

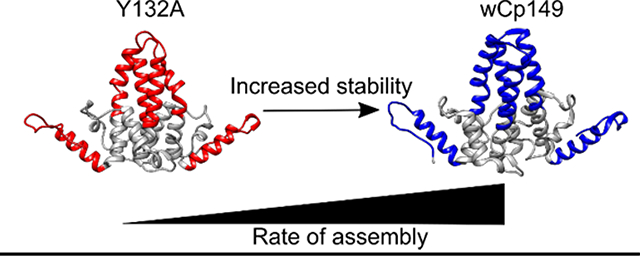
Abstract
While there is an effective vaccine for Human Hepatitis B Virus (HBV), 257 million people have chronic infections for which there is no cure. The assembly process for the viral capsid is a potential therapeutic target. In order to understand the capsid assembly process, we investigated the dimeric building blocks of the capsid. To understand what blocks assembly, we took advantage of an assembly incompetent mutant dimer, Cp149-Y132A, located in the inter-dimer interface. This mutation leads to changes in protein dynamics throughout the structure of the dimer as measured by hydrogen deuterium exchange mass spectrometry (HDX-MS). To further understand how the HBV capsid assembles, the homolog woodchuck HBV (WHV) capsid protein dimer (Cp) was used. WHV is more stable than HBV in HDX-MS and native mass spectrometry experiments. Because the WHV Cp assembles more rapidly into viral capsids than HBV, it was suspected that an increase in stability of the intra-dimer interface and/or in the contact region leads to increased assembly rates. The differences in dynamics when comparing HBV and human Cp149-Y132A as well as the differences in dynamics when comparing the HBV and WHV Cps allowed us to map an allosteric network within the HBV dimer. Through a careful comparison of structure, stability and dynamics using four different capsid protein dimers, we conclude that protein subunit dynamics regulate HBV capsid assembly.
Graphical Abstract
INTRODUCTION
The human pathogen Hepatitis B Virus (HBV) chronically infects 257 million people worldwide and is responsible for around 800,000 deaths each year due to liver failure, hepatocellular carcinoma, and cirrhosis (1). The mature virion consists of a protein studded outer membrane surrounding a nucleocapsid core which contains the gapped dsDNA genome and the reverse transcriptase (2). The capsid assembles with T=4 (Figure 1, panel A) or T=3 symmetry which contain 240 or 180 copies of the core protein (Cp) respectively. T=4 particles are the most prevalent form and are the infectious particle (3–8). The Cp consists of 183 amino acids and serves a variety of functions during the viral lifecycle (9–13). The basic C-terminal domain is transiently exposed to the capsid exterior and is involved in intracellular trafficking through interactions with host cell proteins (14–20). The first 149 amino acids of the Cp (Cp149) make up the assembly domain and are all that are needed for efficient capsid assembly in vitro (Figure 1, panel B, Supplemental movie 1) (3,21–23).
Figure 1.

Structure of human and woodchuck hepatitis B viruses. A) T=4 capsid of human wild type HBV (left) T=4 capsid of the woodchuck wild type HBV (right). B) Subdomains of interest within the homodimer are shown. One monomer of the protein is displayed in blue while the other is yellow. C) Overlay of the mutant hCp149-Y132A crystal structure, which crystalized as a trimer of dimers,(green) over the wild type hCp149 crystal structure (blue) for human HBV in the capsid form (top, PDBIDs 3KXS and 1QGT, respectively). The black arrow indicates the location of the Y132A mutation. Overlay of the wCp149 in the capsid form (purple, PDBID 6EDJ) and wCp149-Y132A, which crystalized as a dimer of dimers, (orange, PDBID 6ECS) for the woodchuck HBV (middle). Overlay of the hCp149 (blue) with the wCp149 (purple, bottom). The sidechain of the mutated residue is displayed. Alpha helices are labeled.
Woodchucks chronically infected with woodchuck hepatitis B virus (WHV) develop hepatocellular carcinomas similar to humans infected with HBV (24). Because of the similarities between WHV and HBV, WHV has been an important model for testing antiviral therapies (24). The capsid proteins of these two viruses share 63% identity, with the highest concentration of differences (23 of 55) in the intra-dimer interface (spike tip) (Supplemental Figure 1; Figure 1, panel C) (25). Woodchuck Cp149 (wCp149) dimers assemble into about half T=4 capsids in vitro, with the other half forming particles larger than T=4 capsids (26). While T=3 capsids are present in wCp149 assembly reactions, they appear less common than in hCp149 assembly reactions (25).
Assembly rates of both hCp149 and wCp149 can be regulated by adjusting dimer concentration, pH, and ionic strength (22,25,27,28). By maintaining a low dimer concentration, low ionic strength, and a near neutral pH, it is possible to study hCp149 and wCp149 proteins in as dimers (27). The ability to study capsid building blocks free in solution, presents an opportunity to develop a deeper understanding of a regulated assembly mechanism. While the tertiary and quaternary structures of WHV and HBV capsids are very similar, the association energy differs. wCp149 assembles at lower ionic strength and temperature than hCp149 and assembly reactions carried out at the same ionic strength show that the formation of wCp149 capsids is more thermodynamically favorable (25). WHV Cp149 assembles more rapidly than hCp149, with a substantially stronger association energy, and a high positive ΔCp, which differs from hCp149’s near zero ΔCp (25,29). The majority of primary sequence differences between WHV and HBV Cps occur in the two helices forming the intra-dimer interface. Nonetheless, these substitutions lead to a difference in assembly; suggesting that allostery may play a role in the assembly process.
An assembly incompetent mutant serves as a tool to connect aspects of protein structure and/or dynamics that modulate capsid assembly. Mutation of tyrosine at position 132 to an alanine (Y132A) in either hCp149 or wCp149 renders the dimer assembly incompetent. The Y132A mutation is located in the inter-dimer contact region (Figure 1, panel C) (30). In HBV, Y132 makes up 10% of the buried surface area when the dimer is assembled into capsids, however HBV Cp149-Y132A can co-assemble with WT hCp149 to form fragile capsids (31). Therefore, it is capable of adopting an assembly competent conformation, though the mutation weakens dimer-dimer interactions. The structures of hCp149-Y132A and hCp149 are similar; however, it is somewhat surprising that the largest differences occur in the spike tip and intra-dimer interface, (Figure 1, panel C)(30) leading us to propose that changes in protein dynamics, both at the site of the mutation and areas distal to the mutation, may be the reason for the functional difference. If changes in dynamics control assembly of dimers into capsids, this suggests that the mutation alters the energy landscape. By comparing the differences in dynamics between hCp149-Y132A and WT hCp149, an understanding of whether these proteins sample different conformations at different rates can start to be determined.
It is suspected that there is an allosteric relationship between the spike tip, at the intra-dimer interface, and the contact region, at the inter-dimer interface, based on biophysical studies of hCp149, wCp149, hCp149-Y132A, and wCp149-Y132A (29,30,32). We sought to provide new evidence for this relationship by directly measuring the intra-dimer interface stability and overall protein dynamics for each of these dimers. In this study, we used hydrogen deuterium exchange coupled to mass spectrometry (HDX-MS), native mass spectrometry (NMS), and differential scanning fluorimetry (DSF) to investigate the biophysical differences between hCp149, wCp149, hCp149-Y132A, and wCp149-Y132A. wCp149 can more readily assemble into capsids than hCp149. This suggests that wCp149 possesses a conformer in its ensemble that is more readily poised for oligomerization. By comparing the hCp149 to the wCp149, we were able to determine that the spike tip and intra-dimer contact region of Cp149 are important functional regions. The assembly incompetent conformation of hCp149-Y132A shows a decrease in stability at the intra-dimer interface. The differences in stability and dynamics at the intra-dimer interface show that there is a connection between the contact region (where the mutation occurs) and this interface, which is a direct effect of the allosteric network that controls the hCp149 protein. This suggests that decreased intra-dimer stability destabilizes a key conformation in the energy landscape, making the free energy of assembly for hCp149-Y132A unfavorable.
METHODS
Purification of HBV and WHV dimers.
Four expression plasmids, pET11a-hCp149, pET11a-hCp149-Y132A, pET11a-wCp149 and pET11a-wCp149-Y132A, were transformed into E. coli BL21(DE3) for protein expression. All proteins were purified based on a previously published protocol (33). Specifically, each protein was purified as described: hCp149 (22,33), hCp149-Y132A (30), wCp149 (25), and wCp149-Y132A (29).
Differential scanning fluorimetry (DSF) of HBV and WHV dimers.
To determine the thermal stability of hCp149, hCp149-Y132A, wCp149, and wCp149-Y132A differential scanning fluorimetry was used. Reactions contained 3–8μg protein in 50 mM ammonium bicarbonate pH 7, as well as 2.5 μL of a 50x stock of SYPRO orange dye (Invitrogen) for a total reaction volume of 30 μL. The samples were loaded into a Rotor-Gene Q instrument (Qiagen) with SYPRO orange absorbance monitored at 570 nm as temperature was ramped from 25°C-95°C at 1°C/minute.
Native mass spectrometry (NMS) of HBV and WHV dimers.
Dimer stocks at 2 mg mL−1 were buffer exchanged into 10 mM ammonium acetate pH 7 using mini-dialysis cassettes (Thermo-Fisher) overnight at 4°C. Buffer exchanged dimers were then diluted 1:10 into 10 mM ammonium acetate pH 7. Buffer exchanged and diluted dimers were then loaded into in house prepared gold coated borosilicate capillaries prepared as outlined in Luo et al.(34) and analyzed using a Waters Synapt G2-Si with the following settings: capillary kV was set to 2.11, sampling cone: 148, source offset: 150, trap collision energy: 20.0–140V, transfer collision energy: 5.0V, source temperature: 60°C, and trap gas: 7.0 mL/minute. Trap collision energy was increased by 10V every 30 seconds from 20V to 140V. Data was analyzed using Waters MassLynx version 4.1.
Hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS) of HBV and WHV dimers.
Dimer (hCp149, hCp149-Y132A, wCp149, and wCp149-Y132A) stocks at 2 mg mL−1 were diluted 1:10 into deuterated reaction buffer (10 mM HEPES, pD 7.5 in D2O). Control samples (time zero) were diluted into non-deuterated reaction buffer. At each time point (0,0.25, 3, 30, 180, and 1800 minutes) 10 μL of the reaction solution was removed and quenched by adding it to 60 μL of 0.75% formic acid (FA, Sigma) and 0.25 mg mL−1 porcine pepsin (Sigma) at pH 2.5 on ice. Each sample was digested for two minutes with vortexing every 30 seconds and then flash frozen in liquid nitrogen. Samples were stored in liquid nitrogen until liquid chromatography mass spectrometry analysis (LC-MS).
LC-MS analysis of HBV dimers was completed on a 1290 UPLC series chromatography stack (Agilent Technologies) coupled directly to a 6538 UHD Accurate-Mass QTOF LC/MS mass spectrometer (Agilent Technologies). Before electrospray−time-of-flight (ESI-TOF) analysis, peptides were separated on a reverse phase (RP) column (Phenomenex Onyx Monolithic C18 column, 100 × 2 mm) at 1 °C using a flow rate of 500 μL/min under the following conditions: 1.0 min, 5% B; 1.0−9.0 min, 5−45% B; 9.0−11.8 min, 45–95% B; 11.80−12.0 min, 5% B; solvent A = 0.1% FA (Sigma) in water (Thermo-Fisher) and solvent B = 0.1% FA in acetonitrile (ACN, Thermo-Fisher). Data were acquired at 2 Hz s−1 over the scan range 50−1700 m/z in positive mode. Electrospray settings were as follows: nebulizer set to 3.7 bar, drying gas at 8.0 L/min, drying temperature at 350 °C, and capillary voltage at 3.5 kV.
Data processing was carried out as outlined in Berry et al.(35) Briefly, peptides were identified using MassHunter Qualitative Analysis version 6.0 (Agilent Technologies), Peptide Analysis Worksheet (PAWs, ProteoMetrics LLC), and Peptide Shaker version 1.16.42 paired with Search GUI version 3.3.16 (Compomics)(36,37). Peptide mapping to the structure was carried out as outlined in van Erp et al. and Berry et al.(35,38) Briefly, nested peptides were resolved. The amount of deuterium taken up per amide was determined by taking the amount of deuterium taken up by the peptide and dividing it by the number of amide hydrogens minus the two N-terminal amides. This amount of deuterium taken up per amide was then divided by the amount of deuterium taken up per amide for the 24-hour time point to determine the percentage of deuterium uptake. The percentage of deuterium uptake was compared between conditions and mapped to the HBV dimer structure.
RESULTS
Differential scanning fluorimetry (DSF) of HBV and WHV dimers.
To determine if differences in the amino acid sequence at the spike tip or a mutation in the contact region alters the overall stability of the dimers, DSF was used. Thermal stability melting curves were generated for the dimers: hCp149, hCp149-Y132A, wCp149, and wCp149-Y132A. In DSF, the sample temperature is slowly raised while recording the fluorescence intensity of a dye that binds to hydrophobic pockets that become exposed as a protein unfolds (39,40). hCp149 had a melting temperature of 68°C (Figure 2), which is consistent with the melting temperature of 67°C for the reduced dimer observed by Wingfield et al. using differential scanning calorimetry(23). The hCp149-Y132A dimer was not significantly different from hCp149 dimer. The melting temperatures of the two types of woodchuck dimer were lower than the human dimers by around 5°C. The similar melting temperatures between the wild type dimers and the mutant dimers indicates that the Y132A mutation has little effect on the overall thermal stability of the dimer. However, the changes in primary structure between WHV and HBV dimers do cause a shift in melting temperature. DSF measurements were repeated across a range of buffer conditions to assure that the differences in melting temperature are intrinsic properties of the dimers themselves and not an artifact of ionic strength or buffer conditions.
Figure 2.

Differential scanning fluorimetry of HBV and WHV dimers. From this experiment it was determined that the two human Cp dimers have similar thermal denaturation temperatures which are higher than the two woodchuck Cp dimers.
Native Mass Spectrometry probes intra-dimer interface stability.
To further investigate the intra-dimer interface stability, a set of native mass spectrometry collisional induced unfolding experiments were performed on each of the dimers. Solutions containing dimers at 0.2 mg mL−1 in 10 mM ammonium acetate were analyzed by direct infusion, producing spectra that contained charge state distributions for both the monomer and the dimer (Figure 3). Low concentrations and low ionic strength conditions were used to eliminate the presence of multimers by limiting dimer-dimer interaction or non-specific clustering in the ionization process (22,25,27,28,41). To probe the relative strength of the intra-dimer interface, the collision energy was increased in 10V increments from 20V to 140V while monitoring a specific dimer charge state (11+). As the energy was increased, the intensity of the peak for the 11+ ion, which only has contributions from the dimeric species, decreased indicating that the dimer was being broken apart into monomers in the mass spectrometer. When the collision energy exceeded 120V the intensity of the 11+ peak was significantly decreased for all dimers, allowing it to be used as a proxy for dimer stability.
Figure 3.

Native mass spectrometry of hCp149 and wCp149. Spectra at 20V collision energy (top) and 120V of collision energy (bottom) for human Cp149. A), woodchuck Cp149. B). The +11 dimer peak is indicated by a red box. As collision energy is increased, the intensity of the dimer decreased. Monomer peaks are indicated by one diamond, dimer peaks by two diamonds, and peaks that have contribution from both monomeric and dimeric species are indicated by three diamonds C) Percentage of the +11 dimer peak intensity left at 120V collision energy. Error bars are representative of the standard deviation of three experiments. Percentages of the dimer +11 peak left at other collision energies can be found in Supplemental Material Figure 5. The intensity of the +11 peak at 120V was divided by the intensity of the +11 peak at 20V. The intensity of the peak is proportional to how much of the dimer species is present in the sample. The +11 peak intensity is a measure of the relative stability of the dimers.
The 11+ dimer peak intensity at 120V was compared to the initial dimer 11+ peak intensity at 20V (Figure 3, panel C). The hCp149-Y132A dimer was the least stable based on the changes in intensity from 20V to 120V followed by the hCp149, the wCp149-Y132A, and finally the wCp149. The Y132A mutation lowered the intra-dimer interface stability for both human and woodchuck dimers. The mutation is distal to the intra-dimer interface, yet, has a clear effect on interface stability. The woodchuck dimers had a stronger intra-dimer interface than the human dimers. This could be due to differences in the amino acid sequence between the human and woodchuck dimers specifically in the intra-dimer interface (Supplemental Figure 1).
HDX-MS of wCp149 and hCp149 dimers.
Because the secondary and tertiary structure of WHV and HBV Cp149 dimers are highly similar (29), we hypothesized that the differences in melting temperature, intra-dimer interface stability, and assembly rates arise from protein dynamics. To investigate the conformational flexibility between the two dimers, we used HDX-MS. Hydrogen deuterium exchange measures the rate of exchange of the amide hydrogen on the protein backbone with solvent (42,43). The exchange rate is inversely related to the stability of hydrogen bonding in the secondary and tertiary structure of the protein (44).
For HDX-MS experiments, each dimer was diluted into deuterated buffer and allowed to exchange for specific lengths of time. Reaction conditions were optimized to maintain the dimeric state of each of the proteins tested. Exchange was quenched with acid followed by pepsin digestion and analysis by LC-MS. Pepsin digestion created a reproducible set of peptides that were used for analysis (Supplemental Figure 2). The amount of deuterium taken up per peptide was determined at 0.25, 3, 30, 180, and 1800 minutes (Supplemental Figure 3).
We found that the intra-dimer interface of the wCp149 spike tip takes up less deuterium than hCp149 dimer (Figure 4). That is, the hydrogen bonding network of the wCp149 spike tip has a more stable intra-dimer interface. To determine if amino acid sequence differences between human and woodchuck proteins in this region were responsible for the observed differences in exchange, we calculated the intrinsic rates of exchange from the sequences of the specific peptides (Supplemental Table 1). The observed rates of exchange at the 15 second time point for hCp149 (84–103) and wCp149 (84–102) differed by greater than three-fold while the intrinsic rates of exchange differed by less than 5%. This indicates that the observed differences are unlikely to be strictly a factor of sequence specific intrinsic exchange and rather are the result of changes in the stability of the hydrogen bonding network in the spike tip. In contrast, the fulcrum and inter-dimer contact regions (helix 5) had greater exchange in wCp149 than the hCp149 dimer (Supplemental Figure 4). The calculated intrinsic rates of exchange in the contact region (helix 5) are similar, giving confidence to the interpretation that this region is more dynamic in the wCp149 dimer than in the hCp149 dimer. However, the calculated intrinsic rate of exchange is different in the fulcrum region between the two proteins, so for the fulcrum it is not possible to confidently assign this to a change in protein stability and dynamics.
Figure 4.

Deuterium uptake in the intra-dimer interface. between hCp149 and wCp149 at the intra-dimer interface. Helix 4a which is comprised of amino acid residues 79–109, is central to stability at the intra-dimer interface (A). The peptide 84–103 in hCp149 or 84–102 in wCp149 is shown in blue. At the three-hour time point, the human peptide takes up 8.8 deuterium (B) whereas the woodchuck peptide takes up 5.1 deuterium (C). Even though the hCp149 peptide is longer by one amino acid residue, the difference is statistically significant with a p-value < 0.01 at all time points. Calculations of the intrinsic rate of exchange are within 5% for the two peptides.
HDX-MS analysis of mutant Cp149.
If an allosteric network connects the inter-dimer interface to the intra-dimer interface, we reasoned there should be differences in dynamics between the mutant Cp149-Y132A dimers and the wild type Cp149 dimers. In support of this hypothesis, we observed by HDX-MS that the Y132A mutation led to differences in exchange that propagated throughout the entire structure (Figure 5, panel A; Supplemental movie 2 and 4). Also, differences in deuterium uptake were not the same between the human and woodchuck dimers. In the HBV dimer, the site of the Y132A mutation was more flexible in the mutant and the chassis and fulcrum were more stable in the mutant. The spike tip showed more exchange at earlier time points in the hCp149 dimer, but at three hours this trend switched and the hCp149-Y132A mutant took up more deuterium overall. This suggests that the spike has at least two populations of amides, fast and slow exchanging, that respond differently to the distal Y132A mutation.
Figure 5.

Mutation at residue 132 alters exchange across Cp149. A. The difference in the amount of deuterium taken up by the wild type and Cp149-Y132A human dimers at 15 seconds (left) and 3 hours (right). Areas where the wild type dimer takes up more deuterium than the Cp149-Y132A mutant dimer are displayed in red. Areas where the mutant Cp149-Y132A dimer takes up more deuterium than the wild type dimer are displayed in blue. Regions with no difference are displayed in white and regions that were not reported in this experiment are displayed in gray. B. The difference in the amount of deuterium taken up by the wild type and Cp149-Y132A woodchuck dimers at 15 seconds (left) and 3 hours (right). The location of the Y132A mutation is shown with an arrow in panel A.
In the wCp149 dimer, the residues near the Y132A mutation had an increase in exchange at early time points, but at later time points shows no difference in exchange when compared to wild type (Figure 5, panel A; Supplemental movies 3 and 5). This shows that the overall secondary structure of this region is not affected by the mutation at position 132, however, hydrogen bonding is less stable in the mutant implying that it is more dynamic than the wild type. The fulcrum and the chassis of the two forms of woodchuck have the same trends as the human forms of the dimer at early time points. At later time points, the chassis has more exchange in the wCp149-Y132A mutant indicating that this region has a less stable secondary structure in the mutant. The spike tip shows more exchange in the wCp149-Y132A mutant than in the wCp149 dimer indicating that the mutation destabilizes the hydrogen bond network on the other side of the protein. An important point is that there are changes in exchange throughout the HBV Cp dimer when the tyrosine at position 132 is mutated to an alanine, consistent with the idea that this is an allosterically regulated protein.
DISCUSSION
Biophysical, structural, and computational studies of the HBV capsid have helped to build a model in which protein dynamics and allostery are critical factors in the assembly and function of the T=4 particle. To date, a detailed model incorporating the behavior of assembly incompetent mutants is lacking. To understand the role of allostery in the capsid assembly process of the HBV Cp149 protein, we compared the melting temperature, the strength of the intra-dimer interface, and protein dynamics of hCp149, hCp149-Y132A, wCp149, and wCp149-Y132A. These four dimers are nearly identical in structure, but perform differently in assembly assays. Both the hCp149-Y132A and wCp149-Y132A mutants are blocked from assembling into capsids. The Y132A mutation, located in the inter-dimer contact region, causes changes throughout the protein including stability of the intra-dimer interface. wCp149 assembles into capsids more rapidly than its human homolog. However, the proteins differ in sequence mainly in the spike tip region. This difference in sequence does not greatly affect the overall structure of the dimer, but it does cause changes to both the dynamics of the dimer and the stability of the intra-dimer interface.
Stability of the HBV intra-dimer interface.
The wCp149 and hCp149 dimers share 63% sequence identity and are structurally very similar (25) (Supplemental Figure 1, Figure 1). Therefore, we were not expecting hCp149 to display 5oC greater thermal transition than wCp149. Most sequence differences are localized to the spike tip. NMS and HDX-MS data show that the intra-dimer interface is more stable in wCp149 than in hCp149. This increase in stability may play a role in the ability of the wCp149 to assemble more readily into capsids than the hCp149. Our data also show that both the hCp149-Y132A and wCp149-Y132A mutations, which are well removed from the spike tip, none-the-less decreases stability at the intra-dimer interface. In both the human and the woodchuck dimers, the collision energy needed to disrupt the dimer in NMS was lower for the mutant (Figure 3). An increase in deuterium exchange was also detected in the mutants in these regions at the three-hour time point (Figure 5). Of the four dimers, hCp149-Y132A was the least stable in NMS and had the most deuterium exchange in the spike region (Figure 3C, Figure 4), which is consistent with a less stable intra-dimer interface and more dynamic spike tip.
In Zhao et al., it was determined that a decrease in stability at the intra-dimer interface caused by the mutation D78S reduced the rate of capsid assembly (32). This mutation, located in the top of the intra-dimer interface, destabilizes helix 4a leading to a more flexible spike tip. This mutation slowed the assembly rate of the capsid showing that destabilization of the intra-dimer interface directly affects assembly kinetics. Together with our data, a model emerges that in order to have productive capsid assembly, the intra-dimer interface must be stable.
If increased stability in the spike tip causes the capsid to assemble more readily, the decrease in stability of this region in the hCp149-Y132A mutant should contribute to the inability of this mutant to assemble into capsids. However, there is a limit to the amount of stability that is beneficial to capsid assembly. Selzer et al. determined that if the intra-dimer interface is stabilized by a disulfide bond between the two C61 residues present in the interface, the rate of capsid assembly is slowed (45). This trend in stabilization was also observed by Zhao et al. when investigating the oxidized wild-type human dimer and the oxidized D78S mutant dimer assembly at differing salt concentrations. These human dimers showed similar assembly kinetics to one another which were both slower than the wild-type hCp149 assembly kinetics, though the difference in rate of assembly decreased at higher ionic strengths (32).Taken together, these data indicate that there is a balance between stability and dynamics in this interface that must be maintained for productive capsid assembly (32,45).
Inter-dimer contact region of WHV and HBV.
The contact region, which mediates dimer-dimer interactions, shows a complex pattern of deuterium exchange. The loop containing residue 132, located within the contact region, takes up more deuterium in both the hCp149-Y132A and wCp149-Y132A mutants than in the WT dimers at early time points (Figure 5). This implies that the mutation causes this region to be more flexible. In the α5 helix (Figure 1C), both the human and the woodchuck Cp149-Y132A mutants take up less deuterium than the respective WT dimers (Figure 5), as the helix becomes more structured upon mutation of Y132. Loss of structure in the loop of the contact region and a gain of structure in the α5 helix may contribute to the inability of the hCp149-Y132A and wCp149-Y132A mutants to assemble into capsids. The loop within the contact region shows similar deuterium exchange between the wCp149-Y132A and the hCp149-Y132A, however, there is more deuterium exchange in the wCp149-Y132A as compared to the hCp149-Y132A in the α5 helix (Supplemental Figure 4). An increase in deuterium exchange is consistent with increased disorder in this region in the WHV Cp149-Y132A as compared to the HBV Cp149-Y132A. While we cannot be certain that the observed exchange rates in HDX were not partially caused by differences in intrinsic exchange rates due to changes in primary amino acid sequence, our results are supported by the observations made by Zhao et al. through limited proteolysis (29). This is also consistent with Hilmer et al., who measured the rate of proteolytic cleavage of the C-terminal region of the hCp149 protein in free dimer and capsid. It was determined that the region near Y132 in the dimer becomes more folded in conditions that favor assembly(46). This implies that stabilization of the contact region is necessary for productive assembly into capsids.
Comparison of dynamics between the hCp149 free dimer and hCp149 capsid
In Bereszczak et al., comparisons between the dynamics of the wild type HBV dimer free in solution and within the capsid show similar hydrogen deuterium exchange patterns in all regions except in the spike tip (residues 89–103) and the contact region (residues 122–140) which show less overall deuterium uptake in the capsid when compared to the free dimer(47). In our data, the spike tip (intra-dimer interface) shows more deuterium incorporation in the hCp149-Y132A mutant than in the WT hCp149 dimer at later time points (Figure 5, panel A). The loss of stability (increased deuterium exchange) in this region may be detrimental to capsid assembly. In the inter-dimer contact region, hCp149-Y132A takes up more deuterium than hCp149 (Figure 5A), indicating that this could be another region where an increase in dynamics blocks capsid assembly. One interpretation of this is that the effect of the Y132A mutation on the spike tip and contact regions is to increase entropy. This must be compensated for during assembly, making the overall assembly process less thermodynamically favorable.
Our data show that changes in capsid assembly occur when the stability ad dynamics of the dimer change. The inter-dimer contact region has increased stability in the capsid and the capsid assembly-competent conformation. This seems logical based on the buried surface area between dimers in this region when assembled. The spike tip also shows an increase in stability in assembly competent dimers. The correlation of the dynamics in the distal spike tip with assembly is characteristic of an allosteric network being present across the Cp149 dimer. Based on our dynamics data, this network spans the whole protein, connecting the inter-dimer interface (contact region) to the intra-dimer interface (spike tip).
An outcome of this work is that a deeper understanding of the capsid assembly process will provide targets for the further development of antivirals that disrupt the infection cycle of HBV. Our work highlights regions of the dimer where altered stability and dynamics lead to either enhanced or diminished capsid assembly. Current assembly directed HBV antivirals act by increasing buried surface area in the inter-dimer contact region generating large aberrant structures (48). An important outcome from our work is that the spike tip or intra-dimer interface represents another target for assembly disruptors.
Conclusions
Dynamic proteins exist as an ensemble of conformers in solution. Modulation of the energy landscape of the ensemble, and thus the relative population, through changes in conformation or dynamics, is the business of allostery (49). To understand how HBV Cp149 assembles into capsids, we have worked to bring allosteric features into the light. The assembly competent conformer of HBV has a stable, but not too stable, intra-dimer interface and a stable inter-dimer interface. Our data strongly suggest that the Y132A mutation alters the ensemble of conformations present in solution. This implies that the hCp149-Y132A dimer does not sample the assembly active conformation. However, the WHV Cp149, which is structurally similar to the HBV Cp149, assembles more rapidly implying that it more frequently samples a capsid assembly competent conformer. HBV dimer is an excellent example of why structural models alone can be insufficient when trying to understand and predict functional outcomes of mutations. By using information about the energy landscape and dynamics of a protein or complex, even sophisticated mechanisms can be elucidated.
Supplementary Material
AKNOWLEGEMENTS
The authors would like to thank J. Thomas and G. Balasubramanian for technical assistance with mass spectrometry. We also acknowledge pioneering work on HBV HDX-MS by N. Movahed. Funding for the Proteomics, Metabolomics and Mass Spectrometry Facility used in this publication was made possible in part by the MJ Murdock Charitable Trust, MSU office of the VPREDGE, and the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM103474. Native mass spectrometry was made possible through an instrumentation grant from the Department of Energy Office of Basic Energy Sciences. We also would like to acknowledge the Montana State University Microfabrication Facility which is supported in part by a National Science Foundation (NSF) National Nanotechnology Coordinated Infrastructure award ECCS-1542210 for help in the preparation of capillaries for native mass spectrometry.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet. The supplemental information includes sequence alignments between HBV and WHV, peptide coverage maps and heat maps displaying the HDX-MS data, an in depth look at the HDX-MS data comparison between HBV and WHV contact regions, representative NMS spectra for the all collision energies used in the NMS experiment, intrinsic rates of deuterium exchange for key regions of the dimers, and supplemental movies showing the HDX-MS data mapped to the HBV and WHV structures.
References
- (1).CDA Foundation’s Polaris Observatory http://cdafound.org/polaris/ (July 9, 2020).
- (2).Seeger C; Mason WS Molecular Biology of Hepatitis B Virus Infection. Virology 2015, 479–480, 672–686. 10.1016/j.virol.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zlotnick A; Cheng N; Conway JF; Booy FP; Steven AC; Stahl SJ; Wingfield PT Dimorphism of Hepatitis B Virus Capsids Is Strongly Influenced by the C-Terminus of the Capsid Protein. Biochemistry 1996, 35 (23), 7412–7421. 10.1021/bi9604800. [DOI] [PubMed] [Google Scholar]
- (4).Bringas R Folding and Assembly of Hepatitis B Virus Core Protein: A New Model Proposal. J. Struct. Biol 1997, 118 (3), 189–196. 10.1006/jsbi.1997.3846. [DOI] [PubMed] [Google Scholar]
- (5).Crowther FA; Kiselev NA; Bottcher B; Berriman JA; Borisova GP; Ose SV; Pumpens P Three-Dimensional Structure of Hepatitis B Virus Core Particles Determined by Electron Cryomicroscopy. Cell 1994, 77, 943–950. 10.1016/0092-8674(94)90142-2. [DOI] [PubMed] [Google Scholar]
- (6).Singh S; Zlotnick A Observed Hysteresis of Virus Capsid Disassembly Is Implicit in Kinetic Models of Assembly. J. Biol. Chem 2003, 278 (20), 18249–18255. 10.1074/jbc.M211408200. [DOI] [PubMed] [Google Scholar]
- (7).Wynne SA; Crowther RA; Leslie AGW The Crystal Structure of the Human Hepatitis B Virus Capsid 1999, 3, 771–780. [DOI] [PubMed] [Google Scholar]
- (8).Stannard LM; Hodgkiss M Morphological Irregularities in Dane Particle Cores. J. Gen. Virol 1979, 45 (2), 509–514. 10.1099/0022-1317-45-2-509. [DOI] [PubMed] [Google Scholar]
- (9).Gazina EV; Fielding JE; Lin B; Anderson DA Core Protein Phosphorylation Modulates Pregenomic RNA Encapsidation to Different Extents in Human and Duck Hepatitis B Viruses. J. Virol 2000, 74 (10), 4721–4728. 10.1128/jvi.74.10.4721-4728.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Porterfield JZ; Dhason MS; Loeb DD; Nassal M; Stray SJ; Zlotnick A Full-Length Hepatitis B Virus Core Protein Packages Viral and Heterologous RNA with Similarly High Levels of Cooperativity. J. Virol 2010, 84 (14), 7174–7184. 10.1128/jvi.00586-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Freund SMV; Johnson CM; Jaulent AM; Ferguson N Moving towards High-Resolution Descriptions of the Molecular Interactions and Structural Rearrangements of the Human Hepatitis B Core Protein. J. Mol. Biol 2008, 384 (5), 1301–1313. 10.1016/j.jmb.2008.10.020. [DOI] [PubMed] [Google Scholar]
- (12).Alexander CG; Jurgens MC; Shepherd DA; Freund SMV; Ashcroft AE; Ferguson N Thermodynamic Origins of Protein Folding, Allostery, and Capsid Formation in the Human Hepatitis B Virus Core Protein. Proc. Natl. Acad. Sci 2013, 110 (30), E2782–E2791. 10.1073/pnas.1308846110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Seitz S; Urban S; Antoni C; Böttcher B Cryo-Electron Microscopy of Hepatitis B Virions Reveals Variability in Envelope Capsid Interactions. EMBO J 2007, 26 (18), 4160–4167. 10.1038/sj.emboj.7601841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li HC; Huang EY; Su PY; Wu SY; Yang CC; Lin YS; Chang WC; Shih C Nuclear Export and Import of Human Hepatitis B Virus Capsid Protein and Particles. PLoS Pathog 2010, 6 (10). 10.1371/journal.ppat.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kann M; Schmitz A; Rabe B Intracellular Transport of Hepatitis B Virus. World J. Gastroenterol 2007, 13 (1), 39–47. 10.3748/wjg.v13.i1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chen C; Wang JCY; Pierson EE; Keifer DZ; Delaleau M; Gallucci L; Cazenave C; Kann M; Jarrold MF; Zlotnick A Importin β Can Bind Hepatitis B Virus Core Protein and Empty Core-Like Particles and Induce Structural Changes. PLoS Pathog 2016, 12 (8), 1–25. 10.1371/journal.ppat.1005802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Selzer L; Kant R; Wang JCY; Bothner B; Zlotnick A Hepatitis B Virus Core Protein Phosphorylation Sites Affect Capsid Stability and Transient Exposure of the C-Terminal Domain. J. Biol. Chem 2015, 290 (47), 28584–28593. 10.1074/jbc.M115.678441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Yeh CT; Liaw YF; Ou JH The Arginine-Rich Domain of Hepatitis B Virus Precore and Core Proteins Contains a Signal for Nuclear Transport. J. Virol 1990, 64 (12), 6141–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Liao W; Ou JH Phosphorylation and Nuclear Localization of the Hepatitis B Virus Core Protein: Significance of Serine in the Three Repeated SPRRR Motifs. J. Virol 1995, 69 (2), 1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kann M; Sodeik B; Vlachou A; Gerlich WH; Helenius A Phosphorylation-Dependent Binding of Hepatitis B Virus Core Particles to the Nuclear Pore Complex. J. Cell Biol 1999, 145 (1), 45–55. 10.1083/jcb.145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gallina A; Bonelli F; Zentilin L; Rindi G; Muttini M; Milanesi G A Recombinant Hepatitis B Core Antigen Polypeptide with the Protamine-like Domain Deleted Self-Assembles into Capsid Particles but Fails to Bind Nucleic Acids. J. Virol 1989, 63 (11), 4645–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ceres P; Zlotnick A Weak Protein-Protein Interactions Are Sufficient to Drive Assembly of Hepatitis B Virus Capsids. Biochemistry 2002, 41 (39), 11525–11531. 10.1021/bi0261645. [DOI] [PubMed] [Google Scholar]
- (23).Wingfield PT; Stahl SJ; Williams RW; Steven AC Hepatitis Core Antigen Produced in Escherichia Coli: Subunit Composition, Conformation Analysis, and in Vitro Capsid Assembly. Biochemistry 1995, 34 (15), 4919–4932. 10.1021/bi00015a003. [DOI] [PubMed] [Google Scholar]
- (24).Tennant BC; Gerin JL The Woodchuck Model of Hepatitis B Virus Infection. ILAR J 2001, 42 (2), 89–102. 10.1093/ilar.42.2.89. [DOI] [PubMed] [Google Scholar]
- (25).Kukreja AA; Wang JC-Y; Pierson E; Keifer DZ; Selzer L; Tan Z; Dragnea B; Jarrold MF; Zlotnick A Structurally Similar Woodchuck and Human Hepadnavirus Core Proteins Have Distinctly Different Temperature Dependences of Assembly. J. Virol 2014, 88 (24), 14105–14115. 10.1128/jvi.01840-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Pierson EE; Keifer DZ; Kukreja AA; Wang JCY; Zlotnick A; Jarrold MF Charge Detection Mass Spectrometry Identifies Preferred Non-Icosahedral Polymorphs in the Self-Assembly of Woodchuck Hepatitis Virus Capsids. J. Mol. Biol 2016, 428 (2), 292–300. 10.1016/j.jmb.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Asor R; Selzer L; Schlicksup CJ; Zhao Z; Zlotnick A; Raviv U Assembly Reactions of Hepatitis B Capsid Protein into Capsid Nanoparticles Follow a Narrow Path through a Complex Reaction Landscape. ACS Nano 2019. 13 (7), 7610–7626. 10.1021/acsnano.9b00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Asor R; Schlicksup CJ; Zhao Z; Zlotnick A; Raviv U Rapidly Forming Early Intermediate Structures Dictate the Pathway of Capsid Assembly. J. Am. Chem. Soc 2020, 142 (17), 7868–7882. 10.1021/jacs.0c01092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhao Z; Wang JC-Y; Gonzalez-Gutierrez G; Venkatakrishnan B; Asor R; Khaykelson D; Raviv U; Zlotnick A Structural Differences between the Woodchuck Hepatitis Virus Core Protein in the Dimer and Capsid States Are Consistent with Entropic and Conformational Regulation of Assembly. J. Virol 2019, 93 (14), 1–16. 10.1128/jvi.00141-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Packianathan C; Katen SP; Dann CE; Zlotnick A Conformational Changes in the Hepatitis B Virus Core Protein Are Consistent with a Role for Allostery in Virus Assembly. J. Virol 2010, 84 (3), 1607–1615. 10.1128/JVI.02033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bourne CR; Katen SP; Fulz MR; Packianathan C; Zlotnick A A Mutant Hepatitis b Virus Core Protein Mimics Inhibitors of Icosahedral Capsid Self-Assembly. Biochemistry 2009, 48 (8), 1736–1742. 10.1021/bi801814y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhao Z; Wang JC-Y; Segura CP; Hadden-Perilla JA; Zlotnick A The Integrity of the Intradimer Interface of the Hepatitis B Virus Capsid Protein Dimer Regulates Capsid Self-Assembly. ACS Chem. Biol 2020. 10.1021/acschembio.0c00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zlotnick A; Lee A; Bourne CR; Johnson JM; Paul L In Vitro Screening for Molecules That Affect Virus Capsid Assembly. Nat. Protoc 2007, 2 (3), 490–498. 10.1016/j.bbi.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Luo ML; Jackson RN; Denny SR; Tokmina-Lukaszewska M; Maksimchuk KR; Lin W; Bothner B; Wiedenheft B; Beisel CL The CRISPR RNA-Guided Surveillance Complex in Escherichia Coli Accommodates Extended RNA Spacers. Nucleic Acids Res 2016, 44 (15), 7385–7394. 10.1093/nar/gkw421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Berry L; Patterson A; Pence N; Peters J; Bothner B Hydrogen Deuterium Exchange Mass Spectrometry of Oxygen Sensitive Proteins. Bio-Protocol 2018, 8 (6), 1–16. 10.21769/BioProtoc.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Vaudel M; Burkhart JM; Zahedi RP; Oveland E; Berven FS; Sickmann A; Martens L; Barsnes H PeptideShaker Enables Reanalysis of MS-Derived Proteomics Data Sets: To the Editor. Nat. Biotechnol 2015, 33 (1), 22–24. 10.1038/nbt.3109. [DOI] [PubMed] [Google Scholar]
- (37).Barsnes H; Vaudel M SearchGUI: A Highly Adaptable Common Interface for Proteomics Search and de Novo Engines. J. Proteome Res 2018, 17 (7), 2552–2555. 10.1021/acs.jproteome.8b00175. [DOI] [PubMed] [Google Scholar]
- (38).Van Erp PBG; Patterson A; Kant R; Berry L; Golden SM; Forsman BL; Carter J; Jackson RN; Bothner B; Wiedenheft B Conformational Dynamics of DNA Binding and Cas3 Recruitment by the CRISPR RNA-Guided Cascade Complex. ACS Chem. Biol 2018, 13 (2), 481–490. 10.1021/acschembio.7b00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Rayaprolu V; Kruse S; Kant R; Movahed N; Brooke D; Bothner B Fluorometric Estimation of Viral Thermal Stability. Bio-Protocol 2014, 4 (15). 10.21769/bioprotoc.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Niesen FH; Berglund H; Vedadi M The Use of Differential Scanning Fluorimetry to Detect Ligand Interactions That Promote Protein Stability. Nat. Protoc 2007, 2 (9), 2212–2221. 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- (41).Chen F; Gulbakan B; Weidmann S; Fagerer SR; Ibanez AJ; Zenobi R Applying Mass Spectrometry to Study Non-Covalent Biomolecule Complexes. Mass Spectrom. Rev 2015, 35 (1), 48–70. 10.1002/mas.21462. [DOI] [PubMed] [Google Scholar]
- (42).Konermann L; Pan J; Liu Y-H Hydrogen Exchange Mass Spectrometry for Studying Protein Structure and Dynamics. Chem. Soc. Rev 2011, 40 (3), 1224–1234. 10.1039/C0CS00113A. [DOI] [PubMed] [Google Scholar]
- (43).McAllister RG; Konermann L Challenges in the Interpretation of Protein H/D Exchange Data: A Molecular Dynamics Simulation Perspective. Biochemistry 2015, 54 (16), 2683–2692. 10.1021/acs.biochem.5b00215. [DOI] [PubMed] [Google Scholar]
- (44).Skinner JJ; Lim WK; Bédard S; Black BE; Englander SW Protein Dynamics Viewed by Hydrogen Exchange. Protein Sci 2012, 21 (7), 996–1005. 10.1002/pro.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Selzer L; Katen SP; Zlotnick A The Hepatitis B Virus Core Protein Intradimer Interface Modulates Capsid Assembly and Stability. Biochemistry 2014, 53 (34), 5496–5504. 10.1021/bi500732b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hilmer JK; Zlotnick A; Bothner B Conformational Equilibria and Rates of Localized Motion within Hepatitis B Virus Capsids. J. Mol. Biol 2008, 375 (2), 581–594. 10.1016/j.jmb.2007.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bereszczak JZ; Watts NR; Wingfield PT; Steven AC; Heck AJR Assessment of Differences in the Conformational Flexibility of Hepatitis B Virus Core-Antigen and e-Antigen by Hydrogen Deuterium Exchange-Mass Spectrometry. Protein Sci 2014, 23 (7), 884–896. 10.1002/pro.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Stray SJ; Bourne CR; Punna S; Lewis WG; Finn MG; Zlotnick A A Heteroaryldihydropyrimidine Activates and Can Misdirect Hepatitis B Virus Capsid Assembly. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (23), 8138–8143. 10.1073/pnas.0409732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hilser VJ; Wrabl JO; Motlagh HN Structural and Energetic Basis of Allostery. Annu. Rev. Biophys 2012, 41 (1), 585–609. 10.1146/annurev-biophys-050511-102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.