

Abstract
Quantitative co-localization analysis, combined with other in vivo and in vitro techniques, can provide valuable information about the interaction and cooperative function of two proteins. Here we describe in detail the technique of quantitative co-localization analysis of two enamel matrix proteins, amelogenin and ameloblastin, in developing mouse enamel.
Keywords: Enamel, Enamel matrix proteins, Amelogenin, Ameloblastin, Immunolabeling, In vivo quantitative co-localization, Manders’ coefficient
1. Introduction
Quantitative co-localization analysis of enamel matrix proteins was first used in our laboratory by Gallon et al., in 2013 [1], to visualize and quantify the co-localization of amelogenin and enamelin proteins in vivo. Previous work had shown that amelogenin and enamelin interact in vitro [2, 3]. Co-localization between the two proteins in developing mouse molars provided evidence that these proteins also have cooperative function in vivo (Fig. 1). Hence, combining co-localization analysis with in vitro functional analysis was shown to be useful for elucidating protein-protein interactions.
Fig. 1.
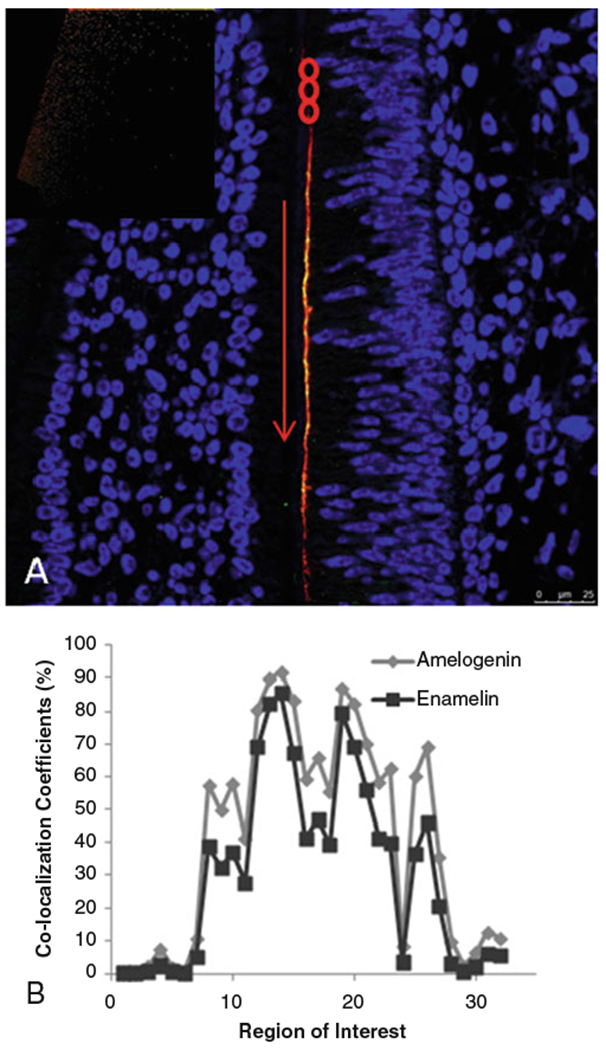
Confocal image of postnatal day 1 mouse mandibular molar showing (a) regions of interest (red circles) selected for quantitative co-localization analysis along the secretory face of ameloblasts; (b) graph of co-localization coefficients from ROIs shown in a. Inset—scatterplot showing distribution of red and green pixels. Reproduced with permission from Ref. [1]
We have further improved and applied this combination of techniques to describe interaction between amelogenin and ameloblastin (Fig. 2) [4, 5]. We added fluorescence resonance energy transfer (FRET) microscopy to the analysis to demonstrate close proximity between two enamel extracellular matrix proteins (Fig. 2c). FRET uses a pair of fluorophores such that the emission wavelength of one is the excitation wavelength of the other (e.g., FITC and TRITC donor-acceptor pair). The efficiency of energy transfer is inversely proportional to the sixth power of the distance between them. The distance between two fluorophores has to be less than ~10 nm to achieve a positive FRET signal. Using this technique, Mazumder et al. [5] showed that amelogenin and ameloblastin proteins not only co-localize but are also within interacting distance of one another.
Fig. 2.
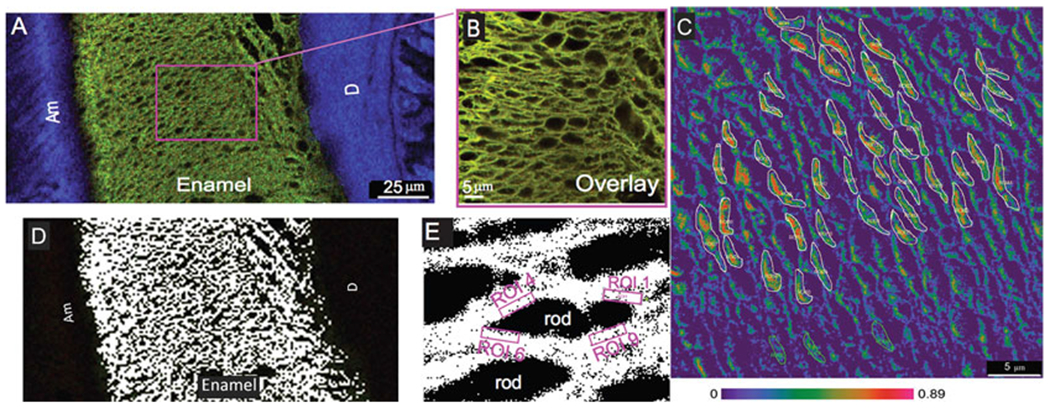
Quantitative co-localization of ameloblastin and amelogenin in P8 mouse mandibular molar (a, b) transverse section. Co-localization between amelogenin (green) and ameloblastin (red) is revealed by overlapping signals resulting in yellow staining. (b) Co-localization pattern of amelogenin and ameloblastin in sagittal section. (d, e) White pixels exhibiting actual co-localization in the confocal images after background correction within thickness of the enamel and around the rod sheaths. (c) Amelogenin-ameloblastin in situ FRET efficiency displayed as an absolute range from highest (red 0.89) to lowest (purple 0.0). Small regions of interest (ROIs) were chosen to obtain the FRET efficiency values around the rod sheaths (areas drawn with white lines). Am ameloblasts, D dentin. Reproduced with permission from Ref. [5]
The most commonly used measure of enamel matrix protein co-localization is the Manders’ co-localization coefficient [6] described later in this chapter. One of the key benefits of Manders’ co-localization coefficient over other coefficients (like Pearson’s co-localization coefficient [7] or Manders’ overlap coefficient [6]) is that it calculates the fraction of each protein that co-localizes with the other protein in a given area [8 ] irrespective of its intensity. This is important, because the matrix proteins tagged by fluorophores may not be evenly distributed within the enamel.
In this chapter, we will describe the technique of immunolabeling developing mouse enamel for amelogenin and ameloblastin proteins followed by co-localization analysis, using postnatal day 5 mouse mandibles as a model. This technique can be modified to immunolabel postnatal day 8 teeth or other mineralized tissues.
2. Materials
2.1. For Paraff Embedding of Mouse Mandibles
4% paraformaldehyde (PFA): Heat 350 mLdeionized (dI) water to 60 °C. Add 20 g PFA and 50 mL 10 × PBS (see below). Adjust pH to 7.4 with 5 N NaOH and bring final volume to 500 mL. Filter using 0.45 μm sterile disposable filters (Nalgene™ Rapid-Flow™), and store at 4 °C (see Note 1).
10% EDTA with 0.1% glutaraldehyde in PBS: Dissolve 50 g disodium salt of EDTA, 50 mL 10x PBS, and 0.5 mL glutaraldehyde in 300 mL dI water. Adjust pH to 8.5 using 5 N NaOH (see Note 2), and bring final volume to 500 mL. Filter using 0.45 μm sterile disposable filters and store at 4 °C.
10× Phosphate-buffered saline (PBS): Dissolve 80 g NaCl, 2 g KCl, 14.4 g Na2HPO4, and 2.4 g KH2PO4 in 800 mL dI water. Adjust pH to 7.4 with 1 M NaOH or HCl, and bring final volume to 1 L (see Note 3).
Ethanol: increasing concentrations from 25% to 100%.
Xylene.
Paraffin wax.
Vacuum-sealed hot air oven.
Plastic base molds and mounting rings.
Hot plate.
Forceps, etc. for handling tissue.
2.2. For Immunohistochemical Labeling
Citrate buffer: Dissolve 1.47 g sodium citrate dihydrate in 400 mL of dI water. Adjust pH to 6.0 with 1 M HCl and bring final volume to 500 mL. Add 0.05% Tween 20 to the Coplin jar each time before use.
Tris-buffered saline (TBS): Dissolve 2.42 g Tris and 9.0 g NaCl in 800 mL dI water. Adjust pH to 7.5 with 1 M HCl and bring final volume to 1 L.
0.3% Hydrogen peroxide: Dilute 0.1 mL of 30% H2O2 in 10 mL dI water.
1% Bovine Serum Albumin (BSA): Dissolve 0.1 g BSA in 10 mL TBS. Store at 4 °C.
Antibody dilution solution: 0.1% BSA, 0.3% Triton X-100 in TBS. Dissolve 0.01 g BSA and 30 μL Triton-X100 in 10 mL TBS. Store at 4 °C.
Primary antibody: Anti-full-length amelogenin (host: chicken; from Dr. Malcolm Snead’s lab), diluted 1:1000 in antibody dilution solution.
Primary antibody: Anti-ameloblastin M-300 (host: rabbit; commercially available from Santa Cruz Biotechnology Inc., Santa Cruz CA), 1:500 diluted in antibody dilution solution.
Secondary antibodies: Bovine anti-chicken conjugated with FITC (Santa Cruz Biotechnology Inc., Santa Cruz CA) and donkey anti-rabbit conjugated with Alexa 594 (Thermo Fisher Scientific Inc., USA), each 1:100 in TBS.
Mounting media: Vectashield with DAPI (Vector Laboratories Inc., MS).
Positively charged microscope slides (like Superfrost™ Plus, Thermo Fisher Scientific Inc., USA) and coverslips.
3. Methods
3.1. Tissue Preparation
In this protocol we use postnatal day 5 (P5) mouse mandibles for the convenience of relatively quick demineralization and tissue processing time. The incisor and molar enamel are co-labeled for amelogenin and ameloblastin using FITC and Alexa 594, respectively, visualized using confocal microscopy.
Fixation: Euthanize and dissect the heads of P5 mice following the appropriate Institutional Animal Care and Use Committee guidelines. Place the heads in 4% PFA immediately for 24–48 h in a cold room (4 °C) on a rocker. For proper fixation, about 15 times the volume of the fixative to tissue is preferred. Remove the heads from PFA the following day, and dissect out the mandibles carefully under a dissecting microscope without damaging the structures of the developing teeth (see Note 1).
Decalcification: Transfer the dissected mandibles to 10% EDTA with 0.1% glutaraldehyde in PBS. For P5 mandibles, up to 5 days of decalcification at 4 °C of decalcification is necessary. The EDTA solution may be changed once every 48 h.
Washing: After decalcification, gently remove the tissue from EDTA, and place in PBS for washing (see Note 4). Wash the tissue in 1 × PBS for 1–2 h with at least four changes of the buffer at 4 °C.
-
Dehydration: Dehydrate the tissue using increasing grades of ethanol as below (see Note 5).
Concentration Changes Time 25% Two to three changes 30 min each 50% Two to three changes 30 min each 70% Two to three changes 30 min each 95% Two to three changes 30 min each 100% Two to three changes 30 min each Clearing: Clear the ethanol from the tissue using two to three changes of xylene, 30 min each (see Note 6).
Paraffin infiltration: After the third change of xylene, place the tissue in 50% xylene-paraffin mix for 30 min in a 60 °C vacuum-sealed oven. Then continue with 100% paraffin for three changes, 30 min each in a 60 °C vacuum-sealed oven. The vacuum will draw out the xylene and paraffin will replace it. Tissue can be left in paraffin in the oven overnight.
Embedding: Select an appropriately sized tissue base mold for your tissue. Place it on a 60 °C hot plate and fill it halfway with molten paraffin wax. Position your tissue in the desired orientation. Remove from the hot plate and let the paraffin solidify partially. Now place the mounting ring on the base mold, and fill it with liquid paraffin all the way to the top. Allow to completely set, preferably overnight. Store at 4 °C (see Note 7).
Sectioning: Place paraffin blocks on ice 15–20 min prior to cutting to make sectioning easier. Cut 5–7-μm-thick sections using a microtome, and place them on positively charged microscope glass slides. Allow to dry on a hot plate overnight. Store prepared slides at 4 ° C (see Note 8).
3.2. Double Labeling
Antigen retrieval: The paraffin needs to be completely removed for the antibodies to be able to attach to their respective protein antigens. To remove paraffin, fill one Coplin jar with citrate buffer with Tween 20. Place it in a water bath set at 60 °C with two jars of dI water. Once the buffer is warm, place the slides in the buffer and leave overnight. Next morning, remove the slides from the buffer, and transfer to one of two water-filled Coplin jars; wash by dipping each slide ten times. Move the slides to the second water jar, and let them cool to room temperature. Rinse the slides with TBS for 30 min (see Note 9).
Blocking: Cover the exposed tissue with 0.3% hydrogen peroxide for 30 min. This helps in reducing the autofluorescence and background from endogenous peroxidase in the tissue. Discard the H2O2, blot excess from the edges of the slide, and wash the slides with TBS for at least 30 min. Then cover the tissue with 1% BSA to block nonspecific binding of antibodies.
Primary antibody. Blot the slides using Kimwipes to remove the BSA. For the co-labeling technique, both the primary antibodies are prepared in the same solution (see Note 10). Dilute anti-full-length amelogenin antibody and anti-M300 ameloblastin antibody 1:1000 and 1:500 in antibody dilution solution. Prepare a moist chamber for overnight primary antibody incubation. Place the slides in the moist chamber, and cover the tissues with about 150–200 μL primary antibody solution (see Note 11). Incubate overnight at room temperature in the moist chamber.
Secondary antibody: Wash slides for 30 min in TBS after blotting to remove the primary antibody solution. Secondary antibody selection is particularly important in case of double labeling as none of the species should cross-react with each other. We use bovine anti-chicken antibody conjugated with FITC (green) for amelogenin and donkey anti-rabbit antibody conjugated with Alexa 594 (red) for ameloblastin; however, any other appropriate pair of species and fluorophores can be used. Dilute the secondary antibodies 1:100 in TBS in the dark. Incubate the slides with secondary antibodies in a dark moist chamber for 3–4 h.
Nuclear stain and coverslip: It is crucial to note that every step after this must be done in the dark. Cover the Coplin jars in aluminum foil to keep the slides in the dark while washing. Wash the slides in TBS in the dark for at least 30 min. Place a drop of Vectashield with DAPI (Vector Laboratories Inc., Burlingame CA) on the slides, and place an appropriately sized coverslip to cover the tissue (see Note 12). Let the mounting medium set for at least 30 min. Coverslips can be further sealed using a clear nail varnish and dried for 15 min.
Visualization: The slides are visualized using a Leica SP8 confocal microscope with 63 – oil immersion lens, or similar equipment. Co-localization patterns within the developing incisor are analyzed with Leica Application Suite (LAS) X version 1.8.1.13759 or equivalent.
3.3. Co-localization Analysis
Before starting co-localization analysis, adjust the threshold and background correction values. Keep these constant throughout the entire analysis, across all the images/regions of interest (ROIs) to be compared. Amelogenin and ameloblastin are known to be secreted in different quantities, with amelogenin being more abundant than ameloblastin. Therefore, it is important to take into account the difference in the amount of each fluorophore that will occur due to difference in the amount of the proteins. Manders’ co-localization coefficient [6] factors in this difference in the number of pixels of each channel in each selected ROI. Simply, it calculates the ratio of the sum of co-localizing pixels of each channel to the total pixels in the ROI. Hence it generates two coefficients for two channels being considered for co-localization (Fig. 3). Manders’ coefficients M1 and M2 are given by the following equations [6]:
Fig. 3.
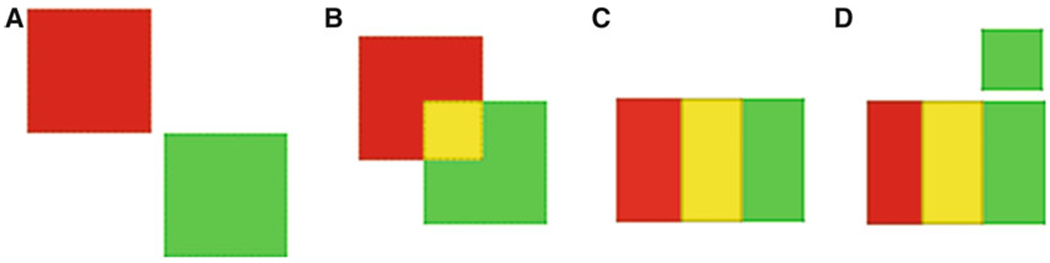
Schematic diagram illustrating Manders’ co-localization coefficient. A, M1 = 0.0, M2 = 0.0; B, M1 = 0.250, M2 = 0.250; C, M1 = 0.500, M2 = 0.500; D, M1 = 0.500, M2 = 0.400 [9]
where Ri,coloc = Ri if Gi > = 0 and Ri,coloc = 0 if Gi = 0, and Gi,coloc = Gi if Ri > 0 and Gicoloc = 0 if Ri = 0 [6].
For amelogenin and ameloblastin co-localization, we selected oval ROIs 3x5 μm in diameter within ameloblasts and at the secretory face of ameloblasts. The Leica Application Suite software provides Pearson’s correlation, overlap coefficient, co-localization rate, co-localization area, ROI area, foreground area, background area, mean intensity ROI, mean intensity co-localization, intensity sum ROI, and intensity sum co-localization for each channel in every ROI. Using these data, Manders’ co-localization coefficients M1 and M2 can be calculated, and graphs can be plotted similar to Fig. 1b (see Note 13).
4 Notes
All steps involving paraformaldehyde must be done carefully under a chemical/fume hood with appropriate personal protective equipment, taking care not to inhale PFA fumes. Make sure to cover the flask with aluminum foil while stirring PFA solution if the stirring is done outside the hood.
EDTA will not dissolve until the pH of solution reaches 8.5. Sometimes it may be necessary to use NaOH pellets instead of 5 N solution to raise the pH to 8.5. Stirring for several hours or overnight at pH 8.5 may also be needed. We add glutaraldehyde to the EDTA solution to prevent protein loss during demineralization.
10× PBS can be autoclaved and kept at 4 ° C for long-term storage. Dilute with dI water for use.
Demineralized tissue is very delicate. Take care not to hold the tissue by the developing incisors or molars to maintain their microstructure.
Tissue can be left in the second change of 70% ethanol indefinitely.
Xylene is toxic and flammable. Work with xylene under a chemical hood with all appropriate personal protective equipment. Discard used xylene in designated chemical waste containers. Dehydration, clearing, and paraffin embedding steps can be automated by using a spin tissue processor like Thermo Scientific™ STP 120.
It takes some practice to be able to position the mandibles correctly in paraffin without giving rise to any bubbles. A convenient trick is to keep a spirit lamp at hand and warm the tips of the instruments used to position the sample. This prevents the wax from suddenly solidifying around the cold metal tips of the instruments, causing bubbles as they are withdrawn. It is a good idea to practice with some old/non-essential samples before starting with actual experimental tissue.
Start with thicker sections initially as they are easier to handle. Thickness of sections can be reduced to 5 μm with practice. Take care while positioning the sections that the tips of incisors and cusps of molars are not folded/torn.
Do not shock the tissue with varying temperatures of buffers. If the buffers have been previously prepared and stored at 4 °C, make sure to bring them to room temperature before use.
It is important to test the antibodies for cross-reactivity before co-labeling using Western blotting. This chapter describes the antibodies that are tried and tested in our lab; however, you can use any antibodies that work best for your protein of choice and do not cross-react with each other.
The volume of primary antibody solution depends upon the number of tissue sections on each slide; enough should be prepared to cover all the sections. Make the desired amount, and then calculate the appropriate quantity of antibody to be diluted. For example, if you have two slides with four to five sections each, you might need 200 μL antibody solution per slide. Therefore, prepare 400 μL which will need 0.4 μL anti-amelogenin and 0.8 μL anti-ameloblastin primary antibody.
Alternately, DAPI can be added with secondary antibody if it is available as a solution. The coverslip can be mounted using glycerol.
To make calculations easier, data generated by the Leica Application Suite software are first exported in a statistical software like Microsoft Excel. M1 and M2 are calculated by dividing intensity sum co-localization with intensity sum ROI for each channel. These figures can then be conveniently plotted in a line graph to determine the co-localization of two channels. A helpful resource to understand various co-localization coefficients can be found at the Scientific Volume Imaging B.V. website https://svi.nl/ColocalizationCoefficients.
References
- 1.Gallon V, Chen L, Yang X, Moradian-Oldak J (2013) Localization and quantitative co-localization of enamelin with amelogenin. J Struct Biol 183(2):239–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan D, Du C, Sun Z, Lakshminarayanan R, Moradian-Oldak J (2009) In vitro study on the interaction between the 32kDa enamelin and amelogenin. J Struct Biol 166(1):88–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang X, Fan D, Mattew S, Moradian-Oldak J (2011) Amelogenin-enamelin association in phosphate buffered saline. Eur J Oral Sci 119:351–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mazumder P, Prajapati S, Lokappa SB, Gallon V, Moradian-Oldak J (2014) Analysis of co-assembly and co-localization of ameloblastin and amelogenin. Front Physiol 5:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazumder P, Prajapati S, Bapat R, Moradian-Oldak J (2016) Amelogenin-ameloblastin spatial interaction around maturing enamel rods. J Dent Res 95(9):1042–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manders E, Verbeek F, Aten J (1993) Measurement of co-localization of objects in dual-colour confocal images. J Microsc 169(3):375–382 [DOI] [PubMed] [Google Scholar]
- 7.Pearson K (1896) Mathematical contributions to the theory of evolution. III. Regression, heredity, and panmixia. Philos Trans Royal Soc London Series A 187:253–318 [Google Scholar]
- 8.Dunn KW, Kamocka MM, McDonald JH (2011) A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300(4):C723–C742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huygens Support Wiki: Scientific Volume Imaging (Hilverum, The Netherlands) [The SVI-wiki is a rapidly expanding public knowledge resource on 3D microscopy, image restoration (deconvolution), visualization and analysis. Based on the wiki principle, it is open to contributions from registered visitors]. https://svi.nl/ColocalizationCoefficients