascular nicotinamide adenine dinucleotide phosphate oxidases, Trends Cardiovasc. Med 18 (2008) 15–19. [DOI] [PubMed] [Google Scholar]
- [7].Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL, Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II, Circ. Res 90 (2002) 1205–1213. [DOI] [PubMed] [Google Scholar]
- [8].Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK, Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways, Circ. Res 88 (2001) 888–894. [DOI] [PubMed] [Google Scholar]
- [9].Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK, Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production, Free Radic. Biol. Med 45 (2008) 1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kodali VK, Thorpe C, Oxidative protein folding and the Quiescin-sulfhydryl oxidase family of flavoproteins, Antioxid. Redox Signal 13 (2010) 1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Heckler EJ, Alon A, Fass D, Thorpe C, Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis, Biochemistry. 47 (2008) 4955–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hoober KL, Joneja B, White HB 3rd, Thorpe C, A sulfhydryl oxidase from chicken egg white, J. Biol. Chem 271(1996) 30510–30516. [DOI] [PubMed] [Google Scholar]
- [13].Coppock D, Kopman C, Gudas J, Cina-Poppe DA, Regulation of the quiescence-induced genes: quiescin Q6, decorin, and ribosomal protein S29, Biochem. Biophys. Res. Commun 269 (2000) 604–610. [DOI] [PubMed] [Google Scholar]
- [14].Ilani T, Alon A, Grossman I, Horowitz H, Kartvelishvily E, Cohen SR, Fass D, A secreted disulfide catalyst controls extracellular matrix composition and function, Science 341 (2013) 74–76. [DOI] [PubMed] [Google Scholar]
- [15].Simper D, Mayr U, Urbich C, Zampetaki A, Prokopi M, Didangelos A, Saje A, Mueller M, Benbow U, Newby AC, Apweiler R, Rahman S, Dimmeler S, Xu Q, Mayr M, Comparative proteomics profiling reveals role of smooth muscle progenitors in extracellular matrix production, Arterioscler. Thromb. Vasc. Biol 30 (2010) 1325–1332. [DOI] [PubMed] [Google Scholar]
- [16].Benayoun B, Esnard-Fève A, Castella S, Courty Y, Esnard F, Rat seminal vesicle FAD-dependent sulfhydryl oxidase. Biochemical characterization and molecular cloning of a member of the new sulfhydryl oxidase/quiescin Q6 gene family, J. Biol. Chem 276 (2001) 13830–13837. [DOI] [PubMed] [Google Scholar]
- [17].Zanata SM, Luvizon AC, Batista DF, Ikegami CM, Pedrosa FO, Souza EM, Chaves DF, Caron LF, Pelizzari JV, Laurindo FR, Nakao LS, High levels of active quiescin Q6 sulfhydryl oxidase (QSOX) are selectively present in fetal serum, Redox Rep. 10 (2005) 319–323. [DOI] [PubMed] [Google Scholar]
- [18].Israel BA, Jiang L, Gannon SA, Thorpe C, Disulfide bond generation in mammalian blood serum: detection and purification of quiescin-sulfhydryl oxidase, Free Radic. Biol. Med 69 (2014) 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sung HJ, Ahn JM, Yoon YH, Na SS, Choi YJ, Kim YI, Lee SY, Lee EB, Cho S, Cho JY, Quiescin Sulfhydryl Oxidase 1 (QSOX1) Secreted by Lung Cancer Cells Promotes Cancer Metastasis. Int. J. Mol. Sci 19 (2018) pii: E3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mebazaa A, Vanpoucke G, Thomas G, Verleysen K, Cohen-Solal A, Vanderheyden M, Bartunek J, Mueller C, Launay JM, Van Landuyt N, D'Hondt F, Verschuere E, Vanhaute C, Tuytten R, Vanneste L, De Cremer K, Wuyts J, Davies H, Moerman P, Logeart D, Collet C, Lortat-Jacob B, Tavares M, Laroy W, Januzzi JL, Samuel JL, Kas K, Unbiased plasma proteomics for novel diagnostic biomarkers in cardiovascular disease: identification of quiescin Q6 as a candidate biomarker of acutely decompensated heart failure, Eur. Heart J 33 (2012) 2317–2324. [DOI] [PubMed] [Google Scholar]
- [21].Caillard A, Sadoune M, Cescau A, Meddour M, Gandon M, Polidano E, Delcayre C, Da Silva K, Manivet P, Gomez AM, Cohen-Solal A, Vodovar N, Li Z, Mebazaa A, Samuel JL, QSOX1, a novel actor of cardiac protection upon acute stress in mice, J. Mol. Cell. Cardiol 119 (2018) 75–86. [DOI] [PubMed] [Google Scholar]
- [22].Katchman BA, Ocal IT, Cunliffe HE, Chang YH, Hostetter G, Watanabe A, LoBello J, Lake DF, Expression of quiescin sulfhydryl oxidase 1 is associated with a highly invasive phenotype and correlates with a poor prognosis in Luminal B breast cancer, Breast Cancer Res. 15 (2013) R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Borges BE, Appel MH, Cofré AR, Prado ML, Steclan CA, Esnard F, Zanata SM, Laurindo FR, Nakao LS, The flavo-oxidase QSOX1 supports vascular smooth muscle cell migration and proliferation: Evidence for a role in neointima growth, Biochim. Biophys. Acta 1852 (2015) 1334–1346. [DOI] [PubMed] [Google Scholar]
- [24].Portes KF, Ikegami CM, Getz J, Martins AP, de Noronha L, Zischler LF, Klassen G, Camargo AA, Zanata SM, Bevilacqua E, Nakao LS, Tissue distribution of quiescin Q6/sulfhydryl oxidase (QSOX) in developing mouse, J. Mol. Histol 39 (2008) 217–225. [DOI] [PubMed] [Google Scholar]
- [25].Raje S, Glynn NM, Thorpe C, A continuous fluorescence assay for sulfhydryl oxidase, Anal. Biochem 307 (2002) 266–272. [DOI] [PubMed] [Google Scholar]
- [26].Liang CC, Park AY, Guan JL, In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro, Nat. Protoc 2 (2007) 329–333. [DOI] [PubMed] [Google Scholar]
- [27].Lo HM, Tsai YJ, Du WY, Tsou CJ, Wu WB, A naturally occurring carotenoid, lutein, reduces PDGF and H2O2 signaling and compromised migration in culture vascular smooth muscle cells, J. Biomed. Sci 19 (2012) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Aebi H, Catalase in vitro, Methods Enzymol. 105 (1984) 121–126. [DOI] [PubMed] [Google Scholar]
- [29].Crouch RK, Gandy SE, Kimsey G, Galbraith RA, Galbraith GM, Buse MG, The inhibition of islet superoxide dismutase by diabetogenic drugs, Diabetes 30 (1981) 235–341. [DOI] [PubMed] [Google Scholar]
- [30].Beckman JS, Minor RL Jr, White CW, Repine JE, Rosen GM, Freeman BA, Superoxide dismutase and catalase conjugated to polyethylene glycol increases endothelial enzyme activity and oxidant resistance, J. Biol. Chem 263 (1988) 6884–6892. [PubMed] [Google Scholar]
- [31].Gimenez M, Schickling BM, Lopes LR, Miller FJ Jr, Nox1 in cardiovascular diseases: regulation and pathophysiology, Clin. Sci. (Lond) 130 (2016) 151–165. [DOI] [PubMed] [Google Scholar]
- [32].Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, Roushc WR, Brown SJ, Bokoch GM, Rosen H, A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in huma colon cancer cells, ACS Chem. Biol 5 (2010) 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fernandes DC, Manoel AH, Wosniak J Jr, Laurindo FR, Protein disulfide isomerase overexpression in vascular smooth muscle cells induces spontaneous preemptive NADPH oxidase activation and Nox1 mRNA expression: effects of nitrosothiol exposure, Arch. Biochem. Biophys 484 (2009) 197–204. [DOI] [PubMed] [Google Scholar]
- [34].Gimenez M, Veríssimo-Filho S, Wittig I, Schickling BM, Hahner F, Schürmann C, Netto LES, Rosa JC, Brandes RP, Sartoretto S, De Lucca Camargo L, Abdulkader F, Miller FJ Jr, Lopes LR, Redox Activation of Nox1 (NADPH Oxidase 1) Involves an Intermolecular Disulfide Bond Between Protein Disulfide Isomerase and p47(phox) in Vascular Smooth Muscle Cells, Arterioscler. Thromb. Vasc. Biol 39 (2019) 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T, Requirement for generation of H2O2 for platelet-derived growth factor signal transduction, Science. 270 (1995) 296–299. [DOI] [PubMed] [Google Scholar]
- [36].Weber DS, Taniyama Y, Rocic P, Seshiah PN, Dechert MA, Gerthoffer WT, Griendling KK, Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration, Circ. Res 94 (2004) 1219–1226. [DOI] [PubMed] [Google Scholar]
- [37].Wang Z, Castresana MR, Newman WH, Reactive oxygen species-sensitive p38 MAPK controls thrombin-induced migration of vascular smooth muscle cells, J. Mol. Cell. Cardiol 36 (2004) 49–56. [DOI] [PubMed] [Google Scholar]
- [38].Schröder K, Helmcke I, Palfi K, Krause KH, Busse R, Brandes RP, Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells, Arterioscler. Thromb. Vasc. Biol 27 (2007) 1736–1743. [DOI] [PubMed] [Google Scholar]
- [39].Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassègue B, Griendling KK, Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation, Arterioscler. Thromb. Vasc. Biol 29 (2009) 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zimmerman MC, Takapoo M, Jagadeesha DK, Stanic B, Banfi B, Bhalla RC, Miller FJ Jr, Activation of NADPH oxidase 1 increases intracellular calcium and migration of smooth muscle cells, Hypertension. 58 (2011) 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kim JS, Huang TY, Bokoch GM, Reactive oxygen species regulate a slingshot-cofilin activation pathway, Mol. Biol. Cell 20 (2009) 2650–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Maheswaranathan M, Gole HK, Fernandez I, Lassègue B, Griendling KK, San Martín A, Platelet-derived growth factor (PDGF) regulates Slingshot phosphatase activity via Nox1-dependent auto-dephosphorylation of serine 834 in vascular smooth muscle cells, J. Biol. Chem 286 (2011) 35430–35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cameron JM, Gabrielsen M, Chim YH, Munro J, McGhee EJ, Sumpton D, Eaton P, Anderson KI, Yin H, Olson MF, Polarized cell motility induces hydrogen peroxide to inhibit cofilin via cysteine oxidation, Curr. Biol 25 (2015) 1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Al Ghouleh I, Rodríguez A, Pagano PJ, Csányi G, Proteomic analysis identifies an NADPH oxidase 1 (Nox1)-mediated role for actin-related protein 2/3 complex subunit 2 (ARPC2) in promoting smooth muscle cell migration, Int. J. Mol. Sci 14 (2013) 20220–20235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Patterson C, Ruef J, Madamanchi NR, Barry-Lane P, Hu Z, Horaist C, Ballinger CA, Brasier AR, Bode C, Runge MS, Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47(phox) may participate in forming this oxidase in vitro and in vivo, J. Biol. Chem 274 (1999) 19814–19822. [DOI] [PubMed] [Google Scholar]
- [46].Shi M, Yang H, Motley ED, Guo Z, Overexpression of Cu/Zn-superoxide dismutase and/or catalase in mice inhibits aorta smooth muscle cell proliferation, Am. J. Hypertens 17(2004) 450–456. [DOI] [PubMed] [Google Scholar]
- [47].Li PF, Dietz R, von Harsdorf R, Differential effect of hydrogen peroxide and superoxide anion on apoptosis and proliferation of vascular smooth muscle cells, Circulation. 96 (1997) 3602–3609. [DOI] [PubMed] [Google Scholar]
- [48].Menshikov M, Plekhanova O, Cai H, Chalupsky K, Parfyonova Y, Bashtrikov P, Tkachuk V, Berk BC, Urokinase plasminogen activator stimulates vascular smooth muscle cell proliferation via redox-dependent pathways, Arterioscler. Thromb. Vasc. Biol 26 (2006) 801–807. [DOI] [PubMed] [Google Scholar]
- [49].Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD, Cell transformation by the superoxide-generating oxidase Mox1, Nature. 401 (1999) 79–82. [DOI] [PubMed] [Google Scholar]
- [50].Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH, Decreased blood pressure in NOX1-deficient mice, FEBS Lett. 580 (2006) 497–504. [DOI] [PubMed] [Google Scholar]
- [51].Sarsour EH, Venkataraman S, Kalen AL, Oberley LW, Goswami PC, Manganese superoxide dismutase activity regulates transitions between quiescent and proliferative growth, Aging Cell. 7 (2008) 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wosniak J Jr, Santos CX, Kowaltowski AJ, Laurindo FR, Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells, Antioxid. Redox Signal 11 (2009) 1265–1278. [DOI] [PubMed] [Google Scholar]
- [53].López-Acosta O, de Los Angeles Fortis-Barrera M, Barrios-Maya MA, Ramírez AR, Aguilar FJA, El-Hafidi M, Reactive Oxygen Species from NADPH Oxidase and Mitochondria Participate in the Proliferation of Aortic Smooth Muscle Cells from a Model of Metabolic Syndrome, Oxid. Med. Cell. Longev 2018 (2018) 5835072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.