

Abstract
We report a concise, stereocontrolled synthesis of the neuro- toxic sesquiterpenoid, (–)-picrotoxinin (1, PXN). The brevity of the route owes to regio- and stereoselective formation of the [4.3.0] bicyclic core by incorporation of a symmetrizing geminal dimethyl group at C5. Dimethylation then enables selective C–O bond formation in multiple intermediates. A series of strong bond (C–C and C–H) cleavages convert the C5 gem-dimethyl group to the C15 lactone of PXN.
Graphical Abstract
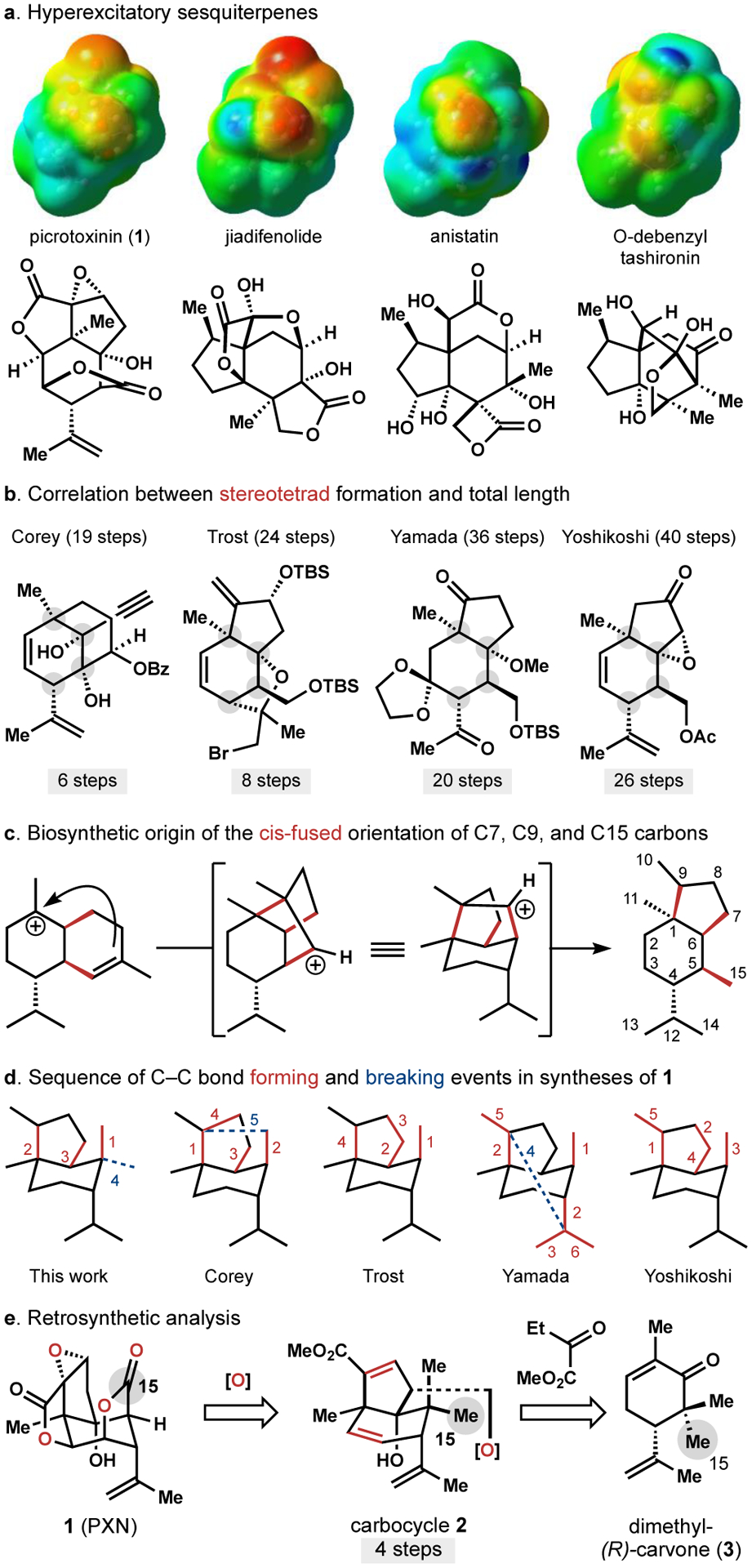
Picrotoxinin (1, PXN) is the flagship member of the picrotoxane family of natural products and continues to attract considerable attention from the synthesis community1–2,3,4,5,6,7,8 due to its stereochemically-dense polyoxygenated structure and its use as a tool compound in neuroscience.9–,1,11 Picrotoxin (PTX), which consists of an equimolar mixture of PXN and its less-active C12 hydrate, picrotin (PTN), can exhibit useful therapeutic properties: chronic dosing of Down’s syndrome model mice (Ts65Dn) normalizes memory performance by reducing overactivity of GABAergic neurons.12 However, the therapeutic window of PTX is narrow: lethal convulsion through hyperexcitatory GABAA receptor antagonism occurs at low dose (LD50 = 2 mg/kg, rat, I.P.).13 In contrast, GABAAR antagonists like bilobalide14 can share the therapeutic properties, target, and binding site of PXN yet avoid acute toxicity.15 Our group has identified ‘neurotrophic’ sesquiterpenes jiadifenolide16 and O-debenzyltashironin17 as sharing the hyperexcitatory effects of convulsant GABAAR antagonists anisatin17 and PXN, yet jiadifenolide displays no convulsive signature in mice (Figure 1a).15,18 A short synthetic route might allow interrogation of analogs of PXN that similarly reduce its toxicity yet still antagonize GABAA receptors.19
Figure 1.
Chemical background and synthetic plan.
The seminal work of Corey,1,2 Yamada,3 Yoshikoshi,4 and Trost5–,6,7 illustrated the difficulty of the contiguous stereotetrad of 1 (Figure 1b). Intermolecular formation of this stereo-dense motif is challenged by the cis-fused orientation of the C7, C9, and C15 carbons, which arises biosynthetically by an anti-Markovnikov cationic cyclization of a cadinyl cation and oxidative cleavage (Figure 1c).20,21 Corey1 and Yamada3 employed intramolecular cyclization/C–C oxidative cleavage steps to overcome this problem, while Trost5–,6,7 leveraged torsional strain with a small nucleophile to set the C7/C15 stereodiad and a classic palladium-cataly-zed cycloisomerization to make the C7/C9 junction. Yet all syntheses concede some C–C disconnections within and about the [4.3.0]-bicyclononane, rather than directly accessing the core by disconnections solely between the [4.3.0] ring junctions (Figure 1d). We found brevity of stereotetrad formation in the literature to correlate with overall synthesis length (Figure 1b). Disconnections solely between the junctions of the bicyclic core should then promote a shorter synthesis of 1.
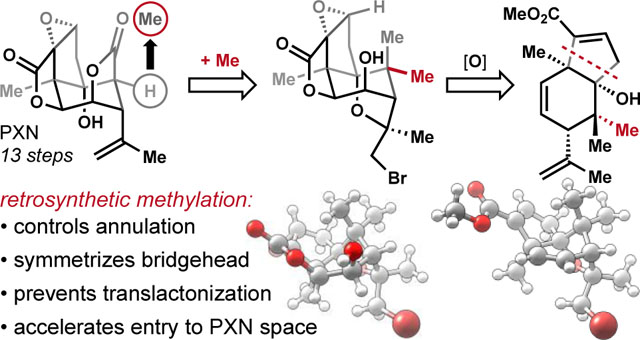
With this strategic goal in mind, we encoded the oxidations of 1 with alkenes to arrive at carbocycle 2, which might derive from (R)-carvone1,3,5,22 via annulation of methyl-2-oxo-butanoate (Figure 1e). Most oxidation patterns were embedded into starting materials,23 with the exception of the C15 carboxylate. Our decision to decrease C15 to the methyl oxidation state was informed by problems encountered in the literature4–,5,6,7,9–,10,11 with C10/C15 translactonization and intramolecular epoxide opening at higher oxidation states of C15. However, we quickly discovered that a single methyl group on carvone led to the incorrect stereoisomer (1314, see Figure 2a). Instead, we found that geminal dimethylation enabled efficient synthesis of 2 in only 4 steps. The challenge then became discovery of a late-stage, stereoselective, cleavage of a strong C–C bond—a counterintuitive24 but, in this case, enabling tactic. Here we report its successful implementation in a concise synthesis of 1 (Scheme 1).
Figure 2.
Importance of the extra methyl group in 2.
Scheme 1.
Synthesis of (–)-picrotoxinin (PXN, 1).
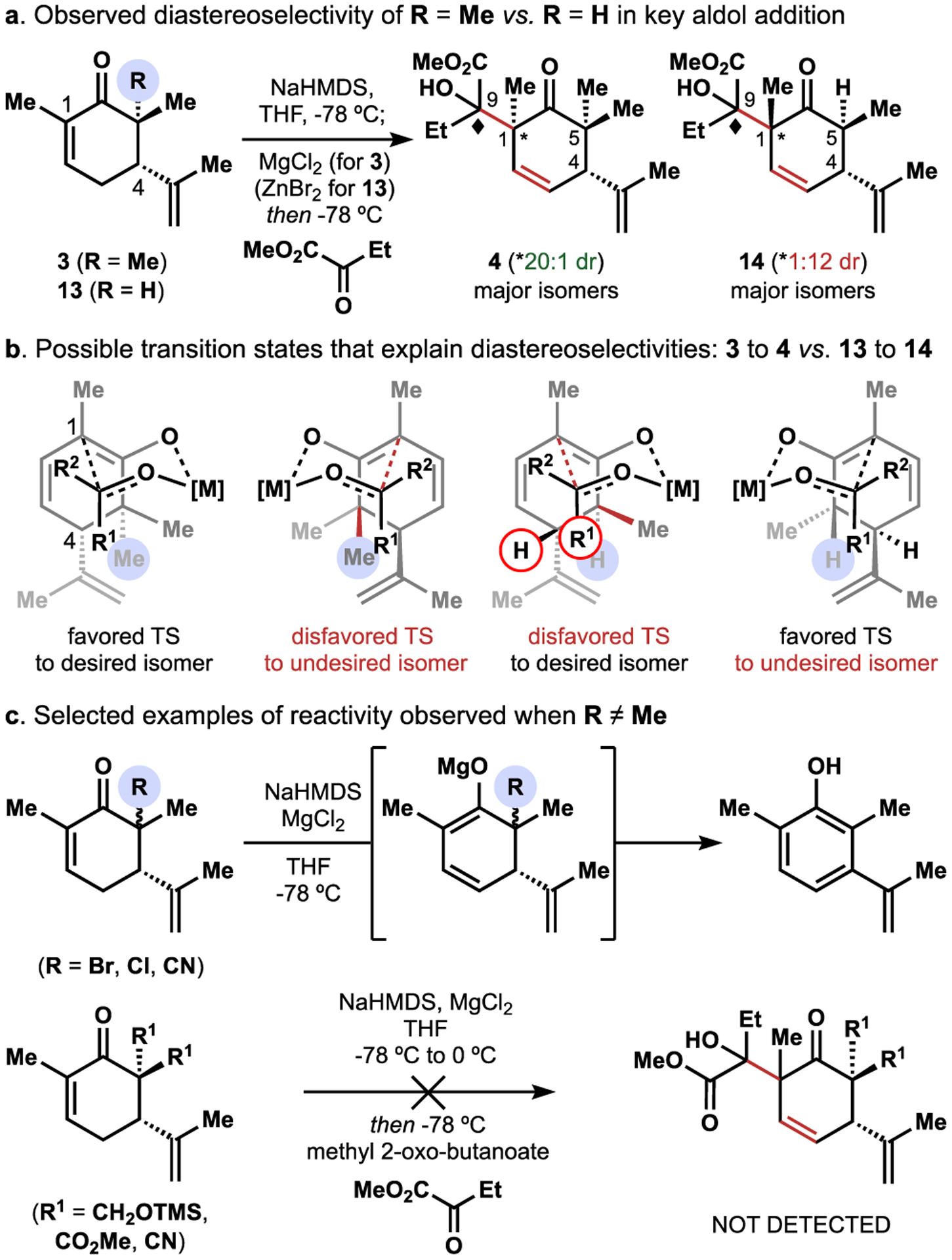
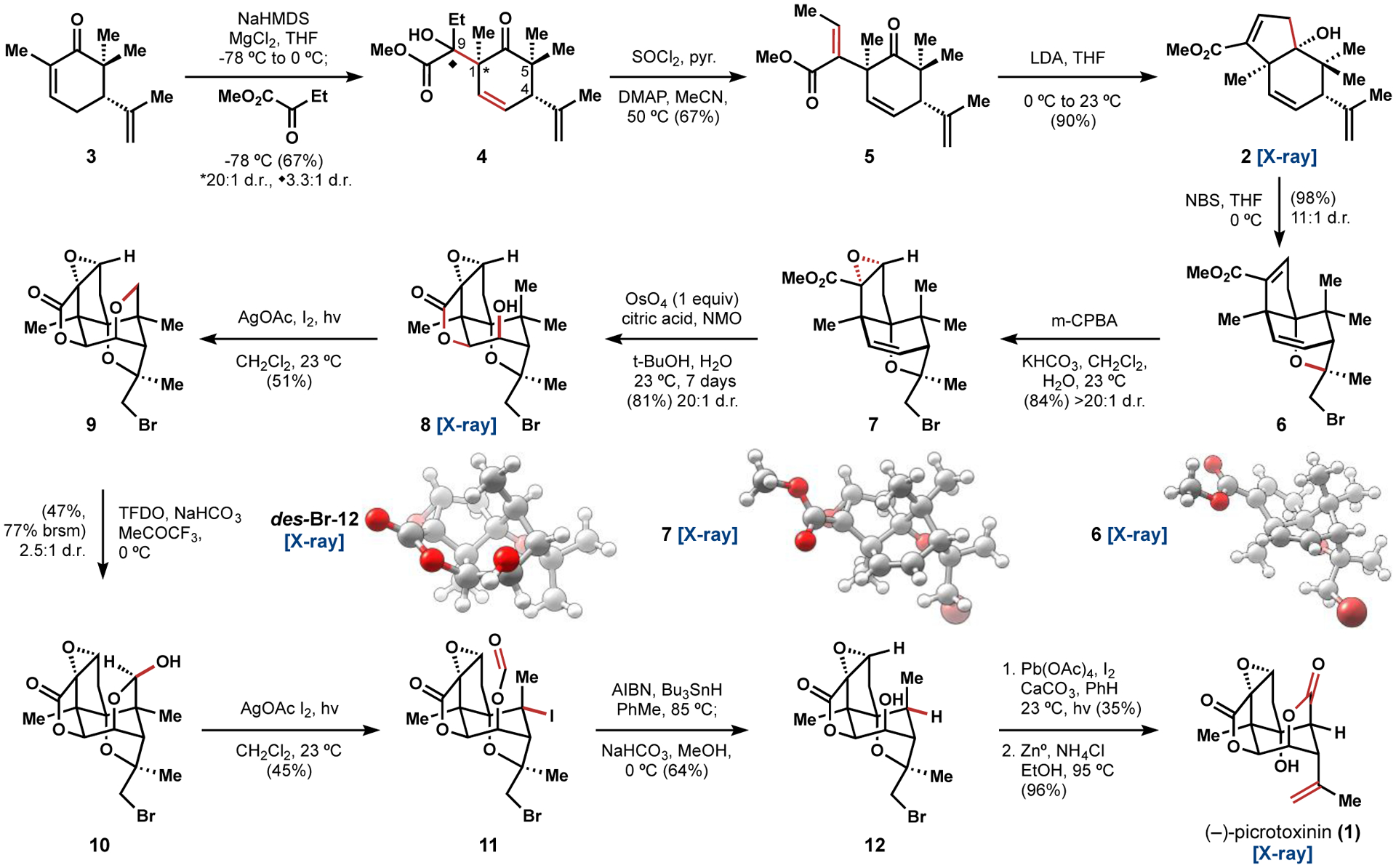
Dimethylation of (R)-carvone was achieved in one25 or two26 steps, although the latter procedure was employed on 30g (200 mmol) scale. The magnesium enolate of 3 was formed by deprotonation with NaHMDS in the presence of anhydrous MgCl2; subsequent addition of methyl-2-oxobutanoate at −78 °C gave the aldol addition product 4 in 67% yield with excellent diastereoselectivity (>20:1) at C1 and inconsequential 3.3:1 diastereoselectivity at C9. Use of lithium, sodium, potassium, or zinc enolates gave diminished to no yield of 4. The reaction was quenched at −78 °C to avoid retro-aldol decomposition that occurs above −20 °C. This unusual aldol reaction occurs with high regio- and diastereoselectivity to form a quaternary carbon (C1) and a neopentyl alcohol (C9). Our working model posits an efficient relay of stereochemical information from the C4 stereocenter to C1 by avoidance of a 1,3-diaxial interaction between the axial C5 methyl group and methyl-2-oxobutanoate in the aldol addition transition state (Figure 2b). In contrast, use of trans-α-methyl-carvone (13, i.e. mono methylation) resulted in a 1:12 diastereomeric mixture favoring the opposite and unproductive diastereomer (14). An extended enolate could not be formed with either (R)-carvone or cis-α-methyl-carvone, and use of Corey’s hydrazone alkylation procedure1 gave no aldol addition product.
Alternative tactics that replaced one of the Me groups with Br, Cl, or CN groups were plagued by poor stereocontrol in formation of the C5 stereocenter and subsequent failure of the aldol addition through proton-transfer, elimination and aromatization pathways (Figure 2c). Symmetrical substitution at C5 with silylhydroxymethylene (R3SiOCH2-),6 methyl ester, or nitrile groups required multiple steps for installation and the aldol addition still failed. Since the inclusion of an extra C5 methyl group enabled installation of all 15 carbon atoms of the picrotoxinin skeleton with the correct regio- and stereochemistry in just two steps and without need for C5 stereocontrol, we continued forward with a plan to excise the extra C5 methyl group at a late stage—a risky, but ultimately successful decision.
Neopentyl alcohol 4 was converted to 5 by a SOCl2-induced elimination.27 These conditions proved uniquely able to eliminate both diastereomers of the sterically congested C9 alcohol 4. A vinylogous intramolecular 5-exo-trig aldol addition reaction yielded 2 in 90% yield upon treatment of 5 with LDA at 0 °C and warming to 23 °C. This reaction failed with the alkene derived from 14 due to competitive deprotonation and epimerization pathways.
Facile and scalable access to 2 allowed extensive interrogation of the remaining alkene oxidations. First, bromoetherification1,5,9 with NBS proved entirely selective for the isopropene group and delivered an 11:1 diastereomeric mixture of 6. This dual-purpose bromoetherification served to protect the Δ12,13 isopropenyl alkene and lock the conformation of 2 to promote lactonization at C10 and directed oxidation of the C5 methyl groups. Epoxidation of 6 initially suffered poor diastereocontrol under nucleophilic epoxidation conditions (e.g. alkali metalperoxides) and low conversion with electrophilic epoxidation reagents (e.g. DMDO, trifluoroperacetic acid). Although mCPBA alone was insufficient to react with 6, we found that use of KHCO3 with mCPBA in a biphasic mixture of CH2Cl2 and H2O at 23 °C afforded 7 with high diastereoselectivity in 84% yield. We had anticipated that dihydroxylation of 7 might be facile by analogy with Yoshikoshi’s OsO4/pyridine oxidation of a similar substrate,4 but no more than 30% conversion could be obtained under these conditions (stoichiometric OsO4, pyridine). We eventually found that addition of citric acid to prevent off-pathway osmium sequestration28 enabled full conversion of 7 to 8. Steric congestion about the Δ2,3 alkene of 7, however, slowed conversion such that one equivalent of OsO4 still required 7 days to elicit an 81% yield.29 This drawback was mitigated by excellent diastereoselectivity (>20:1) at C2 and C3 and spontaneous lactonization at C10. For comparison, the strong oxidant dimethyldioxirane reacted exclusively with the electron-deficient Δ8,9 alkene in 6 to provide 7, which did not react further.
Intermediate 8 set the stage to explore gem-dimethyl modification, including C–C bond cleavage. Geminal dimethyl groups predominate in terpenoids as a result of their biosynthesis from polyprenyl- (dimethylallyl) pyrophosphates.30 Modification of gem-dimethyls, including their excision, can be effected with iron-oxo enzymes to produce biologically active scaffolds (Figure 3a).31 Similar demethylations have not been employed in chemical synthesis since abiotic routes are not often constrained by biosynthetic building blocks, and retrosynthetic addition of an extra carbon-bound methyl group is seldom simplifying.32 In this example we found an exception to the rule.
Figure 3.
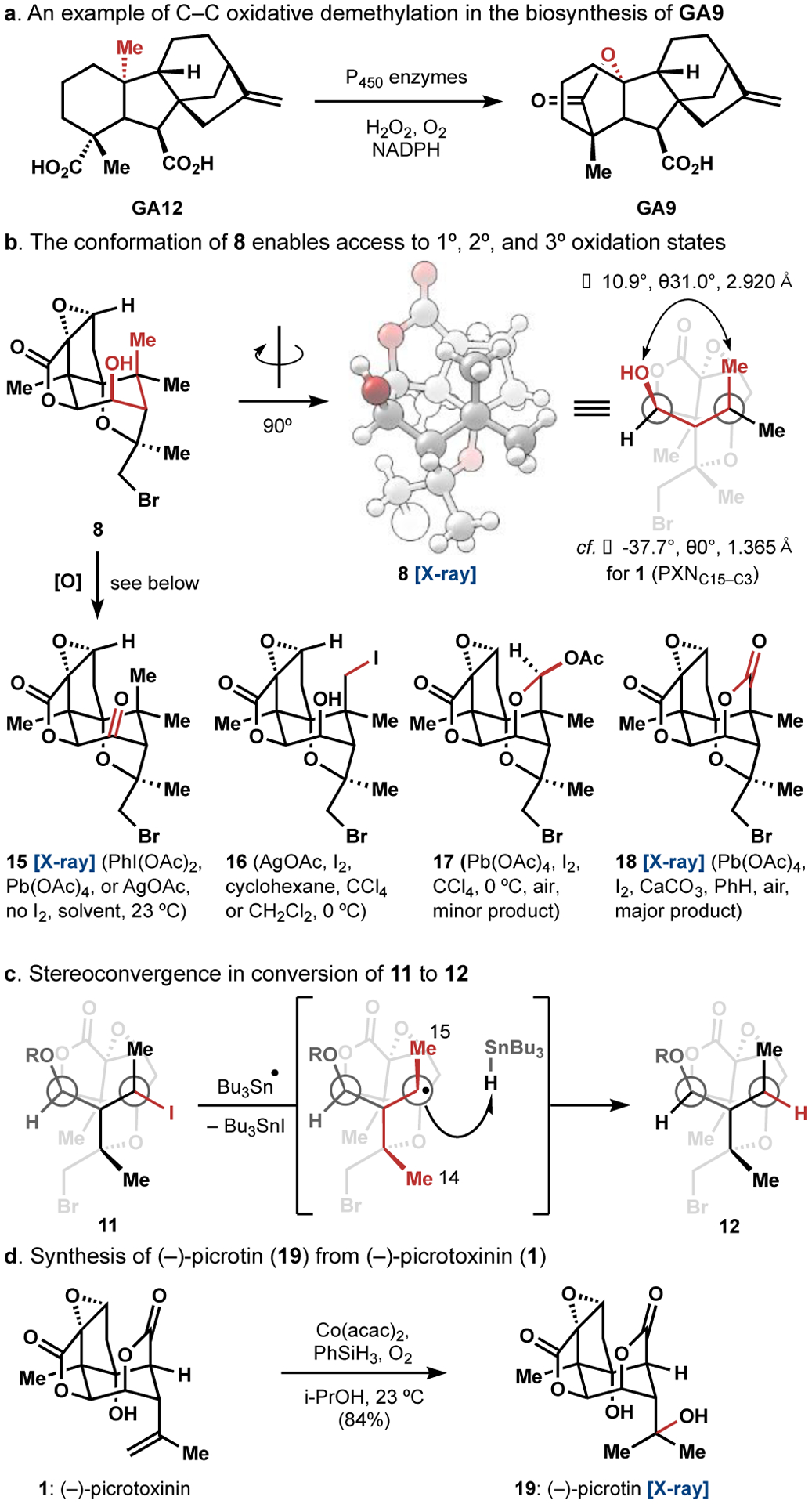
Further details and synthesis of (–)-picrotin (19).
Molecular modeling, which was later confirmed by analysis of the crystal structure of 8, indicated that the strain conferred upon the cyclohexane core by the two fused and one bridged pentacycles causes the C3 alcohol to tilt about 11° away from a parallel orientation to the C15 methyl group, with a dihedral angle (θ) of 31° (Figure 3b). This subtle shift in conformation places the C3 alcohol oxygen 2.920 Å (x-ray) away from the axial methyl group such that ether formation is slow due to torsional strain in the transition state (cf. ∠−37.7°, θ0°, 1.365 Å (x-ray) for the C15 lactone C–O bond of PXN (1)). Consequently, it was possible to directly access the primary (ether 9 or iodide 16), secondary (acetal 17), or tertiary (lactone 18) oxidation states of the axial methyl group in 8 (Figure 3b). These oxidation states were accessible by generating IOAc33 with different reagents and temperatures, although acetal 17 was never formed as a major product (Figure 3b). Thus, use of AgOAc/I2 in methylene chloride at 23 °C under ambient light provided the ether 9 in 51% yield (Scheme 1), whereas the 1° iodide 16 was obtained at 0 °C in cyclohexane as the major product (Figure 3b). Notably, ketone 15 formed readily in the absence of iodine and was observed as a persistent byproduct. Treatment of 9 with TFDO at 0 °C generated hemiacetal 9 as a 2.5:1 diastereomeric mixture, a distribution which may be attributed to the outward-facing C–H bond being both less sterically hindered and experiencing better hyperconjugative donation from the C3 ether oxygen than its inward-facing counterpart. The same conditions for formation of 16 (AgOAc, I2, CH2Cl2, 23 °C) applied here led to Suárez fragmentation34 in 10 of the adjacent strong C–C bond to form 11 as a single stereoisomer. The tertiary iodide of 11 was removed with AIBN/Bu3SnH to form a single isomer of 12 after cleavage of the formyl group in a basic work-up. A plausible explanation for this stereochemistry is that Bu3SnH is too large a hydrogen atom donor for hydrogen atom transfer (HAT) to occur at the concave face of C5. A 1,3-diaxial interaction between the C15 and C14 methyl groups in the transition state for HAT at the concave face would further destabilize this pathway (Figure 3c). Finally, use of Pb(OAc)4/I2 in benzene with CaCO3 at 23 °C under an aerobic atmosphere led directly to formation of the C15 lactone. Reduction with zinc cleaved the bromoether linkage of 12 to deliver (–)-picrotoxinin (1). Conversion to (–)-picrotin (19) occurred in one step and 84% yield by a Mukaiyama hydration,35 which had not been reported previously.2,4,6,7
Geminal dimethylation of carvone at C5 expedited forward entry to the carbocyclic core of PXN but revised our initial retrosynthesis, amounting to a ‘nonsense’ methylation transform (1 ⟹ 20 or 8, see Figure 4) in search of a forward solution. The complexity of 1 versus 20 was not diminished by methylation since information content was added and no stereocenter was removed (Cm = 468 vs. 480 mcbits).36 Symmetrization of C5 in intermediate targets like 8, however, greatly simplified entry into chemical space very close to 1. Interestingly, 5-methyl-picrotoxinin (20) retained modest antagonism of the GABAA receptor (IC50 = 9 μM; vs. [3H] TBOB @ rat cerebral cortex) and slightly improved upon the aqueous stability of 1 at pH 8, more than halving the pseudo-first order rate constant.37 Ongoing biological studies intended for this manuscript that leveraged quick entry into picrotoxinin chemical space (8 steps to 20) have been delayed by recent events.
Figure 4.
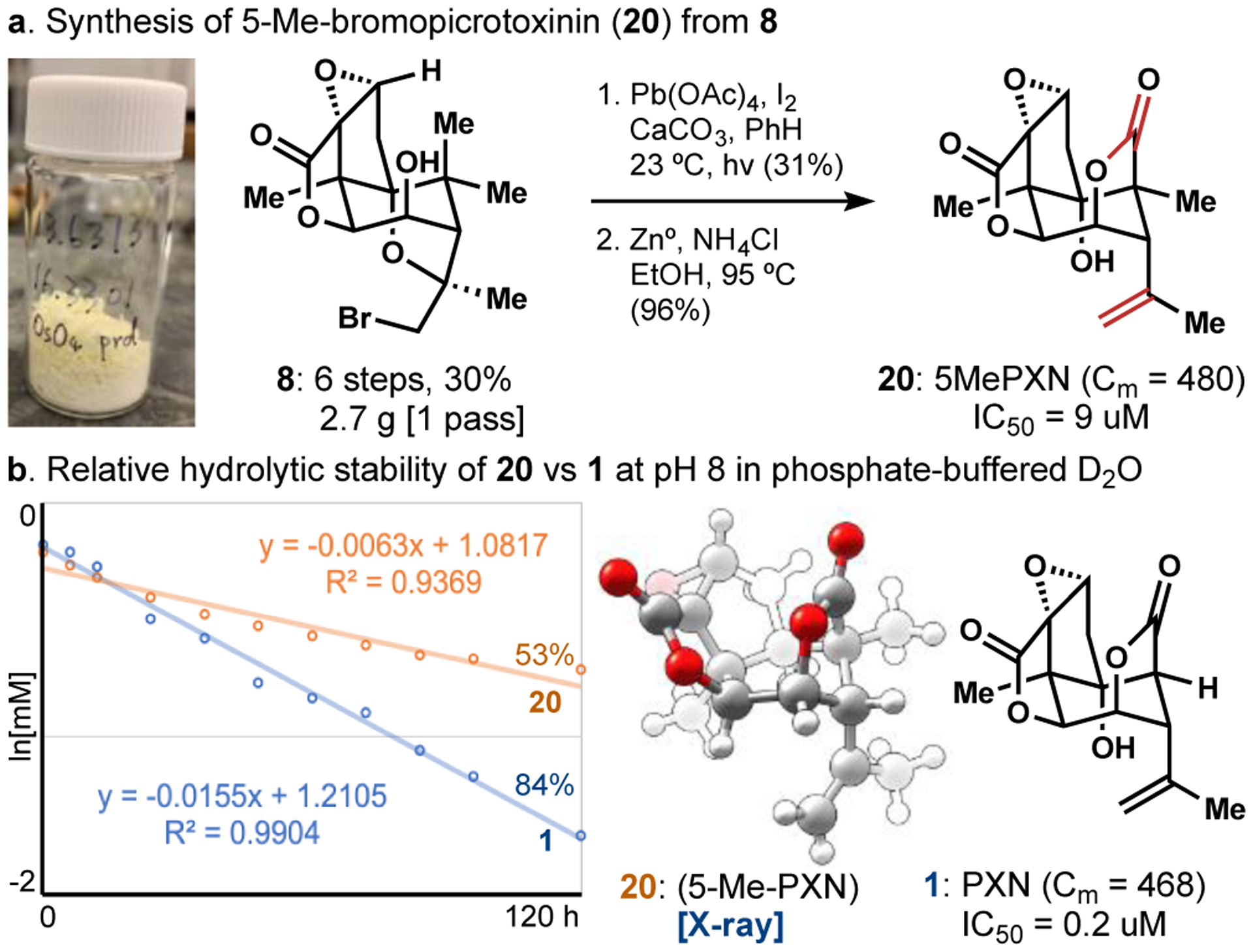
5-Methyl-picrotoxinin (20) is equal in complexity, more stable and less antagonizing than picrotoxinin (1).
In summary, we disclose a concise synthesis of (–)-picrotoxinin (1) via incorporation of a symmetrizing gem-dimethyl moiety that allows efficient annulation to form the [4.3.0]-bicyclononane core. The key stereotetrad was accessed in only 4–5 steps from (R)-carvone and correlated to an overall short synthesis. The facile, stereoselective annulation to form 2 benefitted from symmetrizing dimethylation, allowing stereochemical relay from the C4 β-isopropene of carvone and obviating the need for stereocontrol at C5. High oxidation states in the starting materials were encoded by unsaturation and leveraged to access 1 in the shortest sequence to date. This route provides the first example, to our knowledge, of an oxidative C–C demethylation sequence applied in total synthesis. We aim to use this short entry into PXN chemical space to continue our probe of selectivity within the ligand-gated ion channel (LGIC) superfamily of receptors.
Supplementary Material
Acknowledgements
Generous support was provided by the National Institutes of Health (5R35GM122606), the Natural Sciences and Engineering Research Council of Canada (PGS D3 to S.W.M.C.), JITRI (JITRI Fellowship to G. T.), and the Beckman Foundation (A.O. Beckman PDF to M.W.L.). We thank Dr. L. Pasternack and Dr. D.-H. Huang for NMR assistance, Dr. J. Chen and Brittany Sánchez for HRMS measurements, and Dr. Arnie Rheingold, Dr. Curtis Moore, and Dr. Milan Gembicky for X-ray crystallographic analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website: https://doi.org/10.1021/jacs.0c05042
Materials and methods; details related to synthesis and experiments; X-ray data; NMR spectra
REFERENCES
- 1.Corey EJ; Pearce HL Total synthesis of picrotoxinin. J. Am. Chem. Soc 1979, 101, 5841. [Google Scholar]
- 2.Corey EJ; Pearce HL Total synthesis of picrotin. Tetrahedron Lett. 1980, 21, 1823. [Google Scholar]
- 3.Niwa H; Wakamatsu K; Hida T; Niiyama K; Kigoshi H; Yamada M; Nagase H; Suzuki M; Yamada K Stereocontrolled total synthesis of (–)-picrotoxinin and (+)-coriamyrtin via a common isotwistane intermediate. J. Am. Chem. Soc 1984, 106, 4547. [Google Scholar]
- 4.Miyashita M; Suzuki T; Yoshikoshi A, Stereoselective total synthesis of (–)-picrotoxinin and (–)-picrotin. J. Am. Chem. Soc 1989, 111, 3728. [Google Scholar]
- 5.Trost BM; Krische MJ, A general strategy for the asymmetric synthesis of the picrotoxanes. J. Am. Chem. Soc 1996, 118, 233. [Google Scholar]
- 6.Trost BM, Haffner CD; Jebaratnam DJ; Krische MJ; Thomas AP The palladium-catalyzed enyne cycloisomerization reaction in a general approach to the asymmetric syntheses of the picrotoxane sesquiterpenes. Part I. First-generation total synthesis of corianin and formal syntheses of picrotoxinin and picrotin. J. Am. Chem. Soc 1999, 121, 6183. [Google Scholar]
- 7.Trost B; Krische MJ Palladium-catalyzed enyne cycloisomerization reaction in an asymmetric approach to the picrotoxane sesquiterpenes. 2. Second-generation total syntheses of corianin, picrotoxinin, picrotin, and methyl picrotoxate. J. Am. Chem. Soc 1999, 121, 6131. [Google Scholar]
- 8.For a very recent approach, see:; Cao J; Thor W; Yang S; Zhang M; Bao W; Zhu L; Yang W; Cheng Y-K; Lee C-S Synthesis of the tricyclic picrotoxane motif by an oxidative cascade cyclization. Org. Lett 2019, 21, 4896. [DOI] [PubMed] [Google Scholar]
- 9.Porter LA Picrotoxinin and related substances. Chem. Rev 1967, 67, 441. [DOI] [PubMed] [Google Scholar]
- 10.Coscia CJ Picrotoxin. Cyclopentanoid Terpene Derivatives” 1968, pp. 147–201. [Google Scholar]
- 11.GoÖssinger E Picrotoxanes. Progress in the Chemistry of Organic Natural Products, vol. 93, 2010, Springer. [Google Scholar]
- 12.Fernandez F; Morishita W; Zuniga E; Nguyen J; Blank M; Malenka RC; Garner CC Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci 2007, 10, 411. [DOI] [PubMed] [Google Scholar]
- 13.Picrotoxin; MSDS No. sc-202765 [Online]; Santa Cruz Biotechnology, Inc.: Santa Cruz, CA, December 23, 2008. http://datasheets.scbt.com/sc-202765.pdf (accessedAug 5, 2019). [Google Scholar]
- 14.Baker M; Demoret R; Ohtawa M; Shenvi RA Concise asymmetric synthesis of (–)-bilobalide. Nature, 2019, 575, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Witkin JM; Shenvi RA; Li X; Gleason SD; Weiss J; Wakulchik ML; Ohtawa M; Martinez MD; Schkeryantz JM; Carpenter TS; Lightstone FC; Cerne R Pharmacological characterization of the neurotrophic sesquiterpene jiadifenolide reveals a non-convulsant signature and potential for progression in neurodegenerative disease studies. Biochem. Pham 2018, 155, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu H-H; Martinez MD; Shenvi RA An eight-step gram-scale synthesis of (–)-jiadifenolide. Nature Chem. 2015, 7, 604. [DOI] [PubMed] [Google Scholar]
- 17.Ohtawa M; Krambis M; Cerne R; Schkeryantz J; Witkin JM; Shenvi RA Synthesis of (–)-11-O-debenzoyltashironin: Neurotrophic sesquiterpenes cause hyperexcitation. J. Am. Chem. Soc 2017, 139, 9637. [DOI] [PubMed] [Google Scholar]
- 18.Ng CC; Duke RK; Hinton T; Johnston GAR Effects of bilobalide, ginkgolide B and picrotoxinin on GABAA receptor modulation by structurally diverse positive modulators. Eur. J. Pharm 2017, 806, 83. [DOI] [PubMed] [Google Scholar]
- 19.For the generation of analogs from isolated picrotoxinin, see:; a) Kriscshe MJ; Trost BM Transformations of the picrotoxanes: the synthesis of corianin and structural analogs from picrotoxinin. Tetrahedron 1998, 54, 7109; [Google Scholar]; b) Jarboe CH; Porter LA; Buckler RT Structural Aspects of Picrotoxinin Action. J. Med. Chem 1968, 11, 729; [DOI] [PubMed] [Google Scholar]; c) Shirai Y; Hosie AM; Buckingham SD; Holyoke CW; Baylis HA; Sattelle DB Actions of picrotoxinin analogues on an expressed, homo-oligomeric GABA receptor of Drosophila melanogaster. Neurosci. Lett 1995, 189, 1. [DOI] [PubMed] [Google Scholar]
- 20.Edwards OE; Douglas JL; Mootoo B Biosynthesis of dendrobine. Can. J. Chem 1970, 48, 2517. [Google Scholar]
- 21.Maimone TJ; Baran PS Modern synthetic efforts toward biologically active terpenes. Nature Chem. Biol 2007, 3, 396. [DOI] [PubMed] [Google Scholar]
- 22.Brill ZG; Condakes ML; Ting CP; Maimone TJ Navigating the chiral pool in the total synthesis of complex terpene natural products. Chem. Rev 2017, 117, 11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demoret RM; Baker MA; Ohtawa M; Chen S; Lam C-C; Forli S; Houk K; Shenvi RA Synthesis and Mechanistic Interrogation of Ginkgo biloba Chemical Space en route to (–)-Bilobalide; ChemRxiv DOI: 10.26434/chemrxiv.12132939.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith JM; Harwood SJ; Baran PS Radical retrosynthesis. Acc. Chem. Res 2018, 51, 1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selezneva NK; Gimalova FA; Valeev RF; Miftakhov MS Efficient synthesis of (1R,4S,6R)-4-isopropenyl-1,3,3-trimethyl-7-oxabicyclo[4.1.0]heptan −2-one. Russ. J. Org. Chem 2011, 47, 173. [Google Scholar]
- 26.Srikrishna A; Reddy TJ; Kumar PP Synthesis of taxanes–the carvone approach; a simple, efficient enantioselective synthesis of the functionalized A ring. Chem. Commun 1996, 1369. [Google Scholar]
- 27.Fabrissin S; Fatutta S; Risaliti A Elimination reactions of cis- and trans-8a-hydroxy-2-thiadecalin 2,2-dioxide with thionyl chloride. Evidence for intermediacy of ion pairs. JCS, Perkin Trans 1 1977, 1561.; [Google Scholar]; Margaros I; Vassilikogiannakis G Synthesis of (+)-Zerumin B Using a Regioselective Singlet Oxygen Furan Oxidation. J. Org. Chem, 2008, 73, 2021. [DOI] [PubMed] [Google Scholar]
- 28.Dupau P; Epple R; Thomas AA; Fokin VV; Sharpless KB Osmium catalyzed dihydroxylation of olefins in acidic media: Old process, new tricks. Adv. Synth. Catal 2002, 344, 421. [Google Scholar]
- 29.A referee pointed out: “the following information is worth sharing with the ‘connoisseurs’ among the readership, as it strongly suggests that the presence of the C15 methyl (in the context of the bromoether) is uniquely enabling vis-à-vis modification of the C2-C3 olefin … Specifically, a related alkene-containing bromoether was prepared by Trost. It displayed a PROFOUND lack of reactivity … ‘to stoichiometric quantities of osmium tetroxide at high pressures and temperature (100 C, 16 kbar) or neat bromine.’ Thus, groups larger than the C15 methyl prohibit approach to the concave pi-face of the olefin (and the bromomethyl moiety blocks the convex face). See:; Krische MJ; Trost BM “Total Synthesis of Methyl Picrotoxate via the Palladium Catalyzed Enyne Cycloisomerization Reaction” Tetrahedron 1998, 54, 3693. [Google Scholar]
- 30.Dewick PM (2009) Medicinal Natural Products A Biosynthetic Approach, 3rd ed., John Wiley & Sons, West Sussex, United Kingdom. [Google Scholar]
- 31.Nagel R; Peters RJ Diverging mechanisms: Cytochrome-P450-catalyzed demethylation and γ-lactone formation in bacterial gibberellin biosynthesis. Angew. Chem. Int. Ed 2018, 57, 6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roach JJ; Sasano Y; Schmid CL; Zaidi VK; Stevens RC; Bohn LM; Shenvi RA Dynamic strategic bond analysis yields a ten-step synthesis of 20-nor-salvinorin A, a potent κ-OR agonist. ACS Cent. Sci 2017, 3, 1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.a) Giri R; Yu J-Q Iodine monoacetate. eEROS Encyclopedia of Reagents for Organic SynthesisDOI: 10.1002/047084289X.rn00915, 2008; [DOI] [Google Scholar]; b) Barnett JR; Andrews LJ; Keefer RM Trifluoroacetyl Hypohalites as Aromatic Halogenating Agents. J. Am. Chem. Soc 1972, 94, 6129. [Google Scholar]
- 34.De Armas P; Francisco CG; Suaárez, E. Fragmentation of carbohydrate anomeric alkoxy radicals. tandem β-fragmentation-cyclization of alcohols. J. Am. Chem. Soc. 1993, 115, 8865. [Google Scholar]
- 35.a) Zombeck A; Hamilton DE; Drago RS Novel Catalytic Oxidations of Terminal Olefins by Cobalt(II)-Schiff Base Complexes. J. Am. Chem. Soc 1982, 104, 6782; [Google Scholar]; b) Mukaiyama T; Isayama S; Inoki S; Kato K; Yamada T; Takai T Oxidation-reduction hydration of olefins with molecular oxygen and 2-propanol catalyzed by bis(acetylacetonato)cobalt(II). Chem. Lett 1989, 449; [Google Scholar]; c) Isayama S; Mukaiyama T A new method for preparation of alcohols from olefins with molecular oxygen and phenylsilane by the use of bis(acetylacetonato)cobalt(II). Chem. Lett 1989, 1071. [Google Scholar]
- 36.Böttcher T An Additive Definition of Molecular Complexity. J. Chem. Inf. Model 2016, 56, 462. [DOI] [PubMed] [Google Scholar]
- 37. Slight curvature in the hydrolysis of 20 might indicate greater reversibility.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.