

Abstract
Platelets possess distinct surface moieties responsible for modulating their adhesion to various disease-relevant substrates involving vascular damage, immune evasion, and pathogen interactions. Such broad biointerfacing capabilities of platelets have inspired the development of platelet-mimicking drug carriers that preferentially target drug payloads to disease sites for enhanced therapeutic efficacy. Among these carriers, platelet membrane-coated nanoparticles (denoted ‘PNPs’) made by cloaking synthetic substrates with the plasma membrane of platelets have emerged recently. Their ‘top-down’ design combines the functionalities of natural platelet membrane and the engineering flexibility of synthetic nanomaterials, which together create synergy for effective drug delivery and novel therapeutics. Herein, we review the recent progress of engineering PNPs with different structures for targeted drug delivery, focusing on three areas, including targeting injured blood vessels to treat vascular diseases, targeting cancer cells for cancer treatment and detection, and targeting drug-resistant bacteria to treat infectious diseases. Overall, current studies have established PNPs as versatile nanotherapeutics for drug targeting with strong potentials to improve the treatment of various diseases.
Keywords: Nanomedicine, platelet, cell membrane coating, nanoparticle, targeted delivery
Graphical Abstract
Platelet membrane-coated nanoparticles inherit biofunctions of natural platelets and are capable of adhering to various disease-relevant substrates. They have become drug carriers exceptionally suitable for targeted drug delivery. Herein, we review the recent development of platelet-mimicking nanoparticles with different structures for drug targeting to injured blood vessels, cancer cells, and drug-resistant bacteria.
Introduction
Coating nanoparticles with membranes of natural cells is increasingly explored to acquire cell-like properties for nanoparticle functionalization. This ‘top-down’ biomimetic approach allows nanoparticles to better interact with complex biological microenvironments and has led to a variety of novel nanotherapeutics.[1] For example, nanoparticles coated with membranes of red blood cells (RBC) mimic source cells to intercept incoming toxins. They have been used to absorb and neutralize toxins, preventing cellular targets from toxin attack.[2–4] Nanoparticles coated with cancer cell membranes faithfully present the entire antigenic profile of the source cell and elicit potent anti-cancer immune responses. They have shown promise to become personalized, autologous anti-cancer vaccines.[5, 6] In addition, nanoparticles coated with macrophage membranes simulate the function of source macrophages and concurrently neutralize multiple pro-inflammatory factors, including lipopolysaccharide (LPS) and inflammatory cytokines. They have illustrated a new detoxification strategy for improving sepsis management.[7] Moreover, nanoparticles coated with membranes of CD4+ T cells, lung epithelial cells, or macrophages display the same protein receptors, both identified and unidentified, required by viruses for cellular entry. By acting as decoys of the source cells to intercept and block viral entry, these nanoparticles effectively inhibited the infectivity of critical viruses such as human immunodeficiency virus (HIV) and severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2), thereby holding great potential as new anti-viral therapeutics. [8, 9]
Among emerging applications enabled by cell membrane-coated nanoparticles, targeted delivery of drugs to the disease sites for enhanced therapeutic potency represents a major research area. Drug targeting can be passive, where long-circulating nanoparticles such as those coated with RBC membranes, extravasate through leaky vasculature and accumulate in the surrounding tissue via the enhanced permeability and retention (EPR) effect.[10–12] Alternatively, drug targeting can be active, relying on membrane moieties to bind with protein receptors overexpressed on the target cells or tissues.[13] In this regard, nanoparticles coated with cancer cell membranes engage active targeting through the homotypic binding mechanism common among cancer cells.[5, 6, 14] Neutrophil membrane-coated nanoparticles actively target inflammation through affinity between neutrophil surface antigen and upregulate adhesion markers on inflamed cells.[15] With plentiful source cells to choose from, cell membrane coating technology offers abundant opportunities to achieve targeted drug delivery.
Toward effective drug targeting, nanoparticles coated with platelet membranes (denoted ‘PNPs’) stand out for their unique ability to target both passively and actively. Platelet exhibits a circulation half-life of approximately 30 hours. Such long circulation is attributable in part to their possession of CD47, a ‘marker-of-self’ that interacts with the signal regulatory protein α (SIRPα) on the immune cells and therefore inhibits the immune clearance.[16] For this reason, PNPs circulate long and are capable of passive drug targeting. Platelet also expresses a set of unique surface receptors that dynamically adhere to damaged vasculature, tumor cells, and pathogenic bacteria.[17–19] For example, platelet surface glycoprotein(GP) Ib (GPIb) can bind to exposed collagen of damaged vasculature through von Willebrand factor (vWF) to initiate tissue repair.[20] P-selectin, a cell adhesion molecule upregulated in activated platelets, can bind to P-selectin glycoprotein ligand-1 (PSGL-1) or CD44 overexpressed on tumor cells, allowing for complex and dynamic platelet-tumor cross-talk critical for tumor growth and metastasis.[21, 22] Additionally, platelets bind directly to pathogenic bacteria through GPIb, leading to GPIIb/IIIa-mediated platelet aggregation as a part of host response to remove the bacteria.[23] PNPs inherit these dynamic binding properties from the source platelets for active drug targeting. With such broad and dynamic biointerfacing capabilities, PNPs have become attractive drug carriers for targeted delivery applications.
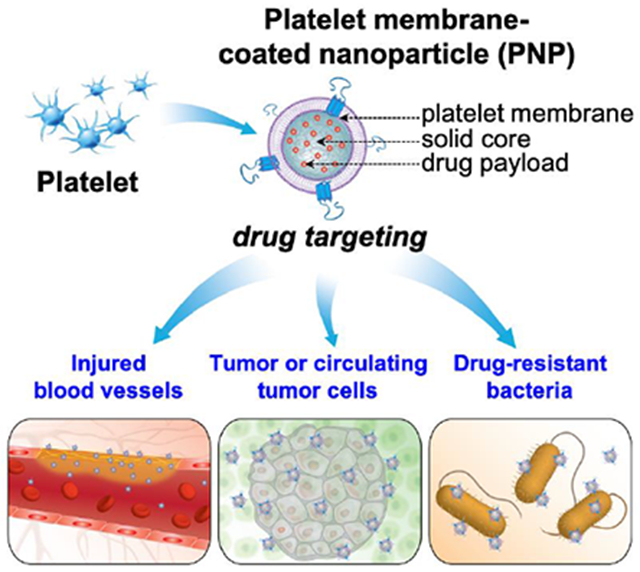
In this article, we review recent advances in developing PNPs as a class of nanomedicine for drug targeting. Specifically, we focus on three areas where PNPs gained the most attention for drug targeting applications: (1) targeting injured blood vessels to treat vascular diseases such as atherosclerosis, angioplasty, myocardial ischemia, ischemic stroke, and pulmonary embolism; (2) targeting tumor or circulating tumor cells (CTCs) for cancer treatment and detection; and (3) targeting drug-resistant bacteria for the treatment of infectious diseases (Figure 1). In each area, we discuss biointerfacing principles underlying specific drug targeting applications. We also highlight the interplay between cell membrane functions and substrate properties, which allows PNPs to adapt the physiological features of each disease for a successful outcome. This review demonstrates that platelet membrane coating is an effective strategy to leverage the multifaceted targeting ability of platelets for targeted drug delivery. We hope that the current development of PNPs will inspire new designs and breakthroughs for more dynamic and effective drug targeting toward better nanotherapeutics in the future.
Figure 1.
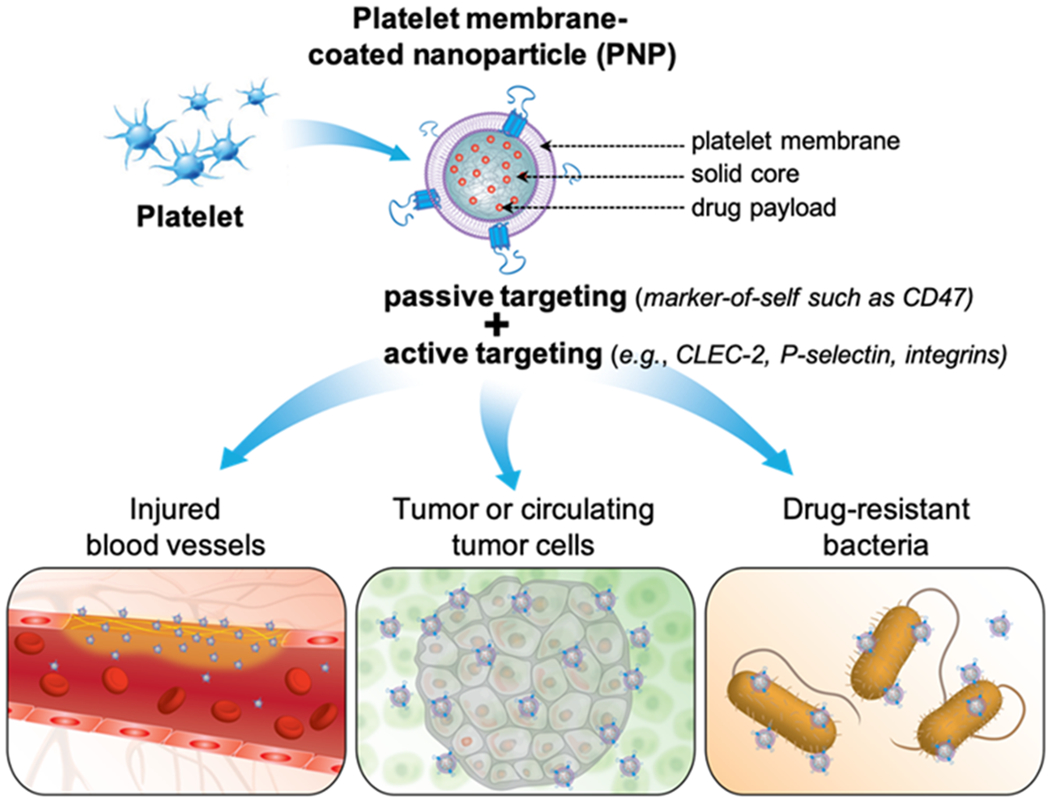
Schematic summary of using platelet membrane-coated nanoparticles (PNPs) for drug targeting. PNPs are made by wrapping membranes derived from natural platelets onto solid nanoparticle cores. PNPs leverage natural markers on the platelet membrane for drug targeting. The mechanism can be passive via markers-of-self such as CD47. It can also be active via surface antigens such as C-type lectin-like receptor 2 (CLEC-2), P-selectin, integrin α6β1, and integrin αIIbβ3. PNPs have been used to target drug payload to injured vasculatures (bottom left), tumor or circulating tumor cells (bottom middle), and drug-resistant bacteria (bottom right).
Targeting injured blood vessels to treat vascular diseases
Platelet adhesion, activation, and aggregation on the exposed subendothelial extracellular matrix (ECM) during the vessel wall injury are essential for hemostasis.[24] In this process, GPIb/V/IX and vWF form complexes that bind to the exposed collagen.[25] Circulating platelets interact with such complexes in part through integrin αIIbβ3, and decelerate. The interactions retain platelets in the proximity of the vessel wall, increase the contact between platelets and collagen, and potentiate further platelet activation cascade for coagulant activity. Such intrinsic ability of platelets to bind with injured vessels through multi-factor receptor-ligand interactions, together with their long-circulating properties, has motivated the use of PNPs for targeted delivery of therapeutics to treat various vascular diseases.
In atherosclerosis, lipids abnormally accumulate at the vascular wall, leading to endothelial activation and leukocyte infiltration.[26] A lipid-rich plaque gradually builds up and may eventually rupture, inducing vascular injury and exposing subendothelial ECM.[27] At the injury site, PNPs mimic platelets to bind with PSGL-1 expressed on activated endothelial cells and infiltrated leukocytes through P-selectin. They also bind with collagen, vWF, and fibrin on the exposed subendothelial ECM via receptors, including GPVI, GPIb/V/IX, and GPIIb/IIIa.[17, 28] Through these binding interactions, PNPs adhere to the injured vessel despite the high shear stress. For example, platelet membrane-coated poly (lactic-co-glycolic acid) (PLGA) nanoparticles adhered better to the plates coated with vWF, collagen, and fibrin than polyethylene glycol (PEG)-conjugated PLGA nanoparticles.[29] The study was conducted with shear stresses varying from 0 to 25 dyn/cm2, the range of physiological shear stresses at the aortic side of the aortic valve.[30] The binding of PNPs showed shear-dependent characteristics: PNPs adhered better to vWF under shear stresses around 20 dyn/cm2, but adhered better to collagen and fibrin under shear stresses lower than 5 dyn/cm2. This shear-dependence of PNPs resembles how natural platelets interact with adhesion factors: under high shear stress, platelets interact with vWF on the subendothelial ECM to decelerate; after slowing down, the platelets then interact with collagen and fibrin for binding.[24] In a mouse model with aortic valve sclerosis, these PNPs showed a higher accumulation at the sclerotic aortic valve than their PEG-conjugated counterparts. Platelet membrane-coated PLGA nanoparticles were also used to deliver rapamycin, an immunosuppressant, to the atherosclerotic plaque for selective induction of macrophage autophagy.[31] This formulation showed better efficacy in reducing the plaque area than PEG-conjugated PLGA nanoparticles. The binding of PNPs with multiple components of atherosclerosis makes them an attractive platform for targeting the disease at various stages. For instance, platelet membrane-coated PLGA nanoparticles were loaded with gadolinium, a magnetic resonance imaging contrast agent for plaque detection.[27] They targeted the established atherosclerotic plaques with prominent collagen deposition, leukocyte infiltration, and endothelial activation (Figure 2). These PNPs also targeted regions featuring early endothelial activation but without plaque development yet. With the ability to target both early and established plaques, they are useful tools to diagnose the different stages of atherosclerosis.
Figure 2.
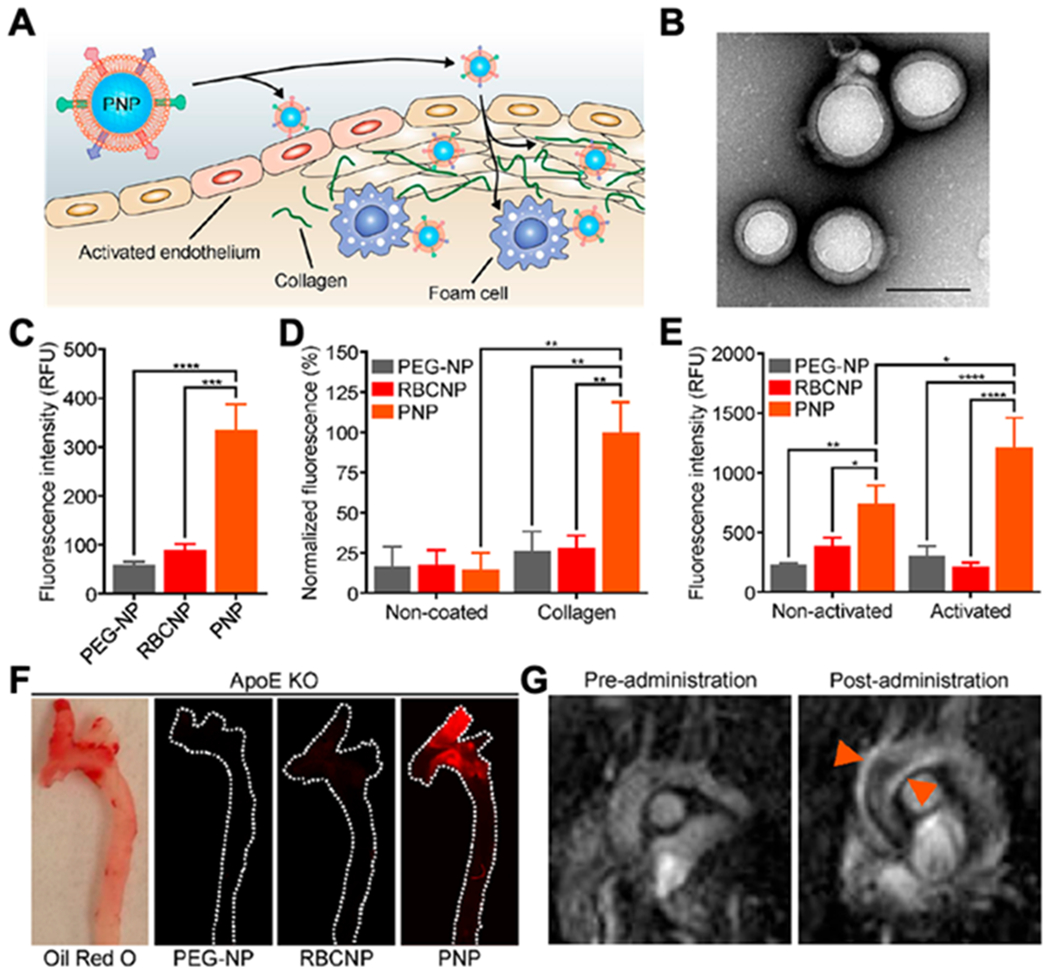
Platelet membrane-coated nanoparticles (PNPs) for targeting and detection of atherosclerosis. (A) Schematic illustration of PNPs targeting different components of the atherosclerotic plaque, including activated endothelium, collagen and foam cells. (B) Transmission electron microscopy (TEM) image of PNPs negatively stained with uranyl acetate (scale bar of 100 nm). (C) Quantification of PLGA nanoparticles functionalized with polyethylene glycol (PEG-NPs), PLGA nanoparticles coated with red blood cell membrane (RBCNPs) or PNPs bound to foam cells measured by flow cytometry (n = 3, mean ± SD). All formulations were made with PLGA cores labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD) dye. (D) Fluorescent quantification of PEG-NPs, RBCNPs, or PNPs bound to non-coated or collagen-coated surfaces (n = 3, mean ± the standard error of the mean). (E) Fluorescent quantification of PEG-NPs, RBCNPs, or PNPs bound to human umbilical vein endothelial cells (HUVEC) in the resting state or after activation with tumor necrosis factor alpha (TNF-α) (n= 3, mean ± SD). (F) Fluorescent imaging of aortic arches from ApoE knockout (ApoE KO) mice fed on a high-fat western diet after intravenous administration with PEG-NPs, RBCNPs, or PNPs (white line represents the physical outline of the aortic arch and red fluorescence represents nanoparticles). Oil Red O staining was used to confirm the presence of plaque in the mice. (G) T1-weighted magnetic resonance imaging (MRI) ofApoE KO mice before and 1 hour after administration with PNPs loaded with gadolinium, an MRI contrast agent (orange arrows point to regions of positive contrast in the aortic arch). The statistical analysis was performed using one-way analysis of variance (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Reproduced with permission. [27] Copyright 2018, The American Chemical Society.
In angioplasty- or stent-induced restenosis, mechanical damages to the endothelium lead to an exposed subendothelial ECM. The exposure then recruits platelets and leukocytes and triggers thrombosis and inflammation. The subsequent cell proliferation and ECM deposition in the intima result in intimal thickening and vessel narrowing.[32] At the disease site, multiple components in the ECM, such as collagen and fibrin, provide binding sites for PNPs.[28] For example, platelet membrane-coated PLGA nanoparticles were shown to selectively bind with the exposed subendothelial ECM of a surgically injured human artery.[33] This selective adhesion occurred in a rat model of angioplasty-induced arterial injury, where PNPs bound rapidly to the injured vessel and retained for 5 days. When loaded with cytotoxic drug docetaxel, this formulation inhibited neointimal growth and reduced restenosis. Platelet membranes were also coated onto dendritic polymer nanoclusters to deliver anti-restenosis drugs.[34] The membrane coating enhanced the colloidal stability of the nanoclusters, making them more dispersible and easier for injection. When loaded with an endothelium-protective epigenetic inhibitor JQ1, this formulation showed better efficacy in mitigating neointimal growth and promoting endothelial recovery than the free drug.
PNPs were also developed to target myocardial ischemia, where a thrombus in the coronary artery occludes the blood flow to the heart.[35] In this disease, tissue hypoxia impairs the endothelial barrier function, leading to vascular injuries and increased vascular permeability. Conventional reperfusion of the ischemic tissue bears risks of inducing oxidative damages and inflammatory response, which together exacerbate tissue injury. In myocardial ischemia, PNPs can adhere to components on the exposed subendothelial ECM for drug targeting.[28] For example, PLGA nanoparticles coated with platelet membranes were shown to target the injured vessel.[36] The membrane was further conjugated with prostaglandin E2 (PGE2) that binds with cardiomyocytes. As a result, the formulation exhibited a dual-targeting ability to the ischemic heart. These nanoparticles encapsulated the whole secretome of cardiac stem cells purified from the cell culture media for delivery. When tested in mice with myocardial ischemia and reperfusion injuries, they showed better efficacy in promoting angiogenesis, repairing the damaged tissue, and restoring cardiac function than various control nanoparticles.
Away from the heart, the thrombus in the artery can block the blood flow to the brain, leading to ischemic stroke. Unless the occluded vessel is rapidly reperfused, ischemic stroke can cause severe neurological damages or death.[37] In this disease, PNPs can target the occluded vessel, where the injury of the vascular wall exposes subendothelial ECM and the fibrin-rich thrombus. For example, ‘nanobubbles’ using platelet membranes to enclose nanometer-scale gas bubbles rather than solid cores were made.[38] In this design, sulfur hexafluoride (SF6) gas was inducted into a suspension of platelet membrane vesicles and trapped by the vesicles when the suspension was sonicated. In a mouse model of ischemic stroke, these ‘nanobubbles” rapidly accumulated in the stroke lesion within 30 min after intravenous administration. Such accumulation promoted microvascular remodeling and recanalized the occluded vessels. The accumulation of SF6 also generated enhanced ultrasound imaging signals useful for timely diagnosis and monitoring of the disease. In another example of treating stroke, dextran polymer nanoparticles loaded with 5-(3,5-dichloro-2-hydroxybenzylamino)-2-hydroxybenzoic acid (ZL006e), a neuroprotectant, were coated with platelet membranes.[39] The membranes were further conjugated with the trans-activator of transcription (Tat), a cell-penetrating peptide, which was linked with recombinant tissue plasminogen activator (rtPA), a thrombolytic drug, through a thrombin-cleavable peptide. Once the nanoparticles bound to the thrombus, a high level of thrombin triggered the cleavage of the peptide linker and released rtPA on-demand. This process also exposed Tat peptide in situ, which then helped the nanoparticles to cross the blood-brain barrier for the delivery of ZL006e to the ischemic brain. As another example, platelet membrane vesicles were co-loaded with magnetic iron oxide (γ-Fe2O3) nanoparticles and L-arginine, a precursor for the biosynthesis of nitric oxide (NO) gas.[40] When intravenously injected into mice with ischemic stroke, these nanoparticles rapidly accumulated in the stroke lesion. The accumulation was due to combined targeting effects via the coated platelet membranes and the external magnetic field. At the disease site, L-arginine was released and internalized by local endothelial cells and platelets to synthesize NO. As a signaling molecule, NO released by endothelial cells induced relaxation of smooth muscle cells and promoted vasodilation while NO produced by platelets inhibited platelet activation and aggregation.[41, 42] These effects worked together and restored the local blood flow in the stroke lesion.
In the lower extremities of the body, thrombi in the deep vein can detach from the vascular wall. They migrate through the bloodstream, lodge in the lung artery, and block the blood flow in the lung, leading to pulmonary embolism.[43] In this case, PNPs target the fibrin of the thrombus via GPIIb/IIIa.[28] Alternatively, PNPs can adhere to the activated platelets in the thrombus, likely via PSGL-1 or GPIb/V/IX, which can both bind to P-selectin on the activated platelets.[44–46] Based on these mechanisms, platelet membrane vesicles encapsulating gold nanorods and urokinase plasminogen activator (uPA), a thrombolytic drug, were developed to treat pulmonary embolism.[47] When intravenously injected into mice with pulmonary embolism, these nanoparticles accumulated at the pulmonary thrombi and released uPA. They successfully induced thrombolysis and resulted in fewer residual thrombi in the lung than bare gold nanorods. In another study, PLGA nanoparticles were coated with platelet membranes, and the membranes were further conjugated with rtPA for targeted delivery (Figure 3).[44] In a mouse model of pulmonary embolism, this formulation targeted the pulmonary thrombus, induced local clot degradation, improved the survival rate of mice, and reduced the risk for bleeding complications compared with free rtPA. Notably, these nanoparticles were also tested in mouse models of mesenteric arterial thrombosis and ischemic stroke. They showed enhanced thrombolysis in mice with mesenteric arterial thrombosis and improved the survival rate of mice with ischemic stroke compared with free rtPA. Therefore, this study demonstrated the versatility of PNPs in targeting various vascular diseases.
Figure 3.
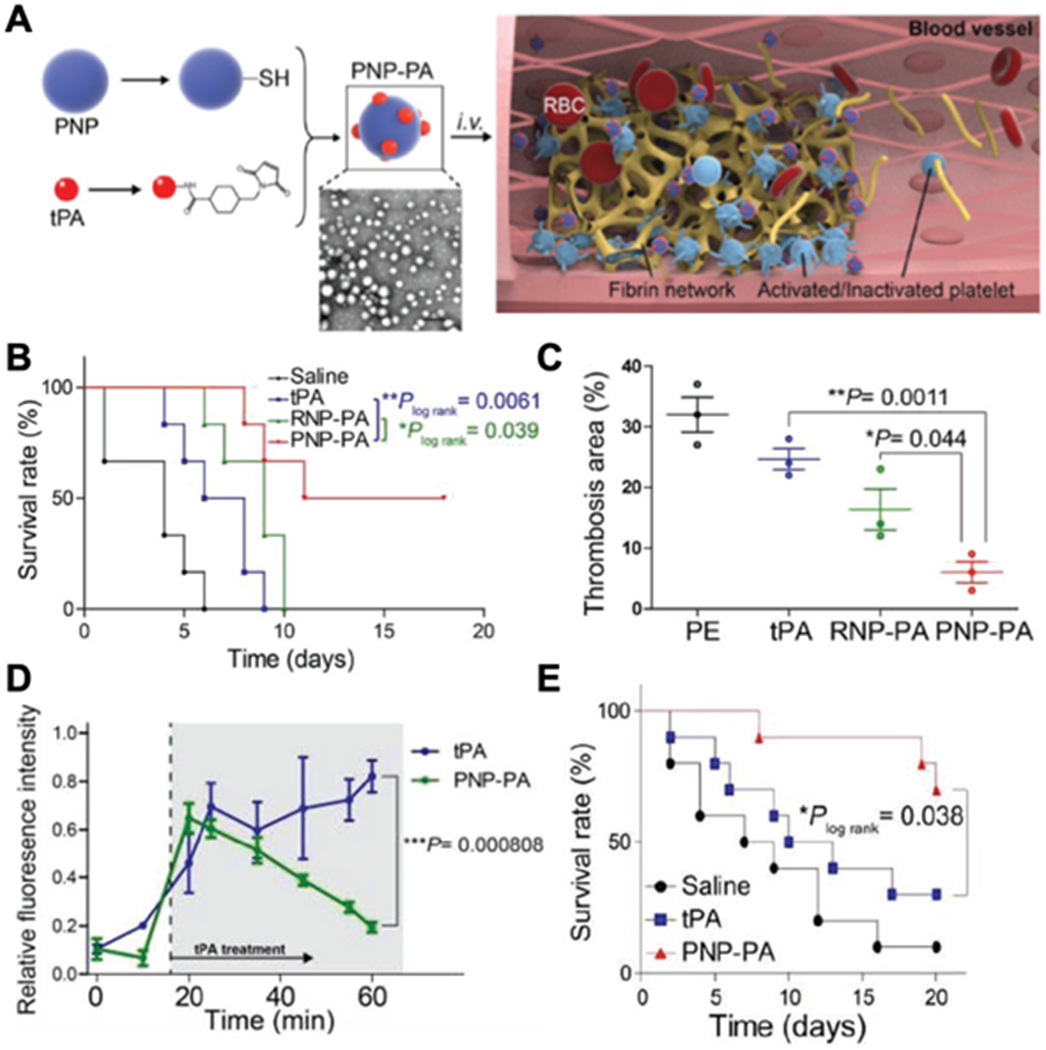
PNP-enabled targeted delivery of recombinant tissue plasminogen activator (tPA) to treat multiple vascular diseases. (A) Schematic illustration of the synthesis of PNPs with tPA conjugated to the particle surface (PNP-PA) and thrombolysis induced by PNP-PA (scale bar, 1 μm). (B) Survival rate of mice with pulmonary embolism (PE) treated with 5mg/kg tPA, 0.5mg/kg PNP-PA or 0.5mg/kg RBC membrane-cloaked PLGA nanoparticles with tPA conjugated to the particle surface (RNP-PA) over 20 days (n = 6, mean ± SD). All dosages were based on tPA content. (C) Quantitative analysis of the thrombotic area of the groups in (B) based on hematoxylin and eosin staining of the lung tissue of the mice (n = 3, mean ± SD). (D) Quantification of the fluorescent intensity of rhodamine 6G-labeled platelets in the thrombus in mice with mesenteric arterial thrombosis treated with 5 mg/kg tPA or 0.5mg/kg PNP-PA (n = 3, mean ± SD). Before establishing the thrombosis disease in mice, rhodamine 6G-labeled platelets were administered into mice for thrombus visualization. (E) Survival rate of mice with transient middle cerebral artery occlusion treated with saline, 1 mg/kg PNP-PA or 1mg/kg tPA (n = 10, mean ± SD). The statistical analysis was performed using Student’s t-test for comparison between two groups and one-way analysis of variance for comparison among multiple groups (*p<0.05, **p<0.01 and ***p<0.001). Reproduced with permission.[44] Copyright 2019, John Wiley & Sons.
Overall, PNPs with the inherent vascular-targeting capability acquired from membrane coating have shown great potential as a versatile delivery platform to treat a wide range of vascular diseases (Table 1). So far, most studies of using PNPs to treat vascular diseases have been conducted in rodent models. Notably, humans and rodents show distinct hemodynamics and disease progression. Optimizing the physicochemical properties of the PNPs, including their size and shape, can help PNPs adapt to the unique hemodynamics in humans.[48, 49] Studies in this regard will aid future clinical translation of PNPs. With continuous development, PNPs are expected to make a high impact on the treatment of vascular diseases.
Table 1.
Summary of PNPs for targeting injured vessels to treat vascular diseases
| Disease | Core | Cargo | Ref. |
|---|---|---|---|
| Atherosclerosis | PLGA nanoparticles | Fluorescent dye | [29] |
| Rapamycin (used as an immunosuppressant) | [31] | ||
| Inserted with lipid-chelated gadolinium (an MRI contrast agent) in the membrane | [27] | ||
| Angioplasty-induced restenosis | PLGA nanoparticles | Docetaxel (a cytotoxic drug) | [33] |
| Poly(amidoamine)-polyvalerolactone nanoclusters | JQ1 (an endothelium-protective epigenetic inhibitor) | [34] | |
| Myocardial ischemia and reperfusion injuries | PLGA nanoparticles | Secretome of cardiac stem cells (promote cardiac repair and regeneration) | [36] |
| Ischemic stroke | Sulfur hexafluoride gas | Sulfur hexafluoride gas | [38] |
| Dextran nanoparticles | ZL006e (a neuroprotectant) and rt-PA (a thrombolytic drug) | [39] | |
| Iron oxide (γ-Fe2O3) nanoparticles | L-arginine (a precursor for biosynthesis of nitric oxide gas) | [40] | |
| Pulmonary embolism | Gold nanorods | Urokinase plasminogen activator (a thrombolytic drug) | [47] |
| PLGA nanoparticles | rtPA | [44] |
Targeting cancer cells for cancer treatment and detection
By presenting ‘markers of self’ such as CD47 on the platelet membranes, PNPs can evade immune clearance, circulate long, and preferentially accumulate at the tumor sites through the EPR effect.[16, 50] But the primary motivation behind their design is the multi-factor and specific binding interactions between the platelets and the cancer cells desirable for active drug targeting.[51] For example, primary tumors recruit platelets and form ‘early metastatic niches’ that promote vascular permeation and tumor extravasation.[52, 53] Tumor cells also form thrombi with platelets for enhanced vascular adhesion and transendothelial migration.[54, 55] During the metastasis, tumor cells shield themselves by attaching to platelets for protection against shear forces and immune attack, therefore increasing the chance of metastasis formation.[56, 57] Tumor cells achieve such binding interactions by overly expressing ligands such as podoplanin, P-selectin PSGL-1, A disintegrin and metalloproteinase domain-containing protein 9 (ADAM-9), and fibrinogen/αvβ3. These ligands bind with platelet receptors, including C-type lectin-like receptor 2 (CLEC-2), P-selectin, integrin α6β1, and integrin αIIbβ3, respectively.[58–61] While tumor cells harness such binding interactions to gain advantages for their metastasis, PNPs can exploit these interactions for active drug targeting.
PNPs have been used to deliver a wide range of anti-cancer drugs for targeted cancer chemotherapy. For example, the platelet membrane was coated onto docetaxel (DTX)-loaded PLGA nanoparticles (denoted ‘PM/PLGA/DTX’).[62] The targeting ability of the nanoparticles was tested by examining the in vivo biodistribution of DTX with mice bearing A549 tumors, a model human lung cancer. The results showed that free DTX rapidly distributed to the tumor 2 h after the injection but was rapidly eliminated with negligible concentration at 12 h. In contrast, when mice were injected with PM/PLGA/DTX and the uncoated control (denoted ‘PLGA/DTX’), DTX concentration at the tumor sites increased gradually, reaching a peak value at 12 h. PM/PLGA/DTX led to higher DTX tumor concentrations at all time points than the uncoated nanoparticles. At 48 h, over half of the DTX was retained, and the concentration was more than twice that in mice injected with uncoated nanoparticles. Due to such targeting effect, PM/PLGA/DTX showed higher inhibition of tumor growth in vivo. Moreover, PNPs have been made with different cores to deliver various anti-cancer compounds for the treatment of cancers in mouse xenograft models of colorectal cancer (HT29 cells),[63] liver cancer (H22 cells),[64] breast cancer (MCF-7 cells),[65] and ovarian cancer (SK-OV-3 cells).[66]
Besides small-molecule anti-cancer compounds, PNPs were also used to target small interfering RNA (siRNA) in vivo to silence tumor-relevant genes (Figure 4). In one design, synthetic siRNAs were loaded inside the porous metal-organic framework (MOF) nanoparticles of zeolitic imidazolate framework-8 (ZIF-8) through an in situ biomineralization method, followed by coating with platelet membranes.[67] The resulting nanoparticles (denoted ‘P-MOF-siRNA’) showed lower immunogenicity than uncoated MOF nanoparticles. A binding test showed a higher affinity of P-MOF-siRNA to SK-BR-3 cells, a human breast cancer cell line, than the RBC membrane-coated control nanoparticles (denoted ‘R-MOF-siRNA’). A cell uptake study showed a six-fold increase of P-MOF-siRNA uptake than R-MOF-siRNA uptake by the cancer cells. The enhanced affinity was attributed to the binding between CD24 and the P-selectin. On nude mice bearing subcutaneous SK-BR-3 tumors, the treatment with P-MOF-siRNA exhibited a better inhibition of tumor growth and a higher overall mouse survival rate than those treated with control nanoparticles. Besides tumor inhibition, the platelet membrane-coated MOFs developed in this study can also load siRNAs against other genes for broader applications.
Figure 4.
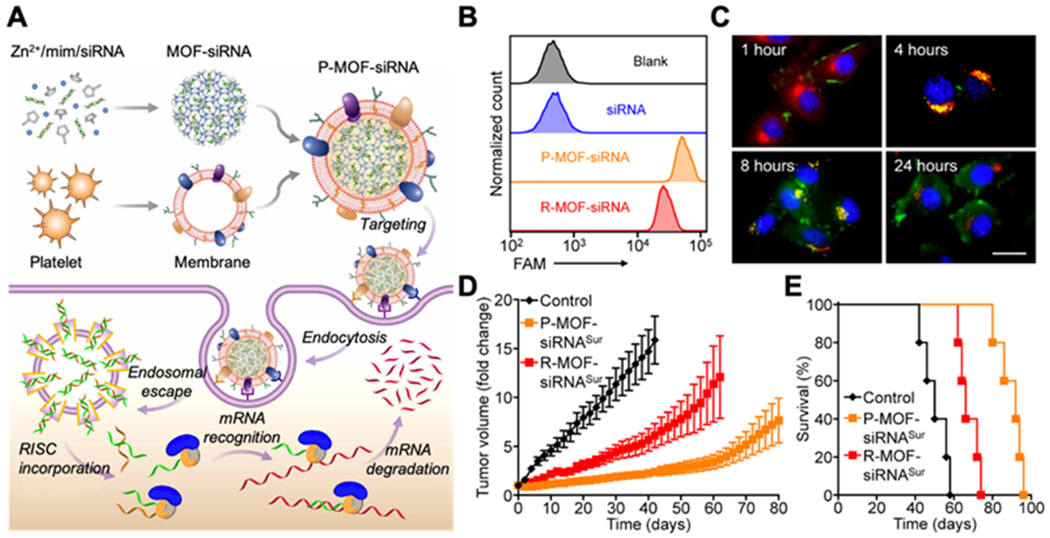
Targeted gene silencing in vivo by platelet membrane-coated metal-organic framework (MOF) nanoparticles. (A) Platelet membrane-coated siRNA-loaded MOF (P-MOF-siRNA) for gene silencing. To fabricate the P-MOF-siRNA formulation, siRNA-loaded MOF (MOF-siRNA) cores are generated by mixing the siRNA payload with Zn2+ and 2-methylimidazole (mim), followed by coating with natural cell membrane derived from platelets. When the P-MOF-siRNA nanoparticles are endocytosed by a target cell, the low pH of the endosomes causes escape of the siRNA into the cytosol. Upon incorporation with RNA-induced silencing complex (RISC), the target mRNA is then recognized and degraded, leading to gene silencing. (B) Uptake of siRNA in SK-BR-3 cells 24 hours after incubation with free siRNA, P-MOF-siRNA, or nanoparticles coated with red blood cell (RBC) membrane (R-MOF-siRNA). (C) Fluorescent visualization of siRNA localization in SK-BR-3 cells 1, 4, 8, and 24 hours after incubation with P-MOF-siRNA (scale bar, 20 μm; siRNA, green; nuclei, blue; endosomes, red). (D) Growth kinetics of SK-BR-3 tumors implanted subcutaneously into nu/nu mice and treated intravenously with P-MOF-siRNASur or R-MOF-siRNASur every 3 days for a total of four administrations (n = 5; mean ± SEM; siRNASur: siRNA against survival). (E) Survival of the mice in (D) over time (n = 5). Reproduced with permission.[67] Copyright 2020, the American Association for the Advancement of Science.
PNPs have also been made to target photosensitizers for cancer photothermal or photodynamic therapies (PTT or PDT).[68] PTT relies on the photothermal agents delivered to the tumor tissue to absorb the energy from the external laser and transform it into heat to ablate the cancer cells. Meanwhile, PDT uses the photosensitizers to generate singlet oxygen upon laser excitation to kill cancer cells. Both therapies are considered minimum invasive for cancer treatment. Recently, verteporfin, a photodynamic agent, was encapsulated into PLGA nanoparticles and coated with platelet membrane (denoted ‘NP-Ver@P’) for cancer PDT.[69] PLGA encapsulation shifted verteporfin absorption peak from 682 nm to 712 nm, making it excitable in deeper tissues. Solar light in 680~730 nm region with a low output energy density of only 0.05 W/cm2 was sufficient to irradiate these nanoparticles. NP-Ver@P combined both passive and active targeting, exhibiting a higher tumor uptake than the RBC membrane-coated counterpart (denoted ‘NP-Ver@R’) capable of passive targeting only. On mice bearing 4T1 breast tumors, the treatment with NP-Ver@P resulted in smaller tumor volumes and a higher survival rate than the treatment with NP-Ver@R or PBS, demonstrating the benefit of platelet membrane coating. In another study, the platelet membrane was coated onto W18O49 nanoparticles for cancer PDT,[70] in which W18O49 served as the PDT agent. Platelet membrane coating protected them from premature oxidation and enhanced tumor accumulation. In a mouse xenograft model of Raji cells, a human lymphoma cell line, these nanoparticles led to higher reactive oxygen species (ROS) concentration at the tumor sites than uncoated nanoparticles. As a result, they generated more heat and showed better inhibition of tumor growth.
As PNPs become popular for cancer drug targeting, they have also been increasingly used to deliver not a single, but multiple agents concurrently for combinatorial cancer treatment. The drug combination can be different chemotherapeutic compounds that target distinct cancer pathways. In a recent study, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and doxorubicin (Dox) were co-encapsulated into nanogel cores and then coated with platelet membrane (Figure 5).[71] In this design, TRAIL-induced apoptosis of tumor cells by binding to their death receptors on the surface.[72] Meanwhile, Dox intercalated into the nuclear DNA of the cancer cells and triggered the intrinsic apoptosis signaling pathway. Together, they generated higher anti-cancer efficacy than single-drug control nanoparticles. Meanwhile, the chemotherapeutic compounds and MRI contrast agents were also combined and encapsulated within PNPs for MRI-guided chemo-chemodynamic combinatorial therapy. For example, hollow MnO2 cores were made and loaded with bufalin, a chemotherapy drug, followed by coating with platelet membranes.[73] In the acidic tumor microenvironment, MnO2 cores decomposed, releasing bufalin and yielding Mn2+ as an MRI contrast agent. In vivo MRI studies revealed a T1 contrast enhancement at the tumor site. The Mn2+ also reacted with endogenous H2O2 and generated hydroxyl radicals (HO·), which boosted bufalin’s anti-cancer efficacy. Additionally, PNPs were also made to combine radio and photothermal agents for anti-cancer treatment.[74] Recently, platelet membranes were coated onto bismuth sulfide nanorods (BNRs). In this design, the nanorod acted as a radiosensitizer upon X-ray irradiation and a photothermal agent upon near-infrared irradiation at a wavelength of 808 nm. In all these examples, PNPs vary in their anti-cancer mechanisms. However, because of the platelet membrane, they were able to target tumors for better efficacy. These studies validate platelet membrane coating as an effective targeting strategy for targeted cancer treatment.
Figure 5.
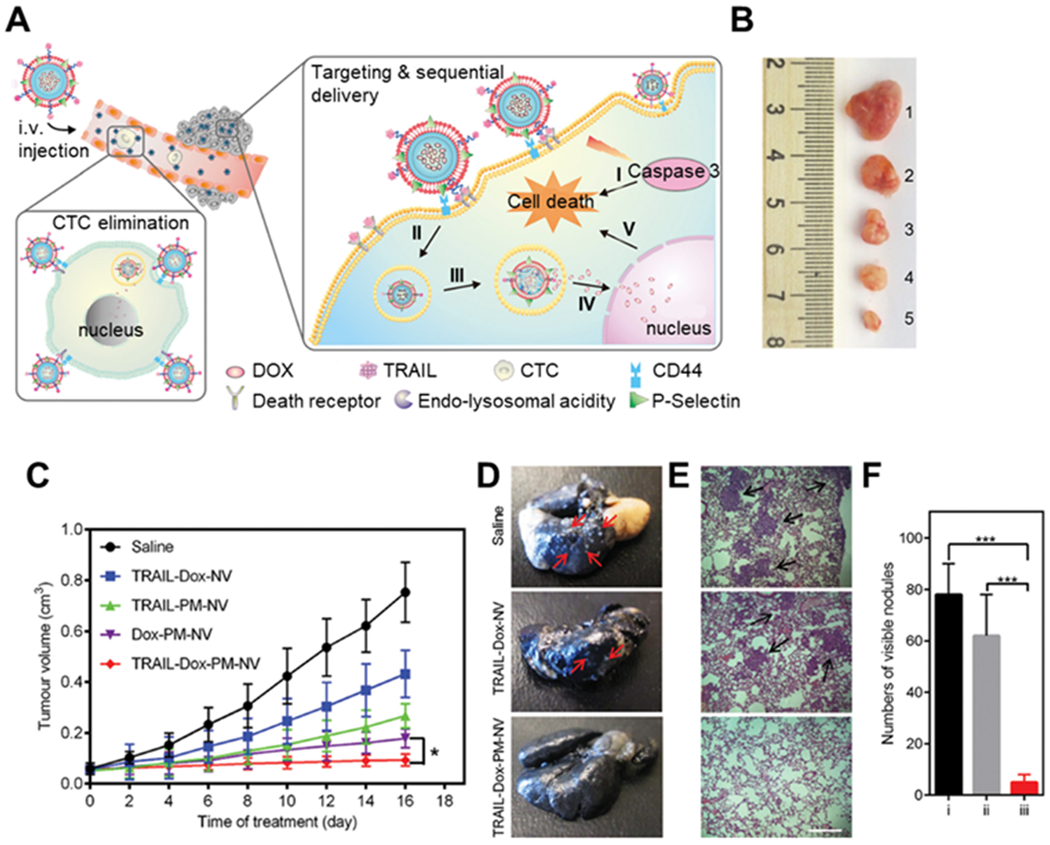
The design of anti-cancer platelet-mimicking nanovehicles. (A) Schematic of drug-loaded platelet membrane (PM)-coated core-shell nanovehicles (PM-NV) for eliminating CTCs and sequential delivery of TRAIL and Dox. TRAIL-Dox-PM-NV captured the CTCs via specific affinity of P-Selectin and overexpressed CD44 receptors and subsequently triggered TRAIL/Dox-induced apoptosis signaling pathways. i) the interaction of TRAIL and death receptors (DRs) to trigger the apoptosis signaling; ii) the internalization of TRAIL-Dox-PM-NV; iii) the dissociation of TRAIL-Dox-PM-NV mediated by the acidity of lyso-endosome; iv) release and accumulation of Dox in the nuclei; v) intrinsic apoptosis triggered by Dox. (B) Representative images of the MDA-MB-231 tumors after treatment with different TRAIL/Dox formulations on day 16 (from top to bottom, 1: saline, 2: TRAIL-Dox-NV, 3: TRAIL-PM-NV, 4: Dox-PM-NV, 5: TRAIL-Dox-PM-NV) at TRAIL dose of 1 mg/kg and Dox dose of 2 mg/kg. (C) The MDA-MB-231 tumor growth curves after intravenous injection of different TRAIL/Dox formulations. Error bars indicate s.d. (n = 5). *p < 0.05 (two-tailed Student’s t -test). (D) Representative images of the lung tissues 8 weeks post intravenous injection with MDA-MB-231 cells and different TRAIL/Dox formulations. Red arrows indicate the visible metastatic nodules. (E) Histological observation of the lung tissues after treatment. The lung sections were stained with H&E. Black arrows indicate the tumor cells. Scale bar: 200 μm. (F) Quantification of visible metastatic nodules. i) Saline; ii) TRAIL-Dox-NV; iii) TRAIL-Dox-PM-NV. Error bars indicate s.d. (n = 3). *** p < 0.001 (two tailed Student’s t-test). Reproduced with permission. [71] Copyright 2015, John Wiley & Sons.
Besides solid tumors, circulating tumor cells (CTCs) have also become a target of PNP-based therapies, including CTC killing for preventing metastasis and CTC capture for early cancer detection. To kill CTCs, researchers conjugated platelet membrane with TRAIL and coated the membrane onto micron-sized silica particles.[75] Mice injected with MDA-MB-23 cells, a human breast cancer cell line, followed by the injection of platelet membrane-coated particles developed less metastasis than those injected with saline buffer, particles without conjugated TRAIL, or soluble TRAIL. For CTC capture, the platelet membrane was hybridized with membranes of leucocytes and then coated onto magnetic nanoparticle cores.[76] In this case, the hybridization with the leucocyte membranes brought in ‘homologous characteristics’ and reduced undesirable binding of nanoparticles with leucocytes if coated with platelet membranes alone. When tested with spiked blood samples, these nanoparticles showed better capture efficiency and higher purity of the capture cells than the same magnetic nanoparticles functionalized with anti-epithelial cellular adhesion molecules (EpCAM).
Overall, recent studies have demonstrated PNPs as a versatile platform for broad cancer targeting (Table 2). Platelet membrane coating can reduce immunogenicity, attenuates macrophage uptake, and enhances pharmacokinetic profiles. These functions all contribute to higher anti-cancer efficacy in vivo. Meanwhile, the successful use of PNPs for CTC detection suggests that the platform may serve as a promising tool for cancer diagnosis.
Table 2.
Summary of PNPs for targeted cancer therapy and CTC capture
| Therapeutic modality | Core | Drug/Active agent | Cancer model | Ref. |
|---|---|---|---|---|
| Chemotherapy | PLGA | docetaxel | A549 mouse xenograft | [62] |
| gelatin nanogels (GN) | irinotecan | HT-29 mouse xenograft | [63] | |
| chitosan oligosaccharide -PLGA copolymer. | bufalin | H22 mouse xenograft | [64] | |
| black phosphorus quantum dots | Hederagenin | n/a | [65] | |
| nanostructured lipid carrier | paclitaxel | n/a | [66] | |
| RNA inference | zeolitic imidazolate framework-8 (ZIF-8) metal-organic frameworks (MOFs) | siRNA | SK-BR-3 mouse xenograft | [67] |
| PTT/PDT | PLGA | verteporfin (a photodynamic agent) | 4T1 mouse xenograft | [69] |
| W18O49 nanoparticles | W18O49 | Raji lymphoma mouse xenograft | [70] | |
| Combinatorial therapy | acrylamide-based nanogels | TRAIL and doxorubicin | Mouse models of MDA-MB-231 xenograft and metastasis | [71] |
| hollow MnO2 NPs | bufalin | H22 mouse xenograft | [73] | |
| mesoporous silica-coated bismuth nanorod (as both a radio sensitizer and a photothermal sensitizer) | n/a | 4T1 mouse xenograft | [74] | |
| CTC capture and killing | synthetic silica particles | TRAIL | Experimental lung metastasis by i.v. injection of MDA-MB-231 cells | [75] |
| Iron oxide magnetic nanoparticles | Dye-conjugated antibody on the surface for detection | MCF-7 cells spiked in whole blood obtained from healthy donors and breast cancer patient blood samples | [76] |
Targeting drug-resistant bacteria to treat infectious diseases
Despite the profound success of antibiotics, the treatment of bacterial infection is increasingly threatened by the emergence of antibiotic resistance.[77–80] Nanoparticles are used to address this challenge by targeting antibiotic payload to the bacteria to enhance drug potency.[81–83] The targeting can be passive, based on the EPR effect at the infection sites. It can also be active through direct binding with the bacteria via electrostatic charge interactions or pathogen-binding ligands on the nanoparticle surfaces. Based on these mechanisms, a variety of nanoparticle platforms have been developed for the targeted delivery of antimicrobials. Meanwhile, many pathogenic bacteria, including several strains of staphylococci and streptococci, rely on the binding interactions with the platelets to promote their colonization and evade the host immune response. The bacteria can bind directly with platelet proteins, such as GPIb,[23, 84–87] GPIIb/IIIa,[88–90] gC1q-R/P32,[91] or the vWF receptor.[92] Alternatively, they can bind indirectly through a plasma-bridging molecule, such as fibrinogen,[93–97] fibronectin,[98] and IgG,[19, 99, 100] that links bacterial and platelet surface receptors. The typical binding mechanisms between various bacteria and platelet are summarized in Table 3. Even under flow conditions, pathogenic bacteria can still engage effective binding with platelet despite the high shear stress.[19] Such common and diverse platelet-bacterium binding interactions have motivated the use of PNPs to harness the inherent bacterial adherence mechanisms for targeted antibiotic delivery to treat bacterial infections.
Table 3.
Summary of direct binding between bacteria and platelet.
| Bacterial pathogens | Bacterial proteins | Platelet receptors and bridging proteins | Reference |
|---|---|---|---|
| Staphylococcus aureus | ClfA | fibrinogen, GPIIb/IIIa, FcγRIIa, IgG, gC1q-R/P32 | [98] |
| ClfB | fibrinogens, GPIIb/IIIa, FcγRIIa, IgG, gC1q-R/P32 | [93] | |
| SpA | GPIb, vWF, gC1q-R/P32 | [91, 92] | |
| FnBPA and FnBPB | fibrinogen, fibronectin, GPIIb/IIIa, FcγRIIa, IgG | [94, 98, 99] | |
| SdrE | complement, IgG | [100] | |
| SraP | GPIb | [86] | |
| IsaB | GPIIb/IIIa | [89] | |
| Staphylococcus epidermidis | SdrG | fibrinogen, GPIIb/IIIa, FcγRIIa, IgG | [88] |
| Streptococcus sanguis | SrpA | GPIb, vWF, FcγRIIa | [23, 85] |
| Streptococcus agalactiae | FbsA | fibrinogen, IgG, GPIIb/IIIa | [96, 97] |
| Streptococcus pyogenes | M protein | FcγRIIa, GPIIb/IIIa, fibrinogen | [95] |
| Streptococcus gordonii | GspB/HsA | GPIb | [84] |
| PadA | GPIIb/IIIa | [90] | |
| Porphyromonas gingivalis | Hgp | GPIb | [87] |
ClfA, B: clumping factor A, B; SpA: surface protein A; FnBPA, B: fibronectin-binding protein A, B; IgG, immunoglobulin G; IsaB: Immunodominant surface antigen B; SdrE, G: serine-aspartate repeat protein E, G; SrpA: serine-rich protein A; FbsA: fibrinogen-binding protein from Streptococcus agalactiae; GspB: glycosylated streptococcal protein B; HsA: hemagglutinin salivary antigen; PadA: platelet adherence protein A; Hgp: high molecular mass gingipain.
Among various bacterial pathogens, Staphylococcus aureus (S. aureus) is a prominent Gram-positive bacterium responsible for a vast range of human skin and wound infections.[101] Over the past few decades, S. aureus has become resistant to a wide range of antibiotics, including the entire beta-lactam class of antibiotics.[102] The extensive use of new antibiotics, including vancomycin, linezolid, tedizolid, daptomycin, ceftaroline, and tigecycline, has accelerated the emergence of resistant S. aureus strains, especially toward vancomycin.[103] Currently, virulent strains of methicillin-resistant S. aureus (MRSA) have become increasingly prevalent, imposing a paramount clinical challenge that threatens public health.[104] Binding to the platelet is one pathway used by MRSA to invade the host. The binding can be directly through bacterial factors such as surface protein A (SpA), serine-rich adhesin for platelets (SraP), or immunodominant surface antigen B (IsaB) with GPIb/vWF on the platelet. It can also be indirectly through fibrinogen-fibrin bridges that link bacterial factors with the platelet receptors such as GPIIb/IIIa, platelet immunoglobulin Fc receptor (FcγRIIa), and gC1q-R/P32.[19, 105]
To exploit natural MRSA-platelet binding for delivery, researchers coated platelet membranes onto PLGA cores and used the nanoparticles to target vancomycin to MRSA bacteria (Figure 6).[33] When incubated with formalin-fixed MRSA, PNPs showed a much higher binding capacity with MRSA than uncoated nanoparticles. Meanwhile, in a mouse model of MRSA bacteremia, mice treated with vancomycin-loaded PNPs had a lower bacterial load across all major organs than those treated with free vancomycin or vancomycin-loaded cores without coating. The results of this study validate the ability of PNPs to target bacteria and enhance antibiotic potency.
Figure 6.
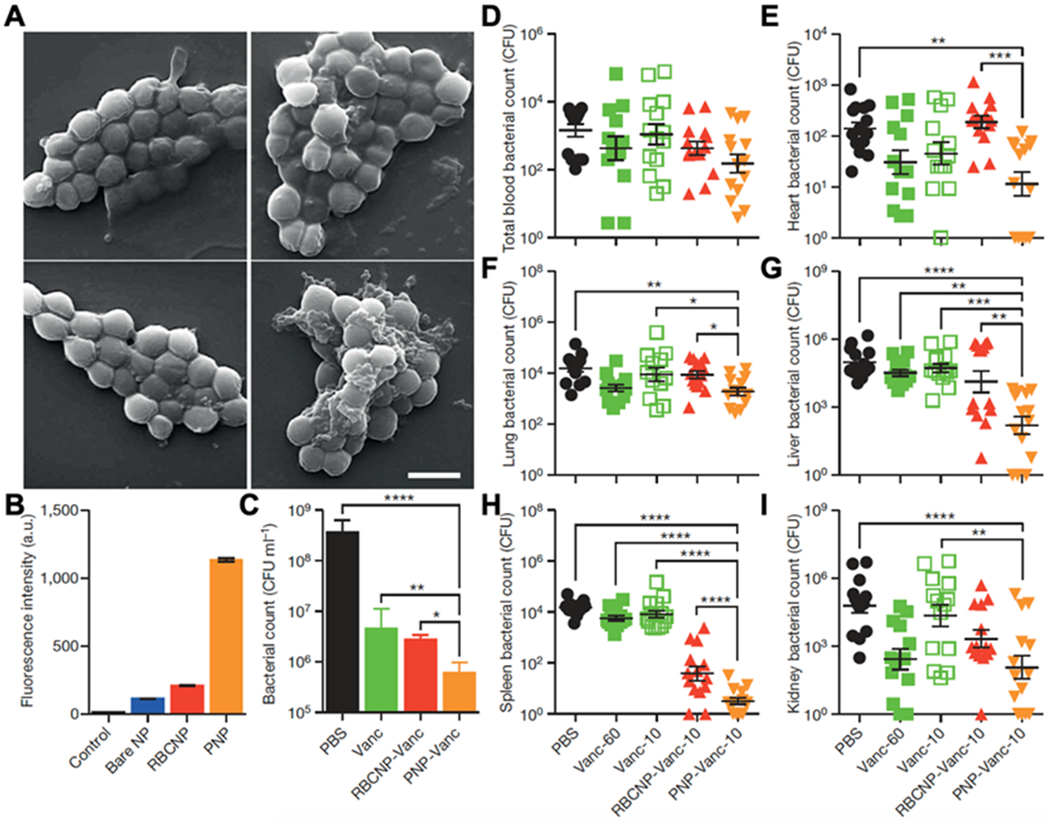
Binding of PNPs to platelet-adhering pathogens for antibiotic delivery. (A) SEM images of MRSA252 bacteria after incubation with PBS (top left), bare NPs (top right), RBCNPs (bottom left), and PNPs (bottom right). Scale bar, 1 mm. (B) Normalized fluorescence intensity of dye-loaded nanoformulations retained on MRSA252 based on flow cytometric analysis. Bars represent means ± s.d. (n = 3). (C) In vitro antimicrobial efficacy of free vancomycin, vancomycin-loaded RBCNPs (RBCNP-Vanc), and vancomycin-loaded PNPs (PNP-Vanc). Bars represent means ± s.d. (n = 3). (D-I) In vivo antimicrobial efficacy of free vancomycin at 10 mg kg-1 (Vanc-10), RBCNP-Vanc-10, PNP-Vanc-10, and free vancomycin at 6 times the dosing (Vanc-60, 60 mg kg-1) was examined in a mouse model of systemic infection with MRSA252. After 3 days of treatments, bacterial loads in different organs including blood (D), heart (E), lung (F), liver (G), spleen (H) and kidney (I) were quantified. Bars represent means ± s.e.m. (n=14). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. Reproduced with permission.[33] Copyright 2015, Springer Nature.
Besides binding with platelet to invade the host, pathogenic bacteria also release virulent toxins to attack platelets and promote infection. For example, MRSA releases α-toxin to induce platelet aggression, which aids bacterial colonization and immune evasion. The mechanism is likely to embed themselves inside the thrombi and therefore escape the immune surveillance.[106, 107] Some strains of Escherichia coli (E. coli) secrete Shiga toxin and LPS, which bind to glycosphingolipid receptors on the platelet. Such binding interactions activate platelet and induce platelet aggregation, leading to ischemic damages.[108] Porphyromonas gingivalis is another pathogen known to use a similar mechanism for immune evasion.[109] On the other hand, Bacillus anthracis secretes lethal toxins (LeTx) and edema toxins (ETx) that inhibit platelet aggregation and therefore promote hemorrhages essential for the infection.[110, 111] Overall, these examples show that platelet is a popular virulence target in various bacterial infections. They have motivated researchers to design PNPs as platelet decoys to intercept and neutralize incoming toxins for anti-bacterial efficacy. This strategy is attractive as it inhibits the virulence critical for bacterial colonization without disrupting bacterial cycles for killing.[112] Therefore, it is less likely to develop resistance than traditional antibiotics.[113] Neutralization agents are commonly developed based on the molecular structure of the virulence factors, therefore requiring customized design for different infections.[114, 115] Given the enormous diversity of bacterial virulence factors, such structure-based approaches are challenged by an overwhelming number of distinctive molecular structures and epitopic targets. PNPs bypass this challenge by presenting the entire platelet protein profile for neutralization regardless of specific binding mechanisms.
In some cases, platelet membranes bind with toxins that also adhere to other cell membranes. For example, α-toxins can insert into RBC membranes for neutralization.[2] LPS can bind with macrophage membranes for detoxification.[7] In other cases, however, platelet membranes show more specific binding interactions with toxins. For example, Shiga toxin bind to platelet membranes via specific glycosphingolipid receptors,[108] where PNPs provide a high specificity to neutralize the toxin. Bound toxins will lose their bioactivity, unable to cause further damages to the cellular targets or tissues before they are eliminated.[2, 116] Researchers recently coated platelet membranes onto synthetic micromotors for biomimicry toxin neutralization. Such platelet membrane-coated motors, namely ‘PL-motors’, offer a controlled motion that enhances both targeting precision and toxin absorption. For example, magnetic helical motors made with Au/Pd alloy were coated with the platelet membrane for neutralizing Shiga toxin.[117] The platelet membrane offered glycosphingolipid receptors for toxin binding. At the same time, the motors provided locomotive power to overcome Brownian motions and high viscosity of the biological fluid. Compared with control motors without platelet membrane coating, PL-motors achieved a higher neutralization efficiency against Shiga toxin and better protection of the cells against the toxin. In another example, instead of using the platelet membrane only, researchers made platelet-RBC hybrid membranes and coated them onto ultrasound-propelled magnetic gold nanowires. The resulting nanorobot, namely ‘RBC-PL-robot’, was tested for neutralizing bacterial toxins (Figure 7).[118] The hybridization of different membranes into a single nanomotor preserved the protein functions of each membrane, providing RBC-PL-robot with multifaceted functionalities. In this case, the RBC membrane absorbed α-toxin of MRSA, while the platelet membrane allowed the pathogens to attach to the motors. As a result, RBC-PL-robots simultaneously removed both bacteria-derived toxins and bacteria.[107, 119] In a hemolysis study, ultrasound-propelled RBC-PL-robot showed higher efficiency of removing circulating α-toxins than uncoated or static nanomotors. Toxin solutions treated with ultrasound-propelled RBC-PL-robot showed a much weaker hemolytic activity than control nanomotors, further validating the strategy of combining nanomotors with the hybrid membrane to improve virulence neutralization.
Figure 7.
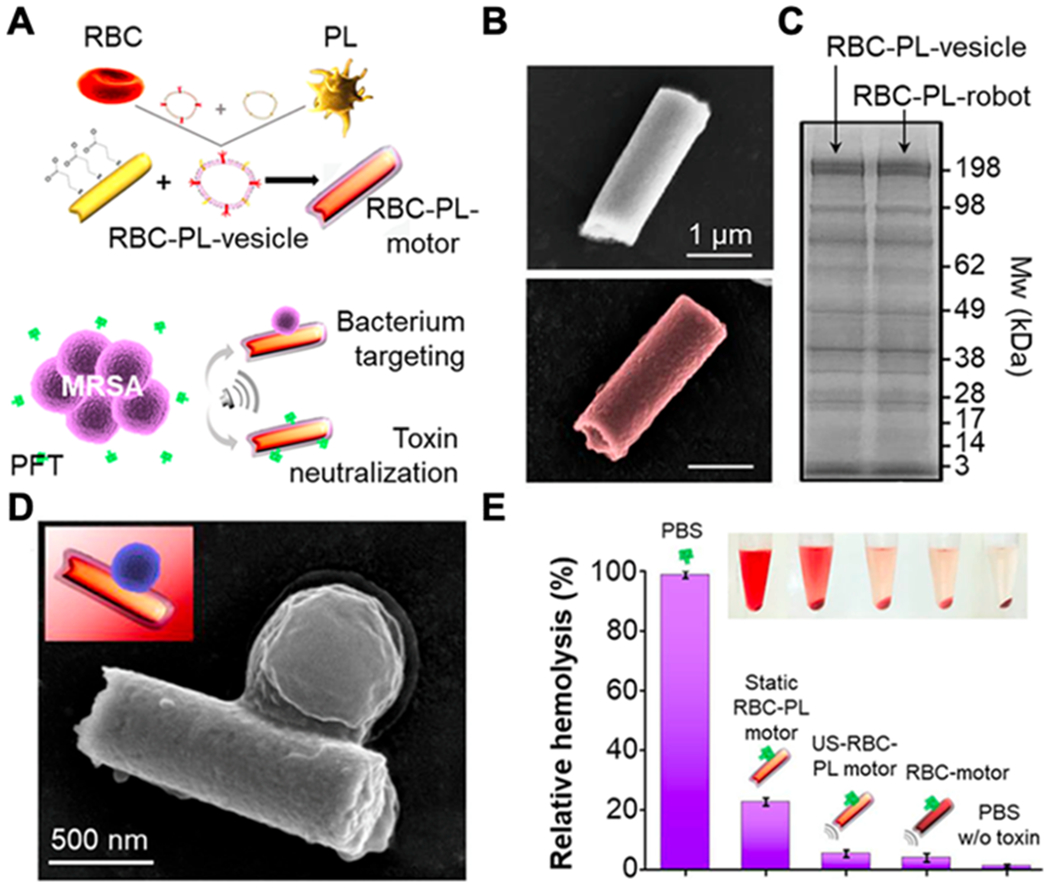
Hybrid biomembrane functionalized motors. (A) Schematic preparation of RBC-PL motors for concurrent removal of pathogens and bacterial toxins. (B) SEM images of a bare motor (top) and an RBC-PL motor (bottom). (C) SDS-PAGE protein analysis on RBC-PL vesicles and RBC-PL motors. (D) SEM image of a MRSA USA300 bacterium attached to an RBC-PL motor. (E) Hemolysis of RBCs after incubation with α-toxin in different solutions (error bars represent SD, n=3; inset, centrifuged RBCs and supernatants). Reproduced with permission. [118] Copyright 2018, The American Association for the Advancement of Science.
Overall, the development of cell membrane-coated nanoparticles, especially PNPs, provides new opportunities for overcoming drug resistance during anti-bacterial treatment. Exciting therapeutic results have been observed in using PNPs as drug carriers and as a drug-free anti-virulence platform. Besides these two applications, PNPs can also be made into toxoid vaccines similar to nanoparticles coated with RBC or macrophage membranes.[116, 120, 121] Like RBC membrane, platelet membrane can also be used for coating and detecting bacteria due to their broad-spectrum absorption of bacteria and bacterium-derived pathogenic factors.[122] With these possibilities, PNPs are expected to generate exciting opportunities for the development of future anti-bacterial therapies.
Summary
Platelets have been a source of inspiration for developing carriers for drug targeting.[123] With the advancement of nanotechnology, nanoparticles have been made to present platelet-like ligands on their surfaces to bind with subendothelial components for drug targeting.[124] Platelet morphology and clotting cascades were also simulated as design cures that may enhance drug targeting.[125, 126] Built upon these achievements, PNPs take the platelet mimicry one step further to truly mimic the behavior of platelets for drug targeting. PNPs inherit the biointerfacing properties of platelet and engage versatile and dynamic drug targeting to various diseases. With such unique capabilities, PNPs have emerged as an attractive nanomedicine platform for targeted drug delivery. In this review, we highlight three areas of drug targeting where PNP-based delivery platforms have drawn significant attention. These include targeting injured blood vessels to treat vascular diseases, targeting cancer cells for cancer treatment and detection, and targeting drug-resistant bacteria to treat infectious diseases. Nanoparticle carriers developed in these areas carried various therapeutic agents to treat distinct diseases. However, they all benefit from platelet membrane coating for drug targeting. Successful outcomes from these studies validate platelet membrane coating as a general and effective strategy to achieve targeted drug delivery.
As PNPs are continually developed for drug targeting, their applications are also expanding. For example, PNPs were made to harness the intrinsic affinity between platelet and inflammation for drug targeting.[127] They were applied to target rheumatoid arthritis, where they preferentially bound to inflamed endothelium.[128] Drug molecules delivered via PNPs accumulated more in joints of mice with collagen-induced arthritis and resulted in a notable anti-arthritic effect. PNPs were also used as platelet decoys to bind with anti-platelet antibodies for neutralization.[129] In a murine model of antibody-induced thrombocytopenia, PNPs neutralized pathogenic antibodies in vivo. Treatment with PNPs preserved circulating platelets and reduced the disease burden. Genetic engineering has recently been applied to modify platelet membranes for targeting CTCs or autoreactive T cells in Type 1 diabetes.[130, 131] Meanwhile, instead of deriving membranes from platelets, platelet-derived extracellular vesicles have also become a membrane source for drug targeting. They offer intrinsic affinity with the site of inflammation and have been used for the delivery of anti-inflammatory agents to pneumonia.[132] Overall, we anticipate PNPs to play significant roles in biomedical research as researchers continue to refine the nanoparticle design and expand their applications in drug targeting and beyond.
Acknowledgements
This work is supported by the National Institutes of Health under Award Number R01CA200574.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Fang RH; Kroll AV; Gao W; Zhang L, Adv. Mater 2018, 30, 1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hu CMJ; Fang RH; Copp J; Luk BT; Zhang L, Nat. Nanotechnol 2013, 8, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen YJ; Chen MC; Zhang Y; Lee JH; Escajadillo T; Gong H; Fang RH; Gao W; Nizet V; Zhang L, Adv. Healthc. Mater 2018, 7, 1701366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pang ZQ; Hu CMJ; Fang RH; Luk BT; Gao W; Wang F; Chuluun E; Angsantikul P; Thamphiwatana S; Lu WY; Jiang XG; Zhang L, ACS Nano 2015, 9, 6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fang RH; Hu CMJ; Luk BT; Gao W; Copp JA; Tai YY; O'Connor DE; Zhang L, Nano Lett. 2014, 14, 2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kroll AV; Fang RH; Jiang Y; Zhou JR; Wei X; Yu CL; Gao J; Luk BT; Dehaini D; Gao W; Zhang L, Adv. Mater 2017, 29, 1703969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thamphiwatana S; Angsantikul P; Escajadillo T; Zhang Q; Olson J; Luk BT; Zhang S; Fang RH; Gao W; Nizet V; Zhang L, Proc. Natl. Acad. Sci. U. S. A 2017, 114, 11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wei X; Zhang G; Ran DN; Krishnan N; Fang RH; Gao W; Spector SA; Zhang L, Adv. Mater 2018, 30, 1802233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang Q; Honko A; Zhou J; Gong H; Downs SN; Vasquez JH; Fang RH; Gao W; Griffiths A; Zhang L, Nano Lett. 2020, 20, 5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Davis ME; Chen Z; Shin DM, Nat. Rev. Drug Discov 2008, 7, 771. [DOI] [PubMed] [Google Scholar]
- [11].Hu CMJ; Zhang L; Aryal S; Cheung C; Fang RH; Zhang L, Proc. Natl. Acad. Sci. U. S. A 2011, 108, 10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luk BT; Fang RH; Hu CMJ; Copp JA; Thamphiwatana S; Dehaini D; Gao W; Zhang K; Li SL; Zhang L, Theranostics 2016, 6, 1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Peer D; Karp JM; Hong S; FaroKhzad OC; Margalit R; Langer R, Nat. Nanotechnol 2007, 2, 751. [DOI] [PubMed] [Google Scholar]
- [14].Min H; Wang J; Qi YQ; Zhang YL; Han XX; Xu Y; Xu JC; Li Y; Chen L; Cheng KM; Liu GN; Yang N; Li YY; Nie GJ, Adv. Mater 2019, 31, 1808200. [DOI] [PubMed] [Google Scholar]
- [15].Zhang Q; Dehaini D; Zhang Y; Zhou JL; Chen XY; Zhang L; Fang RH; Gao W; Zhang L, Nat. Nanotechnol 2018, 13, 1182. [DOI] [PubMed] [Google Scholar]
- [16].Olsson M; Bruhns P; Frazier WA; Ravetch JV; Oldenborg PA, Blood 2005, 105, 3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lievens D; von Hundelshausen P, Thromb. Haemost 2011, 106, 827. [DOI] [PubMed] [Google Scholar]
- [18].Jurasz P; Alonso-Escolano D; Radomski MW, Br. J. Pharmacol 2004, 143, 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fitzgerald JR; Foster TJ; Cox D, Nat. Rev. Microbiol 2006, 4, 445. [DOI] [PubMed] [Google Scholar]
- [20].Cruz MA; Chen JM; Whitelock JL; Morales LD; Lopez JA, Blood 2005, 105, 1986. [DOI] [PubMed] [Google Scholar]
- [21].Kappelmayer J; Nagy B, Biomed Res. Int 2017, 2017, 6138145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Alves CS; Burdick MM; Thomas SN; Pawar P; Konstantopoulos K, Am. J. Physiol. Cell Physiol 2008, 294, C907. [DOI] [PubMed] [Google Scholar]
- [23].Kerrigan SW; Douglas I; Wray A; Heath J; Byrne MF; Fitzgerald D; Cox D, Blood 2002, 100, 509. [DOI] [PubMed] [Google Scholar]
- [24].Chen JM; Lopez KA, Microcirculation 2005, 12, 235. [DOI] [PubMed] [Google Scholar]
- [25].Cosemans JMEM; Schols SEM; Stefanini L; de Witt S; Feijge MAH; Hamulyak K; Deckmyn H; Bergmeier W; Heemskerk JWM, Blood 2011, 117, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Moore KJ; Sheedy FJ; Fisher EA, Nat. Rev. Immunol 2013, 13, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wei X; Ying M; Dehaini D; Su YY; Kroll AV; Zhou JR; Gao W; Fang RH; Chien S; Zhang L, ACS Nano 2018, 12, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ruggeri ZM, Nat. Med 2002, 8, 1227. [DOI] [PubMed] [Google Scholar]
- [29].Yang HB; Song YA; Chen J; Pang ZQ; Zhang N; Cao JT; Wang QZ; Li QY; Zhang F; Dai YX; Li CG; Huang ZY; Qian JY; Ge JB, Int. J. Nanomedicine 2020, 15, 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yap CH; Saikrishnan N; Tamilselvan G; Yoganathan AP, Biomech. Model Mechanobiol 2012, 11, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Song YA; Huang ZY; Liu X; Pang ZQ; Chen J; Yang HB; Zhang N; Cao ZL; Liu M; Cao JT; Li CG; Yang XD; Gong H; Qian JY; Ge JB, Nanomedicine 2019, 15, 13. [DOI] [PubMed] [Google Scholar]
- [32].Chaabane C; Otsuka F; Virmani R; Bochaton-Piallat ML, Cardiovasc. Res 2013, 99, 353. [DOI] [PubMed] [Google Scholar]
- [33].Hu CMJ; Fang RH; Wang KC; Luk BT; Thamphiwatana S; Dehaini D; Nguyen P; Angsantikul P; Wen CH; Kroll AV; Carpenter C; Ramesh M; Qu V; Patel SH; Zhu J; Shi W; Hofman FM; Chen TC; Gao W; Zhang K; Chien S; Zhang L, Nature 2015, 526, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang BW; Chen GJ; Urabe G; Xie RS; Wang YY; Shi XD; Guo LW; Gong SQ; Kent KC, Biomaterials 2018, 178, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Eltzschig HK; Eckle T, Nat. Med 2011, 17, 1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Su T; Huang K; Ma H; Liang H; Dinh PU; Chen J; Shen D; Allen TA; Qiao L; Li Z; Hu S; Cores J; Frame BN; Young AT; Yin Q; Liu J; Qian L; Caranasos TG; Brudno Y; Ligler FS; Cheng K, Adv. Funct. Mater 2019, 29, 1803567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Battaglini D; Robba C; da Silva AL; Samary CD; Silva PL; Dal Pizzol F; Pelosi P; Rocco PRM, Crit. Care 2020, 24, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li MX; Liu Y; Chen JP; Liu TT; Gu ZX; Zhang JQ; Gu XC; Teng GJ; Yang F; Gu N, Theranostics 2018, 8, 4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xu JP; Wang XQ; Yin HY; Cao X; Hu QY; Lv W; Xu QW; Gu Z; Xin HL, ACS Nano 2019, 13, 8577. [DOI] [PubMed] [Google Scholar]
- [40].Li MX; Li J; Chen JP; Liu Y; Cheng X; Yang F; Gu N, ACS Nano 2020, 14, 2024. [DOI] [PubMed] [Google Scholar]
- [41].Gross SS, Nature 2001, 409, 577. [DOI] [PubMed] [Google Scholar]
- [42].Marietta M; Facchinetti F; Neri I; Piccinini F; Volpe A; Torelli G, Thromb. Res 1997, 88, 229. [DOI] [PubMed] [Google Scholar]
- [43].Tarbox AK; Swaroop M, Int. J. Crit. Illn. Inj. Sci 2013, 3, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xu J; Zhang Y; Xu J; Liu G; Di C; Zhao X; Li X; Li Y; Pang N; Yang C; Li Y; Li B; Lu Z; Wang M; Dai K; Yan R; Li S; Nie G, Adv. Mater 2020, 32, 1905145. [DOI] [PubMed] [Google Scholar]
- [45].Frenette PS; Denis CV; Weiss L; Jurk K; Subbarao S; Kehrel B; Hartwig JH; Vestweber D; Wagner DD, J. Exp. Med 2000, 191, 1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Romo GM; Dong JF; Schade AJ; Gardiner EE; Kansas GS; Li CQ; McIntire LV; Berndt MC; Lopez JA, J. Exp. Med 1999, 190, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yang T; Ding XW; Dong LN; Hong C; Ye J; Xiao YF; Wang XL; Xin HB, ACS Biomater. Sci. Eng 2018, 4, 4219. [DOI] [PubMed] [Google Scholar]
- [48].Kelley WJ; Safari H; Lopez-Cazares G; Eniola-Adefeso O, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 2016, 8, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lobatto ME; Fuster V; Fayad ZA; Mulder WJM, Nat. Rev. Drug Discov 2011, 10, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Soto-Pantoja DR; Stein EV; Rogers NM; Sharifi-Sanjani M; Isenberg JS; Roberts DD, Expert Opin. Ther. Targets 2013, 17, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kroll AV; Fang RH; Zhang L, Bioconj. Chem 2017, 28, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Labelle M; Begum S; Hynes RO, Cancer Cell 2011, 20, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Labelle M; Begum S; Hynes RO, Proc. Natl. Acad. Sci. U. S. A 2014, 111, E3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ward Y; Lake R; Faraji F; Sperger J; Martin P; Gilliard C; Ku KP; Rodems T; Niles D; Tillman H; Yin J; Hunter K; Sowalsky AG; Lang J; Kelly K, Cell Rep. 2018, 23, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gay LJ; Felding-Habermann B, Nat. Rev. Cancer 2011, 11, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Borsig L, Expert Rev. Anticancer Ther 2008, 8, 1247. [DOI] [PubMed] [Google Scholar]
- [57].Kanikarla-Marie P; Lam M; Menter DG; Kopetz S, Cancer Metastasis Rev. 2017, 36, 235. [DOI] [PubMed] [Google Scholar]
- [58].Tesfamariam B, Pharmacol. Ther 2016, 157, 112. [DOI] [PubMed] [Google Scholar]
- [59].Foss A; Munoz-Sagredo L; Sleeman J; Thiele W, Clin. Exp. Metastasis 2020, 37, 47. [DOI] [PubMed] [Google Scholar]
- [60].Menter DG; Kopetz S; Hawk E; Sood AK; Loree JM; Gresele P; Honn KV, Cancer Metastasis Rev. 2017, 36, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Schaff M; Tang C; Maurer E; Bourdon C; Receveur N; Eckly A; Hechler B; Arnold C; de Arcangelis A; Nieswandt B; Denis CV; Lefebvre O; Georges-Labouesse E; Gachet C; Lanza F; Mangin PH, Circulation 2013, 128, 541. [DOI] [PubMed] [Google Scholar]
- [62].Chi CL; Li FW; Liu HB; Feng SY; Zhang YJ; Zhou D; Zhang RK, J. Nanopart. Res 2019, 21, 144. [Google Scholar]
- [63].Xu LW; Su TQ; Xu XY; Zhu LD; Shi L, Drug Deliv J. Sci. Technol 2019, 53, 101190. [Google Scholar]
- [64].Wang HJ; Wu JZ; Williams GR; Fan Q; Niu SW; Wu JR; Xie XT; Zhu LM, Nanobiotechnology J 2019, 17, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shang YH; Wang QH; Wu B; Zhao QQ; Li J; Huang XY; Chen WS; Gui R, ACS Appl. Mater. Interfaces 2019, 11, 28254. [DOI] [PubMed] [Google Scholar]
- [66].Bang KH; Na YG; Huh HW; Hwang SJ; Kim MS; Kim M; Lee HK; Cho CW, Cancers 2019, 11, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhuang J; Gong H; Zhou JR; Zhang QZ; Gao W; Fang RH; Zhang L, Sci. Adv 2020, 6, eaaz6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shibu ES; Hamada M; Murase N; Biju V, J. Photochem. Photobiol 2013, 15, 53. [Google Scholar]
- [69].Xu LL; Gao F; Fan F; Yang LH, Biomaterials 2018, 159, 59. [DOI] [PubMed] [Google Scholar]
- [70].Zuo HQ; Tao JX; Shi H; He J; Zhou ZY; Zhang C, Acta Biomater. 2018, 80, 296. [DOI] [PubMed] [Google Scholar]
- [71].Hu QY; Sun WJ; Qian CG; Wang C; Bomba HN; Gu Z, Adv. Mater 2015, 27, 7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Oikonomou E; Pintzas A, BioFactors 2013, 39, 343. [DOI] [PubMed] [Google Scholar]
- [73].Wang HJ; Bremner DH; Wu KH; Gong XR; Fan Q; Xie XT; Zhang HM; Wu JZ; Zhu LM, Chem. Eng. J 2020, 382, 122848. [Google Scholar]
- [74].Chen Y; Zhao GM; Wang S; He YW; Han SL; Du CH; Li SC; Fan ZL; Wang C; Wang JP, Biomater. Sci 2019, 7, 3450. [DOI] [PubMed] [Google Scholar]
- [75].Li JH; Ai YW; Wang LH; Bu PC; Sharkey CC; Wu QH; Wun B; Roy S; Shen XL; King MR, Biomaterials 2016, 76, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Rao L; Meng QF; Huang QQ; Wang ZX; Yu GT; Li A; Ma WJ; Zhang NG; Guo SS; Zhao XZ; Liu K; Yuan YF; Liu W, Adv. Funct. Mater 2018, 28, 1803531. [Google Scholar]
- [77].Willing BP; Russell SL; Finlay BB, Nat. Rev. Microbiol 2011, 9, 233. [DOI] [PubMed] [Google Scholar]
- [78].Spellberg B; Bartlett JG; Gilbert DN, New Engl. J. Med 2013, 368, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Blair JMA; Webber MA; Baylay AJ; Ogbolu DO; Piddock LJV, Nat. Rev. Microbiol 2015, 13, 42. [DOI] [PubMed] [Google Scholar]
- [80].Morens DM; Folkers GK; Fauci AS, Nature 2004, 430, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ma YF; Wang Z; Zhao W; Lu TL; Wang RT; Mei QB; Chen T, Int. J. Nanomedicine 2013, 8, 2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Gao W; Chen YJ; Zhang Y; Zhang QZ; Zhang L, Adv. Drug Del. Rev 2018, 127, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Gao W; Thamphiwatana S; Angsantikul P; Zhang L, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 2014, 6, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bensing BA; Lopez JA; Sullam PM, Infect. Immun 2004, 72, 6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Plummer C; Wu H; Kerrigan SW; Meade G; Cox D; Ian Douglas CW, Br. J. Haematol 2005, 129, 101. [DOI] [PubMed] [Google Scholar]
- [86].Siboo IR; Chambers HF; Sullam PM, Infect. Immun 2005, 73, 2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Naito M; Sakai E; Shi Y; Ideguchi H; Shoji M; Ohara N; Yamamoto K; Nakayama K, Mol. Microbiol 2006, 59, 152. [DOI] [PubMed] [Google Scholar]
- [88].Brennan MP; Loughman A; Devocelle M; Arasu S; Chubb AJ; Foster TJ; Cox D, J. Thromb. Haemost 2009, 7, 1364. [DOI] [PubMed] [Google Scholar]
- [89].Miajlovic H; Zapotoczna M; Geoghegan JA; Kerrigan SW; Speziale P; Foster TJ, Microbiol.-Sgm 2010, 156, 920. [DOI] [PubMed] [Google Scholar]
- [90].Petersen HJ; Keane C; Jenkinson HF; Vickerman MM; Jesionowski A; Waterhouse JC; Cox D; Kerrigan SW, Infect. Immun 2010, 78, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Nguyen T; Ghebrehiwet B; Peerschke EIB, Infect. Immun 2000, 68, 2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].O’Seaghdha M; van Schooten CJ; Kerrigan SW; Emsley J; Silverman GJ; Cox D; Lenting PJ; Foster TJ, FEBS J. 2006, 273, 4831. [DOI] [PubMed] [Google Scholar]
- [93].Walsh EJ; Miajlovic H; Gorkun OV; Foster TJ, Microbiology 2008, 154, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Wann ER; Gurusiddappa S; Hook M, J. Biol. Chem 2000, 275, 13863. [DOI] [PubMed] [Google Scholar]
- [95].Carlsson F; Sandin C; Lindahl G, Mol. Microbiol 2005, 56, 28. [DOI] [PubMed] [Google Scholar]
- [96].Pietrocola G; Schubert A; Visai L; Torti M; Fitzgerald JR; Foster TJ; Reinscheid DJ; Speziale P, Blood 2005, 105, 1052. [DOI] [PubMed] [Google Scholar]
- [97].Schubert A; Zakikhany K; Schreiner M; Frank R; Spellerberg B; Eikmanns BJ; Reinscheid DJ, Mol. Microbiol 2002, 46, 557. [DOI] [PubMed] [Google Scholar]
- [98].Fitzgerald JR; Loughman A; Keane F; Brennan M; Knobel M; Higgins J; Visai L; Speziale P; Cox D; Foster TJ, Mol. Microbiol 2006, 59, 212. [DOI] [PubMed] [Google Scholar]
- [99].Loughman A; Fitzgerald JR; Brennan MP; Higgins J; Downer R; Cox D; Foster TJ, Mol. Microbiol 2005, 57, 804. [DOI] [PubMed] [Google Scholar]
- [100].O’Brien L; Kerrigan SW; Kaw G; Hogan M; Penades J; Litt D; Fitzgerald DJ; Foster TJ; Cox D, Mol. Microbiol 2002, 44, 1033. [DOI] [PubMed] [Google Scholar]
- [101].Kuehnert MJ; Kruszon-Moran D; Hill HA; McQuillan G; McAllister SK; Fosheim G; McDougal LK; Chaitram J; Jensen B; Fridkin SK; Killgore G; Tenover FC, J. Infect. Dis 2006, 193, 172. [DOI] [PubMed] [Google Scholar]
- [102].Chambers HF; Deleo FR, Nat. Rev. Microbiol 2009, 7, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Weigel LM; Clewell DB; Gill SR; Clark NC; McDougal LK; Flannagan SE; Kolonay JF; Shetty J; Killgore GE; Tenover FC, Science 2003, 302, 1569. [DOI] [PubMed] [Google Scholar]
- [104].McKenna M, Nature 2012, 482, 23. [DOI] [PubMed] [Google Scholar]
- [105].Kerrigan SW; Cox D, Cell. Mol. Life Sci 2010, 67, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Bhakdi S; Muhly M; Mannhardt U; Hugo F; Klapettek K; Mueller-Eckhardt C; Roka L, J. Exp. Med 1988, 168, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Wilke GA; Bubeck Wardenburg J, Proc. Natl. Acad. Sci. U. S. A 2010, 107, 13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Proulx F; Seidman EG; Karpman D, Pediatr. Res 2001, 50, 163. [DOI] [PubMed] [Google Scholar]
- [109].Takii R; Kadowaki T; Baba A; Tsukuba T; Yamamoto KJ, Infect. Immun 2005, 73, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Kau JH; Sun DS; Tsai WJ; Shyu HF; Huang HH; Lin HC; Chang HH, J. Infect. Dis 2005, 192, 1465. [DOI] [PubMed] [Google Scholar]
- [111].Alam S; Gupta M; Bhatnagar R, Biochem. Biophys. Res. Commun 2006, 339, 107. [DOI] [PubMed] [Google Scholar]
- [112].Cegelski L; Marshall GR; Eldridge GR; Hultgren SJ, Nat. Rev. Microbiol 2008, 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Rasko DA; Sperandio V, Nat. Rev. Drug Discov 2010, 9, 117. [DOI] [PubMed] [Google Scholar]
- [114].Maura D; Ballok AE; Rahme LG, Curr. Opin. Microbiol 2016, 33, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Beckham KSH; Roe AJ, Front. Cell. Infect. Microbiol 2014, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hu CMJ; Fang RH; Luk BT; Zhang L, Nat. Nanotechnol 2013, 8, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Li JX; Angsantikul P; Liu WJ; de Avila BEF; Chang XC; Sandraz E; Liang YY; Zhu SY; Zhang Y; Chen CR; Gao W; Zhang L; Wang J, Adv. Mater 2018, 30, 1704800. [DOI] [PubMed] [Google Scholar]
- [118].Esteban-Fernández de Ávila B; Angsantikul P; Ramirez-Herrera DE; Soto F; Teymourian H; Dehaini D; Chen YJ; Zhang L; Wang J, Sci. Robot 2018, 3, eaat0485. [DOI] [PubMed] [Google Scholar]
- [119].Cooling LL; Walker KE; Gille T; Koerner TA, Infect. Immun 1998, 66, 4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wang F; Fang RH; Luk BT; Hu CMJ; Thamphiwatana S; Dehaini D; Angsantikul P; Kroll AV; Pang ZQ; Gao W; Lu WY; Zhang L, Adv. Funct. Mater 2016, 26, 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhou JR; Kroll AV; Holay M; Fang RH; Zhang L, Adv. Mater 2020, 32, 1901255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Gong H; Chen F; Huang ZL; Gu Y; Zhang QZ; Chen YJ; Zhang Y; Zhuang J; Cho YK; Fang RNH; Gao W; Xu S; Zhang L, ACS Nano 2019, 13, 3714. [DOI] [PubMed] [Google Scholar]
- [123].Lu YF; Hu QY; Jiang C; Gu Z, Curr. Opin. Biotechnol 2019, 58, 81. [DOI] [PubMed] [Google Scholar]
- [124].Kamaly N; Fredman G; Subramanian M; Gadde S; Pesic A; Cheung L; Fayad ZA; Langer R; Tabas I; Farokhzad OC, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Anselmo AC; Modery-Pawlowski CL; Menegatti S; Kumar S; Vogus DR; Tian LL; Chen M; Squires TM; Sen Gupta A; Mitragotri S, ACS Nano 2014, 8, 11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Simberg D; Duza T; Park JH; Essler M; Pilch J; Zhang LL; Derfus AM; Yang M; Hoffman RM; Bhatia S; Sailor MJ; Ruoslahti E, Proc. Natl. Acad. Sci. U. S. A 2007, 104, 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Boilard E; Blanco P; Nigrovic PA, Nat. Rev. Rheumatol 2012, 8, 534. [DOI] [PubMed] [Google Scholar]
- [128].He YW; Li RX; Liang JM; Zhu Y; Zhang SY; Zheng ZC; Qin J; Pang ZQ; Wang JX, Nano Res. 2018, 11, 6086. [Google Scholar]
- [129].Wei X; Gao J; Fang RH; Luk BT; Kroll AV; Dehaini D; Zhou JR; Kim HW; Gao W; Lu WY; Zhang L, Biomaterials 2016, 111, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Li JH; Sharkey CC; Wun B; Liesveld JL; King MR, J. Control. Release 2016, 228, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Zhang XD; Kang Y; Wang JQ; Yan JJ; Chen Q; Cheng H; Huang P; Gu Z, Adv. Mater 10.1002/adma.201907692 [DOI] [PubMed] [Google Scholar]
- [132].Xia YQ; Ning PB; Wang ZL; Chen XY, Matter 2020, 3, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]