

Abstract
Background
We conducted a phase I clinical trial that infused CCR5 gene–edited CD4+ T cells to determine how these T cells can better enable HIV cure strategies.
Methods
The aim of trial was to develop RNA-based approaches to deliver zinc finger nuclease (ZFN), evaluate the effect of CCR5 gene–edited CD4+ T cells on the HIV-specific T cell response, test the ability of infused CCR5 gene–edited T cells to delay viral rebound during analytical treatment interruption, and determine whether individuals heterozygous for CCR5 Δ32 preferentially benefit. We enrolled 14 individuals living with HIV whose viral load was well controlled by antiretroviral therapy (ART). We measured the time to viral rebound after ART withdrawal, the persistence of CCR5-edited CD4+ T cells, and whether infusion of 10 billion CCR5-edited CD4+ T cells augmented the HIV-specific immune response.
Results
Infusion of the CD4+ T cells was well tolerated, with no serious adverse events. We observed a modest delay in the time to viral rebound relative to historical controls; however, 3 of the 14 individuals, 2 of whom were heterozygous for CCR5 Δ32, showed post-viral rebound control of viremia, before ultimately losing control of viral replication. Interestingly, only these individuals had substantial restoration of HIV-specific CD8+ T cell responses. We observed immune escape for 1 of these reinvigorated responses at viral recrudescence, illustrating a direct link between viral control and enhanced CD8+ T cell responses.
Conclusion
These findings demonstrate how CCR5 gene–edited CD4+ T cell infusion could aid HIV cure strategies by augmenting preexisting HIV-specific immune responses.
REGISTRATION
ClinicalTrials.gov NCT02388594.
Funding
NIH funding (R01AI104400, UM1AI126620, U19AI149680, T32AI007632) was provided by the National Institute of Allergy and Infectious Diseases (NIAID), the National Institute on Drug Abuse (NIDA), the National Institute of Mental Health (NIMH), and the National Institute of Neurological Disorders and Stroke (NINDS). Sangamo Therapeutics also provided funding for these studies.
Keywords: AIDS/HIV
Keywords: Gene therapy, T cells

Introduction
Prolonged HIV-1 infection results in massive CD4+ T cell loss and profound immunodeficiency (1). Even with effective antiretroviral therapy (ART), the resident CD4+ T cell response does not fully recover (2–4). Damage to lymphoid tissue, lack of differentiation to enable improved effector function, and preferential killing of HIV-specific CD4+ T cells all limit the CD4+ T cell response (5). This lack of CD4+ T cell activity impedes the development of HIV functional cure strategies, given the central role that CD4+ T cells play in coordinating and executing effective and sustained immune responses (6, 7). Although HIV eradication approaches are being developed (8), functional cure strategies in which an engineered immune system is designed to control HIV reactivation may be both more attainable and effective, since a functionally cured individual will likely be more resistant to HIV reinfection. Adoptive T cell therapy is a promising approach for restoring CD4+ T cell activity in people living with HIV (PLWH) (9). The first clinical trial that infused ex vivo CD4+ T cells in PLWH took advantage of a T cell manufacturing process that substantially reduced CCR5 expression via a β-chemokine and KLF2-dependent process so that these T cells could be expanded in the absence of ART, and could thus be infused safely in PLWH without augmenting HIV replication (10–14). Interestingly, individuals who received ex vivo–expanded CD4+ T cells had sustained increases in peripheral CD4+ T cell levels, and these studies laid the foundation for future studies that used engineered T cells. However, these infused T cells eventually reacquired susceptibility to HIV once reinfused, limiting their ability to be part of an HIV cure strategy.
A number of strategies to protect infused CD4+ T cells from HIV infection have been devised, including overexpression of restriction factors, use of fusion inhibitors, targeting HIV-coreceptor expression through RNA and genome editing approaches, and targeting HIV through siRNA, dominant-negative proteins, and antisense RNA (15–17). Of these, a handful have been tested in the clinic (17, 18). In short, these studies have shown that it is feasible to manufacture up to 3 × 1010 engineered HIV-1–resistant CD4+ T cells and that infusion of these T cells was well tolerated (15). However, it has been challenging to demonstrate whether these engineered T cells had a durable therapeutic benefit. A previous study that infused up to 1 × 1010 CD4+ T cells rendered CCR5 deficient by adenoviral delivery of CCR5-targeted zinc finger nucleases (ZFNs) showed that an infusion of CCR5 gene–edited T cells led to a survival advantage of the genetically modified cells (19). Additionally, 1 individual who was retrospectively identified to be heterozygous for Δ32 had an undetectable viral load after a 12-week analytical treatment interruption (ATI). Moreover, there was a correlation between the reduction of HIV DNA present in the blood and the number of CCR5-edited alleles (19).
Here, we performed a similarly structured phase I clinical trial involving 14 participants who were chronically infected with HIV and fully suppressed on ART to test improvements in the T cell manufacturing process, determine whether the use of cyclophosphamide as pre-infusion lymphodepletion increased the engraftment of the genetically modified cells, and further explore how the infusion of CCR5 gene–edited T cells may facilitate a functional cure of HIV infection. We confirmed that these infusions can delay viral rebound but have no long-term effect on the viral reservoir. Importantly, we observed that a subset of individuals had improved HIV-specific CD8+ T cell responses that persisted at least 6 months after the resumption of ART. In individuals with improved HIV-specific immune responses, we observed some post-rebound control of HIV replication, and in 1 individual, we observed viral escape in the case of one of these heightened CD8+ T cell responses. Together, our data show the potential of HIV-resistant CD4+ T cells to reinvigorate the endogenous HIV-specific immune response and augment post-viral rebound control of HIV replication.
Results
Study timeline, patient demographics, T cell infusion product characterization, and adverse events.
We conducted a 3-arm, open-label pilot study in chronically infected, antiretroviral HIV–suppressed individuals to determine the safety and antiviral activity of a single infusion of autologous CD4+ T cells edited at the CCR5 gene locus via electroporation of in vitro–transcribed mRNA encoding the CCR5-targeted ZFNs (SB-728mR-T), with or without prior administration of 1 of 2 different doses of cyclophosphamide (CTX) (1 g/m2 or 1.5 g/m2) (Figure 1 and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI144486DS1). Eligibility criteria allowed for participation of individuals 18 years of age or older with documented HIV infection and a known viral set point, a CD4+ T cell count of 450 cells/mm3 or higher, a CD4 nadir of not less than 200 cells/mm3 (Karnofsky’s performance score of 70 or higher), and adequate venous access for apheresis. Participants were also required to be clinically stable on their first or second ART regimen, with no changes within 4 weeks of enrollment, and to have undetectable HIV-1 RNA levels in the 60-day period prior to screening. Subjects in cohorts 2 and 3 were required to have a left ventricular ejection fraction of 40% or higher.
Figure 1. Schematic outlining the timeline and sample collection points for NCT02388594.

The clinical study was divided into 4 steps. During step 1, participants underwent leukapheresis to collect cells for manufacturing, followed by a second leukapheresis and rectal biopsy to serve as baseline samples. In cohorts 2 and 3, participants were treated with a single dose of cyclophosphamide 2 days before receiving modified cells (day –2). Participants entered step 2 on the day of cell infusion (day 0), and the edited cells were allowed to engraft for either 4 weeks (cohorts 2 and 3) or 8 weeks (cohort 1) before the 16-week analytical treatment interruption was initiated in step 3. At the conclusion of the treatment interruption, the participants entered step 4 to be monitored for safety until HIV RNA levels fell below the limit of quantification. Safety laboratory values and HIV viral load were monitored at regular intervals throughout the study.
Cohort 1 included 3 participants who received genetically modified cells but not CTX. This cohort was enrolled first to ensure there were no safety issues associated with using mRNA for CCR5 gene editing, as opposed to adenoviral vectors. Subsequently, participants in cohort 2 (which included only individuals who were WT for the CCR5 genotype) and participants in cohort 3 (individuals with the CCR5 Δ32 heterozygote genotype) were enrolled. The first 3 individuals each in cohorts 2 and 3 received the low dose of CTX (1 g/m2), and the remaining participants received the higher dose of CTX (1.5 g/m2) prior to T cell infusion. Cohort 3 included only those individuals with the CCR5 Δ32 heterozygote genotype, to determine whether these individuals were more likely to have sustained viral control in the absence of ART.
Table 1 summarizes the demographic characteristics of the 14 participants: 3 in cohort 1 (no CTX); 6 in cohort 2 (CTX and CCR5 WT/WT); and 5 in cohort 3 (CTX and CCR5 WT/Δ32 heterozygotes). Ninety-three percent of the participants were male and 7% were female, 57% were African American, 43% were White, and 7% were Hispanic. They had a median age of 44 years (IQR 36–51). The median baseline CD4 count was 693 c/mm3 (IQR 630–1030), and all participants had suppressed HIV replication via ART. Cohorts 2 and 3 received a single dose of CTX two days before the infusion of genetically modified cells. To manufacture CCR5-edited CD4+ T cells, we purified CD4+ T cells from apheresis product, electroporated RNA encoding CCR5 ZFNs, incubated the cells at 30°C for 2 days, as cold shock has been shown to promote gene disruption (20), and expanded the cells using CD3/CD28-coated beads (13). According to the protocol, all participants received a single infusion of 10 billion cells, except participant 204, who received only 6.6 billion cells because of insufficient ex vivo T cell expansion during manufacturing. The percentage of CCR5 editing in the product varied between 10% and 34%, with 20% to 30% of the CCR5 alleles edited in most products. Infusion of SB-728mR-T was safe and generally well tolerated: we observed a total of 31 adverse events in cohort 1 (n = 24 severity grade 1; n = 7 severity grade 2) and 97 adverse events in cohorts 2 and 3 (n = 79 severity grade 1; n = 12 severity grade 2; n = 5 severity grade 3; n = 1 severity grade 4). There were no serious adverse events related to SB-728mR-T infusion. Supplemental Table 1 describes all of the adverse events associated with this clinical trial.
Table 1. Patient demographics and cell manufacturing.

Slight delay in viral rebound after infusion of CCR5 ZFN–treated CD4+ T cells.
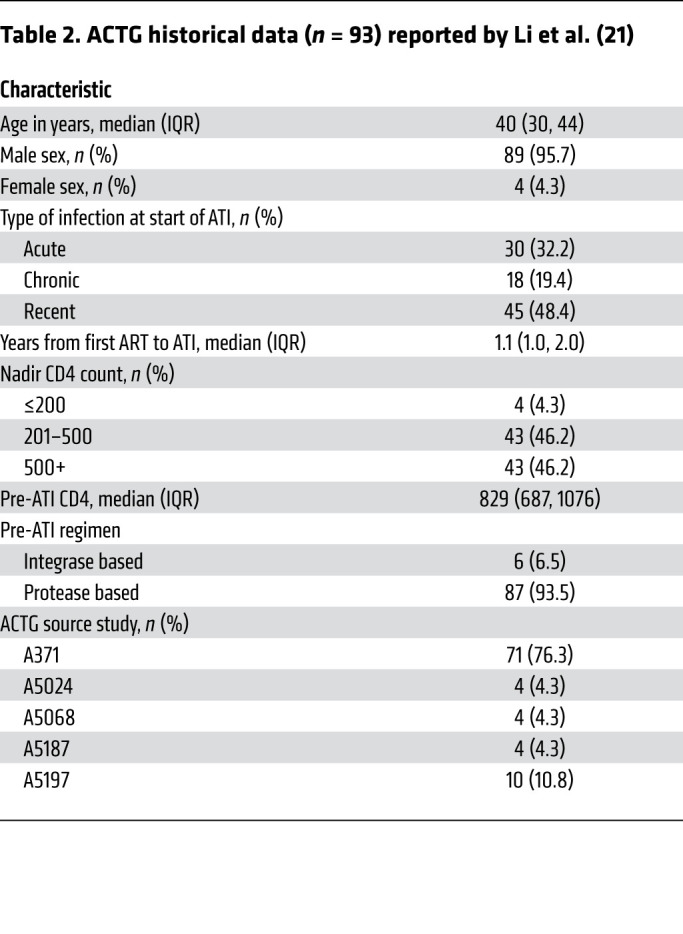
We performed an ATI to determine whether PLWH who received an infusion of SB-728mR-T had a delay in viral rebound after ART was discontinued relative to the AIDS Clinical Trial Group (ACTG) historical controls. This historical control cohort includes 93 ACTG trial participants whose viral rebound data were captured in four ACTG ATI studies without additional interventions (ACTG 371, A5024, A5068, and A5197) (21–25). The historical controls included acutely infected, recently infected, and chronically infected individuals to account for the uncertainty about the length of infection in our participants. The controls were selected on the basis of similar inclusion criteria (Table 2): age 18–65 years (median age, 40 yr; IQR, 30–44); sex (male, 96%; female, 4%); plasma HIV-1 RNA of fewer than 50 copies/mL for at least 12 months before ATI while on combination ART; a CD4+ count at the time of the ATI of more than 500 cells/μL (median, 829; IQR, 687 and 1076); a CD4 nadir of more than 200 cells/μL (96%); and frequent viral load measurements at least until viral rebound occurred. Similar to the historical controls, most participants in our study were on integrase inhibitor or boosted protease inhibitor–based (PI-based) therapy. The participants on non-nucleoside reverse transcriptase inhibitor (NNRTI) therapy, based on the participant’s preference in consultation with the referring physician, either (a) discontinued the NNRTI 7 days prior to discontinuation of the other antiretroviral agents in their regimens or (b) initiated a potent PI-based regimen (with discontinuation of the NNRTI) without altering the nucleosides of the regimen for the last 2 weeks of highly active antiretroviral therapy (HAART), prior to simultaneous interruption of all antiretroviral agents. This was done to account for the long-term pharmacokinetic decay of non-nucleoside reverse transcriptase antiretroviral regimens, which has been associated with delays in virological rebound during ATI studies (21, 26).
Table 2. ACTG historical data (n = 93) reported by Li et al. (21).
In cohort 1, the participants began an ATI 8 weeks after infusion with SB-728mR-T cells. In cohorts 2 and 3, the participants began ATI 4 weeks after infusion with SB-728mR-T cells, similar to our previous ZFN study (27). Most participants rebounded by week 4 of the ATI and reached peak viremia by week 8. We found that the rebound of HIV viremia (HIV RNA >200 copies/mL) was delayed compared with the ACTG historical controls (Figure 2A) when considering all of the participants (Peto-Peto P = 0.01), but there was a lack of statistical power to detect whether any of the cohorts were individually delayed relative to the control group or to any of the other cohorts (Figure 2B). Additionally, pretreatment with CTX (either dose) did not have a detectable effect on the time to virologic rebound (Peto-Peto P = 0.70).
Figure 2. Time to viral rebound during an ATI.

(A) All individuals who received CCR5-edited CD4+ T cells were compared with historical controls for the time to reach a viral load of at least 200 copies/mL. The Peto-Peto P value is shown for the group comparison of the survival curves. (B) Each cohort within the clinical trial was compared with the ACTG control group. Participant 305 was excluded because of continued ART during the ATI. Pt, participant.
Changes in circulating lymphocytes and persistence of CCR5-edited CD4+ T cells.
We tracked the persistence of engineered T cells, total CD4+ and CD8+ T cells, and viral load throughout the duration of the clinical trial (Figure 3). The median (IQR) percentage of CCR5-edited alleles in CD4+ T cells was 17.0% (11.6%, 26.2%) one week after infusion (Table 3). The engraftment of the gene-edited cells varied between cohorts 4 weeks after infusion (Kruskal-Wallis P = 0.03), with a nonsignificant trend toward greater early engraftment in the CTX groups that was significant for the Δ32 group (cohort 3) compared with the control group (cohort 1) (Wald test from linear regression [LR], P = 0.02). Considering the average percentage of CCR5-edited CD4+ T cells during the 16-week ATI, calculated using the participant’s AUC divided by length of the 16-week ATI, we found no detectable differences between groups; however, there was a trend toward greater engraftment in the CTX groups (LR P = 0.09) and in the heterozygous group compared with the control group (LR P = 0.06). We used a simple linear mixed-effects model, with fixed effect for time and a random effect for subject, to examine patterns in the change from baseline based on the available measurements in the period prior to and during each participant’s ATI. Prior to ATI, there was an average (95% CI) change from baseline of 17.0%/week (6.6%, 27.4%) for CD4, without any detectable trends for CD8, which had an average change from baseline of –0.5%/week (–2.9%, 1.9%). During the ATI, participants across all groups experienced an average (95% CI) percentage of decrease in CD4 levels relative to the pre-ATI level of 1.1%/week (0.8%, 1.4%) and an average increase in CD8 of 3.2%/week (2.4%, 4.0%), with no consistent trend detectable in the percentage of CCR5-edited CD4+ T cells, for which there was an average change of –0.6%/week (–3.2%, 1.9%) relative to pre-ATI levels. The overall pattern in the number of CCR5-edited alleles in the CD4+ T cells across the entire study period was similar to that previously described, in that there tended to be a notable increase in gene-edited cells after infusion, followed by an eventual decline in peripheral blood, which may indicate cell death or trafficking into tissues (Figure 2). All participants were advised to restart ART at week 16 of the ATI. However, 3 participants (1 WT/WT [participant 203] and 2 WT/Δ32 CCR5 Δ32 heterozygotes [participants 301 and 303]) with low-level viremia (<1000 copies/mL) elected to extend the ATI. They continued to maintain low-level viremia and restarted treatment 26, 33, and 44 weeks after infusion, respectively (see the individual trajectories of the viral load in Figure 3). One of these individuals had a “low-positive” result for the presence of antiretrovirals in urine on 1 isolated occasion during the ATI. Comparing individuals whose average percentage of CCR5-edited CD4+ T cells during the 16-week ATI was above, versus below, the median, we found no a significant difference in the time to virologic failure (Peto-Peto P = 0.24), with a HR (95% CI) for failure of 1.93 (0.58, 6.37) in the group below the median. Together, these data indicate that some of the individuals who received CCR5 gene–edited CD4+ T cells exhibited limited post-rebound control of viral replication, consistent with previous descriptions of post-rebound controllers (28).
Figure 3. Analysis of CD4+ and CD8+ T cell counts, viral load, and percentage of CCR5 disruption within peripheral CD4+ T cells.

At the indicated time points, CD4+ and CD8+ T cell counts as well as viral load were measured. The percentage of CCR5 disruption was calculated as described in Methods. Individuals who participated in an extended ATI are indicated with a red subject ID.
Table 3. Persistence of CD4+ T cells with disrupted CCR5 alleles.

No change in viral reservoir after infusion of CCR5 gene–edited T cells.
The ultimate goal of HIV cure research is to eliminate the latent viral reservoir. However, more immediate safety concerns focus on whether the use of immunological interventions and ATIs augment the latent reservoir (29). To address these questions, we performed the recently described intact proviral DNA assay (IPDA) (30) to address the intact reservoir levels prior to treatment and at least 6 months after the resumption of ART following ATI. We found no significant difference in the reservoir levels before or after therapy, indicating that infusion of SB-728mR-T cells followed by an ATI had no beneficial effects in reducing the viral reservoir, but also did not significantly increase the size of the intact reservoir, 5′ defective viruses, or 3′ defective viruses (Figure 4, A–C). Of interest, the individuals with CCR5 heterozygosity (cohort 3) had lower reservoir levels. Given the small number of participants in this clinical trial, it is unclear whether this lower reservoir level in CCR5 Δ32 heterozygotes would be observed in a larger cohort of individuals, but the results contrast with other studies that measured the HIV reservoir in this population using less precise methods (31).
Figure 4. Viral reservoir before and after infusion of CCR5-edited CD4+ T cells.

An IPDA was performed before and at least 6 months after the reinitiation ART. (A) Number of integrated HIV genomes with defective 5′ sequences per million CD4+ T cells. (B) Number of integrated HIV genomes with defective 3′ sequences per million CD4+ T cells. (C) Number of integrated HIV genomes with intact viruses per million CD4+ T cells. P values were calculated by paired t test. Blue indicates individuals with CCR5 Δ32 heterozygosity, and green represents CCR5 WT individuals.
Heightened HIV-specific CD8+ T cell responses in individuals with regained control of HIV replication.
Given the delay in viral rebound and the evidence of post-rebound control of HIV replication in a subset of participants who received CCR5 ZFN–treated CD4+ T cells, we investigated the state of HIV-specific CD8+ T cell responses before and after SB-728mR-T infusion. We limited our analysis to the HIVGag response so it would be feasible to do peptide deconvolution to identify the reactive peptides. To study both the breadth and magnitude of the HIVGag T cell response, we used PBMCs isolated prior to SB-728mR-T infusion (apheresis no. 2), as well as PBMCs collected at least 6 months after the reinstitution of ART (apheresis no. 5) to allow the HIV-specific immune response to return to homeostasis after seeing robust levels of antigen during the ATI (Figure 5, A and B, and Supplemental Table 2). Consistent with previous reports analyzing the HIV-specific T cell response before and after ATI (32), most individuals had no noticeable difference in their HIV-specific CD8+ T cell response. However, 3 of the individuals showed impressive differences in both the breadth and magnitude of their HIV-specific CD8+ T cell response (Figure 5B). Interestingly, these were the same individuals who showed some post-rebound control of HIV replication (Figure 3). We also studied the HIVGag CD4+ T cell response and observed no changes before or after ATI (Supplemental Figure 2). We performed deconvolution analysis of the peptide pool data to identify which peptide was being targeted by each heightened response. We then obtained the HLA haplotype of each individual, noticing that none of these individuals harbored protective alleles (33). We then used computer algorithms to determine which HLA allele bound the targeted peptide with the highest affinity. For several of the responses, we were able to identify which allele likely presented the peptide, but for some of the alleles there were no peptide-binding models available (Figure 5C). Together, these findings suggest that infusion of CCR5 ZFN–treated CD4+ T cells reinvigorated the HIV-specific CD8+ T cell responses in a subset of individuals who demonstrated post-treatment control of HIV replication similar to that described in other studies (28).
Figure 5. Enhanced HIVGag-specific CD8+ T cell responses were observed after infusion of CCR5-deficient CD4+ T cells.

(A) Representative flow cytometric plots showing the IFN-γ response to Gag pool number 21 from cells collected from participant 303 during apheresis numbers 2 and 5. (B) Heatmap showing the change in the percentage of IFN-γ+ CD8+ T cells when exposed to the indicated peptide pool between apheresis number 2 and number 5. (C) Deconvolution of peptide pool data shows where within the Gag protein enhanced responses were observed. Patient ID, MHC haplotype, and epitope for each enhanced response greater than 0.5%. Ensemble score estimates peptide affinity for a particular HLA allele, with the highest-affinity allele identified. Green indicates that the allele affinity is under 500 µM, and yellow highlights alleles in which there is no predication model.
Viral escape observed in a heightened CD8+ T cell response.
To better support the hypothesis that the heightened CD8 HIV–specific response observed following the infusion of CCR5-deficient CD4+ T cells was responsible for post-rebound control of HIV replication in some participants, we sequenced HIV-1Gag in virus circulating before and after the observed periods of virus control in the 3 participants. Single-genome sequences of Gag (n = 35, median; n = 6 per participant time point) demonstrated the expected sequence diversity among the participants’ Gag sequences. Comparing each participant’s early and late sequences, however, we detected only 1 conserved change within Gag, which fell within the predicted CTL epitope for participant 303 (Figure 6, A and B), suggesting viral escape from T cell responses against this epitope. To validate the observation that this Gag mutation conferred viral escape, we synthesized both the WT (ERFALNPGL) and mutated (ERFAFNPGL) peptides to interrogate the CD8+ T cell response. Compared with the robust CD8+ T cell response to the WT peptide, we observed a substantially reduced response to the mutated peptide, indicating that this mutation allows viral escape (Figure 6C). Together, these data suggest that this dominant, reinvigorated CD8+ T cell response in subject 303 was sufficiently robust to drive the selection of an HIV escape mutant.
Figure 6. Immune escape was observed following a heightened immune response.

(A) Consensus amino acid sequences, derived by single-genome sequencing (the number of sequences is enumerated on the right), are shown for the predicted CD8+ T cell epitopes within Gag for participants 203, 301, and 303, at early and late time points. In participant 303, the fifth position of the first Gag T cell epitope (highlighted in yellow), changed from lysine (in 5 of 5 sequences) to phenylalanine in (6 of 6 sequences). (B) Highlighter plot of Gag amino acid single-genome sequences for participant 303, demonstrating a change in the CTL epitope from consensus (L) to F. (C) Both WT and L-to-F–mutated peptides from Gag pool number 21 were incubated with PBMCs isolated from participant 303, and intracellular IFN-γ production was measured.
Discussion
To date, the only individuals who have been cured of HIV infection have received a CCR5-deficient reconstituted immune system via bone marrow transplantation (34–36). Although large-scale implementation of this strategy remains impractical, lessons can be learned that would aid in the design of a strategy that could be implemented in healthy PLWH. For one, an HIV cure is possible. Second, rendering cells CCR5 deficient is an effective strategy to block HIV replication. Genome editing provides an avenue by which an HIV-positive individual’s own cells can be made CCR5 defective (37–39), but currently, this can only be done ex vivo, limiting the number of cells that can be rendered CCR5 deficient. Previously, up to 10 billion autologous CD4+ T cells that were treated with CCR5 ZFNs delivered by Ad5f35 vectors were infused into 12 PLWH (19). One individual who was subsequently identified to be a heterozygous for CCR5 Δ32 experienced viral rebound, but this person’s viral load later became undetectable by the end of the 12-week ATI. Moreover, 4 other individuals experienced a more than 10-fold reduction in their viral load after an initial peak was reached, suggesting post-rebound control of HIV replication. The goals of the study described here were to test improvements in the cell manufacturing process, explore strategies to improve the engraftment of CCR5 gene–edited T cells, and uncover mechanisms that may be responsible for limited post-rebound control of HIV replication.
First, we confirmed that infusion of up to 10 billion ZFN gene–edited CD4+ T cells was both safe and feasible. Additionally, 2 of the individuals involved in this clinical study were also participants in the original phase I clinical trial, and T cells with defective CCR5 alleles were still present up to 5 years after the original infusions, confirming the long-term persistence of gene-edited CD4+ T cells, and consistent with previous cohort studies of chimeric antigen receptor T cells (40). Additionally, we observed no unique adverse events in those who received repeated dosing of CCR5 gene–edited CD4+ T cells, suggesting that multiple infusions of these cells is possible and may lead to a higher proportion of HIV-resistant T cells. Interestingly, participant 301 in this study was the Δ32 heterozygotic participant 205 from the original study who appeared to regain control of HIV replication at the end of the ATI. In the current study, we recruited 4 additional participants with Δ32 heterozygosity to determine whether these individuals preferentially benefit from infusion of CCR5 ZFN–treated CD4+ T cells. In general, we observed better early engraftment of the genome-engineered T cells in the Δ32 heterozygotic cohort, but by the end of the 16-week ATI, there were no statistically significant differences (Table 3). Moreover, we observed no differences in the time to viral rebound, despite the much lower HIV reservoir levels in this cohort, casting further doubt on the existence of a relationship between the time to viral rebound and the number of intact, latent viruses (41). However, 2 of the 4 evaluable individuals (1 member of this cohort continued to undergo ART throughout the ATI) became post-rebound controllers, whereas only 1 of 9 of the CCR5 WT individuals became post-rebound controllers. Because the participants with Δ32 heterozygosity already have 1 defective CCR5 allele, there is a greater chance that the introduction of the CCR5 ZFNs would render their T cells fully CCR5 deficient. This suggests that if gene editing were more efficient, the CCR5 WT individuals could resemble individuals heterozygous for CCR5 Δ32 and be more likely to achieve post-rebound HIV control. New technologies to enhance the efficiency of gene editing while maintaining safety and specificity are emerging, including incorporation of ZFN structures into lipid nanoparticles (42) and improvement of ZFN structure algorithms that are able to generate ZFNs with a higher degree of precision, efficiency, and specificity (43) — and these improvements should be considered for future clinical trials. Other gene-editing methods including CRISPR are also in development for HIV (44).
The use of adenovirus-based vectors has been associated with the formation of immune complexes, and this may be undesirable for use in HIV cure strategies (45, 46). To circumvent this potential limitation, we generated mRNA encoding the CCR5 ZFNs and transfected CD4+ T cells by electroporation. We observed similar levels of overall editing, but the types of mutations observed were distinct. The median CCR5 editing of 24% via mRNA delivery was comparable to the 23% observed previously with our Ad5f35 vector (19). Curiously, using Ad5f35 vectors to deliver CCR5 ZFNs, we found that approximately 25% of the edited CCR5 alleles contained a 5 nucleotide duplication, which simplified tracking CCR5 of the edited T cells, as quantitative PCR approaches could be used (37). We did not observe the preponderance of this duplication when mRNA was used to deliver the CCR5 ZFNs, suggesting that the method of delivery affects the DNA repair process. It is unclear why this difference occurred, but it may reflect how delivery of the ZFNs via activation of distinct TLR pathways can affect the activity of various DNA repair mechanisms (47). Given equivalent disruption efficiency, reduced cost, and ease in manufacturing, the production of mRNA-encoding ZFNs is an attractive means to perform genome editing.
Adoptive T cell therapy studies in patients with cancer have demonstrated that lymphodepletion prior to infusion enhances both the engraftment and efficacy of the transferred T cells (48). We asked whether lymphodepletion prior to infusion of T cells from PLWH would have a similar effect. Although treatment of these patients with CTX was well tolerated, the overall effects were at best modest, and we did not observe a statistically significant difference in engraftment between those who received CTX at either dose and those who did not receive CTX. The lack of effect may reflect that the single, relatively low dose of CTX was not sufficiently immunosuppressive to create more space for the infused T cells to engraft, or that lymphodepletion in healthy PLWH does not have the same benefit as that for patients with advanced cancer. Thus, in the absence of greater clarity on how to alter the lymphodepletion protocol to better promote T cell engraftment in PLWH, we do not recommend that CTX be included in any further HIV cure studies as a means to bolster engineered T cell engraftment.
We performed an ATI to determine whether CCR5-deficient T cells could prevent viral rebound. Similar to the first clinical trial, we observed a slight delay when compared with a matched ACTG control cohort. However, because our protocol allowed the ATI to continue for 16 weeks, or until an individual had a viral load of over 100,000 for 3 consecutive weeks, we were able to observe some post-rebound control of HIV replication. Analysis of the HIV-specific immune response showed that all 3 individuals who had post-rebound control of HIV replication also maintained heightened HIVGag immune responses, suggesting a correlation between augmented CD8+ T cell responses and post-treatment control. It is important to emphasize that the apheresis samples from these individuals were collected 6 months after the end of the ATI, as was done for the other participants in the trial. Thus, even though these individuals were exposed to low levels of HIV antigen for a longer period, the T cells studied had not encountered HIV antigen for 6 months, indicating that these improved HIV-specific responses were durable. For 1 of the individuals, we were able to identify an immune escape variant that emerged at the time of viral recrudescence, indicating that the heightened CD8+ T cell response was sufficiently strong to drive selection of an escape mutant. For the other 2 individuals, viral escape could have occurred elsewhere in the virus, or the overall potency of the CD8+ T cell response was insufficiently potent to drive escape. Our studies, of course, do not rule out other activities that CD4+ T cells can bolster like the Ab responses that also might have played a role in the post-rebound control after CD4+ T cell infusion. Nonetheless, the demonstration of viral escape shows the potential of these engineered CD4+ T cells to reinvigorate the native CD8+ T cell, HIV-specific immune response and aid in HIV cure strategies.
Activated CD4+ T cells further mature antigen-presenting cells (APCs) so that they produce higher levels of CD8+ T cell–promoting molecules such as MHC, 4-1BBL, IL-15, CD70, CD86, and IL-12 (49) and also produce molecules such as CD40-L and IL-2 that directly augment CD8+ T cell activity (50, 51). However, CD4+ T cells do not need to recognize the same antigen, protein, or pathogen as the CD8+ T cell but rather just need to share an APC with the CD8+ T cell at the time of activation (52). Accordingly, we did not observe restoration of the HIVGag-specific CD4+ T cell response, suggesting that non-HIVGag, CCR5-edited CD4+ T cells were responsible for restoring function to the HIV-specific CD8+ T cells. This is not surprising, given the paucity of the HIV-specific CD4+ T cells that remain in PLWH (53). Thus, one explanation of why only a subset of individuals experienced a reinvigoration of their HIV-specific CD8+ T cells is that only this individual had an activated CD4+ T cell present to assist at the time of HIV-specific CD8+ T cell activation. While CCR5-competent CD4+ T cells are capable of providing help, they are also targets of HIV infection, which may interfere with their ability to sustain CD4+ T cell help. Thus, we postulate that it is likely that CCR5-edited CD4+ T cells were responsible for this help. Support for this hypothesis may be attained in an ongoing phase I clinical trial that is testing infusion of up to 10 billion CCR5 ZFN–treated, CD4+ CAR–expressing T cells (ClinicalTrials.gov ID: NCT03617198) (54). In the present study, HIV-specific and HIV-resistant CD4+ and CD8+ T cells were coinfused. We recently demonstrated that CD4+ T cells expressing an HIV-specific chimeric antigen receptor (CAR) can augment the function of CD8 CAR (55). Thus, it is likely that these CCR5-edited, CD4+ T cells expressing an HIV CAR will not only be able to augment the function of the CD8+ CAR T cells, but also the natural CD8+ T cell response we observed here. Together, these responses may be able to durably contain HIV replication in a sizeable proportion of individuals and thus become a key component of a functional cure strategy.
Methods
ZFN mRNA production.
The coding sequences for ZFNs 8196 and 8267 (37, 56) were each generated separately in the pIDTSmart vector (Integrated DNA Technologies). Plasmids were digested with BamHI and SpeI restriction enzymes, and the resulting fragments were ligated into the pDA-A.2bg.150A vector. These constructs were introduced into XL Blue Chemically Competent E Coli cells (Stratagene) for generation of a master cell bank. Plasmid DNA was generated from each master cell bank using the QIAfilter Plasmid Giga DNA isolation kit and linearized by SpeI digestion prior to mRNA in vitro transcription (IVT). The mMessage mMachine T7 Ultra Kit (Life Technologies, Thermo Fisher Scientific) was used for preparation of the IVT mRNA according to the procedure indicated by the manufacturer. The RNA was purified using the RNeasy Maxi kit (QIAGEN), and sequences were verified after production. As part of release testing, each mRNA lot was tested for identity (gel electrophoresis for size and sequencing), purity (OD 260:230 ratio >1.8; OD 260:280 ratio >2.0), and sterility (negative bacterial and fungal cultures).
T cell manufacturing.
Autologous peripheral blood lymphocytes were obtained via leukapheresis collection. Leukapheresis products from the participants were enriched for CD4+ cells by depletion of monocytes via counterflow centrifugal elutriation and by magnetically depletion of CD8+ T cells (CliniMACS CD8 Depletion, Miltenyi Biotec). The resulting enriched CD4+ T cells were electroporated with ZFN mRNAs in a closed system using the MaxCyte GT Flow Transfection System. Cells were next incubated at 30°C for 2 days prior to activation with anti-CD3/anti-CD28 mAb–coated paramagnetic beads (Dynabeads CD3/CD28 CTS, 43200D, Thermo Fisher Scientific,). The cells were then expanded and cultured in a closed system at 37°C with X-VIVO 15 media (Lonza) supplemented with the antiretroviral drugs Retrovir and Norvir (Abbott Laboratories). T cell expansion continued after transfer to a WAVE Bioreactor (Cytiva) for additional expansion under perfusion conditions. At the end of the culture period, cells were depleted of magnetic beads, washed, and formulated in infusible cryopreservation media. Prior to release, cells were tested for sentinel viability (≥70%); residual bead numbers (≤100 beads per 3 × 106 cells); bacterial and fungal contamination (no growth by day 7) and mycoplasma (negative) contamination; HIVGag provirus copies (post-expansion numbers not statistically greater than pre-expansion numbers); percentage of CD3/CD45 cells greater than or equal to 80%; endotoxin levels of 1.0 EU/mL or less; and CCR5 gene disruption (detectable).
Measurement of CCR5 disruption by targeted genome sequencing.
CCR5 gene modification efficiency was assessed at Sangamo Therapeutics as previously described (56).
IPDA.
Fifty million cells isolated by apheresis were shipped to Accelevir Diagnostics, where CD4+ T cells were purified by negative selection (Miltenyi Biotec). DNA from 0.5 × 107 to 1 × 107 CD4+ T cells was used to perform the IPDA as previously described (30).
Peptides.
One hundred twenty-three peptides (15 mers with an overlap of 11 amino acids) corresponding to full-length HIV Gag were obtained from the NIH AIDS Reagent Program (catalog 8117). Twenty-five pools, each with 5 peptides, were generated on the basis of their solubility. Some peptides were tested individually. ASRELERFAVNPGLL, ASRELERFALNPGLL, ASRELERFAFNPGLL, ERFAVNPGL, ERFALNPGL, and ERFAFNPGL peptides were synthesized at greater than 95% purity by New England Peptide.
Peptide-binding affinity analysis.
The MHC alleles of the participants were determined by the Clinical Immunology and HLA Immunogenetics Laboratory at the Hospital of the University of Pennsylvania. Peptide-MHC affinities were determined using the average predicted nanomolar affinities from MHCflurry 2.0 (57) and netMHC 4.0a (58) or netMHCpan 4.1b (59), as implemented in the antigen.garnish 2.0 R package (60). Alleles for which there were no prediction models in any of the programs were excluded from the analysis and included HLA-A*02:05, HLA-C*02:02, HLA-C*12:03, and HLA-C*14:03.
Cell stimulation and flow staining.
Cryopreserved human PBMCs or apheresis material was thawed in complete RPMI media (RPMI 1640 supplemented with 10% FCS, 100 U/mL penicillin, 100 μg/mL streptomycin sulfate) and 0.5 U/mL benzonase (MilliporeSigma), followed by incubation at 37°C in a 5% CO2 incubator for 30 minutes. Cells were then resuspended with complete media without Benzonase and rested at 37°C in a 5% CO2 incubator overnight. After resting, at least 0.5 × 106 cells were cultured with each pool or individual peptide at a concentration of 2 mg/mL of each peptide in 100 mL medium containing costimulatory Abs (anti-CD28 [555725] and anti-CD49d [555501], 1 μg/mL, BD Biosciences; GolgiStop [554724], 2 μL/mL, BD Biosciences; GolgiPlug [555029], 1 μg/mL, BD Biosciences; and anti–CD107a-BV421 [328626], BioLegend) for 6 hours at 37°C in a 5% CO2 incubator. Peptide diluent (0.03% DMSO) served as the negative control. PMA (10 ng/mL, MilliporeSigma) and ionomycin (1 μg/mL, MilliporeSigma) stimulation served as a positive control. Following incubation, the cells were washed with PBS and stained with LIVE/DEAD Fixable Aqua (Thermo Fisher Scientific) and then washed with flow buffer (PBS containing 1% BSA and 0.1% sodium azide). The cells were then by incubated with surface Abs against CD3 (SK7, APC-H7, 560176, BD Biosciences); CD14 (M5E2, V500, 561391, BD Biosciences); CD19 (HIB19, V500, 561121, BD Biosciences); CD4 (OKT4, BV785, 317442, BioLegend); and CD8a (RPA-T8, PE-Cy5.5, 35-0088-42, Thermo Fisher Scientific) for 20 minutes, followed by 2 washes with flow buffer. The cells were fixed with Cytofix/Cytoperm reagents (BD Biosciences) according to the manufacturer’s instructions. Following fixation, the cells were washed twice in 1× Perm/Wash Buffer and stained with Abs against intracellular IFN-γ (4S.B3, BV570, 502534, BioLegend) for 20 minutes. Following staining, the cells were washed twice and resuspended in 250 μL FACS buffer for acquisition on an LSRFortessa flow cytometer (BD Biosciences). Data were analyzed with FlowJo software, version 10.
Single-genome HIV-1 Gag sequencing.
Viral RNA (vRNA) from 100–400 μL plasma was extracted using the QIAGEN BioRobot EZ1 Workstation with the EZ1 Virus Mini Kit, version 2.0 (QIAGEN). Eluted vRNA was subsequently used as a template for cDNA synthesis and reverse-transcribed using the reverse primer Gag 3out (5′-CCTACTAATACTGTACCTATAGC-3′) and SuperScript III reverse transcriptase (Invitrogen, Life Technologies, Thermo Fisher Scientific). cDNA was serially diluted in a 96-well plate (Applied Biosystems) to identify the dilution at which fewer than 30% of wells contained PCR amplicons of the correct size. The HIV-1 Gag genome was amplified via nested PCR with primers as follows: first round forward primer Gag 5out (5′-TTTGCCTGTACTGGGTCTCTCTGGTT-3′); second round forward primer Gag 5in (5′-GCTTAAGCCTCAATAAAGCTTGCCTT-3′); first round reverse primer Gag 3out (5′-CCTACTAATACTGTACCTATAGC-3′); and second round reverse primer Gag 3in (5′-AATACTGTATCATCTGCTCCTGTATC-3′). The following conditions were used for the first round of nested PCR: 94°C for 2 minutes; 37× (94°C for 20 seconds; 55°C for 30 seconds; 68°C for 2 minutes); 68°C for 10 minutes. For the second round of nested PCR, the PCR conditions were as follows: 94°C for 2 minutes; 37× (94°C for 20 seconds; 58°C for 30 seconds; 68°C for 2 minutes); 68°C for 10 minutes. Amplicons were sequenced via the MiSeq platform (Illumina). Raw reads were aligned to the HXB2 Gag reference from LANL.gov using Geneious R9. Sequences that contained mixed bases at a frequency of more than 25% per nucleotide position were excluded from further analysis. Nucleotide sequences were translated and codon-optimized via Geneious R9 and submitted to GenBank (MW531185-MW531220).
Statistics.
Descriptive statistics were calculated for baseline characteristics using either the number (percentage) or the median and IQR. The time to virologic failure after the start of the ATI, defined as a viral load of more than 200 copies/mL, was estimated with a Kaplan-Meier curve and compared between groups using the Peto-Peto log-rank test. The percentage of CD4-disrupted cells was summarized at weeks 1, 4, 12, and 16 of the ATI, with the mean (95% CI) calculated for each time point and the 4-week peak, an overall average calculated for the 4-week period (4-week mean), and the overall average calculated as an AUC scaled by the length of the individual’s exact observation window for the 16-week ATI. The AUC was calculated with the trapezoid rule, as implemented by the trapz package in R. Groups levels were compared using the Kruskal-Wallis test and the LR (Wald test). Linear mixed-effects regression models were also fitted to the repeated-measures data to estimate trends in the percentage of change in CD4 and CD8 levels and in the percentage of disrupted CD4 cells for the period preceding the ATI and during the ATI. All statistical tests were 2 sided and done at the 0.05 significance level. Analyses were performed using R software, version 4.02.
Human studies.
This clinical trial was approved by the IRB of the University of Pennsylvania (protocol 820573). The study was conducted between April 2015 and June 2018, with recruitment done through the University of Pennsylvania. Written consent was obtained from all participants prior to their enrollment on the clinical trial. Enrollment was closed at the end of the funding period. This clinical trial is registered at ClinicalTrials.gov (NCT02388594).
Author contributions
PT, JKJ, LT, ALB, SFL, DA, DLP, KJB, CHJ, and JLR conceived and designed research studies. PT, JKJ, ALB, EE, SK, SYN, ANV, CHJ, BLL, DLS, and CHJ performed clinical trial cell manufacturing, recruitment, and oversight. LT, MWR, CRM, HK, AMB, and FM conducted experiments analyzing clinical trial samples. PT, JKJ, PAS, LT, SFL, GL, KJB, and JLR analyzed data. XL and YZ provided reagents. PT, JKJ, PAS, and JLR drafted the manuscript, and all authors contributed to writing of the methods and editing of the manuscript.
Supplementary Material
Acknowledgments
We would like to thank the Penn Center for AIDS Research (CFAR) Virology Core for performing the Δ32 heterozygote screening and the CFAR Immunology Core for specimen processing. We also thank the members of the Clinical Cell and Vaccine Production Facility for manufacturing and release testing, the members of the Translational and Correlative Studies Laboratory for analysis, and the Center for Cellular Immunotherapies Monitoring, Pharmacovigilance, and Clinical Operations teams for their contributions to overseeing the safety and management of this trial. We also wish to thank Gabriela Plesa and Liz Veloso for regulatory guidance and support, Ambar Jimenez, Helen Budworth, and Jason Fontenot for proofreading, and Richard Pollard, Dan Kuritzkes, Judy Lieberman, and Martin Markowitz of the Data and Safety Monitoring Board. The NIH AIDS Reagent Program, Division of AIDS, NIAID provided the following reagent: HIV-1 Con B Gag Peptide Pool (NIAID, DAIDS, catalog 12425). This work was supported by grants from the NIAID, NIDA, NIMH, and NINDS (R01AI104400, to CHJ; UM1AI126620, to JLR) and by a NIH T32 grant (AI007632, to CRM).
Version 1. 02/11/2021
In-Press Preview
Version 2. 04/01/2021
Electronic publication
Version 3. 05/01/2024
Corrected figure 2 as detailed in erratum
Funding Statement
Sangamo funded and performed the next gen sequencing only
Footnotes
Conflict of interest: YZ, BLL, CHJ, and JLR are founders of and hold equity in Tmunity Therapeutics. YZ and JLR receive research support from Tmunity Therapeutics. CHJ reports receiving grants from Tmunity Therapeutics and holds founders stock in Tmunity Therapeutics and DeCART Therapeutics. CHJ also receives personal income from Celldex Therapeutics, Viracta Therapeutics, and WIRB-Copernicus Group as well as royalties from Novartis. BLL is a consultant for Novartis, Terumo, and Lilly Asia Ventures and serves on the scientific advisory boards of Avectas, Patheon/Thermo Fisher Viral Vector Services, Immuneel Therapeutics, Incysus Therapeutics, Ori Biotech, and Vycellix. DA and GL were employees and equity holders of Sangamo Therapeutics.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(7):e144486. https://doi.org/10.1172/JCI144486.
Contributor Information
Pablo Tebas, Email: Pablo.Tebas@pennmedicine.upenn.edu.
Julie K. Jadlowsky, Email: juliejad@pennmedicine.upenn.edu.
Pamela A. Shaw, Email: shawp@pennmedicine.upenn.edu.
Lifeng Tian, Email: Lifeng.Tian@pennmedicine.upenn.edu.
Erin Esparza, Email: eod@pennmedicine.upenn.edu.
Sukyung Kim, Email: Sukyung.Kim@pennmedicine.upenn.edu.
Soe Yu Naing, Email: Soeyunaing94@gmail.com.
Max W. Richardson, Email: maxwr@mail.med.upenn.edu.
Ashley N. Vogel, Email: avogel06@gmail.com.
Colby R. Maldini, Email: colby.maldini@gmail.com.
Hong Kong, Email: hongkong@mail.med.upenn.edu.
Xiaojun Liu, Email: liu.x@utctherapeutics.com.
Simon F. Lacey, Email: simon.lacey@uphs.upenn.edu.
Anya M. Bauer, Email: anyabauer1@yahoo.com.
Felicity Mampe, Email: mampef@pennmedicine.upenn.edu.
Lee P. Richman, Email: leepr@upenn.edu.
Gary Lee, Email: gary.lee@sentibio.com.
Dale Ando, Email: author@deceased.sangamo.
Bruce L. Levine, Email: levinebl@pennmedicine.upenn.edu.
David L. Porter, Email: David.Porter@pennmedicine.upenn.edu.
Yangbing Zhao, Email: zhao.y@utctherapeutics.com.
Don L. Siegel, Email: siegeld@mail.med.upenn.edu.
Katharine J. Bar, Email: BarK@pennmedicine.upenn.edu.
Carl H. June, Email: cjune@upenn.edu.
James L. Riley, Email: rileyj@upenn.edu.
References
- 1.Okoye AA, Picker LJ. CD4(+) T-cell depletion in HIV infection: mechanisms of immunological failure. Immunol Rev. 2013;254(1):54–64. doi: 10.1111/imr.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riou C, et al. Restoration of CD4+ responses to copathogens in HIV-infected individuals on antiretroviral therapy is dependent on T cell memory phenotype. J Immunol. 2015;195(5):2273–2281. doi: 10.4049/jimmunol.1500803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muenchhoff M, et al. Differential pathogen-specific immune reconstitution in antiretroviral therapy-treated human immunodeficiency virus-infected children. J Infect Dis. 2019;219(9):1407–1417. doi: 10.1093/infdis/jiy668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sepako E, et al. Incomplete recovery of pneumococcal CD4 T cell immunity after initiation of antiretroviral therapy in HIV-infected malawian adults. PLoS One. 2014;9(6):100640. doi: 10.1371/journal.pone.0100640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Douek DC, et al. Emerging concepts in the immunopathogenesis of AIDS. Annu Rev Med. 2009;60:471–484. doi: 10.1146/annurev.med.60.041807.123549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riley JL, Montaner LJ. Cell-mediated immunity to target the persistent human immunodeficiency virus reservoir. J Infect Dis. 2017;215(Suppl 3):S160–S171. doi: 10.1093/infdis/jix002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg ES, et al. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science. 1997;278(5342):1447–1450. doi: 10.1126/science.278.5342.1447. [DOI] [PubMed] [Google Scholar]
- 8.Chun TW, et al. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat Immunol. 2015;16(6):584–589. doi: 10.1038/ni.3152. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Y, et al. Challenges and opportunities of using adoptive T cell therapy as part of an HIV cure strategy. Journal Infect Dis. 2021;223(Supplement 1):38–45. doi: 10.1093/infdis/jiaa223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine BL, et al. Antiviral effect and ex vivo CD4+ T cell proliferation in HIV-positive patients as a result of CD28 costimulation. Science. 1996;272(5270):1939–1943. doi: 10.1126/science.272.5270.1939. [DOI] [PubMed] [Google Scholar]
- 11.Carroll RG, et al. Differential regulation of HIV-1 fusion cofactor expression by CD28 costimulation of CD4+ T cells. Science. 1997;276(5310):273–276. doi: 10.1126/science.276.5310.273. [DOI] [PubMed] [Google Scholar]
- 12.Riley JL, et al. Intrinsic resistance to T cell infection with HIV type 1 induced by CD28 costimulation. J Immunol. 1997;158(11):5545–5553. [PubMed] [Google Scholar]
- 13.Levine BL, et al. Large-scale production of CD4+ T cells from HIV-1-infected donors after CD3/CD28 costimulation. J Hematother. 1998;7(5):437–448. doi: 10.1089/scd.1.1998.7.437. [DOI] [PubMed] [Google Scholar]
- 14.Levine BL, et al. Adoptive transfer of costimulated CD4+ T cells induces expansion of peripheral T cells and decreased CCR5 expression in HIV infection. Nat Med. 2002;8(1):47–53. doi: 10.1038/nm0102-47. [DOI] [PubMed] [Google Scholar]
- 15.Leibman RS, Riley JL. Engineering T cells to functionally cure HIV-1 infection. Mol Ther. 2015;23(7):1149–1159. doi: 10.1038/mt.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cillo AR, Mellors JW. Which therapeutic strategy will achieve a cure for HIV-1? Curr Opin Virol. 2016;18:14–19. doi: 10.1016/j.coviro.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Wang CX, Cannon PM. The clinical applications of genome editing in HIV. Blood. 2016;127(21):2546–2552. doi: 10.1182/blood-2016-01-678144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson CW, Kiem HP. Cell and gene therapy for HIV cure. Curr Top Microbiol Immunol. 2018;417:211–248. doi: 10.1007/82_2017_71. [DOI] [PubMed] [Google Scholar]
- 19.Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370(10):901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doyon Y, et al. Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat Methods. 2010;7(6):459–460. doi: 10.1038/nmeth.1456. [DOI] [PubMed] [Google Scholar]
- 21.Li JZ, et al. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS. 2016;30(3):343–353. doi: 10.1097/QAD.0000000000000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volberding P, et al. Antiretroviral therapy in acute and recent HIV infection: a prospective multicenter stratified trial of intentionally interrupted treatment. AIDS. 2009;23(15):1987–1995. doi: 10.1097/QAD.0b013e32832eb285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kilby JM, et al. A randomized, partially blinded phase 2 trial of antiretroviral therapy, HIV-specific immunizations, and interleukin-2 cycles to promote efficient control of viral replication (ACTG A5024) J Infect Dis. 2006;194(12):1672–1676. doi: 10.1086/509508. [DOI] [PubMed] [Google Scholar]
- 24.Jacobson JM, et al. Evidence that intermittent structured treatment interruption, but not immunization with ALVAC-HIV vCP1452, promotes host control of HIV replication: the results of AIDS Clinical Trials Group 5068. J Infect Dis. 2006;194(5):623–632. doi: 10.1086/506364. [DOI] [PubMed] [Google Scholar]
- 25.Schooley RT, et al. AIDS clinical trials group 5197: a placebo-controlled trial of immunization of HIV-1-infected persons with a replication-deficient adenovirus type 5 vaccine expressing the HIV-1 core protein. J Infect Dis. 2010;202(5):705–716. doi: 10.1086/655468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conway JM, et al. Predictions of time to HIV viral rebound following ART suspension that incorporate personal biomarkers. PLoS Comput Biol. 2019;15(7):1007229. doi: 10.1371/journal.pcbi.1007229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tebas P, et al. Antiviral effects of autologous CD4 T cells genetically modified with a conditionally replicating lentiviral vector expressing long antisense to HIV. Blood. 2013;121(9):1524–1533. doi: 10.1182/blood-2012-07-447250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Namazi G, et al. The control of HIV after antiretroviral medication pause (CHAMP) study: posttreatment controllers identified from 14 clinical studies. J Infect Dis. 2018;218(12):1954–1963. doi: 10.1093/infdis/jiy479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salantes DB, et al. HIV-1 latent reservoir size and diversity are stable following brief treatment interruption. J Clin Invest. 2018;128(7):3102–3115. doi: 10.1172/JCI120194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruner KM, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature. 2019;566(7742):120–125. doi: 10.1038/s41586-019-0898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henrich TJ, et al. CCR5-Δ32 heterozygosity, HIV-1 reservoir size, and lymphocyte activation in individuals receiving long-term suppressive antiretroviral therapy. J Infect Dis. 2016;213(5):766–770. doi: 10.1093/infdis/jiv504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macatangay BJC, et al. Therapeutic vaccination with dendritic cells loaded with autologous HIV type 1-infected apoptotic cells. J Infect Dis. 2015;213(9):1400–1409. doi: 10.1093/infdis/jiv582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goulder PJ, Walker BD. HIV and HLA class I: an evolving relationship. Immunity. 2012;37(3):426–440. doi: 10.1016/j.immuni.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hutter G, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360(7):692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 35.Duarte RF, et al. CCR5 Δ32 homozygous cord blood allogeneic transplantation in a patient with HIV: a case report. Lancet HIV. 2015;2(6):e236–e242. doi: 10.1016/S2352-3018(15)00083-1. [DOI] [PubMed] [Google Scholar]
- 36.Gupta RK, et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature. 2019;568(7751):244–248. doi: 10.1038/s41586-019-1027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez EE, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26(7):808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maier DA, et al. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Hum Gene Ther. 2013;24(3):245–258. doi: 10.1089/hum.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z, et al. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4+ T cells from HIV-1 infection. Cell Biosci. 2017;7:47. doi: 10.1186/s13578-017-0174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scholler J, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4(132):132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pannus P, et al. Rapid viral rebound after analytical treatment interruption in patients with very small HIV reservoir and minimal on-going viral transcription. J Int AIDS Soc. 2020;23(2):e25453. doi: 10.1002/jia2.25453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conway A, et al. Non-viral delivery of zinc finger nuclease mRNA enables highly efficient in vivo genome editing of multiple therapeutic gene targets. Mol Ther. 2019;27(4):866–877. doi: 10.1016/j.ymthe.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paschon DE, et al. Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat Commun. 2019;10(1):1133. doi: 10.1038/s41467-019-08867-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu L, et al. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. N Engl J Med. 2019;381(13):1240–1247. doi: 10.1056/NEJMoa1817426. [DOI] [PubMed] [Google Scholar]
- 45.Buchbinder SP, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372(9653):1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perreau M, et al. Activation of a dendritic cell-T cell axis by Ad5 immune complexes creates an improved environment for replication of HIV in T cells. J Exp Med. 2008;205(12):2717–2725. doi: 10.1084/jem.20081786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Menendez D, et al. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011;7(3):1001360. doi: 10.1371/journal.pgen.1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muranski P, et al. Increased intensity lymphodepletion and adoptive immunotherapy--how far can we go? Nat Clin Pract Oncol. 2006;3(12):668–681. doi: 10.1038/ncponc0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shreedhar V, et al. Dendritic cells require T cells for functional maturation in vivo. Immunity. 1999;11(5):625–636. doi: 10.1016/S1074-7613(00)80137-5. [DOI] [PubMed] [Google Scholar]
- 50.Schoenberger SP, et al. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393(6684):480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 51.Williams MA, et al. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441(7095):890–893. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Borst J, et al. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635–647. doi: 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 53.Douek DC, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417(6884):95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 54.Leibman RS, et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. 2017;13(10):1006613. doi: 10.1371/journal.ppat.1006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maldini CR, et al. HIV-resistant and HIV-specific CAR-modified CD4+ T cells mitigate HIV disease progression and confer CD4+ T cell help in vivo. Mol Ther. 2020;28(7):1585–1599. doi: 10.1016/j.ymthe.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DiGiusto DL, et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol Ther Methods Clin Dev. 2016;3:16067. doi: 10.1038/mtm.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Donnell TJ, et al. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 2018;7(1):129–132. doi: 10.1016/j.cels.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 58.Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reynisson B, et al. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. J Proteome Res. 2020;19(6):2304–2315. doi: 10.1021/acs.jproteome.9b00874. [DOI] [PubMed] [Google Scholar]
- 60.Richman LP, et al. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst. 2019;9(4):375–382. doi: 10.1016/j.cels.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.