

Abstract
The chemokine system plays an important role in mediating a proinflammatory microenvironment for tumor growth in hepatocellular carcinoma (HCC). The CXCR6 receptor and its natural ligand CXCL16 are expressed at high levels in HCC cell lines and tumor tissues and receptor expression correlates with increased neutrophils in these tissues contributing to poor prognosis in patients. Availability of pharmacologcal tools targeting the CXCR6/CXCL16 axis are needed to elucidate the mechanism whereby neutrophils are affected in the tumor environment. We report the discovery of a series of small molecules with an exo-[3.3.1]azabicyclononane core. Our lead compound 81 is a potent (EC50 = 40 nM) and selective orally bioavailable small molecule antagonist of human CXCR6 receptor signaling that significantly decreases tumor growth in a 30-day mouse xenograft model of HCC.
Keywords: CXCR6 receptor antagonist, Hepatocellular carcinoma, SK-HEP xenograft model, B-arrestin signaling, Azabicyclononane scaffold
CXCR6 is a seven transmembrane domain G protein–coupled receptor (GPCR) target for the natural ligand, CXCL16, a chemokine that exists in both membrane-anchored and soluble variant forms to regulate leukocyte trafficking during cellular stress. CXCR6 receptor is expressed on many cell types in the immune system including dendritic cells, CD4+ cells, CD8+ cells, natural killer cells and natural killer T cells.1–3 The CXCR6/CXCL16 axis plays a critical role in proinflammatory cytokine expression in liver that may induce CXCR6 immune-dependent mechanism in hepatocarcinogenesis. CXCR6 and CXCL16 are highly expressed in many types of human cancers including hepatocellular carcinoma (HCC),4 a cancer that ranks fifth in frequency worldwide among all malignancies and is the third leading cause of cancer mortality.5 CXCR6 is consistently expressed in all eight hepatoma cell lines reported.6 The expression profile is low in normal hepatocytes, increases in noninvasive HCC cells, and reaches highest levels in invasive HCCs. Upregulation of CXCR6 receptor contributes to a proinflammatory tumor microenvironment that promotes metastasis and has been identified as an independent predictor for increased recurrence and poor survival in patients with HCCs.6,7 In agreement, genetic downregulation of CXCR6 receptor inhibited HCC cell invasion in vitro and in mice subcutaneous xenografts, inhibited tumorigenicity, neutrophil recruitment, angiogenesis, and metastasis of hepatoma cells.6 Verifying the therapeutic impact of CXCR6 in HCC has been hampered by lack of available small molecule probes. Thus, the development of pharmacologically available small molecule antagonists to CXCR6/CXCL16 signaling that reduce inflammation in the liver or modulate the immune response may provide an avenue to ameliorate hepatic tumor progression and metastasis and represent promising therapeutics for HCC and related cancers. We report the discovery of the first potent, selective orally bioavailable small molecule antagonist of the CXCR6 receptor signaling that decreases tumor growth in a mouse xenograft model of HCC.
We previously reported the discovery of an initial series of CXCR6 receptor antagonists represented by probe compound 17 (ML339, Table 1)8 through high throughput screening (HTS) of the NIH Molecular Library Small Molecular Repository (MLSMR), medicinal chemistry and structure-activity relationship (SAR) studies as a first in class small molecule probe. Compound 17 is potent in antagonizing (β-arrestin recruitment and cAMP signaling of the human CXCR6 receptor induced by CXCL16 (IC50 = 0.3 and 1.4 μM, respectively) and is devoid of activity against the CCR6, CXCR5, and CXCR4 human receptors (IC50 > 80 μM).
Table 1.
SAR of Analogs of alternative scaffolds.
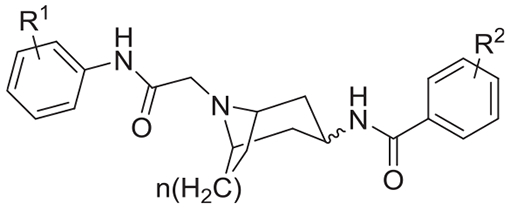 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | n | R1 | R2 | CXCR6: β-arrestin | CXCR6: cAMP | ||
| IC50 (μM) | % response | IC50 (μM) | % response | ||||
| 1Screen hit | 3* | H | 3,4,5-tri-OMe | 5.8 | 65 | 30.6 | 86 |
| 2 | 3 endo | H | 3,4,5-tri-OMe | > 40 | NA | 40.9 | 70 |
| 3 | 2* | H | 3,4,5-tri-OMe | 26 | 63 | > 40 | NA |
| 4 | 2 | H | 3,4,5-tri-OMe | > 40 | NA | > 40 | NA |
| 5 | 0 | H | 3,4,5-tri-OMe | > 40 | NA | > 40 | NA |
| 6 | 3 | H | H | > 40 | NA | > 80 | NA |
| 7 | 3 | 4-F | 3,4,5-tri-OMe | 5.8 | 100 | 30.7 | 90 |
| 8 | 3 | 4-Cl | 3,4,5-tri-OMe | 1.5 | 101 | 14.2 | 93 |
| 9 | 3 | 4-Me | 3,4,5-tri-OMe | 9.5 | 100 | > 40 | NA |
| 10 | 3 | 4-OMe | 3,4,5-tri-OMe | 19 | 76 | > 40 | NA |
| 11 | 3 | 4-Cl | 3,4-di- OMe | > 40 | NA | > 40 | NA |
| 12 | 3 | 4-Cl | 3,5-di-OMe | > 40 | NA | > 40 | NA |
| 13 | 3 | 4-Cl | 3-OMe | > 40 | NA | 14.5 | 87 |
| 14 | 3 | 4-Cl | 4-OMe | > 40 | NA | 5.9 | 94 |
| 15 | 3 | 4-Cl | 3,4-(OCH2O) | > 40 | NA | 5.3 | 84 |
| 16 | 3 | 2-F | 3,4,5-tri-OMe | 0.62 | 103 | 6.5 | 99 |
| 17 ML339 | 3 | 2-Cl | 3,4,5-tri-OMe | 0.14 | 100 | 1.4 | 102 |
| 18 | 3 | 2-Me | 3,4,5-tri-OMe | 0.79 | 104 | 9.3 | 101 |
| 19 | 3 | 2-OMe | 3,4,5-tri-OMe | 2.1 | 102 | 14.7 | 100 |
| 20 | 3 | 2-Br | 3,4,5-tri-OMe | 0.16 | 105 | 0.9 | 104 |
| 21 | 3 | 2-CF3 | 3,4,5-tri-OMe | 0.64 | 101 | 7.9 | 105 |
| 22 | 3 | 2-CN | 3,4,5-tri-OMe | 0.93 | 91 | 7.0 | 105 |
| 23 | 3 | 2-CO2Et | 3,4,5-tri-OMe | 1.6 | 79 | 6.9 | 96 |
| 24 | 3 | 3-F | 3,4,5-tri-OMe | 0.84 | 104 | 8.5 | 103 |
| 25 | 3 | 3-Cl | 3,4,5-tri-OMe | 0.41 | 105 | 4.3 | 97 |
| 26 | 3 | 3-Br | 3,4,5-tri-OMe | 0.35 | 104 | 3.8 | 92 |
| 27 | 3 | 3-Me | 3,4,5-tri-OMe | 1.8 | 102 | 17.9 | 92 |
| 28 | 3 | 3-OMe | 3,4,5-tri-OMe | 5.8 | 96 | 51.0 | 93 |
| 29 | 3 | 2,3-dichloro | 3,4,5-tri-OMe | 0.16 0.12 | 102 96 | 0.41 25.6 | 78 39 |
| 30 | 3 | 2,4-dichloro | 3,4,5-tri-OMe | 0.16 | 105 | 0.9 | 105 |
| 31 | 3 | 2,5-dichloro | 3,4,5-tri-OMe | 0.13 | 99 | 0.7 | 101 |
| 32 | 3 | 2,6-dichloro | 3,4,5-tri-OMe | 38.2 | 57 | > 40 | NA |
| 33 | 3 | 3,4-dichloro | 3,4,5-tri-OMe | 0.35 | 103 | 1.8 | 101 |
| 34 | 3 | 3,5-dichloro | 3,4,5-tri-OMe | 0.31 | 104 | 1.3 | 96 |
| 35 | 3 | 2,5-di Me | 3,4,5-tri-OMe | 0.61 | 105 | 16.5 | 108 |
| 36 | 3 | 2,6-di-Me | 3,4,5-tri-OMe | > 40 | NA | > 40 | NA |
denotes exo configuration. All other entries are in the exo configuration.
From our probe discovery of compound 17, we pursued a hit to lead program under the Florida Translational Research Program (FTRP) and report here an additional lead series distinct from compound 17 with improved potency in cell-based signaling as exemplified by compound 81 (Fig. 1, Table 3). Compound 81 has improved plasma stability and exhibited in vivo activity in a mouse xenograft model of HCC. The synthetic scheme for compound 81 is shown in the supplemental information.
Fig. 1.
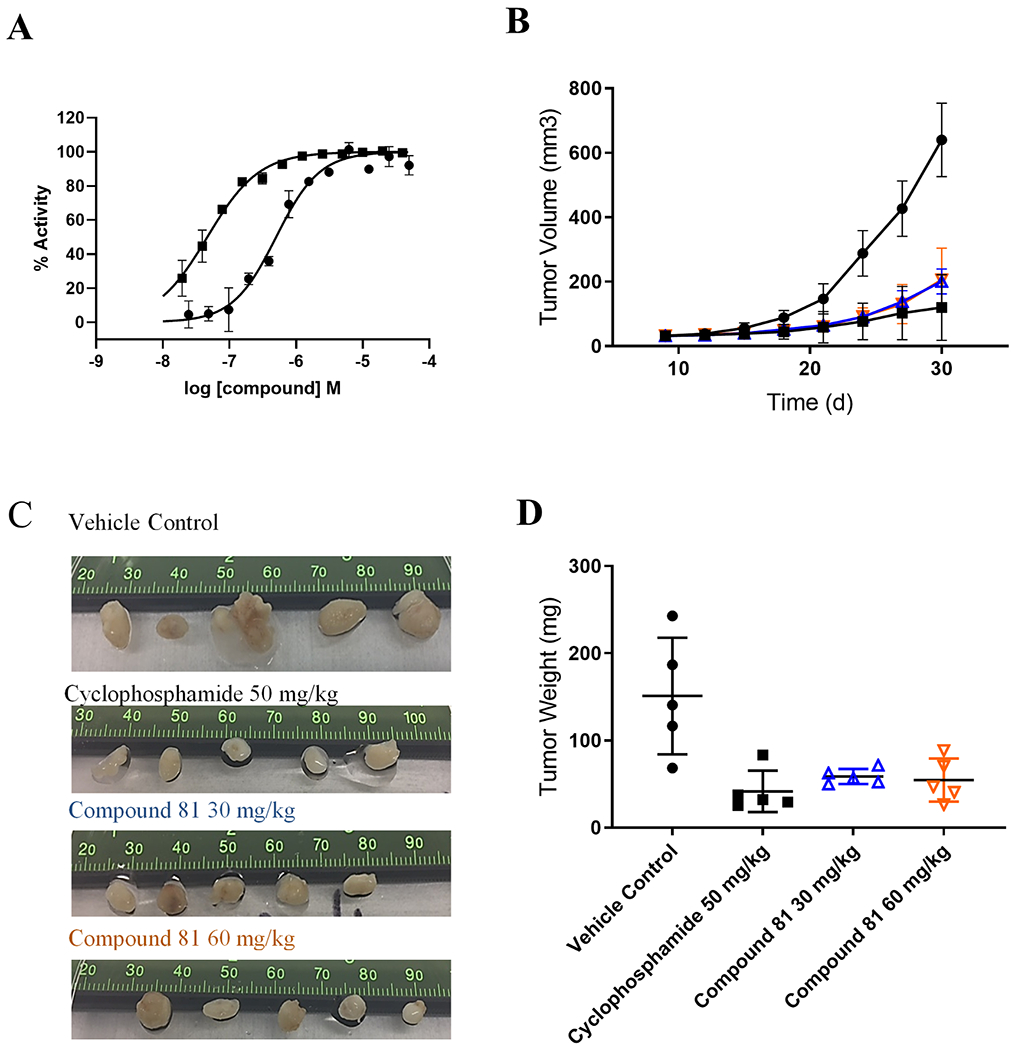
In vitro and in vivo activity of compound 81. (A) Inhibition of B-arrestin recruitment (squares) and forskolin-induced inhibition of CAMP production (circles) was determined using DiscoveRx cells expressing Prolink-tagged hCXCR6 treated with an EC80 concentration of CXCL16 and increasing concentrations of compound 81 (Table 3). (B) SK-HEP-1 induced tumor growth in mice over 30 days (closed circles) was inhibited by 30 mg/kg (blue triangles) and 60 mg/kg dose (red triangles) of compound 81. Cyclophosphamide at 50 mg/kg dose (closed squares) was used as a positive control. (C,D) Tumors were removed from each animal to determine size and weight. Statistical significance of weight of 30 mg/kg and 60 mg/kg treatment groups compared to vehicle-treated was determined by student t-test (P < 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 3.
SAR of benzothiazole analogs.
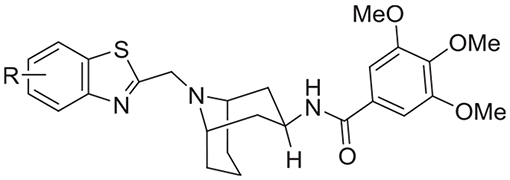 | |||||
|---|---|---|---|---|---|
| Entry | R | CXCR6: β-arrestin | CXCR6: cAMP | ||
| IC50 (μM) | % response | IC50 (μM) | % response | ||
| 73 | 4-Cl | 0.13 | 103 | 0.9 | 95 |
| 74 | 5-Cl | 0.17 | 100 | 3.2 | 90 |
| 75 | 6-Cl | 0.27 | 103 | 14.0 | 83 |
| 76 | 7-Cl | 0.26 | 102 | 8.9 | 93 |
| 77 | 4-F | 0.11 | 103 | 1.9 | 105 |
| 78 | 4-Me | 0.25 | 100 | 4.8 | 86 |
| 79 | 4-OMe | 3.1 | 86 | > 50 | NA |
| 80 | 4-Br | 0.08 | 102 | 1.0 | 91 |
| 81 | 4-CF3 | 0.04 | 103 | 0.5 | 98 |
Unless otherwise noted, compounds referred to in the text are listed in Table 1. The screening hit from the MLSMR library, compound 1, had unassigned stereochemistry for the 3-amino group and was not defined as the endo or exo isomer. Hence synthesis of analogs (Scheme S1) and intial SAR study was designed to address the following key questions; a) is the [3.3.1]azabicyclononane core required for activity, b) is the reverse amide substituent in the exo or endo position in the primary hit compound 1, c) is the exo or endo position essential or preferred, d) is the 3,4,5-trimethoxybenzoyl motif required and e) does substitution of the anilide group appended to the bridgehead nitrogen influence activity? Compound 1 was synthesized from the exo-tert-butyl-3-amino-9-azabicyclo[3.3.1]nonane-9-carboxylate (CAS 1363380-67-9). The resynthesized batch recapitulated the activity of the screening hit and was identical by NMR to the commercial sample of the parent singleton hit and was further characterized by x-ray crystallography (Cambridge Structural Database CCDC 1969634) to unambiguously establish the exo stereochemistry at the 3,4,5-trimethoxybenzamide region of the molecule8 (see Supporting Information).
Compound 2, the 3-endo isomer was obtained from a building block which was a 1:1 mixture of exo/endo isomers, after separation by flash chromatography. Comparison of compounds 1 and 2 showed that for compounds with the [3.3.1] scaffold, the 3-exo stereochemistry was required for activity (compounds 1 and 2). The same trend was noted for the less active [3.2.1] scaffold (compound 3 and 4). The monocyclic 4-aminopiperidine analog (compound 5) was inactive. These data prompted us to next explore aryl substitution at positions R1 and R2 in the anilide and benzamide moieties respectively in the scaffold. The completely unsubstituted analog (Entry 6, R1 = R2 = H) was inactive. Electron withdrawing groups were preferred at the R1 position. The 4-Cl analog was the most active and approximately four-fold better than the 4-H (compounds 1 and 8). The SAR at the benzamide R2 position was explored by varying R2 to 3,4-dimethoxy, 3,5-dimethoxy, 3-methoxy, 4-methoxy, or 3,4-methyleneoxy and keeping R1 = Cl. For these analogous sets, activity was only observed when R2 was 3,4,5-trimethoxy (compounds 8 and 11–15). Substitution at the 2-position was markedly better than at the 4-position, with the 2-OMe and 2-Cl analogs both being 10-fold better than the corresponding 4-OMe and 4-Cl analogs (compounds 7–10 vs 16–19). The 2-Cl was the most potent closely followed by the 2-CN among the electron withdrawing groups at the 2-position (compounds 16–23). At the 3-position the preference for an electron withdrawing group continued but with 2–3 fold lower potency (compounds 24–28). Among the disubstituted analogs at R1 position, analogs with a 2-Cl group were the most potent (compounds 29–34). In contrast 2,6-disubstitution abolished activity possibly by moving the aromatic ring out of plane with respect to the amide group (compounds 32 and 36). The 2-Cl analog (compound 17) met the probe criteria and was scaled-up to be nominated as the probe ML339 under the molecular libraries program.8
Our SAR strategy for further optimization of ML339 to an in vivo active lead for proof of concept studies focused on keeping the exo-[3.3.1] bicycle intact and making broad changes in the anilide group, glycine (acetate) linker and the 3,4,5-trimethoxybenzamide portion of the molecule and then focused SAR follow up around the most potent leads obtained. Demethylation of 3,4,5-trimethoxyphenyl has been linked to poor metabolic stability and pharmacokinetic (PK) properties. Earlier attempts to address this potential liability by removing any of these OMe groups were not productive (compounds 8 and 11–15). Keeping the more potent 2-Cl, R1 substituent in ML339 constant, we systematically screened monomethoxy substituents at the R2 position (compounds 37–39, Supplementry Table 1) and found complete erosion of activity in the β-arrestin assay and only found borderline (EC50 = 15.1 μM) cAMP activity (compound 37, R2 = 2-OMe). Keeping 2-OMe constant at the R2 position, we screened other electron withdrawing groups at the 2-position (R1) and walked the Cl substituent to 3- and 4- position on the anilide group (compound 40–49, Supplementry Table 1). There was no improvement in the cAMP activity except for the 4-Cl (R1) derivative as previously seen (compound 45 vs compounds 11–15). Screening substituents other than methoxy at the R2 position resulted in only a marginal increase in cAMP activity and continued loss of activity in the β-arrestin assay (compound 49). This series of compounds was deprioritized based on its weak activity in the signaling assays.
We next looked at the glycine linker and found that replacement of the amide carbonyl with a flexible methylene group resulted in complete loss of activity in ML339 (compound 50, Supplementry Table 2). A conformationally more rigid linker with gem-dimethyl substitution led to significant or complete loss of activity in cAMP and β-arrestin assay respectively (compound 51, Supplementry Table 2 vs compound 17). Activity in cAMP assay improved 2-fold in 2-methoxybenzamide (R2) vs the 3,4,5-trimethoxybenzamide (compound 51 and 52, Supplementry Table 2). Walking the Cl (R1) substituent or the OMe (R2) around the aryl ring led to completely inactive analogs. This series of compounds was also deprioritized based on its weak activity observed in the cAMP assay.
The potential of toxicity resulting from hydrolysis of the aniline amide in compound 17 under physiological conditions prompted us to explore a series of analogs that replaced the anilide moiety with aryl methylene or heteroaryl methylene groups. Methoxyphenyl at the R position abolished activity (compounds 56–58, Table 2). As before, electron withdrawing substituents seem to retain activity with the 2-chlorobenzyl derivative having the best activity (compound 59, Table 2) albeit at 48-fold lower activity than compound 17. Further exploration of bicyclic aryl and heteroaryl groups led to the 2-benzothiophenyl and 2-benzothiazolyl analogs with significant retention of activity in both β-arrestin and cAMP assays (compound 67–68, Table 2). We recognized that the 2-benzothiazole analog compound 68 was a significant find, as it is a bio-isostere of the anilide group in compound 17. The closely related bio-isosteric benzoxazole and benzimidazole analogs are also active however significantly less potent (compounds 69 and 70, Table 2). Extending the methylene linker by one carbon in compound 71 or a carbonyl linkage in compound 72 led to a complete or 100-fold loss in activity, respectively (Table 2). These results point to specificity of interaction of the target with the 2-benzothiazole moiety.
Table 2.
SAR of analogs of exo-tert-butyl-3-amino-9-azabicyclo[3.3.1]nonane-9-carboxylate.
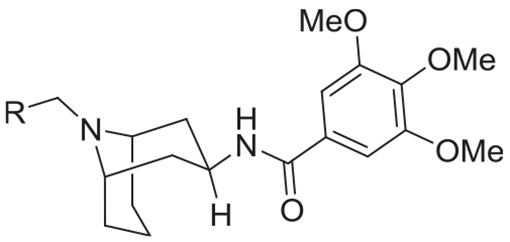 | |||||
|---|---|---|---|---|---|
| Entry | R | CXCR6: β-arrestin | CXCR6: cAMP | ||
| IC50 (μM) | % response | IC50 (μM) | % response | ||
| 56 | 2-methoxyphenyl | > 50 | NA | > 50 | NA |
| 57 | 3-methoxyphenyl | > 50 | NA | > 50 | NA |
| 58 | 4-methoxyphenyl | > 50 | NA | > 50 | NA |
| 59 | 2-chlorophenyl | 6.8 | 99 | 11.5 | 101 |
| 60 | 3-chlorophenyl | 9.5 | 97 | 14.8 | 100 |
| 61 | 4-chlorophenyl | 23.7 | 90 | 38.6 | 58 |
| 62 | 2-napthyl | 3.4 | 94 | 47.3 | 30 |
| 63 | 2-quinolinyl | 6.7 | 88 | 40.3 | 88 |
| 64 | 2-indolyl | 23.7 | 64 | > 50 | NA |
| 65 | 3-indolyl | > 50 | NA | > 50 | NA |
| 66 | 2-imidazo [1,2-a] pyridine | > 50 | NA | > 50 | NA |
| 67 | 2-benzothiophenyl | 0.9 | 93 | 25.0 | 49 |
| 68 | 2-benzothiazolyl | 0.3 | 101 | 7.9 | 85 |
| 69 | 2-benzoxazolyl | 6.9 | 83 | > 50 | 26 |
| 70 | 2-benzimidazolyl | 10.5 | 84 | > 40 | NA |
| 71 | 2-benzothiazolylmethyl | > 50 | NA | > 50 | NA |
| 72 | benzothiazole-2-carbonyl | 12.7 | 75 | > 50 | NA |
In order to further improve the potency of compound 68, we screened a limited set of substituents on the 2-benzothiazole moiety derived from commercially available substituted 2-chloromethylbenzothiazoles (compounds 73–76, Table 3). All chlorobenzothiazole analogs had equipotent or improved activity over compound 68 (Table 2) and the 4-Cl derivative was the most potent in both β-arrestin and cAMP assays (compound 73). Electron withdrawing groups were clearly preferred at the 4-position of the benzothiazole analogs in keeping with the observation for previous anilide series (compounds 73, 77–79, Table 3). Finally, 4-Br and 4-CF3 groups offered significant further improvements in potency especially in the cAMP assay for the 4-CF3 analog (compounds 80 and 81, Table 3). Compound 81 became our lead compound with pIC50 = 7.4 ± 0.1 (IC50 = 40 nM; n = 4) and 6.3 ± 0.2 (IC50 = 540 nM, n = 3) in antagonizing β-arrestin recruitment and forskolin-induced cAMP, respectively (Fig. 1A).
In vitro pharmacological screening showed that the lead compound 81 has good solubility and can achieve concentrations ≥10 × IC50 in aqueous buffer between a pH range of 5.0–7.4 (Supplementry Table 3). Compound 81 also exhibited good PAMPA permeability, high plasma protein binding, excellent stability in mouse and human plasma and had a > 40-fold window for toxicity in human liver microsomes (LC50 ≥ 25 μM). However, compound 81 was almost completely metabolized in human and mouse liver microsomes within 1h. Despite low in vitro microsomal stability, in vivo Rapid Assessment of Compound Exposure (RACE) assay with 10 mg/kg oral dose [DMSO: Tw80: H2O (1:1:8)] achieved good exposure and prompted us to carry out a full DMPK study in mice. Compound 81 had good bioavailability, clearance and exposure (clearance 28.4 mL/min/kg, Vdss 3.32 L/kg, t1/2 1.58 h after a 5 mg/kg intravenous dose, Cmax 798 ng/mL, t1/2 2.82 h, AUC 2047 ng·h/mL after a 10 mg/kg oral dose).
The murine CXCR6 receptor has 74% sequence homology with the human receptor. In addition, CXCL16 has only 46% species homology (https://blast.ncbi.nlm.nih.gov/Blast.cgi). ML339 probe had weak activity in antagonizing β-arrestin recruitment at the mouse CXCR6 receptor (IC50 = 18 μM)8 and compound 81 was devoid of activity against the mouse receptor (IC50 > 40 μM, data not shown) indicating a limitation in validating in vivo activity in animal studies despite the compound’s good bioavailability in mice. Therefore, we investigated human hepatoma cell lines to test functional activity of compound 81 in vitro and in vivo activity.
MicroRNA expression studies indicate CXCR6 expression and protein levels are about 2-fold greater in highly metastatic cells (MHCC97H, HCCLM3, and SK-HEP-1) compared with low metastatic Huh7, Hep3B cells and normal liver cell line L-02.6 These studies also indicated that CXCR6 knockdown substantially inhibited invasive activity of hepatoma cells in vitro but had no impact on cell proliferation and cell-cycle distribution. Intriguingly, CXCL16, in its soluble form or transmembrane full-length form, could prominently promote control SK-HEP-1 and HCCLM3 cell invasion but not CXCR6-knockdown cells. Likewise, downregulation of CXCL16 markedly inhibited invasive activity of hepatoma cells in vitro. These results indicate that the contribution of CXCR6 on HCC cell invasion may be due to CXCR6-CXCL16 signaling and an autocrine loop of CXCL16 production. Thus, we examined SK-Hep-1 invasion using a live cell transwell assay. Compound 73 (Table 3) inhibited invasion while the inactive analog, compound 6 (Table 1), had no effect and compound 81 (Table 3) dose-dependently inhibited migration (IC50 = 2.1 μM) at 72 h without loss in cell viability (Supplementary Fig. 1A–C). Although there was not a significant difference in rate of migration between exogenous CXCL16 and serum control, this provided preliminary data for proof of concept testing in a SK-HEP-1 human xenograft mouse model.
We next evaluated the antitumor potential of compound 81 and the mechanism of CXCR6 inhibition in HCC xenograft model in vivo (Fig. 1B–D). SK-HEP-1 cells were injected subcutaneously into the flank region of female NOD/SCID mice on Day 1 for tumor development followed by oral administration of compound 81 initiated on Day 4 to Day 30. Treatment with the CXCR6 inhibitor at 30 and 60 mg/kg significantly inhibited SK-HEP-1 tumor growth compared to vehicle-treated animals (P < 0.0005 and 0.0002, respectively); cyclophosphamide (50 mg/kg) was used as a positive control (Fig. 1B). No deaths or behavior phenotypes were observed in all mice. Mice weight among four groups differed indistinctly in SK-HEP-1 bearing mice (data not shown). We found that the weights of the excised SK-HEP-1 derived tumors treated with compound 81 at 30 and 60 mg/kg, respectively (59 ± 9 mg and 55 ± 24 mg) were significantly lighter than vehicle-treated tumors (151 ± 66 mg) (P < 0.05) and not significantly different in weight to cyclophosphamide-treated grafts (42 ± 24 mg) (Fig. 1C and D).
We report the discovery of a chemical series distinct from our previously reported probe molecule ML339 represented by compound 81 (Table 3). Compound 81 displays ≥ 10-fold improvement in potency in both the β-arrestin and cAMP assays (IC50 = 40 and 540 nM, respectively), inhibits migration of SK-HEP-1 hepatoma cells in a dose dependent manner and has promising oral DMPK data. Compound 81 is non-toxic to mice (30 and 60 mg/kg) and arrests tumor growth in SK-HEP-1 mouse xenografts to background levels similar to cyclophosphamide in a 30-day study and represents a promising pharmacological agent that may be used in more invasive mouse xenograft models to probe the CXCR6/CXCL16 axis in HCC.
Supplementary Material
Acknowledgements
We thank the cheminformatic and pharmacology cores at Sanford Burnham Prebys for providing compound management and ADME profiling support, respectively. We acknowledge Sang Bae at the University of Florida for providing scientific input and editing of the manuscript. The Molecular Structure Center at the Department of Chemistry, Indiana University, Bloomington graciously recorded and provided the X-ray structures. Finaly, we thank Altogen labs for helpful discussions in designing the xenograft study and WuXi AppTec (Shanghai) for pharmacokinetic data.
Funding
This work was funded by the Florida Translational Research Program (HTL0006), a contract administered by the Florida Department of Health. The funders provided support in the form of supplies and salaries for authors [S.P., P.M.H., R.J.K., E.S., P.R.M., E.H.S, D.D., C.J.M., D.T., L.H.S., S.M.], but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Earlier work focusing on the ML339 probe series was funded by the NIH Molecular Libraries Probe Production Centers network (MLPCN) grant # 1R03MH095589 (cycle 18)
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A.: Supplementary data
Supplementary data to this article can be found online at http://doi.org/10.1016/j.bmcl.2019.126899.
References
- 1.Tabata S, Kadowaki N, Kitawaki T, et al. Distribution and kinetics of SR-PSOX/CXCL16 and CXCR6 expression on human dendritic cell subsets and CD4+ T cells. J Leukoc Biol. 2005;77:777–786. [DOI] [PubMed] [Google Scholar]
- 2.Heydtmann M, Lalor PF, Eksteen JA, Hübscher SG, Briskin M, Adams DH. CXC chemokine ligand 16 promotes integrin-mediated adhesion of liver-infiltrating lymphocytes to cholangiocytes and hepatocytes within the inflamed human liver. J Immunol. 2005;174:1055–1062. [DOI] [PubMed] [Google Scholar]
- 3.Geissmann F, Cameron TO, Sidobre S, et al. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3:e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liang CM, Chen L, Hu H, et al. Chemokines and their receptors play important roles in the development of hepatocellular carcinoma. World J Hepatol. 2015;7:1390–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghouri YA, Mian I, Rowe JH. Review of hepatocellular carcinoma: epidemiology, etiology, and carcinogenesis. J Carcinog. 2017;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao Q, Zhao YJ, Wang XY, et al. CXCR6 upregulation contributes to a proinflammatory tumor microenvironment that drives metastasis and poor patient outcomes in hepatocellular carcinoma. Cancer Res. 2012;72:3546–3556. [DOI] [PubMed] [Google Scholar]
- 7.Sun JJ, Chen GY, Xie ZT. MicroRNA-361-5p Inhibits Cancer Cell Growth by Targeting CXCR6 in Hepatocellular Carcinoma. Cell Physiol Biochem. 2016;38:777–785. [DOI] [PubMed] [Google Scholar]
- 8.Hershberger PM, Peddibhotla S, Sugarman E, et al. Probing the CXCR6/CXCL16 axis: targeting prevention of prostate cancer metastasis. In: Health NIo, ed. Bethesda, MD: National Center for Biotechnology Information (US); 2010-. 2012:25. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.