

Abstract
Background:
Polychlorinated biphenyls (PCBs) are signaling disrupting chemicals that exacerbate nonalcoholic steatohepatitis (NASH) in mice. They are epidermal growth factor receptor (EGFR) inhibitors that enhance hepatic inflammation and fibrosis in mice.
Objectives:
This study tested the hypothesis that epidermal growth factor (EGF) administration can attenuate PCB-related NASH by increasing hepatic EGFR signaling in a mouse model.
Methods:
C57BL/6 male mice were fed a 42% milk fat diet and exposed to Aroclor 1260 () or vehicle for 12 wk. EGF () or vehicle were administered daily for 10 d starting at study week 10. Liver and metabolic phenotyping were performed. The EGF dose was selected based on results of an acute dose–finding study (30 min treatment of EGF at 0.2, 0.02, of via intraperitoneal injection). Hepatic phosphoproteomic analysis was performed using liver tissue from this acute study to understand EGFR’s role in liver physiology.
Results:
Markers of EGFR signaling were higher in EGF-treated mice. –exposed mice had lower hepatic free fatty acids, inflammation, and fibrosis relative to PCB-only exposed mice. EGF-treated mice had higher plasma lipids, with no improvement in hepatic steatosis, and an association with higher LXR target gene expression and de novo lipogenesis. EGF-treated mice showed more severe hyperglycemia associated with lower adiponectin levels and insulin sensitivity. EGF-treated mice had higher hepatic , NRF2, and AhR target gene expression but lower constitutive androstane receptor and farnesoid X receptor target gene expression. The hepatic EGF-sensitive phosphoproteome demonstrated a role for EGFR signaling in liver homeostasis.
Discussion:
These results validated EGFR inhibition as a causal mode of action for PCB-related hepatic inflammation and fibrosis in a mouse model of NASH. However, observed adverse effects may limit the clinical translation of EGF therapy. More data are required to better understand EGFR’s underinvestigated roles in liver and environmental health. https://doi.org/10.1289/EHP8222
Introduction
Nonalcoholic fatty liver disease (NAFLD) represents a pathological spectrum of progressive liver disease ranging from hepatic steatosis to steatohepatitis (NASH, characterized by increased liver injury and inflammation with or without fibrosis) to cirrhosis and hepatocellular carcinoma. NAFLD is the most prevalent liver disease, affecting 30%–40% of the U.S. population and 25% of people worldwide (Younossi et al. 2016). NASH, particularly with fibrosis, is associated with increased liver-related and all-cause mortality (Dulai et al. 2017). Although NAFLD is considered to be the hepatic manifestation of obesity and metabolic syndrome, it may also occur in lean individuals. Environmental chemical exposures may cause NAFLD or modify the severity of diet-induced NAFLD by affecting hepatic lipid metabolism, cell death, inflammation, fibrosis, and carcinogenesis (Wahlang et al. 2019b). At the present time, no medications have been approved by the U.S. Food and Drug Administration for the treatment of NASH. However, a robust therapeutic pipeline exists. Although many lead compounds target nuclear receptors, growth factor therapies are also being investigated (e.g., human growth hormone and analogs of fibroblast growth factors 19 and 21). There are no actively enrolling clinical trials for the treatment of NAFLD associated with specific environmental chemical exposures (www.clinicaltrials.gov). Polychlorinated biphenyls (PCBs) in mice disrupted normal hepatic epidermal growth factor receptor (EGFR) signaling (Hardesty et al. 2018) and were associated with NASH (Wahlang et al. 2019a). Our study investigated epidermal growth factor (EGF) therapy in an animal model of PCB-related NASH, and this paper elucidates the diverse roles of EGFR signaling in normal liver biology.
PCBs are persistent organic pollutants associated with NAFLD in mice (Wahlang et al. 2014b) as well as endocrine, metabolism (Heindel et al. 2017), and signaling disruption (Hardesty et al. 2018). Individual PCB congeners have been classified by their structure–activity relationships as either dioxin- or nondioxin-like, based on their ability to activate the aryl hydrocarbon receptor (AhR). Aroclor 1260 includes dioxin-like PCBs such as PCB-157 but primarily consists of nondioxin-like PCBs such as PCB-181 and PCB-154 (Wahlang et al. 2014a). Both PCB classes have been associated with fatty liver disease in cohort studies (Clair et al. 2018) and animal models (Jin et al. 2020; Wahlang et al. 2019a). In addition to AhR activation, several modes of action affecting hepatic lipid metabolism, cell death/proliferation, inflammation, and fibrosis have been identified for PCBs. These include endocrine (Heindel et al. 2017) signaling disruption (Hardesty et al. 2019a), altered nuclear receptor activities (Hardesty et al. 2019b), and increased oxidative stress (Wahlang et al. 2019a). Aroclor 1260 is an environmentally relevant PCB mixture that was previously commercially produced. It contains the types of higher molecular weight PCB congeners that have bioaccumulated in human adipose tissue (Jensen 1974; McFarland and Clarke 1989). PCBs are lipophilic, and hepatic steatosis markedly increased hepatic PCB levels in mice (Li et al. 2020). In mice, Aroclor 1260 () increased the severity of diet-induced NAFLD (e.g., NASH vs. isolated steatosis) (Wahlang et al. 2014b) and reduced hepatic protein phosphorylation by approximately 25%, consistent with profound phosphoprotein signaling disruption (Hardesty et al. 2019a).
This signaling disruption included the inhibition of EGFR activation in mice (Hardesty et al. 2017). EGFR is a transmembrane receptor tyrosine kinase activated by several ligands, most notably EGF. Despite the fact that most EGF traffics to liver (Jørgensen et al. 1988), the role of EGFR signaling in liver physiology and disease is underinvestigated. PCBs appeared to competitively antagonize the EGFR (Hardesty et al. 2017, 2018), leading to indirect activation of the constitutive androstane receptor (CAR), in a fashion similar to that of the structurally related molecule, phenobarbital (Mutoh et al. 2013). We postulate that the competitive inhibition of EGF signaling by PCBs could contribute to the NASH observed with PCB exposures in mice (Wahlang et al. 2014b). An intriguing possibility is that this potential mode of action could link the endocrine- and metabolism-disrupting effects of PCBs by affecting the downstream cross talk of key hepatic nuclear receptors like CAR and the pregnane xenobiotic receptor (PXR). Although the direct activation of CAR was protective against NASH in mice (Gao et al. 2009), CAR-null mice did not have worse steatohepatitis following PCB exposures (Wahlang et al. 2016). This finding suggests that the broader EGF-sensitive protein phosphorylation cascade upstream of CAR could be critical to PCBs’ mode of action. Limited data support the concept that the EGFR activation could attenuate steatohepatitis (reviewed in Komposch and Sibilia 2015). For example, EGF administration ameliorated ethanol-related steatohepatitis in an animal model (Deaciuc et al. 2002), but it has not been demonstrated for NAFLD. In contrast, EGFR inhibitors can lead to hepatotoxicity (liver enzyme elevation) (Ding et al. 2017; Qian et al. 2020) in cancer patients, similar to PCBs in exposed populations (Clair et al. 2018).
Although PCBs inhibited hepatic EGFR signaling, the potential causality of this mechanism in the NASH associated with PCB exposures has not been proven. The present study tests the hypothesis that EGF administration can attenuate PCB-related NASH by increasing hepatic EGFR signaling in a mouse model. We also aimed to establish whether EGFR activation by its natural ligand, EGF, alters hepatic physiology reversing dystrophy caused by PCBs. In addition, the EGF-sensitive hepatic phosphoproteome and kinome was also investigated. Through a precision medicine approach, this form of growth factor therapy could subsequently be evaluated in clinical trials of subjects with NASH and elevated PCB exposures should this therapy appear safe and effective in vivo.
Methods
Animal Studies
All mouse studies were approved by the University of Louisville Institutional Animal Care and Use Committee, and all animals were treated humanely as outlined in the “ARRIVE” guidelines (Kilkenny et al. 2010; Tilson and Schroeder 2013). Male C57BL/6 mice age 8–10 wk were purchased from The Jackson Laboratory. Mice purchased from The Jackson Laboratory were maintained as a colony with the following breeding scheme: C57Bl/6 males mated with female C57Bl/6 mice. Mice were housed in a temperature- and light-controlled room (12 h of light:12 h of dark) with food and water ad libitum. An acute dose finding study was conducted to identify the EGF dose that would elicit optimal hepatic EGFR phosphorylation at Y1173. Male C57BL/6 mice age 8–10 wk were fed a control diet (Teklad Custom Diet TD.06416, 10.2% kcal from fat) (Envigo) and treated with either saline or EGF (0.2, 0.02, or ) via intraperitoneal (IP) injection ( per treatment group). After 30 min, the mice were euthanized via an IP injection of ketamine () and xylazine (), and whole blood and liver tissue were collected. Whole blood was collected with anticoagulant (EDTA) coated syringes via blood draw from the posterior vena cava. Plasma was prepared (See “Methods: Plasma and Hepatic Lipid Profiling and Plasma Adipocytokine Determination”) for determination of pyruvate and glucose (all treatments). Hepatic proteins were extracted (See “Methods: Western Blot Analysis”) for Western blot analysis (See “Methods: Western Blot Analysis”) (all treatments) and phosphoproteomics (optimal EGF dose and vehicle control only). Phosphorylation of EGFR at Y1173 relative to total EGFR was used as a measure of maximal EGF-stimulated EGFR activity in murine liver at 3 doses of EGF (0.2, 0.02, or ) and saline control. Hepatic EGFR and ERK activity were maximum at of EGF (Figure S1A). Mice were euthanized at 30 min, because this time point previously demonstrated robust liver EGFR activity after EGF IP injection (Yang et al. 2017).
In the chronic animal study, male C57BL/6 mice ( per group, 8–10 wk old) received a single oral gavage of corn oil vehicle or Aroclor 1260 () after 1 wk of high-fat diet (HFD) feeding (42% kcal from fat; Envigo). Mice were fed the HFD ad libitum for the remaining 12 wk of the study. The objective was to determine whether administration of exogenous EGF as a therapy could restore EGFR signaling to attenuate PCB-mediated hepatotoxicity and NASH. At the start of week 10, mice were administered either EGF () via IP injection or saline once a day for 10 d. EGF purchased from EMD Millipore was reconstituted in sterile saline and administered fresh to experimental animals. The therapeutic EGF dose selected () was based on the Y1173-EGFR phosphorylation results from the acute mouse study above. Two days following the last treatment dose, all mice were euthanized via ketamine/xylazine administration. The following abbreviations are subsequently used to denote the various treatment groups used in the chronic exposure mouse study: CS, PCB vehicle control vehicle control (saline); CE, PCB vehicle control ; AS, vehicle control (saline); and AE, .
Dose and Animal Sex Justification
The use of Aroclor 1260 (PCB mixture) was designed to mimic PCB congener bioaccumulation patterns in humans (Jensen 1974; McFarland and Clarke 1989; Wahlang et al. 2014a). The dose of was used to mimic the highest measured lipid adjusted serum concentration of PCBs in humans from the PCB-exposed cohort (Anniston, Alabama), (Pavuk et al. 2014). This dose of Aroclor 1260 was also found to cause steatohepatitis in mice fed an HFD (Wahlang et al. 2014b). It is important to note that this dose was previously shown to inhibit signaling by endogenous EGF in mice (Hardesty et al. 2017; 2019a) and in murine and human cell lines (Hardesty et al. 2017). The PCB congener composition of Aroclor 1260 is presented in Table S1 PCB congener composition. Aroclor 1260 is relevant to human exposures because the PCB congener composition is most similar to the PCB congener profile observed in human fat as summarized previously (Wahlang et al. 2014b) from the original data source (Jensen 1974; McFarland and Clarke 1989). The dose of Aroclor 1260 was demonstrated previously to lead to PCB accumulation in the liver (Hardesty et al. 2019b) and promote steatohepatitis in mice (Wahlang et al. 2014b). Male mice were selected because PCB-exposed men in the Anniston Community Health Survey (ACHS) cohort had a higher prevalence of toxicant associated steatohepatitis (TASH) relative to women (Clair et al. 2018).
Insulin and Glucose Tolerance Test and Measurement of Plasma Pyruvate
In the chronic exposure study, an insulin tolerance test (ITT) was performed during week 11, and a glucose tolerance test (GTT) was performed during week 12. In both cases, serial blood glucose measurements were determined by glucometer (Accu-Check® Aviva) using blood collected from the tail vein. For the ITT and GTT, mice were manually restrained by hand followed by administration of a topical lidocaine and a one-time tail snip from the end of the tail. After the collection of blood, the tail was sanitized with 70% ethanol solution. ITT was performed in mice fasted for 6 h. Insulin ( IP) was administered; blood glucose levels were determined (0, 15, 30, 60, and 120 min postinjection); and the area under the curve (AUC) was calculated (Le Floch et al. 1990). The GTT was performed in a similar fashion but following an 18-h fast and glucose administration ( body weight). Plasma from the acute and chronic animal studies was analyzed to measure pyruvate spectrophotometrically with the BioTek Gen 5™ plate reader in a 96-well plate (NUNC; Thermo-Scientific) format. Absorbance at was measured at baseline and then incubated with of lactate dehydrogenase followed by a second measure at at 5 min to calculate the change in absorbance. Sample concentrations were derived from comparison to a standard curve of pyruvate purchased from Sigma-Aldrich (). Blood glucose was also measured following a 12-h fast prior to sacrifice.
Liver Histological Analysis
Liver tissue from the chronic animal study were fixed with 10% neutral buffered formalin for 7 d and embedded in paraffin. Five-micrometer liver sections were stained with hematoxylin-eosin (H&E) to observe structural changes and steatosis or for chloroacetate esterase (CAE) activity for neutrophil infiltration [Naphthol AS-D Chloroacetate (Specific Esterase) Kit; Sigma-Aldrich]. Hydrated liver sections were stained first in the Naphthol AS-D chloroacetate solution for 15 min at 37°C, rinsed with deionized water, counterstained in hematoxylin solution for 2 min, and finally rinsed with deionized water. Liver tissue sections were stained with picrosirius red to evaluate fibrosis and were imaged with a 20× magnification objective. Each image has a scale bar as a reference point. Next, liver sections (1 section per mouse) were evaluated for each histological stain (H&E, CAE, picrosirius red). All histological slides were examined by light microscopy at a 20× magnification objective and imaged with an Olympus BX43 microscope.
Histological evaluation of steatosis was graded based on the following scale: (), (5%–33%), (34%–66%), and () (Takahashi and Fukusato 2014). Percent area of picrosirius red staining in liver sections was calculated from images at 10× magnification (5 randomized images per mouse liver section) using Image J.
Body Weight and Tissue Weight Measurement
Mice body weights were recorded throughout the study and directly before euthanasia. Whole epididymal adipose tissue was removed at sacrifice and weighed as an index of metabolic syndrome. White adipose tissue weight was normalized to animal body weight.
Plasma and Hepatic Lipid Profiling and Plasma Adipocytokine Determination
After euthanasia and whole blood collection (with EDTA used as the anticoagulant), blood was kept in polypropylene tubes on ice until centrifugation at 4°C at for 15 min. Plasma was then aliquoted and stored at until future analysis. In the chronic animal study, plasma low density lipoprotein (LDL), high density lipoprotein (HDL), triglycerides, glucose, and total cholesterol were measured on the Piccolo® Xpress Chemical Analyzer using a Lipid Panel Plus reagent disc (Abaxis). Plasma free fatty acids (FFAs) were measured with the Free Fatty Acid Quantification Kit purchased from Sigma-Aldrich. Hepatic cholesterol, triglycerides, and FFAs were isolated from liver tissues homogenized in phosphate buffer saline (PBS) followed by lipid extraction using a 2:1 chloroform to methanol separation method (Folch et al. 1957). The lipid extract was dried and suspended in either Infinity Cholesterol Stable Reagent, Infinity Triglyceride (TG) Stable Reagent (Fisher Diagnostics), or FFA assay buffer (Sigma-Aldrich); and the absorbance (Cholesterol and TGs measured at , FFAs measured at ) was measured relative to lipid standard curves. Plasma adiponectin, resistin, tumor necrosis factor alpha (), and plasminogen activator inhibitor-1 (PAI-1) levels were determined using a Milliplex Plasma Adipokine kit on the Luminex® IS 100 instrument (Luminex Corp.).
Real Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Mouse livers from the chronic animal study were homogenized, and total RNA was extracted using RNA-STAT 60 (Tel-Test). RNA purity and quantity were determined with the Nanodrop (ND-1000, Thermo Scientific). cDNA was synthesized from total RNA using the QuantaBio qScript (QuantaBio). Real-time quantitative polymerase chain reaction (RT qPCR) was performed on the Bio-Rad CFX384 Real-Time System using Bio-Rad iTaq™ Universal Probes Supermix with selected TaqMan Gene Expression Assays (ThermoFisher). Bio-Rad Maestro™ software package was used to measure and analyze RT-qPCR data. The TaqMan probes used are listed in the Table S2 TaqMan probes. Technical replicates () were performed for each qPCR reaction. Relative gene expression levels were calculated with target gene normalization to glyceraldehyde 3-phosphate dehydrogenase (Gapdh) using the method (Livak and Schmittgen 2001).
Western Blot Analysis
Mouse liver lysates from the acute and chronic animal studies were homogenized in radioimmunoprecipitation assay (RIPA) buffer [ tissue/mL RIPA supplemented with protease and phosphatase inhibitors (); Sigma-Aldrich]. Silica beads were used to homogenize liver tissue in RIPA buffer via shaking for a 1-min burst on the Mini-Beadbeater instrument (EMD Millipore). Liver homogenates were then centrifuged at 4°C for 5 min at . The liver homogenate supernatants were then aliquoted and stored at until future analysis. Protein concentrations were determined by the bicinchoninic acid protein assay (Sigma-Aldrich). Proteins () were separated on a 7.5% SDS Gel (Bio-Rad), transferred to polyvinylidene difluoride membranes, and then blocked with 5% milk in TBS-T for 2 h at room temperature. Western blots were probed with primary antibodies in 5% BSA in TBS-T (1:1,000 dilution) overnight at 4°C. Western blots were then washed in TBS-T at room temperature followed by incubation with secondary antibodies in 5% milk TBS-T (1:2,000 dilution) at room temperature for 1 h. Western blots were washed in 5% TBS-T at room temperature. Prior to imaging, western blots were incubated in Clarity Max ECL reagent (BioRad) for 5 min. Densitometry bands were quantified using Bio-Rad Image Software. Primary and secondary antibodies are listed in Table S3 “Antibodies.” For each western, a representative figure of the quantitative data (from per group) is presented. For phosphoproteins measured by western blot analysis, antibodies to the phosphorylated protein were used first followed by stripping for 15 min at room temperature with Restore™ Western Blot Stripping Buffer (Thermo Scientific). Blots were then washed with TBS-T and blocked in 5% milk in TBS-T for 1 h at room temperature. Western blots were then probed with antibodies to total protein. Quantification of phosphoproteins are recorded as a ratio of phosphoprotein to total protein.
Liver Phosphoproteomic Analysis
Mouse liver tissues from the acute animal study (saline or EGF ) were homogenized in 1% SDS RIPA buffer. Silica beads were used to homogenize liver tissue in the 1% SDS RIPA buffer via shaking for a 1-min burst on the Mini-Beadbeater instrument (EMD Millipore). Liver homogenates were then centrifuged at 4°C for 5 min at . The liver homogenate supernatants were then aliquoted and stored at until future analysis. Protein concentrations were determined by bicinchoninic acid protein assay. Protein lysates () per sample were prepared and trypsinized using a filter-aided sample prep (FASP) protocol (Wiśniewski et al. 2009). Protein lysates were reduced, followed by the addition of urea and filter centrifugation. Peptides in the filter were incubated overnight at 37°C with Sequencing Grade Modified Trypsin (Promega). From the digested peptides, phosphopeptides were then further enriched with and purified using C18 columns (Pierce) (Murray et al. 2016). Phosphopeptide concentrations were measured by absorbance at via the NanoDrop™ 2000 (Thermo Scientific™) to determine the recovery amount to use for subsequent liquid chromatography and mass spectrometry analysis.
The columns used were an Acclaim™ PepMap™ 100 , nanoViper™ (C18, , ) trap, and an Acclaim™ PepMap™ RSLC , nanoViper™ (C18, , ) separating column (ThermoFisher Scientific). An EASY-nLC 1000 ultra-high performance liquid chromatography (UHPLC) system (ThermoFisher) was used with solvents v/v acetonitrile/0.1% v/v formic acid and v/v acetonitrile/0.1% v/v formic acid as mobile phases. Following injection of of sample onto the trap, separation was accomplished at with a 110-min linear gradient from 0% B to 50% B, followed by a 5 min linear gradient from 50% B to 95% B, and last, a 5-min wash with 95% B. A stainless steel emitter (ThermoFisher) was coupled to the outlet of the separating column. A Nanospray Flex™ source (ThermoFisher) was used to position the end of the emitter near the ion transfer capillary of the mass spectrometer. The ion transfer capillary temperature of the mass spectrometer was set at 225°C, and the spray voltage was set at 1.6 kV.
An Orbitrap Elite – ETD mass spectrometer (ThermoFisher) was used to collect data from the LC eluate. An Nth Order Double Play was created in Xcalibur (version 2.2; Thermo Scientific). Scan event 1 obtained an FTMS MS1 scan (normal mass range; 240,000 resolution, full scan type, positive polarity, profile data type) for the range . Scan event 2 obtained ITMS MS2 scans (normal mass range, rapid scan rate, centroid data type) on up to 20 peaks that had a minimum signal threshold of 5,000 counts from scan event 1. The lock mass option was enabled (0% lock mass abundance) using the polysiloxane peak as an internal calibrant.
The raw data files were analyzed separately with Peaks Studio 7.5 using the UniprotKB mouse reviewed canonical and isoform protein sequences current as of 21 March 2018 and the Denovo, PeaksDB, and PeaksPTM algorithms. Identifications at the 1% FDR threshold from the PeaksDB and PeaksPTM results were loaded into the Peaks Label Free Quantification algorithm.
Data and Statistical Analysis
Western blot densitometry values, RNA expression values, and macromolecule levels were statistically analyzed and graphed using GraphPad Prism version 8 for Macintosh. Outliers were determined via the ROUT test with a . The number of outliers for each measure and each experimental group are included in Table S4 “Outliers.” The data are expressed as box and whisker plots highlighting the median (midline), upper and lower quartiles (box), and upper and lower limits (whiskers). Data were compared using two-way analysis of variance (ANOVA) and was considered statistically significant. Intergroup comparisons were made using a Sidak correction (parametric for Gaussian distributed data, nonparametric for non-Gaussian distributed data); was considered statistically significant. -Values for these comparisons are given in Table S5 “-Values,” and numerical data () for each measure are included in Table S6 “Numerical Data.” Quantified phosphopeptides were statistically compared by unpaired two-tailed -test, with considered statistically significant.
The hepatic phosphopeptides whose abundances were significantly changed by EGF were analyzed in CytoScape to identify hierarchical clusters of phosphorylated proteins regulated by EGFR signaling in the liver. Gene ontology (GO) processes represented by the EGF-sensitive phosphoproteome were identified in CytoScape (Lopes et al. 2010). Kinome analysis was conducted using PhosphoScan Site 4.0 software provided by the Massachusetts Institute of Technology. PhosphoScan Site 4.0 was used to designate kinases that regulated significantly altered phosphopeptides due to EGF treatment.
Reagents
EGF was purchased from EMD Millipore. Aroclor 1260 was purchased from Accustandard. Glucose, pyruvate, LDH, SDS, and urea were purchased from Sigma-Aldrich. H&E, CAE, and picrosirius red staining reagents were purchased from Sigma-Aldrich. Lipid Panel Plus reagent discs were purchased from Abaxis. Triglyceride (TG) and cholesterol Infinity™ Liquid Reagents were purchased from Thermo Scientific. FFA detection kits were purchased from Sigma-Aldrich. The Plasma Cytokine kit was purchased from EMD Millipore. RNA-STAT 60 was purchased from Tel-Test. TaqMan probes were purchased from ThermoFisher. QuantaBio qScript cDNA synthesis reagent was purchased from QuantaBio. BioRad mastermix and Clarity ECL Max reagents were purchased from BioRad. Protease and phosphatase inhibitors and BCA reagent were purchased from Sigma-Aldrich. Antibodies were purchased from Cell Signaling Technology, Abcam, and Santa Cruz Biotechnology. Restore™ Western Blot Stripping Buffer was purchased from Thermo Scientific. Sequencing grade Trypsin was purchased from Promega.
Results
Measures of Hepatic Inflammation and Fibrosis in Mice Fed an Treatment
For all figures (Figures 1–6, Figures S1–S4), significant results are presented with the p-value from the two-way ANOVA and Sidak multiple comparison tests ( effect, effect, effect from two-way ANOVA, and a * denotes significance for individual comparisons by the Sidak multiple comparison test). The numerical data and p-values for every statistical comparison can be found in Tables S5–S6 “-Values” and “Numerical Data,” respectively. All -values reported in the results section of the main text are from the Sidak multiple comparison test. Throughout the results, CS is used to denote control saline mice, CE refers to control EGF mice, AS refers to Aroclor 1260 saline mice, and AE denotes Aroclor 1260 EGF mice. The acute in vivo EGF dose optimization study performed here determined that hepatic EGFR and ERK phosphorylation were maximal after 30 min in mice that received the dose of EGF (Figure S1A). This dose was investigated in the subsequent chronic Aroclor 1260 exposure study (Figure S1B). In that study, EGF-treated mice exposed to Aroclor 1260 had higher hepatic Y1173-EGFR phosphorylation relative to Aroclor 1260 exposed alone mice (Figure S1C).
Figure 1.
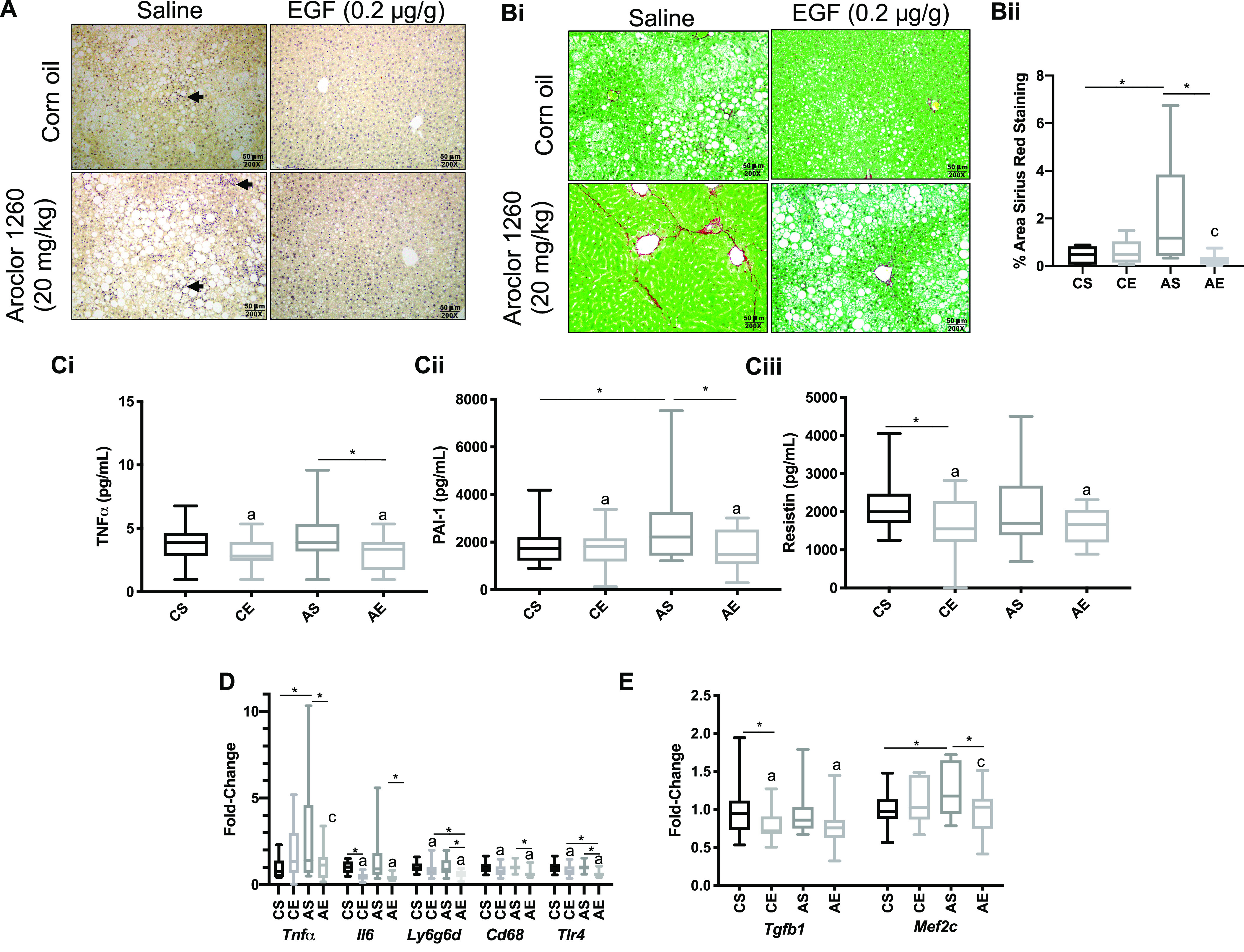
Measures of hepatic inflammation and fibrosis in mice fed an treatment: Male WT C57Bl/6 mice were fed an HFD (12 wk) and exposed to either vehicle or Aroclor 1260 () by a one-time oral gavage at week 1. Aroclor 1260-exposed or vehicle mice were treated (via IP injection) with saline or EGF () daily for 10 d starting at week 10. Mice were fasted for 12 h and euthanized, and tissues were harvested for downstream analyses. (A) Representative image of CAE stained liver section ( per group, 1 liver section per mouse evaluated) (20× magnification objective) (arrows denote necrotic foci). (Bi) Representative image of picrosirius red stained (10× magnification objective) liver sections and (Bii) quantification. One liver section per mouse was evaluated ( per experimental group). (C) Plasma measures by Luminex® of , PAI-1, and resistin. (D) Hepatic qPCR analysis of inflammatory gene marker expression (, Il-6, Ly6g6d, Cd68, Tlr4). (E) Hepatic qPCR analysis of profibrotic genes and Mef2c, and an was used for Figures 1B–F. A two-way ANOVA was used to statistically compare data sets followed by a Sidak correction for intergroup comparisons. A was considered significant. Significance due to EGF is denoted by (a), due to Aroclor 1260 denoted by (b), and due to interaction denoted by (c) for the two-way ANOVA. A was denoted by * for the Sidak multiple comparison test. Data are presented as box and whisker plots for Figure 1 that illustrate the median (midline), upper and lower quartiles (box), and the upper and lower limits (whiskers). All numerical data are presented in Table S6 “Numerical Data” as , and p-values can be found in Table S5 “-Values.” Numeric outliers were identified by ROUT method and removed. The number of outliers are reported in Table S4 “Outliers.” Note: AE, ; ANOVA, analysis of variance; AS, ; CAE, chloroacetate esterase; Cd68, cluster of differentiation 68; CE, ; CS, ; EGF, epidermal growth factor; HFD, high-fat diet; Il-6, interleukin-6; IP, intraperitoneal; Ly6g6d, lymphocyte antigen 6 family member G6D; Mef2c, myocyte enhancer factor 2C; qPCR, quantitative real-time polymerase chain reaction; Tgfb, Transforming growth factor-beta; Tlr4, toll-like receptor 4; , tumor necrosis factor-alpha.
Figure 6.
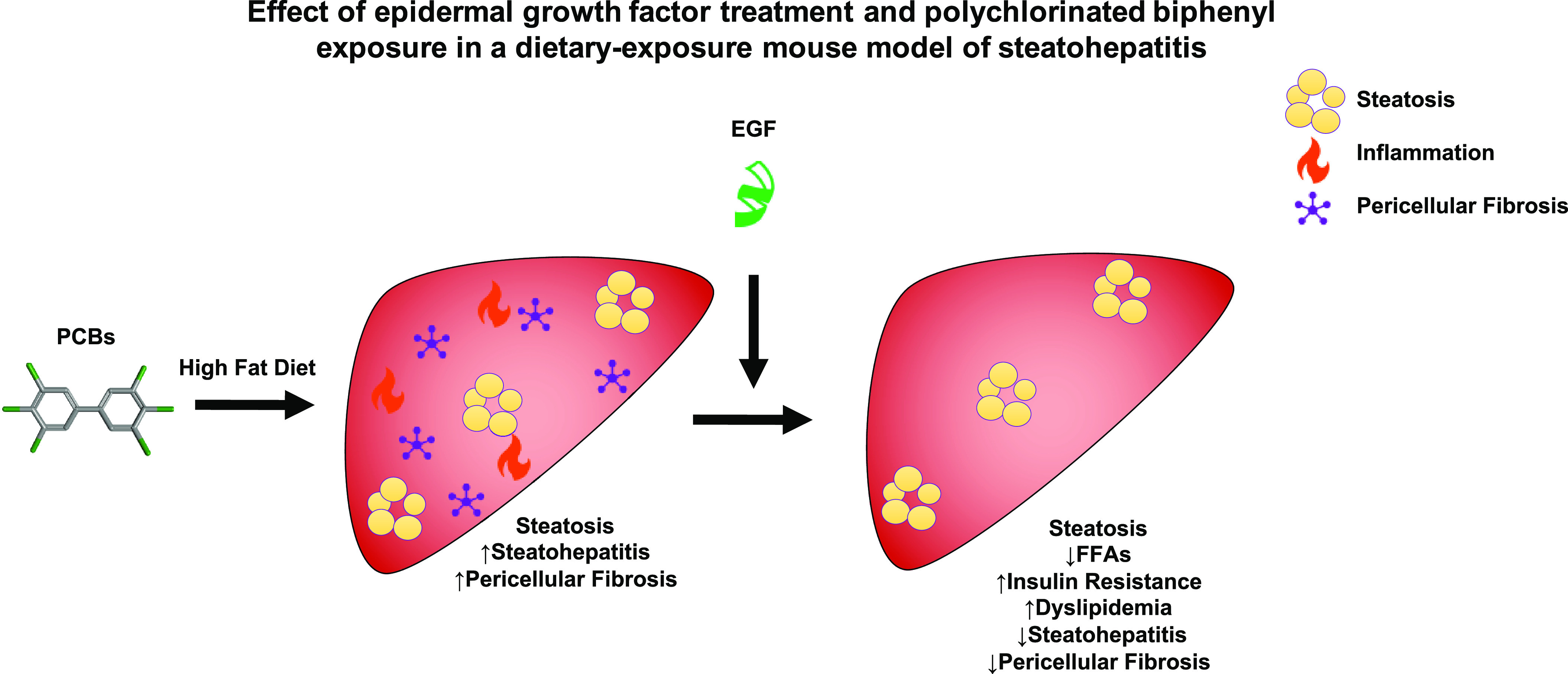
Effect of EGF treatment and PCB exposure in a dietary-exposure mouse model of steatohepatitis: graphical abstract illustrating EGF therapy reducing hepatic inflammation and pericellular fibrosis induced by Aroclor 1260 but only modifying metabolic effects on lipid homeostasis in mice fed an HFD. Note: EGF, epidermal growth factor; HFD, high-fat diet; PCB, polychlorinated biphenyl.
The effects of EGF therapy on hepatic inflammation and fibrosis were then evaluated by histology, liver gene expression, and plasma cytokine levels. EGF-treated mice exhibited qualitatively less histologic inflammation in Aroclor 1260-exposed mice by CAE staining (Figure 1A). EGF-treated mice had less pericellular fibrosis as determined by quantitative picrosirius red staining of liver sections (8.1-fold reduction (AE vs. AS, ) (Figure 1Bi-ii). Plasma proinflammatory cytokines including: (), PAI-1 (), and resistin () were lower in AE vs. AS mice (Figure 1Ci-iii). Hepatic (), Il-6 (), Ly6gd (), Cd68 (), and Tlr4 () gene expression were significantly lower in AE vs. AS mice as well (Figure 1D). AE vs. AS mice had lower hepatic expression of the profibrotic markers, transforming growth factor () () and myocyte enhancer factor 2C (Mef2c) (, Figure 1E).
The Effects of Aroclor 1260 Exposure and EGF Treatment on Lipids in Liver, Blood, and Adipose Tissue in Mice Fed an HFD
The effects of PCB exposures and EGF administration on lipids were characterized in liver, blood, and adipose. In liver, histologic steatosis grade did not differ by treatment group (Figure 2A). Although hepatic triglycerides varied significantly by treatment group (Figure 2Bii), the magnitudes of these changes were small and overall consistent with the liver histology. AE vs. AS mice had lower liver FFA levels (), but higher cholesterol levels (), and no difference in triglycerides (TGs) () (Figure 2Bi and iii).
Figure 2.
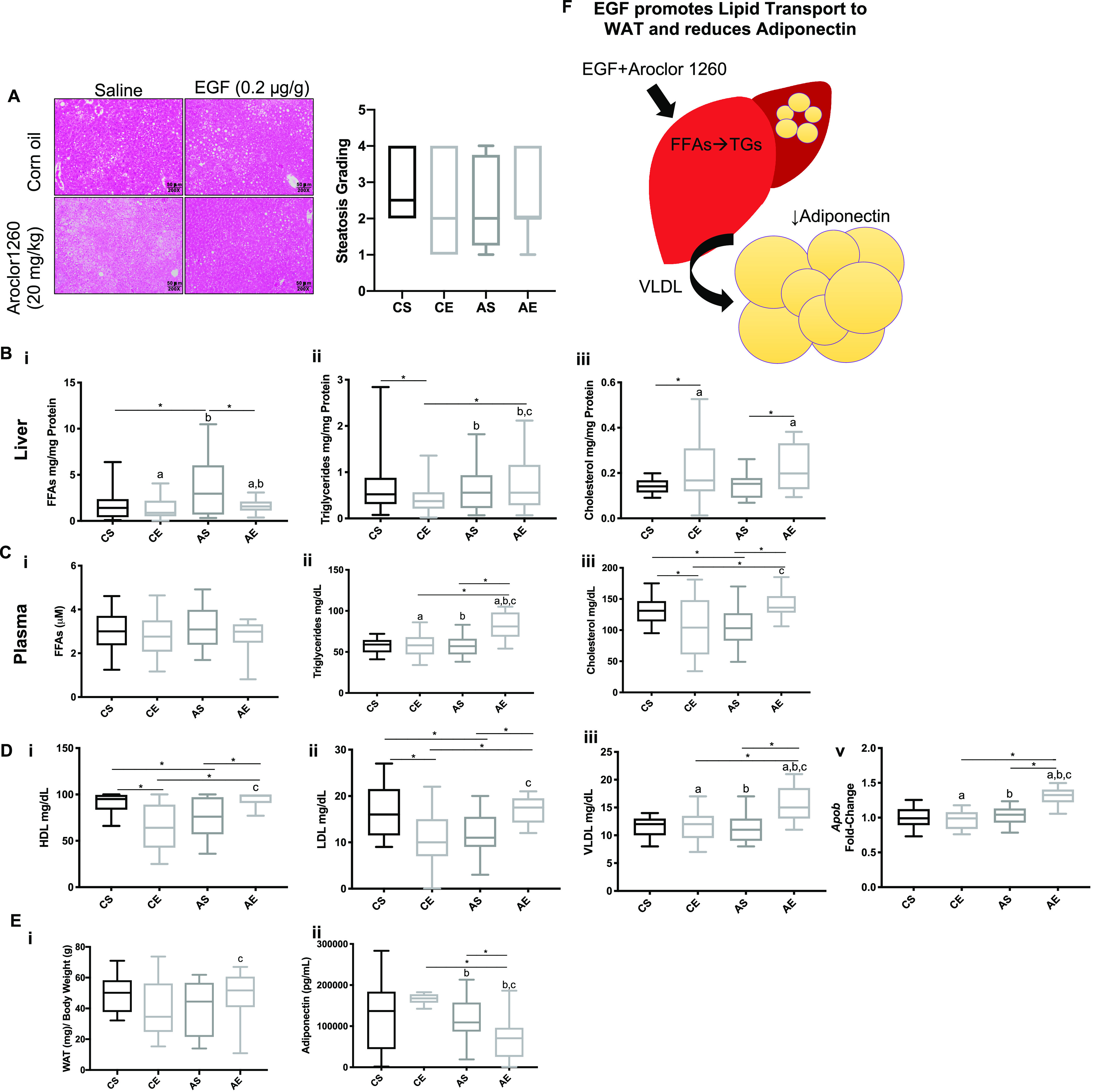
The effects of Aroclor 1260 exposure and EGF treatment on lipids in liver, blood, and adipose tissue in mice fed HFD. Male WT C57Bl/6 mice were fed an HFD (12 wk) and exposed to either vehicle or Aroclor 1260 () by a one-time oral gavage at week 1. Aroclor 1260-exposed or vehicle mice were treated (via IP injection) with saline or EGF () daily for 10 d starting at week 10. Mice were fasted for 12 h and euthanized, and tissues were harvested for downstream analyses. (A) Representative image of H&E staining of liver sections in mice exposed to corn oil and injected with either saline (CS) or EGF () (CE) or gavaged with Aroclor 1260 () and injected with saline (AS) or EGF (AE). One liver section per mouse was evaluated ( per experimental group). Also included is the quantification from the steatosis grading. Steatosis grading was as follows: (), (5%–33%), (34%–66%), and (). (B) Hepatic measures of (Bi) FFAs, (Bii) TGs, and (Biii) cholesterol. (C) Plasma measures of (Ci) FFAs, (Cii) TGs, and (Ciii) cholesterol. (D) Plasma measures of (Di) HDL, (Dii) LDL, and (Diii) VLDL. (Dv) qPCR analysis of hepatic ApoB gene expression in CS, CE, AS, and AE mice. (E) Weight of epididymal WAT normalized to total body weight (Ei). Plasma measures of adiponectin (Ei) in CS, CE, AS, and AE mice. (F) Illustration demonstrating Aroclor 1260-exposed mice treated with EGF have elevated hepatic TGs and plasma VLDL. An was used for Figures 2A-E. A two-way ANOVA was used to statistically compare data sets followed a Sidak correction for intergroup comparisons. A was considered significant. Significance due to EGF is denoted by (a); due to Aroclor 1260 denoted by (b); and due to interaction denoted by (c) for the two-way ANOVA. A was denoted by * for the Sidak multiple comparison test. Data are presented as box and whisker plots for Figure 2 that illustrate the median (midline), upper and lower quartiles (box), and the upper and lower limits (whiskers). All numerical data are presented in Table S6 “Numerical Data” as and p-values can be found in Table S5. Numeric outliers were identified by ROUT method and removed. The number of outliers are reported in Table S4. Note: AE, ; ANOVA, analysis of variance; AS, ; CE, ; CS, ; EGF, epidermal growth factor; FFAs, free fatty acids; HDL, high-density lipoprotein; H&E, hematoxylin & eosin; HFD, high-fat diet; IP, intraperitoneal; LDL, low-density lipoprotein; TGs, triglycerides; VLDL, very low-density lipoprotein; WAT, white adipose tissue.
In plasma, AE vs. AS mice had no difference in FFAs () but higher TGs (), and higher cholesterol levels () (Figure 2Ci–iii). Similarly, AE vs. AS mice had higher HDL (), LDL (), VLDL (), and hepatic apolipoprotein B mRNA (Apob) levels () (Figure 2Di–v).
The epididymal fat pad to body weight ratio was measured to determine the effects of EGF and PCBs on adiposity (Figure 2Ei). This ratio was higher in AE vs. AS mice (), though not significant. Consistent with this observation, AE mice had lower plasma adiponectin levels relative to AS mice () (Figure 2Eii). Body weight gain throughout the study was similar for all groups of mice (Figure S2A). Effects of EGF and Aroclor 1260 on hepatic FFAs and TGs, plasma VLDL, and adiponectin are summarized in Figure 2F.
Effects on Hepatic Gluconeogenesis, Insulin Sensitivity, and Glucose Tolerance in Mice Treated Acutely with EGF Alone or Chronically in HFD-Fed Mice Exposed to Treatment
In the acute EGF administration study presented here, EGF-treated mice had higher fasting blood glucose levels, at 30 min postinjection for all EGF doses tested relative to saline controls (Figure 3Ai). Likewise, EGF-treated mice had lower pyruvate levels by approximately 2.8-fold vs. saline controls after 30 min (Figure 3Aii). Based on these data, acute EGF treatment appeared to enhance hepatic gluconeogenesis leading to fasting hyperglycemia. In the chronic study, EGF-treated mice also had higher fasting blood glucose levels after 6 (Figure S2B), 18 (Figure S2C), and 12-h fasts (Figure 3Bi). AE mice had significantly higher fasting blood glucose levels relative to AS mice (1.4-fold, , AE vs. AS). In contrast to the acute study, this hyperglycemia did not appear to be due to enhanced hepatic gluconeogenesis based on plasma pyruvate levels (Figure 3Bii). AS mice had higher plasma pyruvate levels (AS vs. CS, ) consistent with decreased hepatic gluconeogenesis. Mice treated with EGF or PCBs had no differences in glucose uptake as determined by GTT (Figure 3Ci-ii). Mice treated with EGF in the chronic study had less insulin sensitivity as determined by ITT (Figure 3Di–ii). AE mice had higher glucose AUC by 1.3-fold in the ITT (AE vs. AS, ).
Figure 3.

Effects on hepatic gluconeogenesis, insulin sensitivity, and glucose tolerance in mice treated acutely with EGF alone or chronically in HFD-fed mice exposed to treatment. Male WT C57Bl/6 mice fed a control diet were treated with either saline or EGF at multiple doses (0.002, 0.02, ), followed by euthanasia at 30 min post IP injection, and tissue was harvested for downstream analysis. Male WT C57Bl/6 mice were fed an HFD (12 wk) and exposed to either vehicle or Aroclor 1260 () by a one-time oral gavage at week 1. Aroclor 1260-exposed or vehicle mice were treated (via IP injection) with saline or EGF () daily for 10 d starting at week 10. Mice were fasted for 12 h and euthanized, and tissues were harvested for downstream analyses. (A) Plasma glucose (Ai) and plasma pyruvate (Aii) were measured in mice treated with EGF (0.002, 0.02, ) or saline control for 30 min (acute). (B) Plasma glucose (Bi) and plasma pyruvate (Bii) were measured in mice gavaged with corn oil and injected with either saline (CS) or EGF () (CE) or gavaged with Aroclor 1260 () and injected with saline (AS) or EGF (AE) at the conclusion of the chronic model (12 wk). (Ci-ii) GTTs were performed in the CS, CE, AS, and AE mice as well as (Di-ii) ITTs. An was used for Figures 3Ai–ii, and the red dot is the for saline controls for Figures 3Ai–ii. An was used for Figures 3B–D. A two-way ANOVA was used to statistically compare data sets, followed by a Sidak correction for intergroup comparisons. A was considered significant. Significance due to EGF is denoted by (a); Aroclor 1260 denoted by (b); and due to interaction denoted by (c) for the two-way ANOVA. A was denoted by * for the Sidak multiple comparison test. Data are represented as for Figures 3Ai, 3Aii, 3Ci, and 3Di, but data are presented as box and whisker plots for Figures 3Bii, 3Cii, and 3Dii that illustrate the median (midline), upper and lower quartiles (box), and the upper and lower limits (whiskers). All numerical data are presented in Table S6 “Numerical Data” as , and p-values can be found in Table S5. Numeric outliers were identified by ROUT method and removed. The number of outliers are reported in Table S4. Note: AE, ; ANOVA, analysis of variance; AS, ; CE, ; CS, ; EGF, epidermal growth factor; GTT, glucose tolerance test; HFD, high-fat diet; IP, intraperitoneal; ITT, insulin tolerance test.
Expression of Select Hepatic Nuclear Receptors, AhR, and NRF2 and Their Respective Target Genes in HFD-Fed Mice Exposed to Vehicle or Treatment
Hepatic CAR protein levels were not affected in either EGF-treated or Aroclor 1260-exposed mice (Figure 4Ai). Hepatic Car mRNA expression was higher in both AS vs. CS mice () and AE vs. CE mice () (Figure 4Aii). Expression of the CAR target gene, cytochrome P450 2b10 (Cyp2b10) and CAR/PXR target gene Cyp3a11, were higher in AS vs. CS mice ( and , respectively). Cyp2b10 expression was significantly lower in AE vs. AS mice (). Likewise, there was a trend toward lower Cyp3a11 expression in AE vs. AS mice ().
Figure 4.
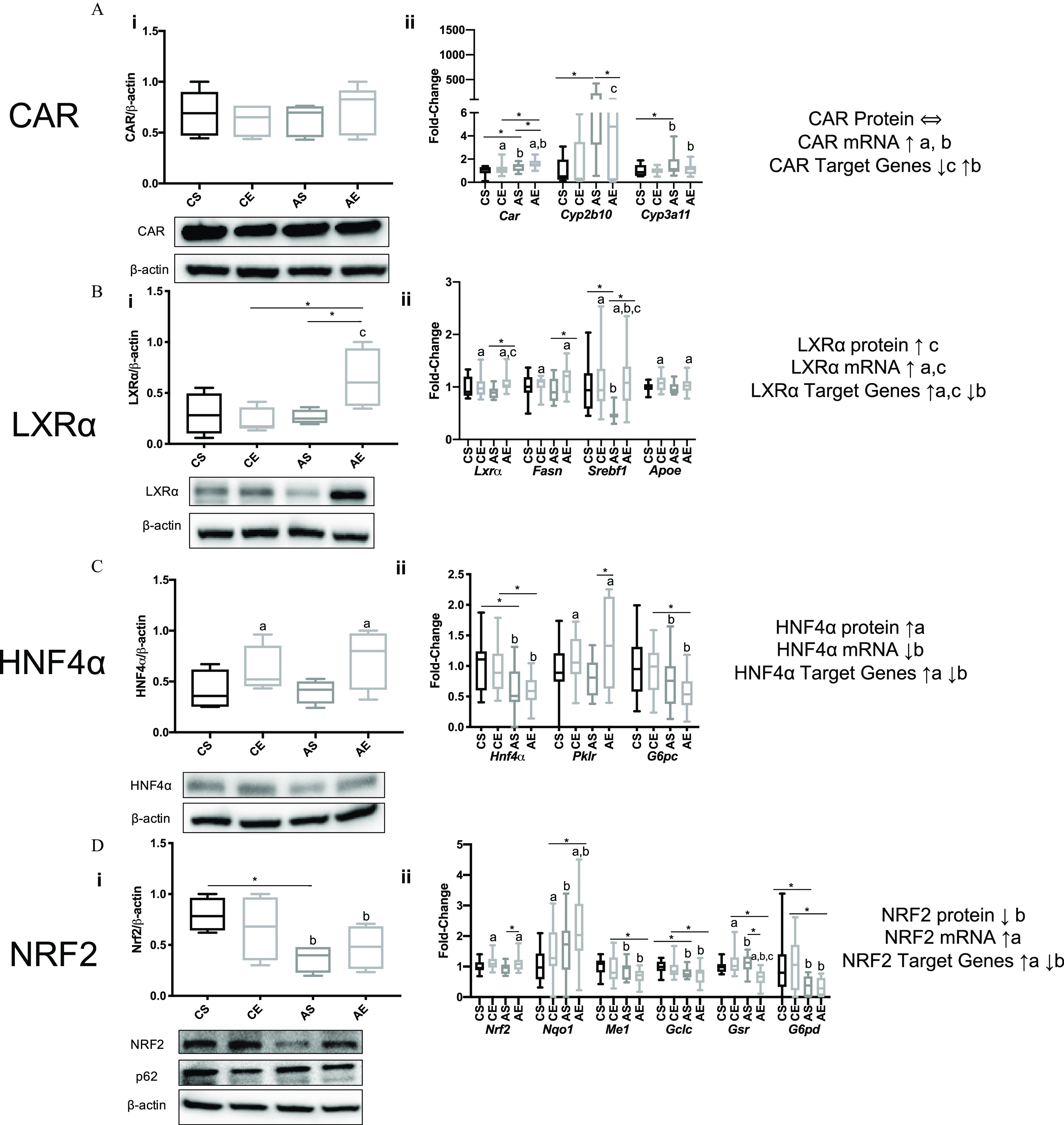
Expression of select hepatic nuclear receptors, AhR, and NRF2 and their respective target genes in HFD-fed mice exposed to vehicle or treatment. Male WT C57Bl/6 mice were fed an HFD (12 wk) Aroclor 1260 () by a one-time oral gavage at week 1. Aroclor 1260-exposed or vehicle mice were treated (via IP injection) with saline or EGF () daily for 10 d starting at week 10. Mice were fasted for 12 h and euthanized, and tissues were harvested for downstream analyses. (Ai–Di) Western blot analysis measuring hepatic (Ai) CAR, (Bi) , (Ci) , (Di) NRF2, and p62 protein expression in mice gavaged with corn oil and injected with either saline (CS) or EGF () (CE) or gavaged with Aroclor 1260 () and injected with saline (AS) or EGF (AE). (Aii–Dii) qPCR analysis measuring hepatic mRNA for (Aii) CAR and CAR/PXR target genes Cyp2b10/Cyp3a11, (Bii) and target genes Fasn, Srebf1, and Apoe, (Cii) and target genes Pklr, and G6pc (Dii) Nrf2 and target genes Nqo1, Me1, Gclc, Gsr, and G6pdh. An was used for the western blot analysis data, and an was used for all the qPCR data. A two-way ANOVA was used to statistically compare data sets followed by a Sidak correction for intergroup comparisons. A was considered significant. A is denoted by * for the Sidak multiple comparison test. Significance due to EGF is denoted by (a), due to Aroclor 1260 denoted by (b), and due to interaction denoted by (c) for the two-way ANOVA. Data are presented as box and whisker plots for Figure 4 that illustrate the median (midline), upper and lower quartiles (box), and the upper and lower limits (whiskers). All numerical data are presented in Table S6 “Numerical Data” as and p-values can be found in Table S5. Numeric outliers were identified by ROUT method and removed. The number of outliers are reported in Table S4. Note: AE, ; ANOVA, analysis of variance; Apoe, apolipoprotein E; AS, ; CAR, constitutive androstane receptor; CE, ; CS, ; Cyp2b10, cytochrome P450 2B10; Cyp3a11, cytochrome P450 3A11; EGF, epidermal growth factor; Fasn, fatty acid synthase; HFD, high-fat diet; , hepatocyte nuclear receptor 4-alpha; Gclc, glutamate-cysteine ligase catalytic subunit; Gsr, glutathione-disulfide reductase; G6pc, glucose-6-phosphatase; G6pdh, glucose-6-phosphate dehydrogenase; IP, intraperitoneal; , liver X receptor-alpha; NRF2, nuclear factor-erythroid 2-related factor 2; Me1, malic enzyme 1; Nqo1, NAD(P)H quinone dehydrogenase 1; Pklr, pyruvate kinase, liver type; PXR, pregnane xenobiotic receptor; p62, sequestosome 1; Srebf1, sterol regulatory element binding transcription factor 1.
Hepatic liver X receptor () regulates lipid synthesis and lipid metabolism, and it was affected by Aroclor 1260 and EGF treatment. protein and message were significantly higher in AE vs. AS mice () (Figure 4Bi–ii). target gene expression [e.g., fatty acid synthase (Fasn), sterol regulatory element binding transcription factor 1 (Srebf1) and Apolipoprotein E (Apoe)] (Figure 4Bii) Fasn () and Srebf1 () mRNA were significantly higher in AE vs. AS mice (Figure 4Bii). AE vs. AS mice had higher LXR target gene expression.
Hepatocyte nuclear factor () and nuclear factor erythroid 2-related factor 2 (NRF2) regulate hepatic metabolism (Watt et al. 2003) and the antioxidant response (Shin et al. 2013), respectively. These EGF-sensitive transcription factors were previously implicated in NASH due to Aroclor 1260 exposure in mice (Hardesty et al. 2019b). protein expression was higher in AE vs. AS mice (; Figure 4Ci), whereas gene expression was lower in AS vs. CS mice (; Figure 4Cii). Expression of the target gene, pyruvate kinase (Pklr), was higher in AE vs. AS mice (), consistent with higher target gene expression, whereas glucose 6 phosphatase (G6pc) was lower in AE vs. CE mice (). Pklr was higher in AE vs. AS mice (). AS vs. CS mice had lower NRF2 protein levels (2.2-fold) (; Figure 4Di) but higher expression of negative regulator p62 (; Figure S3C), whereas AE vs. AS mice had higher Nrf2 expression (; Figure 4Dii). AE vs. CE mice consistently had lower expression of NRF2 target genes including: malic enzyme 1 (Me1, ), glutamate-cysteine ligase catalytic subunit (Gclc, ), glutathione reductase (Gsr, ), and glucose-6-phosphate dehydrogenase (G6pd, ) (Figure 4Dii). With the exception of NADH quinone dehydrogenase 1 (Nqo1), these NRF2 target genes did not differ in AE vs. CE mice.
In a similar fashion, the farnesoid X receptor (FXR) and the AhR were evaluated, AS vs. CS mice had higher Fxr expression and protein levels (Figure S3A). AS vs. CS mice had higher expression of cytochrome P450 family 7A1 (Cyp7a1), which catalyzes the rate limiting step of bile acid synthesis (). AhR target gene expression was higher in AE vs. AS mice as demonstrated by significantly higher Cyp1a1 gene expression () (Figure S3B). EGF treated mice had lower induction of Aroclor 1260-induced hepatic CAR target gene expression and modulated expression of other key transcription factor target genes implicated in PCB-related NASH.
Characterization of the EGF-Sensitive Hepatic Phosphoproteome and Kinome
To the best of our knowledge the in vivo characterization of the EGF-sensitive hepatic phosphoproteome has not been previously been performed. Therefore, to better understand the biology of EGFR signaling in liver, mice were injected with EGF () or saline followed by liver extraction within 30 min and hepatic phosphoproteomic analysis. A total of 723 phosphopeptides were identified, and 263 of those p-peptides were downregulated in EGF treated mice, whereas 159 were significantly enriched ( (Figure 5A). Phosphoprotein abundance and the predicted kinases regulating those phosphosites were used as indirect measures of hepatic kinome activity after EGF administration relative to saline control (Figure 5B). The kinases most robustly regulated by EGF therapy are located to the far right of the graph (or larger node size), with some being positively (red node) and others negatively regulated (blue node) (Figure 5B). Extracellular regulated kinase (ERK), cyclin-dependent kinases (CDKs), and glycogen synthase kinase alpha () had higher activity in EGF-treated mice relative to saline controls. For example, CDK2/12 (227,201-fold/163,201-fold) and (31-fold) were hyperphosphorylated in EGF-treated mice relative to saline controls. In contrast, AMP-activated protein kinase (AMPK) and cAMP-dependent kinase (PKA) had lower activity. Activating phosphorylation sites of PKA (3,298-fold) and AMPK (108,858-fold) were lower in EGF treated mice relative to saline controls. Cluster analysis of the hepatic EGF-sensitive phosphoproteome demonstrated clusters of proteins by functional interactions and homology (Figure 5C). Cluster analysis identified six major protein clusters regulated by EGF in the liver including gene expression, vesicle mediated transport, endocytosis, protein ubiquitination, carbohydrate metabolism, fatty acid metabolism, and long chain fatty acid metabolism (Table 1). Many enzymes involved in glucose metabolism, lipid metabolism, and inflammation were regulated by EGF in the liver (Tables 2–4). It is interesting to note that the pyruvate transporter solute carrier family 16 member 1 (SLC16A1) was phospho-regulated by EGF, as well as glucose transporter 2 (GLUT2). The LDL receptor (LDLR) and acetyl-coA carboxylase (ACC), the rate-limiting enzyme in fatty acid synthesis, were also phospho-regulated by EGF. Receptor interacting serine/threonine kinase 1 (RIPK1), the effector kinase in cytokine mediated cell death, and stabilin-2 (STAB2), the phosphatidylserine receptor, are inflammatory mediators regulated by EGFR signaling. A surprising finding was that myocyte enhancement factor 2C (MEF2C), the hepatic stellate cell (HSC) identity factor, was also regulated by EGFR signaling. Some EGF phosphorylation targets detected were measured in murine liver from the chronic study, including S455-ACLY, S96-KAP2, and Y15-CDK2 (Figure S4).
Figure 5.
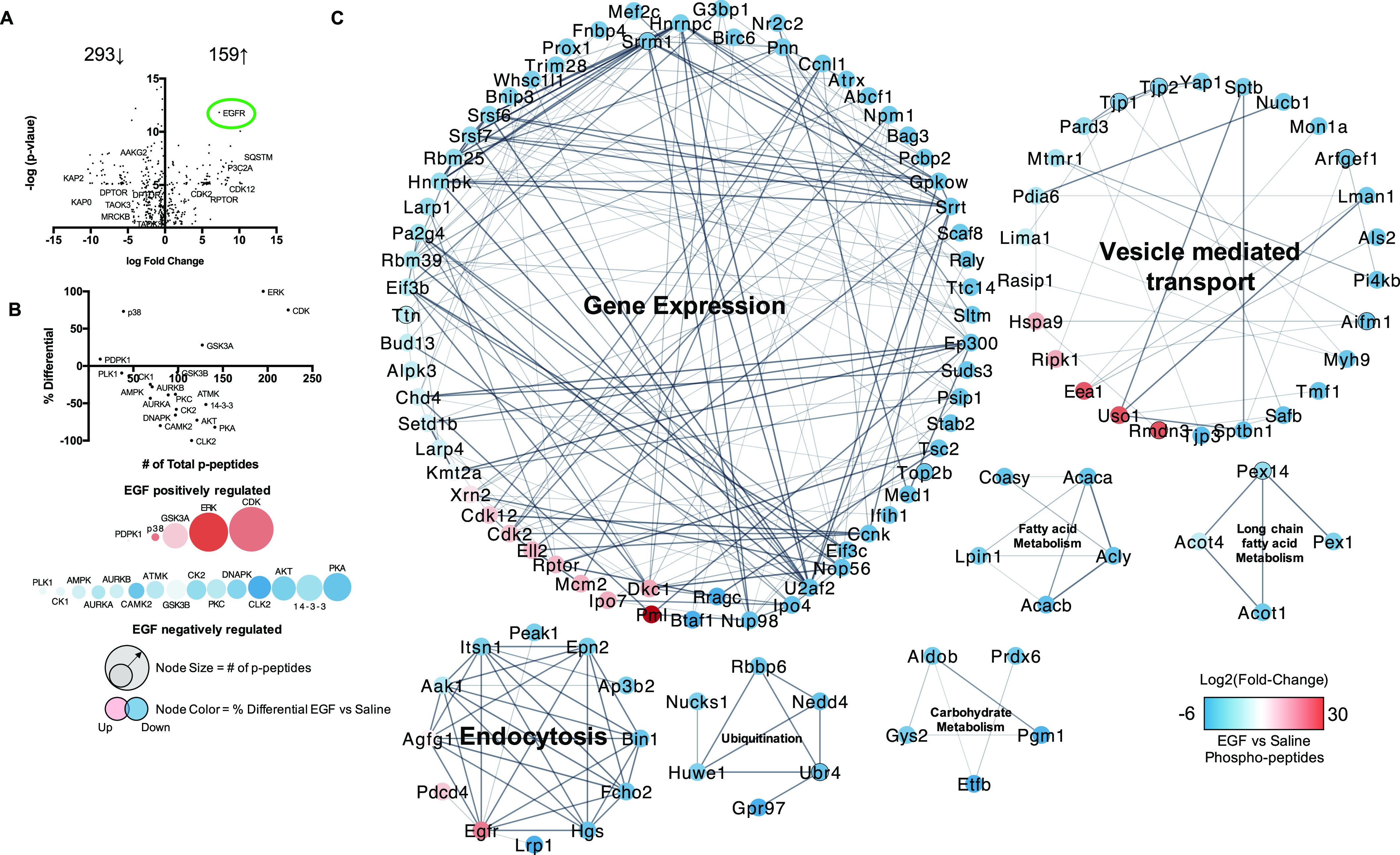
Characterization of the EGF-sensitive hepatic phosphoproteome and kinome: Male WT C57Bl/6 mice fed a control diet were treated with either saline or EGF (), followed by euthanasia at 30 min post IP injection and liver resection for downstream phosphoproteomic analysis. (A) Volcano plot of log 2(fold-change) and -log(p-value) for hepatic phosphopeptides (EGF vs. saline) significantly changed with EGF () IP after 30 min. (B) Kinome analysis based on phosphopeptides significantly regulated by EGF administration in the liver. (C) Cluster and GO processes analysis of the significant EGF-sensitive Phosphopeptides (Clusters numbered 1–7). An was used for the acute phosphoproteomic study, and a was considered significant by a two-tailed t-test. Pathways and processes had to meet an . Note: CE, ; CS, ; EGF, epidermal growth factor; FDR, false discovery rate; GO, gene ontology; IP, intraperitoneal.
Table 1.
Cluster analysis of EGF sensitive hepatic phosphoproteins.
| Cluster ID | Cluster | FDR |
|---|---|---|
| 1 | Gene expression | |
| 2 | Vesicle mediated transport | |
| 3 | Endocytosis | |
| 4 | Protein ubiquitination | |
| 5 | Carbohydrate metabolism | |
| 6 | Fatty acid metabolism | |
| 7 | Long chain fatty acid metabolism |
Note: EGF, epidermal growth factor; FDR, false discovery rate.
Table 2.
EGF-sensitive hepatic phosphoproteins involved in glucose metabolism after EGF treatment.
| Protein | Phosphosite(s) | EGF effect |
|---|---|---|
| GLUT2 | S522 | Decreased |
| GYS2 | S627/T630 | Decreased |
| 6PGL | S591 | Decreased |
| ALDOB | S36/S276/S272 | Decreased |
| PGM1 | S117 | Decreased |
| PGK1 | S203 | Decreased |
| PKLR | S292 | Decreased |
| SLC16A1 | S210/S491/S482 | Decreased/Increased/Decreased |
| PDHA | S293/S232 | Increased/Decreased |
| MDH1 | S241 | Decreased |
| PCK1 | S19 | Increased |
Note: “EGF Effect” refers to the expression of the relevant phosphorylated protein in mice injected with EGF ( IP) vs. those injected with saline followed by liver extraction within 30 min and hepatic phosphoproteomic analysis. 6PGL, 6-phosphogluconolactonase; ALDOB, aldolase B; GLUT2, glucose transporter 2; EGF, epidermal growth factor; GYS2, glycogen synthase 2; IP, intraperitoneal; MDH1, malate dehydrogenase; min, minutes; PCK1, phosphoenolpyruvate carboxykinase 1; PDHA, pyruvate dehydrogenase E1 component; PGK1, phosphoglycerate kinase 1; PGM1, phosphoglucomutase 1; PKLR, pyruvate kinase liver and red blood cell; SLC16A1, solute carrier family 16 member 1.
Table 4.
EGF-sensitive hepatic phosphoproteins involved in inflammation, fibrosis, and cell death.
| Protein | Phosphosite(s) | EGF effect |
|---|---|---|
| RIPK1 | S313 | Increased |
| AIFM1 | S581/S267 | Increased/ Decreased |
| BCL2L13 | T342 | Decreased |
| BCLF1 | S177/S529/S656 | Increased/ Decreased/Decreased |
| BNIP3 | S88 | Decreased |
| PDCD4 | S457 | Increased |
| STAB2 | S2503 | Decreased |
| MEF2C | S285 | Decreased |
Note: “EGF Effect” refers to the expression of the relevant phosphorylated protein in mice injected with EGF ( IP) vs. those injected with saline followed by liver extraction within 30 min and hepatic phosphoproteomic analysis. AIFM1, apoptosis inducing factor mitochondria associated 1; BCL2L13, BCL2 like 13; BCLF1, BCL2 associated transcription factor 1; BNIP3, BCL2 interacting protein 3; EGF, epidermal growth factor; IP, intraperitoneal; MEF2C, myocyte enhancer factor 2c; Min, minutes; PDCD4, programmed cell death protein 4; RIPK1, receptor interacting serine/threonine kinase 1; STAB2, stabilin-2.
Discussion
Receptor-based modes of action have classically been proposed for PCBs in environmental health. Indeed, PCBs, including some nondioxin-like congeners, can activate both human (Wahlang et al. 2014a) and rodent (Wahlang et al. 2014b) CAR and PXR, implying that these receptors could play a mechanistic role in the toxicity of these compounds. Previous studies from our group showed that PCBs exacerbated liver disease induced by high-fat feeding in mice (Wahlang et al. 2014b). However, CAR or PXR null mice fed an HFD still developed steatohepatitis following PCB exposures (Wahlang et al. 2016). Therefore, it appeared that targets other than xenobiotic and endobiotic receptors might play a role in promoting PCB-induced hepatotoxicity. In our more recent studies, we demonstrated that PCBs could interact with another receptor, the EGFR, in NASH in vitro (Hardesty et al. 2017, 2018) and in murine liver (Hardesty et al. 2017, 2019a, 2019b; Jin et al. 2020). Similarly, classes of EGFR inhibitors have been associated with liver injury (Qian et al. 2020) in cancer patients. PCB exposures reduced EGFR signaling by inhibiting EGFR phosphorylation, thereby decreasing downstream protein kinase activity leading to indirect CAR activation in mouse liver (Hardesty et al. 2017, 2019a) and in vitro (Hardesty et al. 2017). However, the causality of this so-called signaling disruption in PCB-associated NASH has not previously been demonstrated. This present research tested the hypothesis that EGF administration can attenuate PCB-related NASH by increasing EGFR signaling in a mouse model. By and large, the data support this hypothesis. EGF administration affected multiple physiological pathways to positively affect key NAFLD end points, but it was also associated with some undesirable metabolic side effects. In addition, the EGF-sensitive hepatic phosphoproteome and kinome were determined. These data support the need for more research investigating the EGFR’s role in NASH and environmental health. Recently, promoter hypermethylation (down-regulation mark) of an EGFR dimerization partner (ERBB3 gene) was associated with dioxin exposures in the ACHS II cohort study (Pittman et al. 2020). This would suggest that environmental chemical exposures may disrupt EGFR signaling through multiple modes of action.
Histologically, NASH is characterized by steatosis, inflammation, and fibrosis (Brunt et al. 2015); and Aroclor 1260-exposed mice fed an HFD developed NASH (Wahlang et al. 2014b). Although EGF-treated mice had lower hepatic FFAs, they did not have improved overall hepatic steatosis. EGF-treated mice did, however, have less PCB-induced hepatic inflammation and fibrosis characteristic of improved NASH. This finding is of interest because NASH, and in particular fibrosis, drives mortality in NAFLD (Angulo et al. 2015). That is why FDA has chosen coprimary end points related to histologic improvement in fibrosis or nonalcoholic steatohepatitis score (NAS) for NAFLD clinical trials (Filozof et al. 2017). Thus, the main finding of the present study is that EGF-treated mice had attenuated PCB-induced hepatic inflammation and fibrosis in NASH. EGF exerted a myriad of anti-inflammatory effects to significantly reduce histological liver inflammation. It reduced plasma levels and liver expression of major proinflammatory cytokines associated with NAFLD such as and PAI-1, while reducing Tlr4 expression. At least in the acute study, EGF regulated numerous phosphoproteins implicated in liver inflammation, injury, and cell death, including RIPK1 and STAB2 (Table 4). RIPK1 has been implicated in PCB-induced neuronal necroptosis (Guida et al. 2017), and STAB2 regulates efferocytosis critical for the resolution of inflammation following cell death (Elliott et al. 2017). At least some target genes of the protective transcription factor, NRF2, were increased in EGF-treated mice in the chronic model. However, not all NRF2 target genes behaved the same way in response to EGF or Aroclor 1260, indicative of transcriptional cross talk or potential off-target effects.
Aroclor 1260 exposure induced pericellular fibrosis, which was not present in combination with EGF cotreated mice, suggesting EGF may induce hepatic resolution of inflammation and or immune cell mediated clearance of dead cells. This in turn would prevent the development of scar tissue (fibrosis) due to excessive hepatocellular death and lack of inflammatory resolution. Previous studies have demonstrated that EGFR is required for efficient liver regeneration, which could explain the lack of fibrosis in EGF-treated mice (Natarajan et al. 2007). An interesting finding is that EGF-treated mice had lower expression of HSC gene markers of activation (e.g., , Mef2c), suggesting a dampened fibrotic response even in Aroclor 1260-exposed mice treated with EGF. In the acute study, HSC activating transcription factor MEF2C had lower phosphorylation at S285 in EGF-treated mice, suggesting EGF may also act on HSCs to diminish HSC activation through diminished MEF2C phosphorylation. This could also be a contributing factor to the decrease in pericellular fibrosis in EGF-treated and Aroclor 1260-exposed mice.
In contrast, EGF’s effects on metabolism in NASH were complex. Cluster analysis of phosphoproteomic data revealed that lipid and carbohydrate metabolism were among the top pathways affected by acute EGF administration (Table 1). Regarding lipid metabolism, EGF-treated mice had lower hepatic FFAs that were higher in chronic Aroclor 1260-exposed mice along with higher plasma VLDL. This impact was counterbalanced by higher target gene expression and higher de novo lipogenesis and cholesterol synthesis in EGF-treated mice. Because more lipids were simultaneously made and exported by liver, steatosis grade did not change significantly. The observed higher liver cholesterol levels observed in EGF-treated mice is consistent with previously published data measuring liver cholesterol in EGFR gain of function mice (Scheving et al. 2014). Phosphoproteomic data demonstrated that acute EGF regulated many proteins involved in lipid metabolism, including ACC and the LDLR (Table 3). Importantly, ACC inhibitors are currently in clinical trials testing for NASH therapeutic efficacy (Loomba et al. 2018). Collectively, AE vs. AS mice had lower hepatic prooxidative FFAs, higher hepatic triglycerides, higher plasma VLDL, and higher white adipose tissue (WAT) weight, suggesting EGF-treated mice had more lipid redistributed from the liver to WAT for storage. Adverse effects included worsened dyslipidemia and a trend toward greater adiposity.
Table 3.
EGF Sensitive hepatic phosphoproteins involved in lipid metabolism.
| Protein | Phosphosite(s) | EGF effect |
|---|---|---|
| ACC | S267/S30/S263 | Decreased |
| ACLY | S455 | Decreased |
| ACACA | S29 | Decreased |
| COASY | S177 | Decreased |
| ACOT1 | S56/S461 | Increased/Decreased |
| LPIN1 | S106 | Decreased |
| OATPB | S115 | Decreased |
| LDLR | S1418 | Decreased |
Note: “EGF Effect” refers to the expression of the relevant phosphorylated protein in mice injected with EGF ( IP) versus those injected with saline followed by liver extraction within 30 min and hepatic phosphoproteomic analysis. ACC, acetyl-CoA carboxylase; ACLY, ATP citrate synthase; ACOT1, acyl-CoA Thioesterase 1; ACSS2, acyl-CoA synthetase short chain family member 2; COASY, coenzyme A synthase; EGF, epidermal growth factor; IP, intraperitoneal; LDLR, LDL receptor; LPIN1, lipin 1; min, minutes; OATPB, solute carrier organic anion transporter family member 2B1.
In addition to effects on lipid metabolism, EGF-treated mice also had altered glucose metabolism. EGF-treated mice, whether acute or chronic, had fasting hyperglycemia. Acute EGF-treated mice had enhanced hepatic gluconeogenesis, whereas chronic EGF treated mice had lower adiponectin levels associated with reduce insulin sensitivity. A previous in vitro study demonstrated that hepatic EGFR signaling was glucogenic through uptake of gluconeogenic substrates such as pyruvate (Rashed and Patel 1991). The present study may be the first to demonstrate acute EGF’s glucogenic effects in vivo. A recent study provides evidence that hepatic EGFR signaling peaks in the inactive state in mice (Robles et al. 2017), suggestive that it may contribute to fasting blood glucose homeostasis. Mice acutely treated with EGF had higher blood glucose and lower pyruvate levels, which was associated with EGF-mediated phosphorylation of glucose metabolizing enzymes in the liver. We were surprised to find that these effects appeared independent of the glucagon-PKA-AMPK signaling pathway because EGF-treated mice had less activation of this pathway. Several specific EGF-sensitive phosphoproteins affecting carbohydrate metabolism were identified such as glucose transporter 2 and phosphoenolpyruvate carboxykinase 1 (Table 2).
Insulin sensitivity was lower in chronic EGF-treated mice contrasting the roles of the structurally related insulin and EGF receptors in hepatic glucose metabolism. Diabetes is perhaps the most significant clinical determinant of NASH disease severity and progression (Younossi et al. 2019), so the observed improvement in NASH severity despite less insulin sensitivity with chronic EGF administration was unexpected. Previously, in vivo characterization of the insulin-sensitive phosphoproteome in the liver identified many key pathways influenced by insulin in mice (Humphrey et al. 2015). Some of the insulin-sensitive kinases (e.g., active AKT and inactive ) contrast the EGF-sensitive kinases found in the acute study presented here, except for AMPK and PKA, which were both inactivated by EGF (Robles et al. 2017) and insulin signaling (Humphrey et al. 2015; Robles et al. 2017) in murine liver. Whether or not these changes persisted contributing to the insulin resistance associated with chronic EGF administration remains to be determined.
Hepatic nuclear receptors are being targeted for NASH therapy (Boeckmans et al. 2019; Patel et al. 2020), and nondioxin-like PCB exposures in mice have been demonstrated to regulate nuclear receptor target gene expression and to influence liver metabolism (Wahlang et al. 2014b, 2016). EGFR is known to regulate many nuclear receptors and transcription factors through protein phosphorylation (Llorens et al. 2011). In this study, chronic EGF treatment modulated the target gene expression of multiple nuclear receptors. EGF-treated mice had lower CAR and FXR target gene expression but higher , , and the related receptor, AhR target gene expression. This work reinforces previous studies by Negishi et al. (Mutoh et al. 2013) demonstrating that EGFR signaling negatively regulated CAR in vitro (Hardesty et al. 2017) and in mouse liver (Hardesty et al. 2017). is a master transcriptional regulator of liver function (Watt et al. 2003). EGFR signaling has previously been shown to increase target gene expression in vitro (Hardesty et al. 2019b); and the present study confirms those results. The impact of dioxins acting through AhR on EGF production and EGFR signaling has been investigated in mice previously (Lin et al. 1991). However, the present study investigated the impact of EGFR activation on AhR target gene expression, and such research does not appear to have previously been reported. Several potential mechanisms could explain the observed regulation of xenobiotic and endobiotic receptors by EGF. These include indirect transcription factor phosphorylation by EGFR effector kinases, alteration in ligand abundance, and cross talk. EGFR signaling may also activate nuclear receptors through indirect activation of retinoid X receptor (nuclear receptor dimerization partner) by increasing the hepatic pool of retinoic acid by phosphoregulation of aldehyde dehydrogenases.
Male mice were used in this study based on the prevalence of suspected TASH in men in the ACHS cohort (Clair et al. 2018). However, a recent study demonstrated that female mice developed exacerbated PCB-mediated TASH associated with decreased hepatic EGFR expression and phosphorylation (Wahlang et al. 2019c). Future studies investigating the responsiveness between male and female mice to EGF treatment in a PCB and dietary TASH model are warranted.
PCBs are signaling (Hardesty et al. 2019a) and metabolism disrupting chemicals (Heindel et al. 2017). PCBs appear to be competitive antagonists of the EGFR (Hardesty et al. 2018). PCB exposures cause more severe NASH in HFD-fed mice (Wahlang et al. 2014b). Because other growth factors are being investigated for NASH therapy, we investigated EGF treatment for Aroclor 1260-induced NASH in a preclinical model. EGF reduced liver FFAs and improved inflammation and fibrosis, thereby validating EGFR inhibition as a causal mode of action for PCBs in a murine model of NASH. Several adverse effects were observed, reducing the enthusiasm for the clinical translation of this research. In this study EGF-treated mice had more severe dyslipidemia and fasting hyperglycemia. These data are summarized in Figure 6. To our knowledge, we performed the first study characterizing the hepatic EGF-sensitive phosphoproteome in vivo. These results demonstrate the potential importance of EGFR signaling in liver physiology. More data are required to better understand the complex and understudied role of the EGFR in environmental health and liver disease. Future studies will use EGFR inhibitors to determine whether the phenotype is similar to PCBs in the context of NAFLD.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institute of Environmental Health Sciences (F31ES028982, R35ES028373, R01ES032189, T32ES011564, P42ES023716, P30ES030283 and R21ES031510), the National Institute of General Medical Sciences (P20GM113226), and the National Institute on Alcohol Abuse and Alcoholism (F32AA027950, P50AA024337). The authors would like to acknowledge the UofL Hepatobiology and Toxicology Center’s a) ’Omics Core for proteomics assistance; and b) Animal Model and Biorepository Core for the support required to develop and analyze the novel animal exposure models used in this study. Likewise, K. Cameron Falkner is acknowledged for his scientific assistance with this manuscript.
For the mass spectrometry studies, LCMS data (.RAW), search engine files (.mgf), peak list files (.mzML), and search results were aggregated into a Peaks data file and deposited in the MassIVE data repository (http://massive.ucsd.edu/) at the Center for Computational Mass Spectrometry at the University of California, San Diego and shared with the ProteomeXchange (www.proteomexchange.org).
References
- Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. 2015. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149(2):389–397.e310, PMID: 25935633, 10.1053/j.gastro.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmans J, Natale A, Rombaut M, Buyl K, Rogiers V, De Kock J, et al. 2019. Anti-NASH drug development hitches a lift on PPAR agonism. Cells 9(1):37, PMID: 31877771, 10.3390/cells9010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, et al. 2015. Nonalcoholic fatty liver disease. Nat Rev Dis Primers 1:15080, PMID: 27188459, 10.1038/nrdp.2015.80. [DOI] [PubMed] [Google Scholar]
- Clair HB, Pinkston CM, Rai SN, Pavuk M, Dutton ND, Brock GN, et al. 2018. Liver disease in a residential cohort with elevated polychlorinated biphenyl exposures. Toxicol Sci 164(1):39–49, PMID: 29684222, 10.1093/toxsci/kfy076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaciuc IV, D’Souza NB, Burikhanov R, Lee EY, Tarba CN, McClain CJ, et al. 2002. Epidermal growth factor protects the liver against alcohol-induced injury and sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res 26(6):864–874, PMID: 12068256, 10.1111/j.1530-0277.2002.tb02616.x. [DOI] [PubMed] [Google Scholar]
- Ding PN, Lord SJ, Gebski V, Links M, Bray V, Gralla RJ, et al. 2017. Risk of treatment-related toxicities from EGFR tyrosine kinase inhibitors: a meta-analysis of clinical trials of gefitinib, erlotinib, and afatinib in advanced EGFR-mutated non-small cell lung cancer. J Thorac Oncol 12(4):633–643, PMID: 28007626, 10.1016/j.jtho.2016.11.2236. [DOI] [PubMed] [Google Scholar]
- Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. 2017. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta-analysis. Hepatology 65(5):1557–1565, PMID: 28130788, 10.1002/hep.29085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MR, Koster KM, Murphy PS. 2017. Efferocytosis signaling in the regulation of macrophage inflammatory responses. J Immunol 198(4):1387–1394, PMID: 28167649, 10.4049/jimmunol.1601520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filozof C, Chow SC, Dimick-Santos L, Chen YF, Williams RN, Goldstein BJ, et al. 2017. Clinical endpoints and adaptive clinical trials in precirrhotic nonalcoholic steatohepatitis: facilitating development approaches for an emerging epidemic. Hepatol Commun 1(7):577–585, PMID: 29404480, 10.1002/hep4.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226(1):497–509, PMID: 13428781, 10.1016/S0021-9258(18)64849-5. [DOI] [PubMed] [Google Scholar]
- Gao J, He J, Zhai Y, Wada T, Xie W. 2009. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J Biol Chem 284(38):25984–25992, PMID: 19617349, 10.1074/jbc.M109.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guida N, Laudati G, Serani A, Mascolo L, Molinaro P, Montuori P, et al. 2017. The neurotoxicant PCB-95 by increasing the neuronal transcriptional repressor rest down-regulates caspase-8 and increases Ripk1, Ripk3 and MLKL expression determining necroptotic neuronal death. Biochem Pharmacol 142:229–241, PMID: 28676433, 10.1016/j.bcp.2017.06.135. [DOI] [PubMed] [Google Scholar]
- Hardesty JE, Al-Eryani L, Wahlang B, Falkner KC, Shi H, Jin J, et al. 2018. Epidermal growth factor receptor signaling disruption by endocrine and metabolic disrupting chemicals. Toxicol Sci 162(2):622–634, PMID: 29329451, 10.1093/toxsci/kfy004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardesty JE, Wahlang B, Falkner KC, Clair HB, Clark BJ, Ceresa BP, et al. 2017. Polychlorinated biphenyls disrupt hepatic epidermal growth factor receptor signaling. Xenobiotica 47(9):807–820, PMID: 27458090, 10.1080/00498254.2016.1217572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardesty JE, Wahlang B, Falkner KC, Shi H, Jin J, Wilkey D, et al. 2019a. Hepatic signalling disruption by pollutant polychlorinated biphenyls in steatohepatitis. Cell Signal 53:132–139, PMID: 30300668, 10.1016/j.cellsig.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardesty JE, Wahlang B, Falkner KC, Shi H, Jin J, Zhou Y, et al. 2019b. Proteomic analysis reveals novel mechanisms by which polychlorinated biphenyls compromise the liver promoting diet-induced steatohepatitis. J Proteome Res 18(4):1582–1594, PMID: 30807179, 10.1021/acs.jproteome.8b00886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, et al. 2017. Metabolism disrupting chemicals and metabolic disorders. Reprod Toxicol 68:3–33, PMID: 27760374, 10.1016/j.reprotox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey SJ, Azimifar SB, Mann M. 2015. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat Biotechnol 33(9):990–995, PMID: 26280412, 10.1038/nbt.3327. [DOI] [PubMed] [Google Scholar]
- Jensen SS. 1974. Structures and levels of most chlorobiophenyls in two technical PCB products and in human adipose tissue. Ambio 3:70–76. [Google Scholar]
- Jin J, Wahlang B, Shi H, Hardesty JE, Falkner KC, Head KZ, et al. 2020. Dioxin-like and non-dioxin-like PCBs differentially regulate the hepatic proteome and modify diet-induced nonalcoholic fatty liver disease severity. Med Chem Res 29:1247–1263, PMID: 32831531, 10.1007/s00044-020-02581-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen PE, Poulsen SS, Nexø E. 1988. Distribution of i.v. administered epidermal growth factor in the rat. Regul Pept 23(2):161–169, PMID: 3266016, 10.1016/0167-0115(88)90024-9. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. 2010. Improving bioscience research reporting: the arrive guidelines for reporting animal research. J Pharmacol Pharmacother 1(2):94–99, PMID: 21350617, 10.4103/0976-500X.72351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komposch K, Sibilia M. 2015. EGFR signaling in liver diseases. Int J Mol Sci 17(1):30, PMID: 26729094, 10.3390/ijms17010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Floch JP, Escuyer P, Baudin E, Baudon D, Perlemuter L. 1990. Blood glucose area under the curve. Methodological aspects. Diabetes Care 13(2):172–175, PMID: 2351014, 10.2337/diacare.13.2.172. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang C, Wang K, Lehmler HJ. 2020. Fatty liver and impaired hepatic metabolism alter the congener-specific distribution of polychlorinated biphenyls (PCBs) in mice with a liver-specific deletion of cytochrome p450 reductase. Environ Pollut 266(pt 1):115233, PMID: 32712482, 10.1016/j.envpol.2020.115233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FH, Clark G, Birnbaum LS, Lucier GW, Goldstein JA. 1991. Influence of the Ah locus on the effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the hepatic epidermal growth factor receptor. Mol Pharmacol 39:307–313, PMID: 1848654. [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta C(T)) Method. Methods 25(4):402–408, PMID: 11846609, 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Llorens F, Hummel M, Pastor X, Ferrer A, Pluvinet R, Vivancos A, et al. 2011. Multiple platform assessment of the EGF dependent transcriptome by microarray and deep tag sequencing analysis. BMC Genomics 12:326, PMID: 21699700, 10.1186/1471-2164-12-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. 2018. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology 155(5):1463–1473, PMID: 30059671, 10.1053/j.gastro.2018.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CT, Franz M, Kazi F, Donaldson SL, Morris Q, Bader GD. 2010. Cytoscape web: an interactive web-based network browser. Bioinformatics 26(18):2347–2348, PMID: 20656902, 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland VA, Clarke JU. 1989. Environmental occurrence, abundance, and potential toxicity of polychlorinated biphenyl congeners: considerations for a congener-specific analysis. Environ Health Perspect 81:225–239, PMID: 2503374, 10.1289/ehp.8981225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray RD, Merchant ML, Hardin E, Clark B, Khundmiri SJ, Lederer ED. 2016. Identification of an RNA-binding protein that is phosphorylated by PTH and potentially mediates PTH-induced destabilization of Npt2a MRNA. Am J Physiol Cell Physiol 310(3):C205–C215, PMID: 26834145, 10.1152/ajpcell.00192.2015. [DOI] [PubMed] [Google Scholar]
- Mutoh S, Sobhany M, Moore R, Perera L, Pedersen L, Sueyoshi T, et al. 2013. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci Signal 6(274):ra31, PMID: 23652203, 10.1126/scisignal.2003705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan A, Wagner B, Sibilia M. 2007. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci USA 104(43):17081–17086, PMID: 17940036, 10.1073/pnas.0704126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. 2020. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology 72(1):58–71, PMID: 32115759, 10.1002/hep.31205. [DOI] [PubMed] [Google Scholar]
- Pavuk M, Olson JR, Sjödin A, Wolff P, Turner WE, Shelton C, et al. 2014. Serum concentrations of polychlorinated biphenyls (PCBs) in participants of the Anniston Community Health Survey. Sci Total Environ 473–474:286–297, PMID: 24374590, 10.1016/j.scitotenv.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman GS, Wang X, Campbell MR, Coulter SJ, Olson JR, Pavuk M, et al. 2020. Dioxin-like compound exposures and DNA methylation in the Anniston Community Health Survey Phase II. Sci Total Environ 742:140424, PMID: 32629249, 10.1016/j.scitotenv.2020.140424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Zhang X, Zhang B, Yan B, Wang L, Gu P, et al. 2020. Tyrosine kinase inhibitor-related hepatotoxicity in patients with advanced lung adenocarcinoma: a real-world retrospective study. Cancer Manag Res 12:3293–3299, PMID: 32494193, 10.2147/CMAR.S237968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashed SM, Patel TB. 1991. Regulation of hepatic energy metabolism by epidermal growth factor. Eur J Biochem 197(3):805–813, PMID: 1903108, 10.1111/j.1432-1033.1991.tb15975.x. [DOI] [PubMed] [Google Scholar]
- Robles MS, Humphrey SJ, Mann M. 2017. Phosphorylation is a central mechanism for circadian control of metabolism and physiology. Cell Metab 25(1):118–127, PMID: 27818261, 10.1016/j.cmet.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Scheving LA, Zhang X, Garcia OA, Wang RF, Stevenson MC, Threadgill DW, et al. 2014. Epidermal growth factor receptor plays a role in the regulation of liver and plasma lipid levels in adult male mice. Am J Physiol Gastrointest Liver Physiol 306(5):G370–G381, PMID: 24407590, 10.1152/ajpgi.00116.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SM, Yang JH, Ki SH. 2013. Role of the Nrf2-are pathway in liver diseases. Oxid Med Cell Longev 2013:763257, PMID: 23766860, 10.1155/2013/763257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Fukusato T. 2014. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 20(42):15539–15548, PMID: 25400438, 10.3748/wjg.v20.i42.15539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilson HA, Schroeder JC. 2013. Reporting of results from animal studies. Environ Health Perspect 121(11–12):A320–A321, PMID: 24284018, 10.1289/ehp.1307676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Falkner KC, Clair HB, Al-Eryani L, Prough RA, States JC, et al. 2014a. Human receptor activation by aroclor 1260, a polychlorinated biphenyl mixture. Toxicol Sci 140(2):283–297, PMID: 24812009, 10.1093/toxsci/kfu083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Song M, Beier JI, Cameron Falkner K, Al-Eryani L, Clair HB, et al. 2014b. Evaluation of aroclor 1260 exposure in a mouse model of diet-induced obesity and non-alcoholic fatty liver disease. Toxicol Appl Pharmacol 279(3):380–390, PMID: 24998970, 10.1016/j.taap.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Hardesty JE, Jin J, Falkner KC, Cave MC. 2019a. Polychlorinated biphenyls and nonalcoholic fatty liver disease. Current Opinion in Toxicology 14:21–28, 10.1016/j.cotox.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Jin J, Beier JI, Hardesty JE, Daly EF, Schnegelberger RD, et al. 2019b. Mechanisms of environmental contributions to fatty liver disease. Curr Environ Health Rep 6(3):80–94, PMID: 31134516, 10.1007/s40572-019-00232-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Jin J, Hardesty JE, Head KZ, Shi H, Falkner KC, et al. 2019c. Identifying sex differences arising from polychlorinated biphenyl exposures in toxicant-associated liver disease. Food Chem Toxicol 129:64–76, PMID: 31026535, 10.1016/j.fct.2019.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Prough RA, Falkner KC, Hardesty JE, Song M, Clair HB, et al. 2016. Polychlorinated biphenyl-xenobiotic nuclear receptor interactions regulate energy metabolism, behavior, and inflammation in non-alcoholic-steatohepatitis. Toxicol Sci 149(2):396–410, PMID: 26612838, 10.1093/toxsci/kfv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Garrison WD, Duncan SA. 2003. Hnf4: a central regulator of hepatocyte differentiation and function. Hepatology 37(6):1249–1253, PMID: 12774000, 10.1053/jhep.2003.50273. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods 6(5):359–362, PMID: 19377485, 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Yang Y-P, Ma H, Starchenko A, Huh WJ, Li W, Hickman FE, et al. 2017. A chimeric EGFR protein reporter mouse reveals EGFR localization and trafficking in vivo. Cell Rep 19(6):1257–1267, PMID: 28494873, 10.1016/j.celrep.2017.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. 2019. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J Hepatol 71(4):793–801, PMID: 31279902, 10.1016/j.jhep.2019.06.021. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. 2016. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64(1):73–84, PMID: 26707365, 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.