

Abstract
Purpose of Review
More than one hundred loci have been identified from human genome-wide association studies (GWAS) for blood lipids. Despite the success of GWAS in identifying loci, subsequent prioritization of causal genes related to these loci remains a challenge. To address this challenge, recent work suggests that candidate causal genes within loci can be prioritized through cross-species integration using genome-wide data from the mouse.
Recent Findings
Mouse model systems provide unparalleled access to primary tissues, like the liver, that are not readily available for human studies. Given the key role the liver plays in controlling blood lipid levels and the wealth of liver genome-wide transcript and protein data available in the mouse, these data can be leveraged. Using co-expression network analysis approaches with mouse genome-wide data, coupled with cross-species analysis of human lipid GWAS, causal genes within lipid loci can be prioritized. Prioritization through both mouse and human along with biochemical validation provide a systematic and valuable method to discover lipid metabolism genes.
Summary
The prioritization of causal lipid genes within GWAS loci is a challenging process requiring a multidisciplinary approach. Integration of data types across species, like the mouse, can aid in causal gene prioritization.
Keywords: GWAS, LDL, Atherosclerosis, Lipid Metabolism
INTRODUCTION
Genome-wide association studies (GWAS) have revolutionized our understanding of the relationship between genotype and phenotype [1]. For more than a decade, GWAS has been utilized with great success to identify loci associated with a variety of complex traits, ranging from height and body mass index (BMI) to blood levels of low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C) and triglycerides (TG) [2–4]. Given the ever-increasing number of human biobanks along with expansion of genetic data, the utilization of GWAS is certainly going to increase. While the success of using GWAS to identify loci for complex traits has been immense, another challenge has emerged; translating these loci into biological function through the identification and validation of the underlying causal genes. As the scientific community enters this post-GWAS era it has become clear that we should focus on the translation of GWAS data into biological discovery [5].
As a result of the ease in analyzing human blood levels of total cholesterol (TC), LDL-C, HDL-C, TG in large human populations, GWAS has been very successful at understanding the genetics of blood lipid levels, which are key contributors to cardiovascular disease [6]. In this regard, numerous GWAS have been conducted in populations exceeding 100,000 individuals as well as in ethnically diverse populations that have identified hundreds of loci associated with individual blood lipid traits [3,7–10]. With these tremendous resources available for human blood lipid traits, a key goal is to prioritize the causal genes that contribute to these loci and understand the biological pathways through which they operate. In this review, we outline current approaches in prioritizing causal genes within loci and present a cross-species integration approach to aid in the discovery of causal genes within lipid loci.
PRIORITIZING CAUSAL GENES WITHIN LIPID LOCI
It is clear that existing and emerging human lipid GWAS datasets represent a significant resource for discovering new genes and biological pathways that can influence lipid metabolism. Despite the success of human GWAS in identifying loci associated with human blood lipid levels of TC, LDL-C, HDL-C, and TG, prioritizing causal genes and understanding the biological pathway through which these genes operate is limited to only a handful of examples [11–13]. The lack of success is likely due to a number of confounding and complicating factors. First, associated loci range in size from kilobases (kb) to megabases (mb) and can contain anywhere from one to more than a dozen genes within the locus. Second, recent studies have demonstrated causal genes may not need be located directly within the linkage disequilibrium (LD) of the locus and may be found outside the locus [14,15]. The best example of this is the FTO locus associated with human BMI and the identified and validated causal gene IRX3 (Iroquois Homeobox 3) being located outside the locus [14,16]. Therefore, due to both locus size and possible causal genes outside the associated locus itself, causal gene prioritization requires identifying possible candidate genes by systematic means.
Early on in the study of human lipid GWAS loci, prioritization and validation of causal genes within a locus region was often performed through a single candidate gene at a time approach. Validation of the candidate causal gene was achieved through generation of a knockout mouse and determining the gene’s influence on lipid metabolism [11]. This one at a time approach was time-consuming and, in some cases, led to negative results. To circumvent this laborious approach, current approaches most often rely on prioritizing causal genes through the integration of expression quantitative trait loci (eQTL). In this approach, genome-wide gene expression is assessed (either by microarray or RNA sequencing technologies) in a specific tissue to determine if genetics can influence the expression of a gene. If a gene is differentially expressed as a function of genetic variants within or close to the gene body, the gene is said to have a cis eQTL [17]. Identifying genes within or near a locus that show significant cis eQTL have greatly improved our ability to prioritize causal genes. For instance, the prioritization and identification of Sortilin1 (SORT1) at the chromosome 1p13 locus, which is associated with LDL-C levels and myocardial infarction, was done through integration of liver eQTL data [12]. Ultimately, this integration of liver eQTL data led to the validation of SORT1 as a modifier of lipid metabolism through a specific single nucleotide polymorphism (SNP) that altered binding of the transcription factor C/EBP, and thereby altered expression of SORT1. Another successful example of integrating liver eQTLs is the identification and validation of the Tetratricopeptide repeat domain protein 39B (TTC39B) gene that is associated with HDL-C [13]. The approach of integrating human liver eQTL data for causal gene prioritization is the current standard for identifying candidate causal genes within and near human GWAS lipid loci (Figure 1).
Figure 1: Commonly used method for prioritization of human lipid GWAS data for biological discovery.
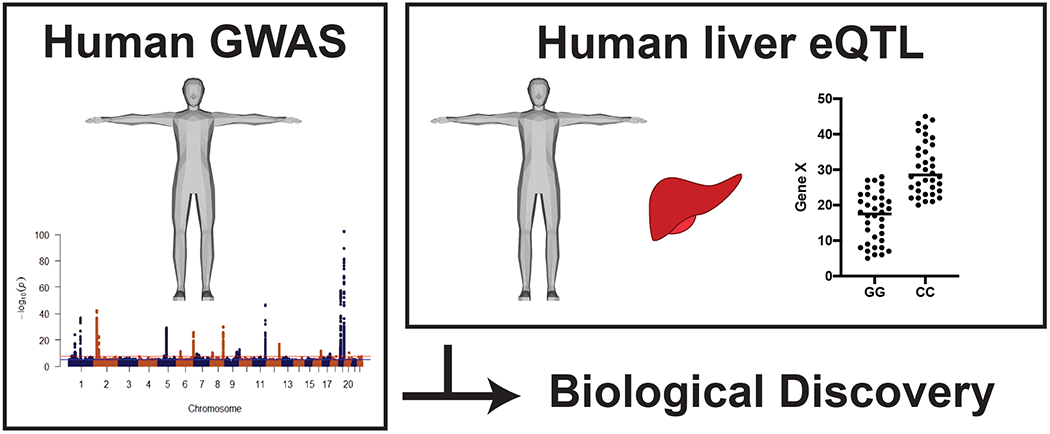
Schematic of the current model for prioritizing human lipid GWAS data which involves the identification of a human liver cis eQTL for a gene within a locus of interest.
Integrating human liver eQTL datasets as a method to prioritize causal genes within lipid loci has accelerated gene discovery, however, there are drawbacks to exclusively relying on this method. First, this method relies on identifying a liver cis eQTL for a specific gene within or near the locus and discards all other genes. While the availability of liver eQTL datasets has increased in recent years, the size of these datasets is still small in comparison to other readily accessible tissues, such as skin or adipose. The smaller sample size of liver eQTL datasets limits the statistical power to identify significant cis eQTL genes [17–19]. Another issue with relying on liver cis eQTL prioritization is that human livers are collected under diverse conditions from during surgical procedures to post-mortem collection. This variability in collection certainly can have implications for quality of downstream gene expression analysis. Therefore, the lack of a significant liver cis eQTL should not be the exclusive way through which prioritization of a causal genes is achieved. In this review we highlight recent work that utilizes mouse liver genome-wide data, to aid in the prioritization of causal genes within human lipid GWAS loci for biological discovery [20]. Ultimately, the goal of this work is to serve as an additional tool to speed up translation of human lipid loci to causal gene prioritization and validation.
MOUSE LIVER DATASETS AND CO-EXPRESSION ANALYSIS
The liver is central to metabolism as it has a critical role in the sensing and proper response to nutrient status. Due to this central role in metabolism, it is a major regulator of systemic lipid metabolism and blood lipid levels [21] [22]. In the fed state, the liver utilizes excess nutrients in the de novo synthesis of cholesterol and fatty acids for export to extrahepatic tissues [21]. The liver also has a key role in the sensing of serum lipid levels and modulating hepatic lipid metabolism in response [22]. These synthesis and sensory pathways are primarily regulated by the transcription factors Sterol Regulatory Element Binding Protein 1 (SREBP1) and Sterol regulatory element binding protein 2 (SREBP2) [21]. Dysregulation of either the lipid biosynthetic or sensory pathways can lead to altered blood lipids, primarily LDL-C and TG, that contribute to cardiovascular disease [6,23,24]. Uncoincidentally, modulation of the biosynthetic and sensory pathways in the liver has been of primary importance while developing pharmaceuticals that can lower blood TG and LDL-C levels [25]. Given the central role the liver plays in regulating blood lipid levels of LDL-C and TG it is likely that many of the causal genes within human lipid GWAS loci operate through the liver. With the limitations of gathering and performing gene expression studies in human liver samples, an alternative approach is to utilize the rich resources of genome-wide gene expression and protein data publicly available in the mouse.
The use of model organisms, such as the mouse, allow for collection of high-quality liver genome-wide data with relative ease in comparison to humans. Furthermore, the ability to perform tightly controlled experiments under uniform environmental conditions make the mouse an ideal model to use in studies to understand the liver. Another advantage of the mouse is the development of genetic reference populations, such as the hybrid mouse diversity panel (HMDP) and the diversity outbred (DO) population, which demonstrate genetic variation similar to that found in human populations [26–28]. These mouse genetic reference populations serve as an ideal platform to understand how genetic variation can influence complex traits and provide access to genome-wide liver transcript and protein data. Upon publication, these genome-wide transcript and protein datasets are publicly deposited and available for researchers across the world to take advantage of. Unfortunately, while this data is deposited and available for all, these rich datasets are more often than not never used or looked at again.
One way to utilize genome-wide transcript and protein data in the mouse is to perform module-based network analyses. These analyses provide a powerful statistical tool to identify genes that share similar co-expression patterns in complex datasets. Given the richness of publicly available data from mouse genetic reference populations, module-based networks are ideal for identifying modules (co-expressed groups) of genes that enrich in specific biological processes [26–29]. One widely used method to perform module-based network analysis is weighted gene co-expression network analysis (WGCNA) [30]. The WGCNA method generates modules based on co-expression patterns found within the inputted dataset. After modules are generated, the modules can then be enriched for specific biological processes using gene ontology (GO) [31,32]. Once a module has been assigned a specific biological function, one is able to develop hypotheses for potential novel functions of genes identified within the module. Ultimately, this approach allows for unbiased analysis of genome-wide transcript and protein data and creation of modules that can be used for downstream analysis.
CROSS-SPECIES PRIORITIZATION OF CAUSAL LIPID GENES
Given the wealth of publicly available human lipid GWAS data and mouse liver genome-wide data available, recent work sought to integrate these datasets together and develop a systematic approach to prioritize causal lipid genes within human lipid loci [20]. In this report, the authors began by taking advantage of the power of WGCNA to generate modules of co-expressed genes from complex datasets. Applying WGCNA to 12 distinct genome-wide liver datasets (transcript and protein) from multiple mouse genetic reference populations they were able to identify a module of genes significantly enriched for the GO term “cholesterol biosynthetic process” in 11 out of the 12 datasets. Of note, one contributing factor to the high reproducibility of this cholesterol enriched module is likely related to the transcriptional regulation of lipid and cholesterol metabolism genes. In the liver, lipid and cholesterol related genes are regulated by the common transcription factors, SREBP1 and SREBP2 [21]. Within the cholesterol enriched module many of the genes are known to play a role in cholesterol metabolism, however, many have no known role. To systematically prioritize genes, replication was used across the 11 datasets and allowed for 2,435 genes to be prioritized down to 112 genes, which showed strong replication across the modules enriched for the GO term “cholesterol biosynthetic process”.
After prioritization of genes from the mouse liver datasets, genes were then cross-referenced against human lipid GWAS data for TC, LDL-C, HDL-C, and TG [3]. After cross-referencing the 112 replicated genes, 48 genes were identified to have a SNP within or near the gene associated below a 5% false discovery rate (FDR). Of note, given the multiple datasets through which candidate genes are filtered in the mouse liver network data, there is a high probability that these genes would likely contribute to cholesterol or lipid metabolism. As a result, the stringent threshold employed in standard human GWAS of 5x10−8 was lowered to a 5% false discovery rate (FDR) threshold. Other studies have also lowered the 5x10−8 threshold when integrating data from multiple sources [33,34]. Out of the 48 genes identified to have associations with human lipid blood lipid traits, 25 genes were prioritized which have no known prior role in lipid or cholesterol metabolism. These 25 genes represent candidate causal genes within human lipid loci that have not yet been investigated in scientific research [20]. Taken together, this systematic method of combining human lipid GWAS data with mouse liver genome-wide data provides a valuable method for prioritizing candidate causal genes within human GWAS data (Figure 2). To highlight the power of this cross-species analysis between mouse and human data, Li et. al., systematically identified and validated Sestrin1 (SESN1) as a gene associated with TC in humans. Through functional testing and human lipid GWAS replication in independent datasets, SESN1 was found to be the only gene out of the 25 prioritized to pass all replication criteria. Validation of Sesn1 in multiple mouse models demonstrated that it regulates blood cholesterol levels. Further biological testing indicated that Sesn1 operated in a liver-specific manner and is necessary for feedback regulation of cholesterol biosynthesis. This approach emphasizes the usefulness of cross-species analysis for prioritizing causal genes within lipid loci and validating the biological pathway through which they operate.
Figure 2: Cross-species method of prioritization of human GWAS data for biological discovery.
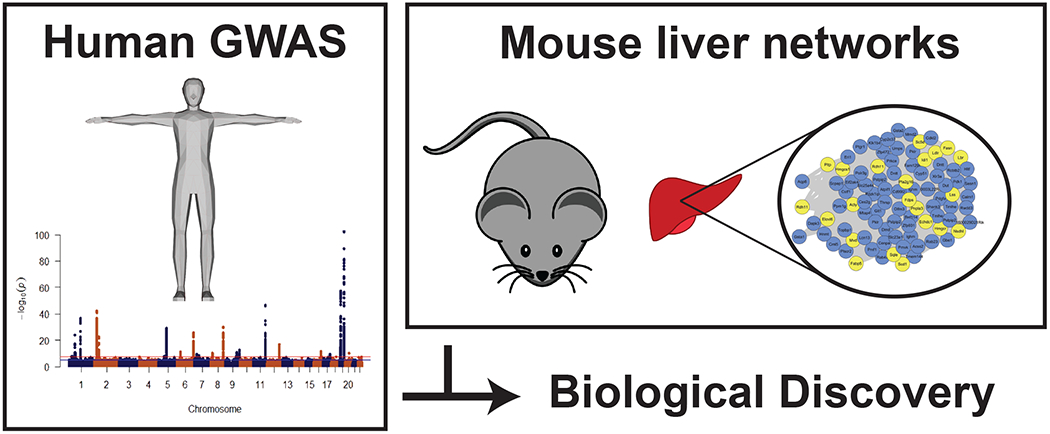
Schematic of a method for prioritizing human lipid GWAS data using mouse liver networks to identify genes involved in lipid metabolism which can be cross-referenced to human lipid gwas data to identify novel genes involved in regulating human lipid metabolism.
CONCLUSION
Prioritization and identification of causal genes within human lipid GWAS loci has lagged due to the complex nature of the majority of lipid loci. This demonstrates that unique approaches to aid in prioritizing causal genes within lipid loci are necessary. The most common method of causal gene prioritization involves human liver eQTL datasets to identify significant cis eQTL genes within a locus followed by validation studies (Figure 1). Here we overviewed a method for prioritizing causal genes within lipid loci through the cross-species integration with mouse genome-wide liver network data (Figure 2). This approach has the advantage of prioritizing genes relevant to lipid and cholesterol metabolism in the mouse first and then cross-referencing them against lipid loci. This approach was successfully utilized to identify and validate Sesn1 as a regulator of cholesterol biosynthesis while also prioritizing numerous other candidate genes for follow up study [20]. Similar approaches have been used by other labs to identify novel modifiers of bone density or schizophrenia risk suggesting that cross-species analysis can be used in other tissues and biological systems [35–37]. It is clear that there is still plenty of biological wealth to be mined from publicly available human GWAS and mouse genome-wide data and novel methods such as the one outlined here will allow us to begin to harness these resources.
Key Points.
Human lipid GWAS has been very successful at identifying genetic loci associated with blood lipids, which are a key contributing factor to the development of cardiovascular disease.
Current methodologies for identifying the causal genes at human lipid GWAS loci have been slow and many of the loci remain uncharacterized.
Publicly available liver genome wide transcriptomic and proteomic data from the mouse can be harnessed and integrated with human lipid GWAS to identify new genes that regulate lipid metabolism.
This method has been used to identify and subsequently validate the role of Sestrin1 (SESN1) in feedback regulation of hepatic cholesterol biosynthesis.
Acknowledgements
Financial support and sponsorship
This work was supported in part by NIH HL147097 awarded to BWP.
Footnotes
Conflicts of interest
There are no conflicts of interest.
Literature cited
- 1.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J: 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 2017, 101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J ’an, Mägi R, et al. : Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010, 42:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. : Discovery and refinement of loci associated with lipid levels. Nat Genet 2013, 45:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Human lipid GWAS paper from Global Lipid Genetics Consortium (GLGC) that identifies more than 100 lipid loci.
- 4.Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM, et al. : Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet 2018, 27:3641–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallagher MD, Chen-Plotkin AS: The Post-GWAS Era: From Association to Function. Am J Hum Genet 2018, 102:717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hegele RA: Plasma lipoproteins: genetic influences and clinical implications. Nat Rev Genet 2009, 10:109–121. [DOI] [PubMed] [Google Scholar]
- 7.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. : Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann TJ, Theusch E, Haldar T, Ranatunga DK, Jorgenson E, Medina MW, Kvale MN, Kwok P-Y, Schaefer C, Krauss RM, et al. : A large electronic-health-record-based genome-wide study of serum lipids. Nat Genet 2018, 50:401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, Saleheen D, Emdin C, Alam D, Alves AC, et al. : Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet 2017, 49:1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, Gagnon DR, DuVall SL, Li J, Peloso GM, et al. : Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet 2018, 50:1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bauer RC, Sasaki M, Cohen DM, Cui J, Smith MA, Yenilmez BO, Steger DJ, Rader DJ: Tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPα. J Clin Invest 2015, 125:3809–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, et al. : From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 2010, 466:714–719.* Key paper that demonstrates the causal gene at 1p13 locus associated with blood cholesterol levels is SORT1.
- 13.Hsieh J, Koseki M, Molusky MM, Yakushiji E, Ichi I, Westerterp M, Iqbal J, Chan RB, Abramowicz S, Tascau L, et al. : TTC39B deficiency stabilizes LXR reducing both atherosclerosis and steatohepatitis. Nature 2016, 535:303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. : Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014, 507:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, Emdin CA, Hilvering CRE, Bianchi V, Mueller C, et al. : A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell 2017, 170:522–533.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Claussnitzer M, Dankel SN, Kim K-H, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, et al. : FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med 2015, 373:895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.GTEx Consortium: The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013, 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Innocenti F, Cooper GM, Stanaway IB, Gamazon ER, Smith JD, Mirkov S, Ramirez J, Liu W, Lin YS, Moloney C, et al. : Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet 2011, 7:e1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. : Mapping the genetic architecture of gene expression in human liver. PLoS Biol 2008, 6:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Votava JA, Zajac GJM, Nguyen JN, Leyva Jaimes FB, Ly SM, Brinkman JA, De Giorgi M, Kaul S, Green CL, et al. : Integrating Mouse and Human Genetic Data to Move beyond GWAS and Identify Causal Genes in Cholesterol Metabolism. Cell Metab 2020, 31:741–754.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** A study demonstrating the cross-species integrative approach by which mouse liver expression data can be used to prioritize human lipid GWAS genes. This approach was used to validate Sestrin 1 as a modulator of cholesterol feedback inhibition.
- 21.Horton JD, Goldstein JL, Brown MS: SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002, 109:1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dietschy JM, Turley SD, Spady DK: Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res 1993, 34:1637–1659. [PubMed] [Google Scholar]
- 23.Abifadel M, Varret M, Rabès J-P, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, et al. : Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003, 34:154–156. [DOI] [PubMed] [Google Scholar]
- 24.Brown MS, Goldstein JL: A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232:34–47. [DOI] [PubMed] [Google Scholar]
- 25.Pinkosky SL, Newton RS, Day EA, Ford RJ, Lhotak S, Austin RC, Birch CM, Smith BK, Filippov S, Groot PHE, et al. : Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat Commun 2016, 7:13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, et al. : Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 2013, 17:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parks BW, Sallam T, Mehrabian M, Psychogios N, Hui ST, Norheim F, Castellani LW, Rau CD, Pan C, Phun J, et al. : Genetic architecture of insulin resistance in the mouse. Cell Metab 2015, 21:334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chick JM, Munger SC, Simecek P, Huttlin EL, Choi K, Gatti DM, Raghupathy N, Svenson KL, Churchill GA, Gygi SP: Defining the consequences of genetic variation on a proteome-wide scale. Nature 2016, 534:500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett BJ, Farber CR, Orozco L, Kang HM, Ghazalpour A, Siemers N, Neubauer M, Neuhaus I, Yordanova R, Guan B, et al. : A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res 2010, 20:281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langfelder P, Horvath S: WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. : Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000, 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gene Ontology Consortium: The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 2021, 49:D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Tucker NR, Rizki G, Mills R, Krijger PH, de Wit E, Subramanian V, Bartell E, Nguyen X-X, Ye J, et al. : Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Important paper that demonstrates integration of data can yield biological insight below the traditional human GWAS threshold of 5x10−8.
- 34.Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, Zeng L, Ntalla I, Lai FY, Hopewell JC, et al. : Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet 2017, 49:1385–1391. [DOI] [PubMed] [Google Scholar]; * Important paper that challenges the traditional human GWAS threshold of 5x10−8 and demonstrates the utility of using false discovery rate, instead.
- 35.Ashbrook DG, Cahill S, Hager R: A Cross-Species Systems Genetics Analysis Links APBB1IP as a Candidate for Schizophrenia and Prepulse Inhibition. Front Behav Neurosci 2019, 13:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calabrese G, Bennett BJ, Orozco L, Kang HM, Eskin E, Dombret C, De Backer O, Lusis AJ, Farber CR: Systems genetic analysis of osteoblast-lineage cells. PLoS Genet 2012, 8:e1003150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calabrese GM, Mesner LD, Stains JP, Tommasini SM, Horowitz MC, Rosen CJ, Farber CR: Integrating GWAS and Co-expression Network Data Identifies Bone Mineral Density Genes SPTBN1 and MARK3 and an Osteoblast Functional Module. Cell Syst 2017, 4:46–59.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]