

Abstract
Allosteric regulation of protein functions is ubiquitous in organismal biology, but the principles governing its evolution are not well understood. Here we discuss recent studies supporting the large-scale existence of latent allostery in ancestor proteins of superfamilies. As suggested, the evolution of allostery could be driven by the need for specificity in paralogs of slow evolving protein complexes with conserved active sites. The same slow evolution is displayed by purifying selection exhibited in allosteric proteins with somatic mutations involved in cancer, where disease-associated mutations are enriched in both orthosteric and allosteric sites. Consequently, disease-associated variants can be used to identify druggable allosteric sites that are specific for paralogs in protein superfamilies with otherwise similar functions.
Introduction
Allostery is a protein regulatory mechanism where perturbations to a site distal from the active site induce conformational and/or functional change. Allostery is induced when information is transferred between a distal allosteric site and the orthosteric site [1••,2]. The perturbations at the allosteric site can be due to multiple factors [3], commonly in the form of either covalent or non-covalent binding of a molecule, or due to environmental factors such as light absorption or high-pressure [4–7]. Allosteric regulation has been extensively studied due to its role in a wide range of biological processes [8–17]. The concept of allostery was discovered five decades ago, after which several mechanisms have been proposed relating allosteric binding to conformational change of protein structure or protein dynamics [1••]. Recent ensemble-based techniques used to study allosteric mechanisms are well outlined in a recent review [4].
While these theories address the physical mechanisms of allostery, their relationship to molecular evolution remains less clear. Some suggest allosteric modulation of extant proteins arose in ancestor proteins with a latent capacity for allosteric modulation [18,19]. Alternately, allosteric proteins could have arisen from non-allosteric predecessors that bind the same orthosteric ligand [20,21]. Importantly, the existence of allostery can impose epistatic (non-additive) effects on mutations, which were evaluated for potential allosteric networks in PDZ domains using statistical covariation of residues [22]. Such work suggests that conditional neutrality mediates evolutionary adaptation of PDZ domains and that this allosteric network has its origins in the capacity of proteins to adapt to binding alternate ligand classes [23]. While the presence of allosteric networks in PDZ domains has since been challenged (reviewed in Ref. [24]), the hypothesis that local changes in phase and structure can lead to long range negative epistasis in a model system of ligand binding supports this idea of conditional neutrality [25].
Allosteric sites are diverse, in contrast to active sites that are conserved [26]. Accordingly, allosteric modulators can achieve a high level of selectivity for individual proteins in large superfamilies [27]. Furthermore, allosteric modulators do not compete with substrate/ligands that bind to the active site, but instead, work together to induce allosteric activity [26]. These properties offer the potential for allosteric sites to serve as drug targets, allowing development of safer medicines [26]. Moreover, certain mutations at allosteric sites do not hinder catalytic activity of the protein [28], although they might profoundly influence evolution. As such, allosteric mutations have shown to be deleterious and disease-causing in many cases [29–31], supporting their roles in fitness. This review summarizes current views of allosteric evolution driven by the need for specificity in extant paralogs and the existence of latent allostery in ancestors of large superfamilies. Lastly, this work highlights how studying allosteric mutations in the context of evolution has proven useful for identifying both disease-associated genes and drug targets.
Large-scale existence of latent allostery in ancestors of protein superfamilies
It is unknown whether the acquisition of allostery results from natural selection of allosteric regulation over non-regulated paralogs, or whether it stems from existing co-evolving residues. Elastic network models (ENMs) can access much larger conformational ensembles than more intensive all-atom molecular dynamics methods [32]. Abrusan et al. investigate the hypothesis that allostery is an evolutionary mechanism to regulate slow-evolving homomeric protein complex specificity, where ligand binding sites are made up of residues from multiple chains [1••]. Expanding on previous analysis of homologous protein complexes with binding sites composed of residues from either multiple chains (MBS) or single chains (SBS) [33•], they analyze differences in residue correlated motion between MBS and SBS complexes from the Allosteric Database [34]. Using ENMs they find MBS complexes more likely to have residue communities (i.e. networks of residues with correlated motions) spanning multiple chains, whereas those in SBS complexes tend to be within single chains. In silico alanine mutation scanning of interface residues by FoldX [35] found that interface mutations in MSB communities weaken the binding energy significantly more than mutations from SBS communities. Taken together, these results suggest that the type of ligand-binding site is intimately linked to the strength of allosteric communication. The authors suggest that for paralogous proteins, binding site similarity drives allosteric evolution.
As the ensemble model of allostery has gradually come to replace canonical symmetry model [36], more focus has turned to studying relationships between protein dynamics and allosteric signal transmission. The ‘intrinsic dynamics’ of a protein, or its native conformational ensemble under physiological conditions, can be used to probe residue motion types accompanying allostery. Zhang et al. modeled protein intrinsic dynamics on a large scale using SignDy [37••], which uses an ENM simulation pipeline designed to compare correlated motions between equivalent positions of homologs (from structural superposition and sequence alignment). The intrinsic dynamics of three structurally distinct superfamilies (LeuT, PBP-1, and Tim Barrel) shows that each retains conserved ‘global’ modes. Interestingly, these global modes have been shown in some cases to correspond to the structural rearrangements induced by ligand binding. Higher frequency modes do not show the same level of similarity between families but are distinctly conserved within sequence families from these CATH superfamily groups. This differentiation between conservation of higher frequency modes may provide some mechanism by which homologous proteins that share global modes may become subject to differing allosteric mechanisms.
Allosteric mutations contribute to evolutionary fitness and disease
Mutations contribute to genetic variation within a population, providing material for evolution. Advantageous mutations undergo positive selection and get fixed within a population and increase fitness [38,39]. Deleterious mutations decrease fitness and can lead to disease [40,41]. Furthermore, distal mutations of >15Å from the active site of a protein have been associated with networks of coupled residues that alter function [42,43•,44–47]. Because of this interconnection between mutations, disease, and allostery, current research has focused on studying disease-associated mutations in allosteric dysregulation. Shen et al. [48••] systematically investigated cancer-based regulation perturbed by somatic mutations at allosteric sites. Their dataset included 574 manually curated proteins from the Allosteric Database [49], out of which 74 proteins had experimentally validated allosteric sites and PDB structures. The evolutionary rate [50] and dN/dS ratio of human-mouse orthologous gene products were generally lower for allosteric proteins, suggesting a purifying selection.
To study the relationship between allosteric mutations and disease, the authors designed a pipeline to identify variants in allosteric sites among over 47 000 somatic missense mutations from 6958 pairwise tumor-normal matched pairs across 33 cancer types. They mapped 1990 predicted deleterious and 2461 known disease-causing variants on 74 allosteric structures and showed their enrichment at both allosteric and orthosteric sites, highlighting the potential role of allostery in disease pathology. The authors developed a statistical approach to predict cancer-associated allosteric genes, AlloDriver, using pan-cancer data. Known BRAF, HRAS, and AKT1, and four others, and unknown GCK and SERPINC1 cancer-associated proteins exhibited-enriched somatic missense mutations in their allosteric sites. Extending analysis to individual cancer types, they identify 20 known and 15 novel cancer proteins, suggesting allosteric dysregulation as an important factor influencing tumorigenesis. These results not only provide allosteric examples for further experimental investigation of fitness but can also help with developing novel targeted cancer therapies.
Evolutionary selective pressure on allosteric sites is used for drug development
Selective pressures are factors that contribute to variant selection and adaptation within a population. Over time, evolution occurs as a result of these selective pressures. In relation to protein sequence, selective pressures provide the basis for conservation among homologs [51]. Allosteric sites tend to be less conserved than the active sites they regulate [26,52,53], which has increased the popularity of their use as drug targets [26,53,54•]. Selective pressure is also expressed through rare variants in the population, and studying these can identify uncharacterized regulatory areas that can be exploited for drug development [54•].
Agne et al. [55••] used rare variants to identify a putative allosteric site in Caspase-6 (Casp6) and developed inhibitors to target the site. Casp6 functions in cell-apoptosis and is activated by a unique self-cleavage of its L2 loop that stabilizes the substrate-binding pockets [56]. Casp6 is allosterically inhibited by phosphorylation at S257 that prevents the L2 loop from attaining an active conformation for self-cleavage [57,58]. Similarly, zinc binding at another allosteric site (via K36, E244 and H287) locks Casp6 in a non-canonical inactive conformation [59]. The authors investigated four rare natural variants in Casp6 that lead to missense mutations. Three of these (A34, E35 and A109) line up a well-defined pocket 30 Å from the catalytic site [55••,56]. In an active VEID-bound CASP6 dimer, the binding pocket is near a zinc binding site, a phosphorylation site from the adjacent subunit, and the L2 cleavage loops of both subunits (Figure 1a). Being unconserved and identified by a prediction tool as potentially druggable, the authors pursued in silico screening for allosteric inhibitors targeting the site (Figure 1b), which likely transmits a signal to the active site of the adjacent subunit through conformational change of L2, and disorder-to-order transition of the active site ‘loop bundle’ (Figure 1a and c).
Figure 1.
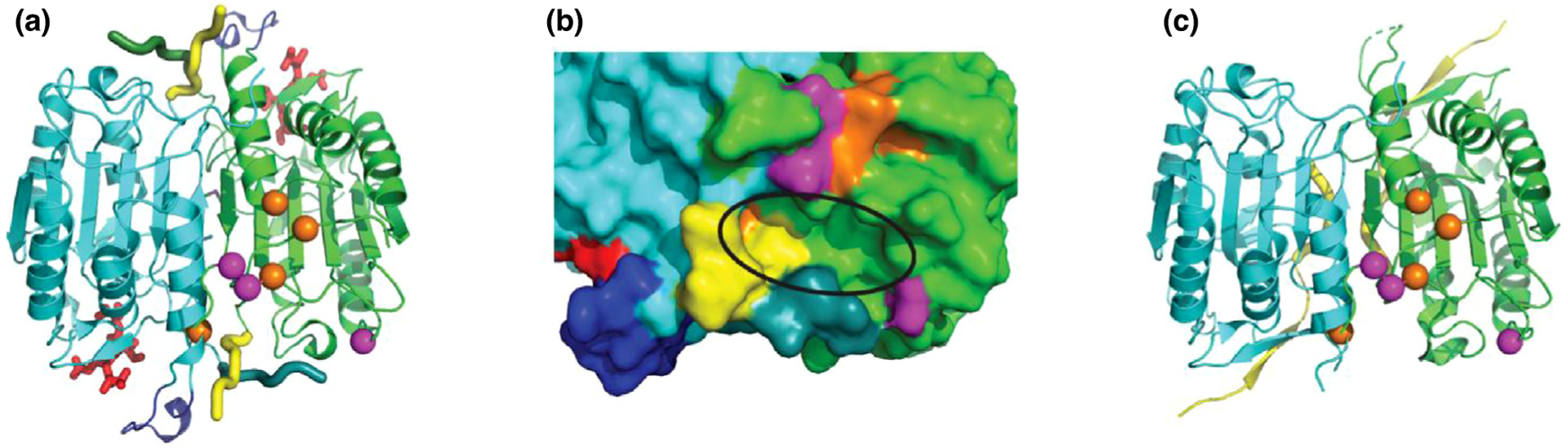
Casp6 allostery and activation.
(a) Casp6 dimer (PDB 3OD5; green and cyan chains) in active conformation with VEID (red sticks) bound in the active site. Loop2′ C-terminus (yellow tube) interacts with Loop2 N-terminus (dark cyan/green tube) and stabilizes the active site ‘loop bundle’ (blue cartoon). Allosteric phosphorylation (S257) and Zinc binding residues (K36, E244, and H287; orange spheres) are near two of the rare variant residues (A34E and E35K; magenta spheres) depicted in the green subunit. (b) Zoom of allosteric pocket (black ellipse) surface in active conformation formed by L2 loops, parts of allosteric sites, and variant residue (A109T; magenta). (c) Conformation change of Loop2 (yellow) and disordered active site ‘loop bundle’ in inactive zymogen (PDB 3NR2).
In silico screening followed by fluorescence-based Casp6 enzyme assays found two inhibitory compounds (S10G and C13) with low micromolar Ki (inhibitor constant). Kinetic analysis showed that they non-competitively inhibited Casp6 activity, suggesting binding to an allosteric site. Although the predicted molecular binding model (C13) and binding evaluation using Hydrogen-Deuterium Exchange Mass Spectrometry (S10G) hinted at the allosteric pocket, future experimental structures are required for conformation. Despite this drawback, reports of known of allosteric regulation by residues around this binding pocket and the structural involvement of L2 loops suggests that this site could be allosteric, and the work provides a successful example of using rare variants to discover drug target sites.
Conclusion
Allosteric regulation could be an inherent property of biological polymer molecules, or could have evolved from non-allosteric proteins (Introduction and Figure 2). The degree to which ‘latent’ or potential allosteric sites exist in ancestor proteins is still a matter of discussion. Recently developed methods to study protein dynamics are rein-forcing the notion of latent allostery in protein superfamilies [1••,32,37••]. Many known allosteric mechanisms involve protein oligomerization [20,60,61]. Molecular dynamics studies of protein complexes described by Abrusan et al. suggest that active site similarity in paralogous proteins could be the driving force behind the evolution of allostery [1••], which would allow selective control of similar active sites through divergent allosteric sites (Figure 2). Their observation of residue communities spanning across dimeric interfaces and their key role in interaction suggest that monomer to homodimer equilibrium represents the latent allosteric property of the ancestor. Similarly, the discovery by Zhang et al. of conserved global modes in various protein superfamilies suggests that their common ancestors possessed similar intrinsic dynamics [37••]. In these cases, conserved global modes likely transmit allosteric communication between active sites and allosteric sites displaying less conserved high frequency modes. The key roles of allosteric residues are also revealed in the investigation of somatic mutations by Shen et al. [48••]. They describe purifying selection for allosteric proteins and find enriched disease mutations at allosteric sites, suggesting that signal transmission between allosteric and orthosteric sites in extant proteins contributes to fitness. Such key roles of allosteric sites in disease have led to their use in developing novel drug therapies [48••,62], but also for discovering novel allosteric sites that can be exploited for drug development in the work by Agne et al. [55••]. Their use of rare variants to identify an allosteric pocket in Casp6 highlights the advantage of using allosteric site specificity in drug development.
Figure 2.
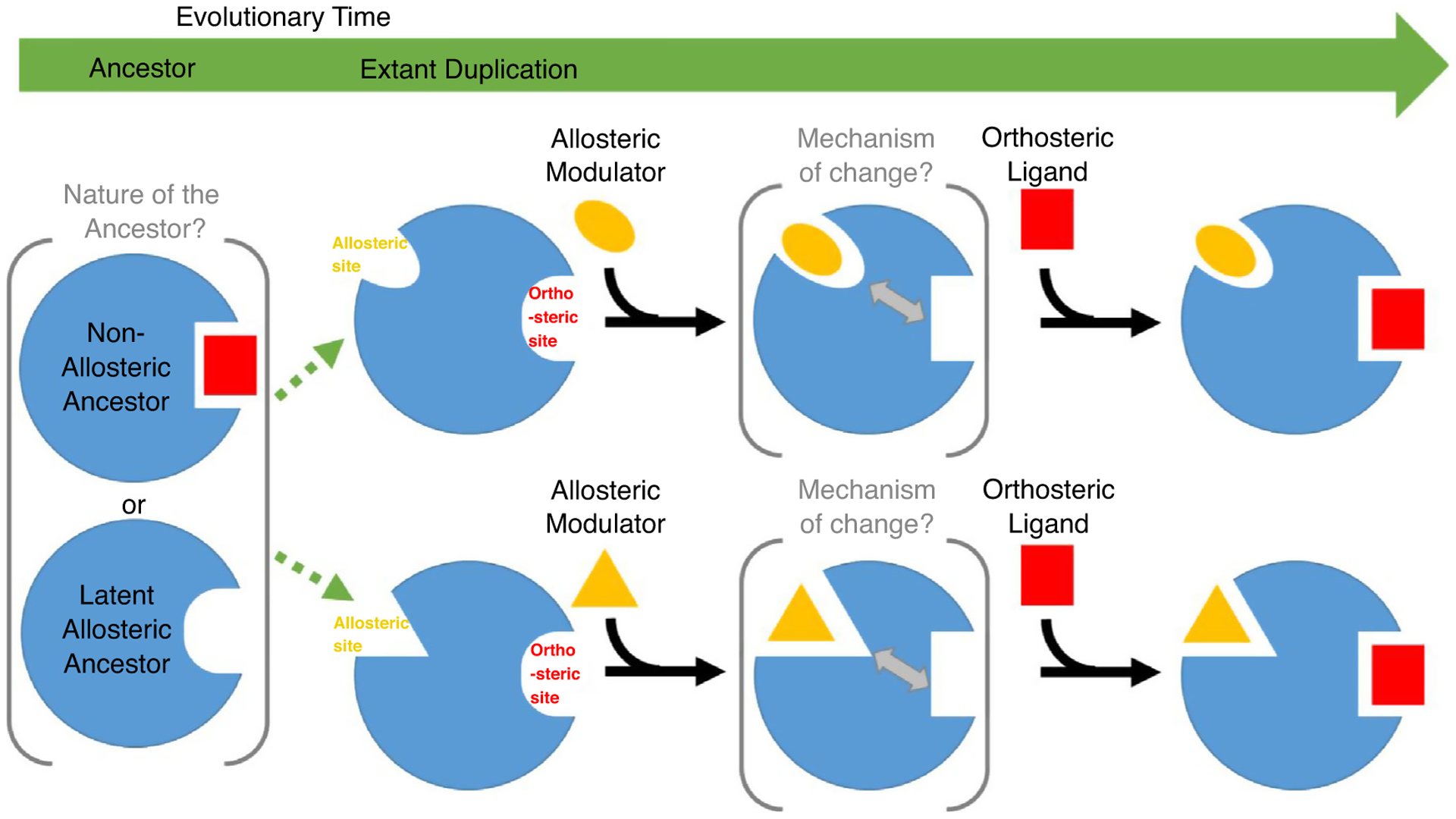
Evolution of allostery.
Ancestral proteins that are either non-allosteric with similar active site ligands or have latent allosteric properties duplicate and diverge (green dotted arrows) to acquire divergent allosteric sites. Allosteric modulators induce change at the conserved active site through several possible mechanisms guiding communication between sites.
Acknowledgements
This work was supported by the National Institutes of Health [GM127390 to NVG] and the Welch Foundation [I-1505 to NVG].
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Abrusan G, Marsh JA: Ligand-binding-site structure shapes allosteric signal transduction and the evolution of allostery in protein complexes. Mol Biol Evol 2019, 36:1711–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Using elastic-network models, the authors perform a comprehensive survey of known allosteric protein complexes and identify differences in residue communities where binding sites are formed from multiplechains, as opposed to residues solely from a single chain.
- 2.Greener JG, Sternberg MJ: Structure-based prediction of protein allostery. Curr Opin Struct Biol 2018, 50:1–8. [DOI] [PubMed] [Google Scholar]
- 3.Berezovsky IN, Guarnera E, Zheng Z, Eisenhaber B, Eisenhaber F: Protein function machinery: from basic structural units to modulation of activity. Curr Opin Struct Biol 2017, 42:67–74. [DOI] [PubMed] [Google Scholar]
- 4.Guarnera E, Berezovsky IN: On the perturbation nature of allostery: sites, mutations, and signal modulation. Curr Opin Struct Biol 2019, 56:18–27. [DOI] [PubMed] [Google Scholar]
- 5.Williamson MP, Kitahara R: Characterization of low-lying excited states of proteins by high-pressure NMR. Biochim Biophys Acta Proteins Proteom 2019, 1867:350–358. [DOI] [PubMed] [Google Scholar]
- 6.Takayanagi M, Kurisaki I, Nagaoka M: Non-site-specific allosteric effect of oxygen on human hemoglobin under high oxygen partial pressure. Sci Rep 2014, 4:4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strickland D, Moffat K, Sosnick TR: Light-activated DNA binding in a designed allosteric protein. Proc Natl Acad Sci U S A 2008, 105:10709–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR: Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Revi Mol Cell Biol 2017, 18:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ozorowski G, Pallesen J, de Val N, Lyumkis D, Cottrell CA, Torres JL, Copps J, Stanfield RL, Cupo A, Pugach P et al. : Open and closed structures reveal allostery and pliability in the HIV-1 envelope spike. Nature 2017, 547:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thal DM, Glukhova A, Sexton PM, Christopoulos A: Structural insights into G-protein-coupled receptor allostery. Nature 2018, 559:45–53. [DOI] [PubMed] [Google Scholar]
- 11.Li J, White JT, Saavedra H, Wrabl JO, Motlagh HN, Liu K, Sowers J, Schroer TA, Thompson EB, Hilser VJ: Genetically tunable frustration controls allostery in an intrinsically disordered transcription factor. eLife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan Y, Cross PJ, Jameson GB, Parker EJ: Exploring modular allostery via interchangeable regulatory domains. Proc Natl Acad Sci U S A 2018, 115:3006–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capdevila DA, Braymer JJ, Edmonds KA, Wu H, Giedroc DP: Entropy redistribution controls allostery in a metalloregulatory protein. Proc Natl Acad Sci U S A 2017, 114:4424–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lakhani B, Thayer KM, Hingorani MM, Beveridge DL: Evolutionary covariance combined with molecular dynamics predicts a framework for allostery in the MutS DNA mismatch repair protein. J Phys Chem B 2017, 121:2049–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kroll K, Holland CK, Starks CM, Jez JM: Evolution of allosteric regulation in chorismate mutases from early plants. Biochem J 2017, 474:3705–3717. [DOI] [PubMed] [Google Scholar]
- 16.Campitelli P, Guo J, Zhou HX, Ozkan SB: Hinge-shift mechanism modulates allosteric regulations in human Pin1. J Phys Chem B 2018, 122:5623–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su CT-T, Lua W-H, Ling W-L, Gan SK-E: Allosteric effects between the antibody constant and variable regions: a study of IgA Fc mutations on antigen binding. Antibodies 2018, 7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reynolds Kimberly A, McLaughlin Richard N, Ranganathan R: Hot spots for allosteric regulation on protein surfaces. Cell 2011, 147:1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coyle SM, Flores J, Lim WA: Exploitation of latent allostery enables the evolution of new modes of MAP kinase regulation. Cell 2013, 154:875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peracchi A, Mozzarelli A: Exploring and exploiting allostery: models, evolution, and drug targeting. Biochim Biophys Acta 2011, 1814:922–933. [DOI] [PubMed] [Google Scholar]
- 21.Mas-Droux C, Biou V, Dumas R: Allosteric threonine synthase. Reorganization of the pyridoxal phosphate site upon asymmetric activation through S-adenosylmethionine binding to a novel site. J Biol Chem 2006, 281:5188–5196. [DOI] [PubMed] [Google Scholar]
- 22.Lockless SW, Ranganathan R: Evolutionarily conserved pathways of energetic connectivity in protein families. Science 1999, 286:295–299. [DOI] [PubMed] [Google Scholar]
- 23.Raman AS, White KI, Ranganathan R: Origins of allostery and evolvability in proteins: a case study. Cell 2016, 166:468–480. [DOI] [PubMed] [Google Scholar]
- 24.Gautier C, Laursen L, Jemth P, Gianni S: Seeking allosteric networks in PDZ domains. Protein Eng Des Sel 2018, 31:367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson ED, Grishin NV: Long-range epistasis mediated by structural change in a model of ligand binding proteins. PLoS One 2016, 11:e0166739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu S, Shen Q, Zhang J: Allosteric methods and their applications: facilitating the discovery of allosteric drugs and the investigation of allosteric mechanisms. Acc Chem Rese 2019, 52:492–500. [DOI] [PubMed] [Google Scholar]
- 27.Lu S, Zhang J: Small molecule allosteric modulators of G-protein-coupled receptors: drug-target interactions. J Med Chem 2019, 62:24–45. [DOI] [PubMed] [Google Scholar]
- 28.Yang J-S, Seo SW, Jang S, Jung GY, Kim S: Rational engineering of enzyme allosteric regulation through sequence evolution analysis. PLoS Comput Biol 2012, 8:e1002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuetz AK, Kay LEJE: A dynamic molecular basis for malfunction in disease mutants of p97/VCP. eLife 2016, 5: e20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nussinov R: Allosteric modulators can restore function in an amino acid neurotransmitter receptor by slightly altering intramolecular communication pathways. Br J Pharmacol 2012, 165:2110–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nussinov R, Tsai C-JJC: Allostery in disease and in drug discovery. Cell 2013, 153:293–305. [DOI] [PubMed] [Google Scholar]
- 32.Clarke D, Sethi A, Li S, Kumar S, Chang RWF, Chen J, Gerstein M: Identifying allosteric hotspots with dynamics: application to interand intra-species conservation. Structure 2016, 24:826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abrusan G, Marsh JA: Ligand binding site structure influences the evolution of protein complex function and topology. Cell Rep 2018, 22:3265–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]; • A comparison of ligand-binding homomeric protein complexes where the binding site is formed from multiple-chains as opposed to a single chain. Multiple bioinformatics resources were used to leverage known protein structures and their binding sites.
- 34.Shen Q, Wang G, Li S, Liu X, Lu S, Chen Z, Song K, Yan J, Geng L, Huang Z et al. : ASD v3.0: unraveling allosteric regulation with structural mechanisms and biological networks. Nucleic Acids Res 2016, 44:D527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schymkowitz JW, Rousseau F, Martins IC, Ferkinghoff-Borg J, Stricher F, Serrano L: Prediction of water and metal binding sites and their affinities by using the Fold-X force field. Proc Natl Acad Sci U S A 2005, 102:10147–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monod J, Changeux JP, Jacob F: Allosteric proteins and cellular control systems. J Mol Biol 1963, 6:306–329. [DOI] [PubMed] [Google Scholar]
- 37.Zhang S, Li H, Krieger JM, Bahar I: Shared signature dynamics tempered by local fluctuations enables fold adaptability and specificity. Mol Biol Evol 2019, 36:2053–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The authors detail the methodology of their new implementation of elastic network models for study of homologous proteins. As an example, they publish comparative analysis of intrinsic dynamics of three protein families.
- 38.Naganathan AN: Modulation of allosteric coupling by mutations: from protein dynamics and packing to altered native ensembles and function. Curr Opin Struct Biol 2019, 54:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loewe L, Hill WG: The population genetics of mutations: good, bad and indifferent. Philos Trans R Soc Lond B Biol Sci 2010, 365:1153–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charlesworth B: The effects of deleterious mutations on evolution at linked sites. Genetics 2012, 190:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crow JF: The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 2000, 1:40–47. [DOI] [PubMed] [Google Scholar]
- 42.Doshi U, Holliday MJ, Eisenmesser EZ, Hamelberg D: Dynamical network of residue–residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc Natl Acad Sci U S A 2016, 113:4735–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vu PJ, Yao X-Q, Momin M, Hamelberg D: Unraveling allosteric mechanisms of enzymatic catalysis with an evolutionary analysis of residue–residue contact dynamical changes. ACS Catal 2018, 8:2375–2384 [Google Scholar]; •Dynamic changes in the form of contact formation and breaking were studied in cyclophin isoforms by comparing substrate-free and the cisbound states. Changes among residues occurred at a site 15 Å away from the active site, suggesting the allosteric nature of cyclophilins.
- 44.Holliday MJ, Camilloni C, Armstrong GS, Vendruscolo M, Eisenmesser EZ: Networks of dynamic allostery regulate enzyme function. Structure 2017, 25:276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L, Goodey NM, Benkovic SJ, Kohen A: Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proc Natl Acad Sci U S A 2006, 103:15753–15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Tharp S, Selzer T, Benkovic SJ, Kohen A: Effects of a distal mutation on active site chemistry. Biochemistry 2006, 45:1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Isogai S, Deupi X, Opitz C, Heydenreich FM, Tsai CJ, Brueckner F, Schertler GF, Veprintsev DB, Grzesiek S: Backbone NMR reveals allosteric signal transduction networks in the beta1-adrenergic receptor. Nature 2016, 530:237–241. [DOI] [PubMed] [Google Scholar]
- 48.Shen Q, Cheng F, Song H, Lu W, Zhao J, An X, Liu M, Chen G, Zhao Z, Zhang J: Proteome-scale investigation of protein allosteric regulation perturbed by somatic mutations in 7,000 cancer genomes. Am J Hum Genet 2017, 100:5–0020 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Somatic missense mutations in cancer genomes were studied to find that deleterious mutations are enriched at allosteric sites. Predicting significantly mutated proteins with AlloDriver helped find known and novel proteins that cause allosteric dysregulation, further imposing the role of allosteric mutations in tumorigenesis.
- 49.Huang Z, Mou L, Shen Q, Lu S, Li C, Liu X, Wang G, Li S, Geng L, Liu Y et al. : ASD v2.0: updated content and novel features focusing on allosteric regulation. Nucleic Acids Res 2014, 42: D510–6 (Database issue). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bezginov A, Clark GW, Charlebois RL, Dar VU, Tillier ER: Coevolution reveals a network of human proteins originating with multicellularity. Mol Biol Evol 2012, 30:332–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Przytycka TM, Jothi R, Aravind L, Lipman DJ: Differences in evolutionary pressure acting within highly conserved ortholog groups. BMC Evol Biol 2008, 8:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu S, He X, Ni D, Zhang J: Allosteric modulator discovery: from serendipity to structure-based design. J Med Chem 2019, 62:6405–6421. [DOI] [PubMed] [Google Scholar]
- 53.Lu S, Huang W, Wang Q, Shen Q, Li S, Nussinov R, Zhang J: The structural basis of ATP as an allosteric modulator. PLoS Comput Biol 2014, 10:e1003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tubeleviciute-Aydin A, Zhou L, Sharma G, Soni IV, Savinov SN, Hardy JA, LeBlanc AC: Rare human Caspase-6-R65W and Caspase-6-G66R variants identify a novel regulatory region of Caspase-6 activity. Sci Rep 2018, 8:4428. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Studying natural single amino acid polymorphisms in Casp6 revealed that variants Casp6-G66R and Casp6-R65W have lesser proteolytic activity, which also helped in finding a novel regulatory area within Casp6.
- 55.Tubeleviciute-Aydin A, Beautrait A, Lynham J, Sharma G, Gorelik A, Deny LJ, Soya N, Lukacs GL, Nagar B, Marinier A et al. : Identification of allosteric inhibitors against active caspase-6. Sci Rep 2019, 9:5504. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Active Caspase6 (Casp6) inhibition is important as it is known to accumulate before the onset of Alzheimer’s disease. Visualizing the positions of four natural variants of Casp6 (Casp6-A34E, Casp6-E35K, Casp6-A109T) on X-ray crystallography structures along with molecular modelling helped identify an allosteric binding pocket that can be targeted for drug development.
- 56.Wang XJ, Cao Q, Liu X, Wang KT, Mi W, Zhang Y, Li LF, LeBlanc AC, Su XD: Crystal structures of human caspase 6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep 2010, 11:841–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Velazquez-Delgado EM, Hardy JA: Phosphorylation regulates assembly of the caspase-6 substrate-binding groove. Structure 2012, 20:742–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao Q, Wang XJ, Liu CW, Liu DF, Li LF, Gao YQ, Su XD: Inhibitory mechanism of caspase-6 phosphorylation revealed by crystal structures, molecular dynamics simulations, and biochemical assays. J Biol Chem 2012, 287:15371–15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Velazquez-Delgado EM, Hardy JA: Zinc-mediated allosteric inhibition of caspase-6. J Biol Chem 2012, 287:36000–36011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anderson BW, Liu K, Wolak C, Dubiel K, She F, Satyshur KA, Keck JL, Wang JD: Evolution of (p)ppGpp-HPRT regulation through diversification of an allosteric oligomeric interaction. eLife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stanger K, Steffek M, Zhou L, Pozniak CD, Quan C, Franke Y, Tom J, Tam C, Krylova I, Elliott JM et al. : Allosteric peptides bind a caspase zymogen and mediate caspase tetramerization. Nat Chem Biol 2012, 8:655–660. [DOI] [PubMed] [Google Scholar]
- 62.Chen B, Altman RB: Opportunities for developing therapies for rare genetic diseases: focus on gain-of-function and allostery. Orphanet J Rare Dis 2017, 12:61. [DOI] [PMC free article] [PubMed] [Google Scholar]