

Abstract
The NH3-mediated selective catalytic reduction (NH3-SCR) of NOx over Cu-ion-exchanged chabazite (Cu-CHA) catalysts is the basis of the technology for abatement of NOx from diesel vehicles. A crucial step in this reaction is the activation of oxygen. Under conditions for low-temperature NH3-SCR, oxygen only reacts with CuI ions, which are present as mobile CuI diamine complexes [CuI(NH3)2]+. To determine the structure and reactivity of the species formed by oxidation of these CuI diamine complexes with oxygen at 200 °C, we have followed this reaction, using a Cu-CHA catalyst with a Si/Al ratio of 15 and 2.6 wt% Cu, by X-ray absorption spectroscopies (XANES and EXAFS) and diffuse reflectance UV-Vis spectroscopy, with the support of DFT calculations and advanced EXAFS wavelet transform analysis. The results provide unprecedented direct evidence for the formation of a [Cu2(NH3)4O2]2+ mobile complex with a side-on μ-η2,η2-peroxo diamino dicopper(II) structure, accounting for 80–90% of the total Cu content. These [Cu2(NH3)4O2]2+ are completely reduced to [CuI(NH3)2]+ at 200 °C in a mixture of NO and NH3. Some N2 is formed as well, which suggests the role of the dimeric complexes in the low-temperature NH3-SCR reaction. The reaction of [Cu2(NH3)4O2]2+ complexes with NH3 leads to a partial reduction of the Cu without any formation of N2. The reaction with NO results in an almost complete reduction to CuI, under the formation of N2. This indicates that the low-temperature NH3-SCR reaction proceeds via a reaction of these complexes with NO.
1. Introduction
The selective catalytic reduction of NOx by ammonia (NH3-SCR) to nitrogen and water is the basis for the current technology for NOx abatement in the exhaust of lean-burn heavy-duty and passenger vehicles. This technology has already resulted in significant improvements of exhaust gas emissions from diesel vehicles. Catalysts based on Cu-ion exchanged chabazite (Cu-CHA) are very effective for this reaction and are commonly applied today. These catalysts feature a high activity around 200 °C, a good selectivity for N2 formation, and excellent thermal stability in the harsh conditions of exhaust after-treatment systems.1,2
The NH3-SCR reaction is a redox reaction following the equation 4NO + 4NH3 + O2 → 4N2 + 6H2O. The NH3-SCR activity of Cu-CHA catalysts is due to the capability of the Cu ions to reversibly change the oxidation state between CuI and CuII.3−5 In the NH3-SCR reaction cycle, CuII is reduced to CuI, followed by a reoxidation of the CuI to restore the CuII. The reaction cycle can be performed stepwise, by alternating a reduction in a mixture of NO and NH3, and an oxidation in a mixture of NO and O2.4,6−9 For the reduction half-cycle, there is converging evidence that the reduction of CuII by NO and NH3 at around 200 °C results in the formation of linear [CuI(NH3)2]+ complexes. These complexes are weakly bound to the zeolite, and therefore mobile.3,6,10,11
In the oxidation half cycle, the CuI species reacts with O2 to form a CuII species, and at low temperatures, O2 exclusively reacts with the CuI species. Therefore, the reaction of O2 with the linear [CuI(NH3)2]+ complexes is an essential step in the NH3-SCR reaction cycle at low temperature.12 To complete the activation and dissociation of the O2 molecule, four electrons are required. As a single CuI is capable of delivering only one electron (no evidence for CuIII formation has ever been reported in the numerous studies on NH3-SCR), this means that other electron sources are required, which can be other CuI ions, NO or other reaction intermediates.4−6,13,14 Following these thoughts, it has been shown that the dissociation of O2 becomes easier when a single O2 molecule interacts with two CuI ions simultaneously to form Cu-pairs.3,4,7,15,16 Combining this with the mobility of the linear [CuI(NH3)2]+ complexes, a reaction mechanism has been worked out where Cu-pair formation is facilitated by diffusion of the [CuI(NH3)2]+ complexes inside the zeolite.4,13 Such a mechanism involving the formation of Cu-pairs is supported by the observation that the NH3-SCR rate at low temperature is, for low Cu contents, proportional to the square of the Cu content in the catalyst.4,12,17,18 At higher temperatures, the [CuI(NH3)2]+ complexes decompose,10,19 and the CuI is then expected to lose its mobility. As a result, the formation of Cu-pairs, and therefore also the activation of O2, becomes more difficult. This seems to be the reason for the often observed decrease in NOx conversion with increasing temperatures around 300 °C, which separates the low- and high-temperature regimes for Cu-CHA catalysts.4 At high temperatures, the reaction may occur on isolated ZCuI sites (where Z indicates coordination to zeolite oxygens in the proximity of an Al exchange site), possibly mediated by the formation of Cu-nitrate and Cu-nitrite species,2,6,15,16,20 which then further reacts with ammonia to yield N2 and H2O.
In a model where the formation of Cu-pairs is facilitated by diffusion of [CuI(NH3)2]+ complexes, the actual active center for the activation O2 is not directly associated with a specific site or location of the Cu in the zeolite. In a fresh Cu-CHA material, the positive Cu ions balance the negative charges in the zeolite framework induced by the Al substitution; a CuII ion is anchored by either a single framework Al atom, as a Z[CuII(OH)] species, or by two framework Al atoms, to yield a Z2CuII species. These Cu species are then located either in a double 6-membered ring or an 8-membered ring. Upon exposure to a reaction gas for NH3-SCR, which contains NO, NH3, O2 and H2O, these Z[CuII(OH)] and Z2CuII species become solvated by NH3 leading to the formation of the mobile [CuI(NH3)2]+ complexes. These complexes are able to diffuse to about 9 Å away from their anchor point in the time scale of catalytic turnover, which enables the Cu-pair formation necessary for the activation of O2.4,17 This means that the original location of the Cu ions does not immediately affect the reactivity of the Cu, but the different local environments of the Cu ions may affect the solvation of the Cu by ammonia. Furthermore, Density Functional Theory (DFT) calculations have shown that the formation of Cu pairs from two [CuI(NH3)2]+ complexes becomes more difficult in areas with two Al atoms located close to each other.21
The structures that are formed after the reaction of an O2 molecule and two [CuI(NH3)2]+ complexes are Cu complexes containing two Cu-centers bridged by oxygen; the general formula of these complexes is [Cu2(NH3)4O2]2+. Efforts to determine the structure of this complex have mainly been based on DFT calculations. Figure 1 shows three possible structures of the [Cu2(NH3)4O2]2+ complexes, which differ in the way the oxygen molecule is bound to the Cu, and whether dissociation of the O–O bond takes place or not. The stability of these structures calculated with DFT depends on the functional chosen in the calculation.22 Calculations on ammonia-ligated Cu2O2 cores in the gas phase have shown how the predicted stability of different structures depends on the method (DFT vs post-Hartree–Fock), due to the differences in the description of the electron correlation contribution to the core conformation.23,24 More recent DFT calculations on O2 activation and dissociation by [CuI(NH3)2]+ complexes in CHA also showed a strong dependence of the calculated stabilities and structural parameters on the selected functional. DFT calculations using a PBE functional, with or without van der Waals corrections, often result in the bis-μ-oxo diamino dicopper(III) complex17,22 (Figure 1c), which implies that the reaction of O2 with [CuI(NH3)2]+ complexes results in the dissociation of the O–O bond. With a HSE06 hybrid functional, one finds a μ-η2,η2-peroxo diamino dicopper(II) complex (Figure 1b), in which the O2 molecule binds with the Cu-centers in a side-on configuration without dissociation of the O–O bond.22 Including a Hubbard-U term of 6 eV in DFT (DFT+U) with a PBE functional, and van der Waals corrections results in a correct prediction of the dissociation of the O–O bond in Cu2O2 complexes in enzymes with a structure similar to those expected in the Cu-CHA zeolite.22 Calculations of the [Cu2(NH3)4O2]2+ complex with that method pointed to the formation of the μ-η2,η2-peroxo diamino dicopper(II) complex (Figure 1b),22 just like the HSE06 functional. Clearly, experimental data for the structure of the [Cu2(NH3)4O2]2+ complex are needed to resolve this issue.
Figure 1.

Pictorial representation of the [Cu2(NH3)4O2]2+ complexes proposed in ref (17): (a) trans-μ-1,2-peroxo diamino dicopper(II) (end-on), (b) μ-η2,η2-peroxo diamino dicopper(II) (side-on), and c) bis-μ-oxo diamino dicopper(III). Atom color code: Cu, green; H, white; O, red; N, blue.
In this work, we present a spectroscopic investigation of the structure and reactivity of the [Cu2(NH3)4O2]2+ complexes formed upon reaction of O2 with the mobile [CuI(NH3)2]+ complexes in a Cu-CHA catalyst for NH3-SCR. The available X-ray absorption spectroscopy data for Cu-CHA at the end of the transient O2 oxidation experiment are not conclusive on the detailed structure of the [Cu2(NH3)4O2]2+ complexes.17 The concept that dissociation of O2 requires two solvated CuI ions also opens the question whether Cu pairs play a role beyond O2 activation. Therefore, we also investigate the reactivity of the [Cu2(NH3)4O2]2+ complexes toward NO and NH3. These goals have been pursued by applying X-ray absorption spectroscopy (XAS), both near-edge (XANES) and extended range (EXAFS), with a well-established operando setup,6,10,25 and diffuse reflectance ultraviolet–visible–near-infrared (DR UV-Vis-NIR) spectroscopy19,26 using a Cu-CHA catalyst. DFT calculations were used for optimization of the [Cu2(NH3)4O2]2+ structures illustrated in Figure 1, which were used as input for the interpretation of the EXAFS data. To enhance the sensitivity of EXAFS to multicopper moieties, we applied wavelet transform (WT) analysis, giving unprecedented insights in the formation and separation of the solvated Cu-pairs in the NH3-SCR reaction cycle over Cu-CHA catalysts.
2. Experimental Section
The catalyst used in this study was a Cu-CHA material with Si/Al = 15 with a Cu content of 2.6 wt%, corresponding to Cu/Al = 0.5, a Cu density of around 0.4 Cu/1000 Å3 (0.3 Cu per chabazite cage) and a theoretical mean Cu–Cu distance of 16.6 Å. At this Cu density, about 90% of CuI ions can be oxidized by O2, forming the [Cu2(NH3)4O2]2+ complexes.17 The steps depicted in Scheme 1 have been used to form these complexes and to study the reactivity toward NO and NH3 experimentally. These steps are as follow:
-
1.
pretreatment in O2 at 400 °C;
-
2.
reduction in a mixture of 1000 ppm of NO and 1000 ppm of NH3 at 200 °C to form [CuI(NH3)2]+;4,6,17,27
-
3.
oxidation of the [CuI(NH3)2]+ complexes by O2 at 200 °C to form the [Cu2(NH3)4O2]2+ complexes;17
-
4.
reaction of the [Cu2(NH3)4O2]2+ complexes with NO or NH3 at 200 °C
Scheme 1. Experimental Steps Followed by XAS and UV-Vis-NIR DR Spectroscopies.
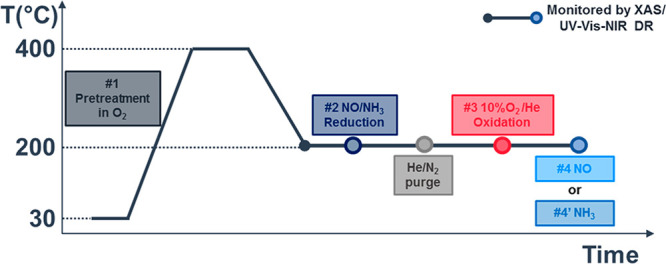
Step 1, pretreatment at 400 °C in pure O2; step 2, reduction in 1000 ppm of NO/1000 ppm of NH3/He; step 3, oxidation in 10% O2/He; step 4, reaction with 1000 ppm of NH3/He or step 4′, with 1000 ppm of NO/He. Total gas flow: 100 mL/min for XAS, 50 mL/min for UV-Vis. The purge step was carried out in He for XAS and in N2 for UV-Vis.
The steps in this protocol were followed independently by XAS coupled to an online mass spectrometer for a qualitative effluent gas analysis (BM23 beamline of the European Synchrotron Radiation Facility),28 and by DR UV-Vis-NIR. In this Article, we present the development of spectra with time under isothermal conditions, until a steady-state was observed. The DR UV-Vis-NIR spectra are reported as relative reflectance (R%), to avoid artifacts due to the use of the Kubelka–Munk function.26 We refer to the Supporting Information (SI), section 1, for more experimental details and description of the equipment used.
3. Results
3.1. Oxidation of [CuI(NH3)2]+ Complexes
3.1.1. Oxidation and Coordination State of Cu Ions
The linear [CuI(NH3)2]+ complexes are the starting point of the experiment discussed in this section. These complexes are formed by exposure of the pretreated Cu-CHA catalyst (step 1) to a NO/NH3 mixture at 200 °C (step 2). The presence of this state is indicated by a characteristic rising-edge peak at ∼8982.5 eV in the Cu K-edge XANES spectrum, corresponding to the 1s→4p transition of linearly coordinated CuI centers (Figure 2a, blue curve) and validated by EXAFS fitting based on the [CuI(NH3)2]+ model (see SI, section 2.2). Upon reaction of this linear [CuI(NH3)2]+ complex with a mixture of 10% O2 in He, this distinct peak at 8982.5 eV disappears almost completely, the XANES resembles that observed after pretreatment in O2 (Figure 2a, red and gray dashed lines) and the dipole-forbidden 1s→3d transition of CuII becomes visible at 8977.3 eV (Figure 2b). These changes clearly indicate that the linear [CuI(NH3)2]+ complex is oxidized by O2. After 30 min in O2/He, a small feature at 8982.5 eV is still recognized, but the spectra do not change any more. This indicates, that not all [CuI(NH3)2]+ complexes are oxidized; in agreement with earlier observations17 about 10–20% of the [CuI(NH3)2]+ complexes do not react, based on qualitative XANES analysis. The mass spectrometer connected to the cell did not detect any NH3 desorption during the oxidation, indicating that the Cu does not lose the NH3 ligands in the oxidation process.
Figure 2.
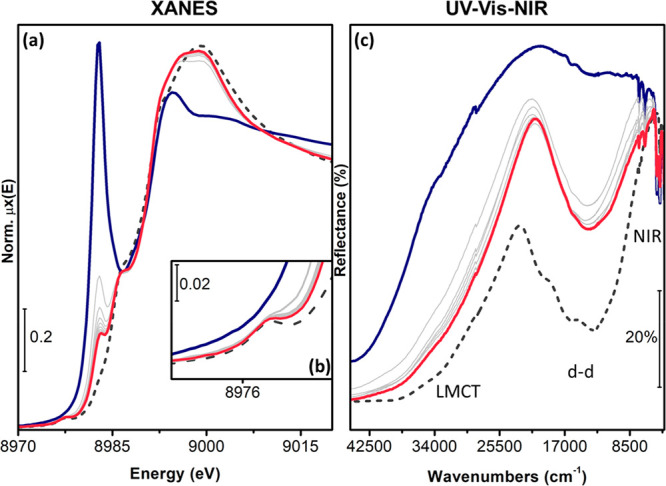
Evolution of the Cu K-edge XANES (a) and DR UV-Vis-NIR spectra (c) of Cu-CHA during exposure to 10% O2/He at 200 °C after reduction in NO/NH3/He at 200 °C. Inset (b) reports a magnification of the pre-edge peak arising from the CuII 1s →3d transition. Dark blue thick line: after NO/NH3/He exposure; red thick line: final spectrum after exposure to 10% O2; gray thin lines: intermediates; dark gray dashed line: Cu-CHA pretreated in O2, by heating in O2 up to 400 °C and subsequently cooling in O2 down to 200 °C prior to XAS data collection, step 1.
This evidence points to the presence of different, NH3-solvated CuII species with respect to the framework-coordinated CuII ions (fw-CuII) known to be present in the catalyst after pretreatment in O2.7,25,29−31 To strengthen such indications, we initially analyzed by linear combination fit (LCF) the XANES after oxidation in O2 (step 3) using the spectra obtained at step 1 (pretreated in O2) and at step 2 (reduced in NO/NH3) as references for CuII and CuI components, respectively (see SI, Figure S11). Overall, estimates for CuI and CuII percentages are in reasonable agreement with qualitative analysis. It is also evident that the rising edge peak at 8982.5 eV diagnostic of CuI species is excellently reproduced, indicating that the CuI component, i.e., linear [CuI(NH3)2]+, is the same at both steps 2 and 3. However, significant discrepancies between experimental and LCF curve are found when considering the rising-edge region where CuII 1s→4p transitions typically occurs, as well as the shape and energy position of the white-line peak. These discrepancies translate into a structured residual function, with well-defined maxima and minima well above the noise level, further indicating that two spectroscopically distinguishable CuII species are present at steps 1 and 3.
In the UV-Vis-NIR spectra, the oxidation of the linear [CuI(NH3)2]+ complex is visible as follows. Due to the d10 closed-shell configuration of CuI, the typical ligand-field d-d transitions are absent, and the spectrum is dominated by a ligand-to-metal charge transfer (LMCT) transition observed in the range 30 000–45 000 cm–1 (blue curve in Figure 2c).26,32 The oxidation of CuI to CuII by O2 is reflected in the development of an intense d-d absorption centered at 13 850 cm–1 and a red-shift of the LMCT transitions, from ca. 35 000 to 25 000 cm–1 (arbitrarily measured at R = 60%). The features at 6515 and 4970 cm–1 in the NIR region are due to the overtones and combination modes of NH3 and NH4+, confirming that the CuII species after oxidation still contains the NH3 ligands. The spectrum of the Cu-CHA catalyst pretreated in O2 (dark gray dashed curve) is reported for comparison, showing that the coordination geometry of the CuII species formed by oxidation of [CuI(NH3)2]+ is different with respect to that of the variety of framework-coordinated monomeric/multimeric ions (fw-CuII, such as Z[CuII(OH)]/Z[CuII(OO*)] etc.) responsible for the typical Cu-CHA “quadruplet” (complex absorption in the d-d region with components at 20 000, 16 350, 13 300, and 10 600 (sh) cm–1).26,33−36
3.1.2. Structure of the [Cu2(NH3)4O2]2+ Complexes
To determine the precise structure of the CuII complex formed by the oxidation of the linear [CuI(NH3)2]+ species with O2, we have analyzed the Fourier-transformed (FT) EXAFS spectra; Figure 3a shows these data without phase correction. The first-shell peak is almost doubled in intensity after O2 interaction (from blue to red), indicating that the coordination number of Cu in the CuII complexes is higher than in the linear [CuI(NH3)2]+ complex. In the next shell, the unstructured feature observed for the mobile [CuI(NH3)2]+ complex evolves toward a broad scattering feature peaking at ca. 2.4 Å. Even though this feature is close to the second-shell EXAFS signature of framework-coordinated CuII ions after pretreatment in O2 (dashed gray line), it can be clearly distinguished, indicating that, at least a substantial part of the CuII complex is still mobile in the cage. Finally, a third peak around 3.2 Å develops, which could correspond to contributions from a Cu–Cu scattering in the CuII complexes shown in Figure 1. All these features in the FT-EXAFS are consistent with the formation of [Cu2(NH3)4O2]2+ complexes. However, a more detailed analysis is required to rule out re-coordination of CuII ions to zeolite framework and, once this possibility is excluded, to identify the precise [Cu2(NH3)4O2]2+ structure, based on the three possibilities shown in Figure 1.
Figure 3.
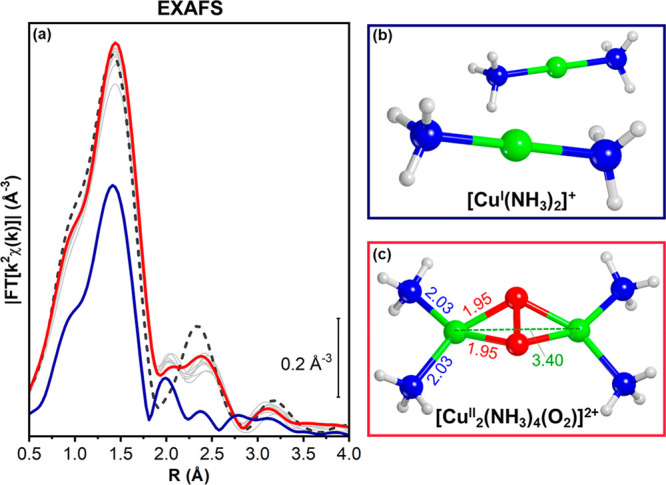
(a) Phase-uncorrected k2-weighted FT-EXAFS curves during exposure of the Cu-CHA catalyst to NO/NH3/He (dark blue thick line), followed by 10% O2 in He (gray thin lines; red thick line: final spectrum; dark gray dashed line: pretreatment in O2 by heating in O2 up to 400 °C and subsequently cooling in O2 down to 200 °C prior to XAS data collection, step 1. (b) Illustration of [Cu(NH3)2]+ and (c) μ-η2,η2-peroxo diamino dicopper(II) (side-on) complexes. Atom color code: Cu, green; H, white; O, red; N, blue. Part (c) also report selected DFT bond distances in Å.
To this aim, we have first performed quantitative EXAFS analysis of the spectra obtained at step 1 (gray dashed line in Figure 3). The adopted fitting model is based on structural characteristics conserved for most fw-CuII species previously proposed to form in Cu-CHA upon pretreatment in O2.7,25,29−31,34 Importantly, it accounts for a distinctive scattering contribution of charge-balancing Al atoms (Alfw) located at ca. 2.75 Å range from the CuII center, while maintaining a certain degree of flexibility to account for fractional contribution from multicopper species (additional information can be found in SI, section 2.1). As detailed in the SI, the fit resulted in a good level of reproduction of experimental EXAFS spectrum for the pretreated catalyst at step 1, with physically meaningful values for all the refined parameters, as well as for their fitting errors (see SI, Figure S2 and Table S2).
A second stage of our analysis consisted in a test EXAFS fit of the experimental spectrum of the [Cu(NH3)2]+ complexes oxidized in O2 (step 3, red solid line in Figure 3), using exactly the same model based on fw-CuII, which guaranteed a successful fit for the EXAFS of the pretreated catalyst (see SI, section 2.3). While the numerical agreement between best-fit and experimental curve was formally satisfactory, this was achieved at the expense of the physical meaning of the optimized parameters (e.g., Debye–Waller factors as high as 0.1 Å2) as well as of their accuracy (unphysically high fitting errors). Such inconsistencies most severely affect the Cu–Alfw coordination shell, representing a diagnostic contribution for the large majority of fw-coordinated CuII species proposed so far in the CHA framework. Consistently with the LCF XANES results in SI, Figure S11, the failure of this test EXAFS fit strongly supports the spectroscopically detectable diversity of CuII species formed in Cu-CHA at steps 1 and 3. The state of the catalyst at step 3 is clearly not consistent with fw-coordinated CuII species, thus paving the way to deeper structural analysis considering mobile [CuII2(NH3)4O2]2+ complexes.
To determine the precise structure of the [Cu2(NH3)4O2]2+ complexes, first we have optimized the structures in the gas phase (that is not including the zeolite effect in the calculations) for the three complexes shown in Figure 1 by DFT, including the three main structures proposed in the literature by different authors: NH3-solvated side-on22 and end-on4 peroxides as well as the bis(μ-oxo)-dicopper core.17 Then, these structures were used as input to fit the observed EXAFS features. A detailed description of the DFT and fitting procedures and results can be found in the SI, section 2.4. Note that, due to the difficulty in determining the relative stabilities of the [Cu2(NH3)4O2]2+ complexes by DFT,22−24 we have only used the DFT calculations to provide reasonable structures for the [Cu2(NH3)4O2]2+ complexes as input for fitting the EXAFS data.
The difference in the structures of the [Cu2(NH3)4O2]2+ complexes in Figure 1 lays in the Cu2O2 cores. The Cu2O2 cores differ in the Cu–Cu, Cu–O, and O–O bond lengths, and in the oxidation state of the Cu. Table 1 contains a comparison of our calculated values for the different possible [Cu2(NH3)4O2]2+ complexes and the known values for similar Cu2O2 cores from enzymes, where the precise structure is determined experimentally.37−41 From the table, we observe that our calculated values agree well with the experimentally determined values for enzymes, which validates our structural models derived from DFT calculations for further EXAFS analysis.
Table 1. Experimental and Calculated Structural Parameters of Cu2O2 Cores.
| bond length (Å) |
|||||
|---|---|---|---|---|---|
| Cu–Cu | Cu–O | O–O | Cu oxidation state | ref | |
| trans (end-on) μ-1,2-peroxo dicopper(II) | 4.36 | – | 1.43 | 2+ | (38,41) |
| 4.149–4.300 | 1.860–2.113 | 1.270–1.449 | This worka | ||
| (side-on) μ-η2,η2-peroxo dicopper(II) | 3.52–3.59 | 1.97–1.98 | 1.37–1.54 | 2+ | (38,39,42) |
| 3.059–3.597 | 1.910–1.983 | 1.416–1.508 | this worka | ||
| bis-μ-oxo dicopper(III) | 2.74–2.83 | 1.80–1.83 | 2.29–2.37 | 3+ | (37−39,42) |
| 2.644–2.872 | 1.772–1.870 | 2.265–2.359 | this worka | ||
| EXAFS best fit | 3.40 ± 0.05 | 1.911 ± 0.009 | – | – | this work |
DFT calculations on zeolite-free molecular complexes with M06-HF-D and M06-L-D functionals.
Fitting the EXAFS data with the (end-on) trans μ-1,2-peroxo diamino dicopper(II) or bis-μ-oxo diamino dicopper(III) models (Figure 1a,c) lead unsatisfactory results (see SI, section 2.4), and therefore, the observed EXAFS features are not consistent with these structures. Using the side-on μ-η2,η2-peroxo diamino dicopper(II) complex (Figure 1b and Figure 3c) to fit the EXAFS data, we find a Cu–O distance of 1.911 ± 0.009 Å, a Cu–N distance of 2.06 ± 0.02 Å, and a Cu–Cu distance of 3.40 ± 0.05 Å. These results are in excellent agreement with values calculated by DFT, and also match the values for a similar Cu2O2 core in enzymes (Figure 3c and Table 1). Furthermore, the EXAFS fit based on the μ-η2,η2-peroxo diamino dicopper(II) complex also revealed intense multiple scattering contributions in the 2.5–3.5 Å range—mostly triangular scattering paths involving the two O atoms of the peroxo group and quasi-collinear paths across the N(NH3)–O(peroxo) diagonal. This points to the fact that the majority of the formed [Cu2(NH3)4O2]2+ complexes are characterized by the μ-η2,η2-peroxo diamino dicopper(II) structure depicted in Figure 3c.
Having identified the [Cu2(NH3)4(O2)]2+ complex as μ-η2,η2-peroxo diamino dicopper(II), we further refined the individual contributions of this complex and the unreacted [CuI(NH3)2]+ complexes by multi-component EXAFS fitting (see SI, section 2.4.3 for details on the procedure). We find that 16 ± 8% of Cu is still present as [CuI(NH3)2]+, indicating that not all [CuI(NH3)2]+ are oxidized by O2. The amount of unreacted [CuI(NH3)2]+ complexes we find here is in good agreement with the estimate based on the qualitative XANES analysis presented above, and with theoretical predictions based on a limited mobility of the [CuI(NH3)2]+ complexes at the studied Cu density.17
3.1.3. Validating the Structural Dynamics of Cu Species by EXAFS Wavelet Transform (WT) Analysis
The DFT-guided EXAFS fitting results allow us to define a consistent experimentally based model for the [Cu2(NH3)4(O2)]2+ complexes previously proposed.4,17 A conclusive assignment of the features observed in conventional Fourier transform (FT) EXAFS spectra, however, is hindered by the simultaneous presence of various types of atomic neighbors surrounding the Cu absorber, especially in the high-R region. If two or more types of elemental neighbors and/or scattering interactions are localized at close distances around the absorber, their contributions in the direct space R overlap and often become indistinguishable. For the Cu-CHA zeolite studied here, these potentially include single scattering paths from framework Al/Si/O in zeolite-coordinated Cu moieties as well as Cu in multicopper species, which can be coordinated to the zeolite or mobile.29 The intense multiple scattering paths involving first-shell O/N neighbors in the proposed μ-η2,η2-peroxo diamino dicopper(II) moiety mentioned above are also expected to fall in this R-space range.
To resolve this, it is possible to exploit the fact that the contributions from different elemental neighbors appear at different locations in k-space, because the backscattering amplitude factor F(k) strongly depends on the atomic number Z. Figure 4a shows the F(k) curves associated with the elements relevant in this work, namely O, N, Al, Si, and Cu. It is clear that signals produced by heavier atoms, such as Cu, are localized at higher k values with respect to lighter atoms. On this background, a WT analysis allows for a better discrimination of the nature of the scattering contributions around the absorber, compared to the classical FT analysis.30,43−47 The WT analysis results in a 2D representation of the EXAFS, simultaneously revealing the signal features in both R- and k-space. Then, one can visually resolve the scattering contributions originating from atomic neighbors having enough Z-contrast in their F(k) functions. A more detailed description of the WT analysis technique is given in the SI, section 3.1.
Figure 4.

(a) Backscattering amplitude factors associated with the elements present in the system under study. Moduli of EXAFS WTs magnified in the 2–4 Å R-space range for the following reaction steps: (b) step 1, pretreatment in O2; (c) step 2, reduction in NO/NH3/He; (d) step 3, oxidation in 10% O2/He; (e) step 4, exposure to NO/He or (f) step 4′, exposure to NH3/He. For all the WTs a common intensity scale is employed. Part (d) also shows the corresponding EXAFS spectra in k-space and R-space (conventional FT) in the relevant ranges, as well as the k-space range characteristic of low-Z (O/N, Si/Al) and high-Z scatterers (Cu). All spectra measured at 200 °C.
Figure 4b–f reports the moduli of EXAFS WTs for the key reaction steps explored in this work, measured in steady-state conditions at 200 °C. Here, EXAFS WTs are magnified in the 2–4 Å R-space range, where signal interpretation with conventional FT-EXAFS approach is mostly complicated by overlapping contributions. Corresponding full-range EXAFS-WTs are reported and discussed in detail in the SI, section 3.3.
In the relevant R-space range, EXAFS WT for the spectrum collected in O2 at 200 °C after pretreatment in O2 at 400 °C (Figure 4b), clearly splits in two lobes. The first sub-lobe, localized in the k range 1–5 Å–1 and R range 2–2.8 Å is associated with the framework atoms: O, Si and Al. The second one, localized at higher k values (i.e., 6–8 Å–1), is principally related to Cu–Cu contributions in oxygen-bridged Cu dimers or, more in general, multicopper moieties. WT analysis further validate the previously mentioned EXAFS fitting results for step 1 based on a prototypical fw-CuII model (see SI, section 2.1), confirming the simultaneous presence of Cu–Al, Cu–O/Si, and Cu–Cu scattering contributions in the high-R EXAFS range for the pretreated catalyst. The presence of Cu-oxo dimeric/polymeric cores in oxygen activated Cu-CHA catalysts represents a novelty with respect to previous literature in the context of the NH3-SCR reaction.25 This aspect has been recently established by different authors studying the nature of Cu-oxo species in Cu-CHA for the direct methane to methanol conversion.26,29,30,34,35,43,47 While from the EXAFS fit in SI, section 2.1 an average Cu–Cu coordination number NCu = 0.5 ± 0.3 is estimated, a more precise identification of the nature and amount of these dimeric/polymeric structures in the oxygen activated Cu-CHA is outside the scope of this manuscript and probably beyond the possibilities of the technique. However, the contrast between F(k) function for Cu and for the rest of relevant elements in the system is sufficient to reveal the presence of (a fraction) of multicopper moieties.
Indeed, the EXAFS backscattering amplitude factors in Figure 4a show that the F(k) functions of lighter elements, such as O/N and Si/Al, have maxima at around 3–4 Å–1, while for Cu, the position of the main peak significantly shifts to a k-value of around 7 Å–1. These differences lead to the observed lobe splitting, enabling an unambiguous, visual discrimination of contributions stemming from Cu or framework atomic contributions. Due to the substantial overlap of the related backscattering amplitude functions, it is not possible to discriminate by means of WT among O/N and Si/Al contributions.
A WT analysis of the EXAFS spectrum for the mobile [CuI(NH3)2]+ complexes (Figure 4c), which is obtained after exposure of the catalyst pretreated in O2 to the NO/NH3 gas mixture at 200 °C (step 2), shows a complete reduction and mobilization of the Cu ions in the system.6,10,17,19,48 The second-shell peak in the conventional FT-EXAFS disappears (Figure 3a and SI, Figure S6) and all the high-R features are substantially decreased in the corresponding WT map in Figure 4c. Even though the sub-lobe at k = 7 Å–1 associated with the Cu–Cu signal is completely lost at this step, a low-intensity feature is still visible in the k-space range 1–5 Å–1, which is most likely due to multiple scattering paths involving the first-shell N ligands in the linear [CuI(NH3)2]+ moieties.
The crucial step for the activation of oxygen is the isothermal oxidation of the [CuI(NH3)2]+ complexes, and the corresponding WT analysis is shown in Figure 4d. The most important observation here is the appearance of a sub-lobe at around k = 7 Å–1 indicating a contribution of Cu–Cu scattering. The presence of this feature unambiguously demonstrates the formation of a Cu complex containing more than one Cu atom. This agrees well with the formation of [Cu2(NH3)4O2]2+ complexes, and is in line with our EXAFS results (section 3.1.2) and earlier work.4,16−18 The second sub-lobe occurs in the same k-space range observed in the EXAFS WT after pretreatment in O2. However, we note a different morphology of the WT along the R direction in the two probed states, indicating that there is a difference in Cu–Cu coordination after pretreatment in O2 or oxidation of [CuI(NH3)2]+ at 200 °C. After oxidation of [CuI(NH3)2]+ (step 3, Figure 4d), the WT intensity is rather localized in R-space. It peaks at ca. 2.8 Å in the phase-uncorrected R-axis, pointing to a uniform Cu–Cu interatomic distance around 3.5 Å. This indicates, that the reaction of [CuI(NH3)2]+ with O2 results in a well-defined structure, compatible with the side-on μ-η2,η2-peroxo diamino dicopper(II) complex shown in Figure 1b and Figure 3c. In contrast, after heating in O2 at 400 °C and subsequent cooling to 200 °C in O2 (step 1, Figure 4b), a broader intensity distribution in R-space is observed in the k-space region characteristic of Cu–Cu scattering, in agreement with the presence of more heterogeneous multicopper species in the pretreated Cu-CHA catalyst.29,34,35
Figure 4 also reports the EXAFS-WTs related to the reactivity of the formed μ-η2,η2-peroxo diamino dicopper(II) complexes with the key SCR reactants, NO (step 4, Figure 4e) and NH3 (step 4′, Figure 4f). In both cases, the sub-lobe at k = 7 Å–1 is clearly lost, providing direct structural evidence for the cleavage of dicopper cores upon separate exposure to NO or NH3 at 200 °C. A moderately intense sub-lobe is instead still visible in the low-k range, characteristic of low-Z scatterers. As argued before, this feature most likely stems from multiple scattering contributions involving N and O atoms.
Finally, to comparatively assess the presence of Cu–Cu scattering contributions throughout the investigated reaction steps, we computed the power density function ΦR of the WT representation.45 This quantity was obtained integrating the square of the modulus of the WT over the R-range 2–4 Å, that should contain the Cu–Cu signal contribution. ΦR is given by the following expression:
| 1 |
where Rmin = 2 Å, Rmax = 4, and Wψ(k,R) is the wavelet transform representation of the EXAFS signal depending on the mother function used (see SI, section 3.1 for details).
Figure 5 presents the results of these calculations, summarizing the above observations about EXAFS WTs. A common first peak, for all the steps, is localized in the 0.0–5.5 Å–1 range: it corresponds to the WT low-k sub-lobe, collectively accounting for the contributions from O, N, Si and Al atoms. The second main peak is present only in catalyst pretreated in O2 (curve 1) and after [CuI(NH3)2]+ reaction with O2 (curve 3). The position of this peak exactly corresponds to the maximum of the Cu backscattering amplitude function shown in Figure 4a, clearly indicating the formation of [Cu2(NH3)4(O2)]2+ complexes.
Figure 5.
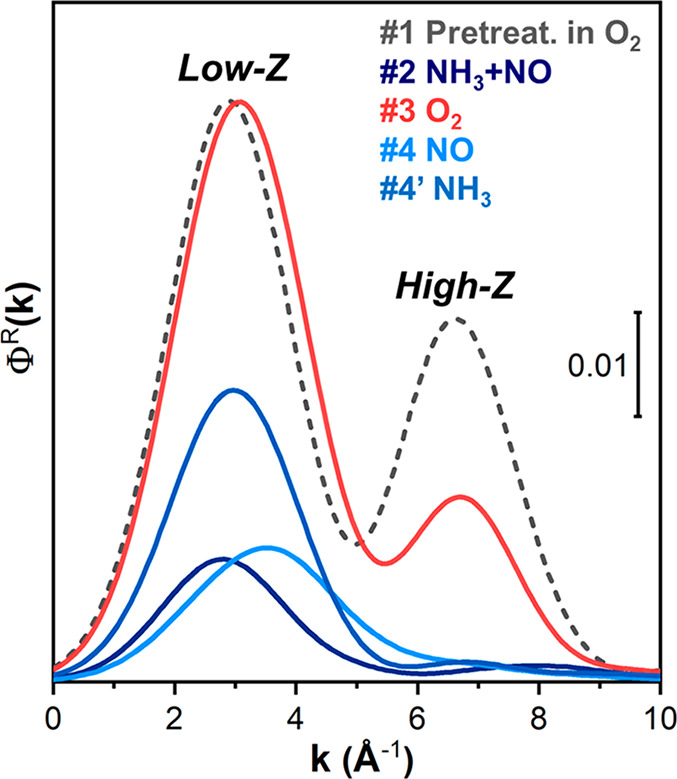
Density power function ΦR calculated for the WT representations showed in Figure 4b–f. k-Space ranges diagnostic for light-Z scatterers (O/N, Si/Al) and high-Z scatterers (Cu), based on backscattering amplitude factors F(k) reported in Figure 4a, are indicated.
3.2. Reactivity of [Cu2(NH3)4(O2)]2+ Species toward NH3 and NO
To determine the reactivity of μ-η2,η2-peroxo diamino dicopper(II) complexes toward NO and NH3, which are the main reactants in NH3-SCR, we have exposed them to NO and NH3 separately, and in a 1:1 mixture. Exposure to a mixture of NO and NH3 results in a complete restoration of a CuI oxidation state as [CuI(NH3)2]+ with formation of the N2 product, confirming the NH3-SCR reaction and the reversibility of the oxidation of the [CuI(NH3)2]+ species (see SI, Figure S12 and qualitative mass spectrometry analysis in SI, Figure S16). This observation provides experimental evidence that it is possible to close the NH3-SCR reaction cycle between the above-discussed and identified “homogeneous-like” μ-η2,η2-peroxo diamino dicopper(II) and [Cu(NH3)2]+ complexes.
3.2.1. Reactivity toward NH3: Structural Changes
As shown by EXAFS-WT analysis above (Figure 4f), exposure of the μ-η2,η2-peroxo diamino dicopper(II) complexes to NH3 results in the separation of the copper centers. No significant N2 evolution is observed during this transformation (see SI, Figure S17). In this section, we focus on the structural changes of the resulting Cu complexes.
Figure 6 reports the XAS and UV-Vis-NIR spectra for the μ-η2,η2-peroxo diamino dicopper(II) complexes during exposure to NH3 at 200 °C. The characteristic CuI transition at ∼8982.5 eV for the linear [CuI(NH3)2]+ appears, but does not reach the intensity observed in the fully reduced catalyst (dashed dark-blue curve in Figure 6a). The intensity of the 1s→3d pre-edge peak (Figure 6c) decreases, corroborating that some reduction of the Cu takes place. In the corresponding UV-Vis-NIR spectra, the intense d-d transition, which appears at around 14 000 cm–1 (Figure 6d) becomes less intense and shifts from 13 800 to 14 400 cm–1, without substantial change in the peak shape. The fact that this peak does not disappear completely, indicates that a part of the Cu remains in the CuII state. The shift is in agreement with a change in the ligands bonding the CuII ions, affecting the d-d orbital splitting.19,32,34,49 This is also testified by the progressive consumption of the LMCT absorption between 27 000 and 31 000 cm–1, which is consistent with the disappearance of the peroxo group in the diamino dicopper(II) complex.26,34,35 Thus, exposure of the μ-η2,η2-peroxo diamino dicopper(II) complexes to NH3 leads to a change in the ligands of the Cu and to a partial reduction to a CuI species.
Figure 6.

Exposure to NH3/He at 200 °C of Cu-CHA catalyst after step 3. (a) Cu K-edge XANES spectra. (b) Phase-uncorrected k2-weighted FT EXAFS curves. (c) Magnification of CuII 1s →3d transition. (d) UV-Vis-NIR DR spectra. Red thick line: spectrum collected in step 3 (same as Figures 2 and 3); blue thick line: after NH3/He exposure; gray thin lines: intermediates; dashed dark blue line : step 2 (NO/NH3 at 200 °C after pretreatment in O2, same as dark blue in Figures 2 and 3). (e) Illustration of Cu species proposed to be formed upon reaction of the μ-η2,η2-peroxo diamino dicopper(II) complexes with NH3. Atom color code: Cu, green; H, white; O, red; N, blue.
The UV-Vis features described above only indicate the changes in the coordination sphere of CuII ions, CuI ions being essentially silent. On the other hand, XANES and EXAFS give average information on all copper species formed in this reaction step. A decrease in the average number of ligands surrounding Cu ions is indicated by the reduced intensity of the EXAFS first shell peak (Figure 6b). The intensity, shape and energy position of the XANES features suggest the presence of a mixture of [CuI(NH3)2]+ and [CuII(NH3)3(X)]+ complexes, with a geometry similar to the [CuII(NH3)3(OH)]2+ species, as reported earlier.19,32 A XANES linear combination fit (see SI, Figure S13) indicates that the fractions of [CuI(NH3)2]+ and [CuII(NH3)3(X)]+ at equilibrium are 65% and 35%, respectively. Considering that we have 16% CuI and 84% CuII before exposure to NH3 (see section 3.1), this means that approximately 58% of the amount of the copper ions in the μ-η2,η2-peroxo diamino dicopper(II) complexes are reduced to CuI.
3.2.2. Reactivity toward NH3: Interpretation
The results described above indicate that NH3 is reacting with the μ-η2,η2-peroxo diamino dicopper(II) complexes, breaking the copper dimer and reducing a consistent fraction of the CuII ions to CuI. No N2 is observed during this reaction, in agreement with the fact that direct oxidation of ammonia (eq 2), only occurs on Cu-CHA at higher temperature.50
| 2 |
The XANES and UV-Vis spectra (light blue curves in Figure 6a,d) are consistent with the presence of CuII ions in a pseudo-square-planar geometry, similar to the [CuII(NH3)3(OH)] + species predicted by Paolucci et al. and experimentally observed by Borfecchia et al.7,19 We could thus hypothesize that the corresponding CuII ions are in the form of a superoxo amino [CuII(NH3)3(OO*)]+ complex, as depicted in Figure 6e. This geometry is consistent with the relatively high intensity of the low-k sub-lobe in EXAFS WT data (Figure 4f and curve 4′ in Figure 5), related to multiple scattering contributions from N/O ligand atoms.
The superoxo [CuII(NH3)3(OO*)]+ complex could be formed by a one-electron transfer from the bridged peroxo group in the μ-η2,η2-peroxo diamino dicopper(II) complexes to one of the CuII ions, with consequent formation of CuI ions and of the superoxo ligand. The resulting CuI ions are thus stabilized as [CuI(NH3)2]+. This could be rationalized with eq 3, which should result in the reduction of 50% of the CuII ions in the dimer to CuI.
| 3 |
Our XANES linear combination fitting indicates a higher efficiency of the CuII-to-CuI reduction with NH3 (ca. 58% of CuII reduced) with respect to what is expected on the basis of eq 3. This could be related to a further reduction of the superoxo [CuII(NH3)3(OO*)]+ complexes by the available NH3 present in the system, with formation of [CuI(NH3)2]+. Interestingly, DFT simulations using the M06-HF-D3 functional predict that reaction 3 is exothermic at the experimental conditions. The computed variation of internal energy is −4.6 kJ/mol; see SI for further details. Despite the clear indications provided by the linear combination fitting and DFT simulations, the associated uncertainties are too high to use them as an ultimate proof for the reaction 3, so this may be investigated in further works. Interestingly, the reaction tentatively proposed in (3) could provide clues as to the origin of the observed NH3-inhibition effect and negative apparent NH3 rate orders, observed by different authors.12,51,52
3.2.3. Reactivity toward NO: Experimental Evidence
The reaction of the μ-η2,η2-peroxo diamino dicopper(II) complexes with NO results in the separation of the copper centers, as shown by EXAFS-WT analysis (see above, Figure 4e). This is accompanied by some formation of N2, as monitored by online mass spectrometry (see SI, Figure S18). Due to the used experimental setup, the acquired mass spectrometry data are not accurate enough to be used for quantitative considerations. This section provides information about the dynamics of the reaction and summarizes the main experimental findings.
Figure 7 reports the XAS and UV-Vis-NIR spectra collected when contacting the μ-η2,η2-peroxo diamino dicopper(II) complexes formed in step 3 with NO at 200 °C. The changes in the pre-edge features at the Cu K-edge in XANES reveals a fast and effective CuII-to-CuI transformation. The characteristic 1s→4p transition at ∼8982.5 eV reappears in the spectrum (Figure 7a), and the weak 1s→3d transition at 8977.3 eV, indicative of a CuII species, disappears (Figure 7c). To illustrate the differences in the formation of CuI species in the reactions of the μ-η2,η2-peroxo diamino dicopper(II) complexes with NO and NH3, we have compared the temporal evolution of the 1s→4p transition at ∼8982.5 eV in the two cases (Figure 7f). These data show that the reduction of the μ-η2,η2-peroxo diamino dicopper(II) complex is faster and more efficient with NO than with NH3, since the CuI peak reaches about 76% of the intensity observed for the fully reduced reference state, i.e., [CuI(NH3)2]+, as formed in step 2 (dark blue dashed line in Figure 7a) after only 5 min, stabilizing at about 84% after 35 min. In the case of reduction in NH3, the intensity is about 43% after 5 min, and reaches about 62% after 35 min. We note that the intensity of the CuI rising-edge peak can be affected by the geometry of Cu complexes.43 However, even though these data cannot be used to obtain precise kinetics of the reaction, they clearly show a difference in the reduction behavior in the two cases.
Figure 7.

Exposure to NO/He at 200 °C of Cu-CHA catalyst after step 3. (a) Cu K-edge XANES spectra. (b) Phase-uncorrected k2-weighted FT EXAFS curves. (c) Magnification of CuII 1s→3d transition. (d) Initial and (e) subsequent evolution of DR UV-Vis-NIR spectra. Red thick line: spectrum collected in step 3 (same as Figures 2 and 3); light blue thick line: after of NO/He exposure; gray thin lines: intermediates; dashed dark blue line: step 2 (NO/NH3 at 200 °C after pretreatment in O2, same as dark blue in Figures 2 and 3); orange thick line is the final spectrum in the panel (d) and initial one in the panel (e). (f) Temporal evolution of the intensity of the CuI 1s →4p transition at ∼8982.5 eV during exposure to NO or NH3 after step 3. (g) Illustration of Cu species proposed to be formed upon reaction with NO. Atom color code: Cu, green; H, white; O, red; N, blue; Si, yellow; Al, pink.
In the DR UV-Vis-NIR spectra, the reaction with NO is visible in the symmetrical d-d absorption at around 13850 cm–1, corresponding to the CuII sites in a pseudo-square-planar geometry in the μ-η2,η2-peroxo diamino dicopper(II) complex. In the first minutes of NO exposure, the intensity reaches a minimum (Figure 7d, from red to orange curve), followed by the development of a complex absorption with maxima at 20 000, 16 350, 13 300, and 10 600 cm–1 (shoulder), associated with CuII in a different local environment (Figure 7e, from orange to light blue). These new features resemble the typical “quadruplet”, as often observed in Cu-CHA samples after pretreatment in O2 (see Figure 2 and SI, Figure S14), which are assigned to the formation of a variety of monomeric and multimeric framework-coordinated CuII ions, such as Z[CuII(OH)]/Z[CuII(OO*)] sites etc.26,33,34 This indicates the formation of some oxidic CuII species during the reaction. The LMCT absorption between 27 000 and 31 000 cm –1 (Figure 7d), related to the bridged peroxo groups in the μ-η2,η2-peroxo diamino dicopper(II) complex, also shows a rapid decrease in intensity in the reaction with NO. The subsequent change in the geometry of remaining CuII sites is reflected in a small blue-shift in the LMCT position (Figure 7e). We note that the XANES data do not reveal the presence of a CuII species after reaction with NO, suggesting that the observed CuII fraction remains below the XAS detection limit under our experimental conditions, which is estimated at around 10% of the total Cu content. This would then indicate that DR UV-Vis is very sensitive to the formation of this oxidic CuII species, due to the strong influence of the local geometry on the corresponding extinction coefficient.
The structure of the CuI ions formed during the reaction of μ-η2,η2-peroxo diamino dicopper(II) complex with NO is different from those observed after reduction in NO/NH3 at 200 °C or in the reaction with NH3. The shape of the XANES rising-edge and white-line peaks clearly differ from those of the linear [Cu(NH3)2]+ complexes, as indicated by the light blue and dotted dark blue curves in Figure 7a, and those of ligand-free, framework-coordinated ZCuI.25 Overall, the Cu K-edge XANES resembles that observed after desorption of NH3 (see SI, Figure S15), which has been assigned to framework-coordinated linear CuI amino complexes, Z[CuI(NH3)].19 This assignment is supported by the NH3/NH4+ vibrational modes still present in the NIR region (Figure 7d and e) and by the decrease observed in the first-shell peak in EXAFS (Figure 7b), indicating a change from a four- to a two-fold coordination of Cu (Figures 4e and 7g). The broadening of the second and third shell regions can be moreover connected to the relatively high degree of freedom (in terms of bond length and angles) of the proposed Z[CuI(NH3)] entities with respect to [Cu(NH3)2]+ or “bare” ZCuI species.6,10,19
3.2.4. Reactivity toward NO: Interpretation
The results in Figure 7d show that NO reaction causes a fast disaggregation of the side-on μ-η2,η2-peroxo diamino dicopper(II) complexes. The CuII species are almost completely reduced to CuI, while the bridging peroxo groups are consumed and N2 is formed. The formation of N2 is fast (see SI, Figure S18), indicating that μ-η2,η2-peroxo diamino dicopper(II) complexes are very reactive toward NO. These results provide experimental support for the conclusion from DFT calculations, that NO facilitates the dissociation of the O–O bond in oxygen.4,13,15,16
According to our interpretation of the XANES results (Figure 7a), the CuI species after reaction with NO consists of framework-coordinated Z[CuI(NH3)] moieties, implying that each Cu ion in the dimer loses only one amino ligand during the reduction. Starting from the μ-η2,η2-peroxo diamino dicopper(II) complexes, the NH3-SCR reaction requires stoichiometrically two NH3 molecules per Cu for a complete reduction of the CuII,8 and therefore the second NH3 molecule is a nonligand NH3. In a recent DFT study, it is proposed that the NH3-SCR reaction proceeds via the decomposition of HONO and H2NNO intermediates to N2 and H2O, over Brønsted NH4+(or H+/NH3(g)) sites.13 This would be a way to include a second nonligand NH3 molecule in the SCR cycle, but such a role of Brønsted sites in the NH3-SCR cycle still needs experimental verification.53
Even though the XANES results point to a complete reduction of the CuII, a minor fraction of CuII is still present after reaction with NO, which remains below the detection limit of XANES under our experimental conditions. The presence of these CuII moieties in the XAS experiment is indicated by the small amount of N2 that is formed upon adding NH3 to the NO feed after step 4, to restore the fully reduced state consisting of [Cu(NH3)2]+ complexes (SI, Figure S19). The formation of N2 indicates that some reduction of CuII takes place, thus proving that the reduction with NO alone was not complete. We expect the amount of this residual CuII fraction to depend on the Cu content and Si/Al ratio of the Cu-CHA material.
The features in the UV-Vis observed at 20 000, 16 350, 13 300, and 10 600 (sh) cm–1 after the reaction with NO is completed resemble the UV–Vis “quadruplet”, that is often observed for CuII in Cu-CHA and other small pore zeolites.26,33−36,54 These features have been assigned to CuII species attached to the zeolite framework, such as Z[CuII(OH)], Z[CuII(OO*)], framework-coordinated Cu dimers with O/OH bridging moieties, or even larger Cu clusters.34 This suggests that the CuII species that remains after reaction of the μ-η2,η2-peroxo diamino dicopper(II) complex with NO is attached to the zeolite framework as well. We also note that the high intensity of these features is comparable to that observed in fully oxidized Cu-CHA samples (see SI, Figure S14), despite the low fraction of CuII. This puzzling finding could be related to the fact that this spectroscopic feature is the result of a variety of CuII ions with similar but not identical local environments affecting the d-splitting,55,56 as recently predicted by Li et al.34
4. Conclusions
We have studied the activation of oxygen over the mobile linear [Cu(NH3)2]+ complexes in a Cu-CHA catalyst for NH3-SCR (Si/Al ratio = 15 and 2.6 wt% Cu), which is a crucial step in the NH3-SCR reaction, by combining X-ray absorption spectroscopy, and diffuse reflectance UV-Vis-NIR spectroscopy.
The reaction of the linear [Cu(NH3)2]+ complexes with O2 at 200 °C results in the formation of a side-on μ-η2, η2-peroxo diamino dicopper(II) complex ([Cu2(NH3)4O2]2+) as shown in Figure 1b, indicating a reaction of O2 with a pair of [Cu(NH3)2]+ complexes. About 84% of the Cu present is oxidized by O2, the remaining 16% stays present as linear [Cu(NH3)2]+ species. The structure of the [Cu2(NH3)4O2]2+ complex also indicates that an O–O bond is retained in this reaction. We have also successfully applied a wavelet transform analysis of the EXAFS data to identify Cu–Cu scattering contributions after the reaction with O2, providing unprecedented direct spectroscopic evidence for the formation of Cu-pairs in the NH3-SCR reaction.
The [Cu2(NH3)4O2]2+ complexes show a different reactivity toward NH3, NO, or a mixture of NO and NH3 at 200 °C. The reaction with a mixture of NO and NH3 leads to a complete reduction of the [Cu2(NH3)4O2]2+ complexes with evolution of N2, and the linear [Cu(NH3)2]+ complexes are restored, confirming that the [Cu2(NH3)4O2]2+ complex plays a role in the NH3-SCR reaction cycle.
In the reaction of the [Cu2(NH3)4O2]2+ complex with NH3, in the absence of NO, about 58% of the CuII species is reduced to CuI, while no N2 is formed. The [Cu2(NH3)4O2]2+ complexes dissociate and a variety of mononuclear Cu complexes is formed, consisting of CuI and CuII species with NH3 and oxidic ligands.
In the reaction with NO, some N2 is formed, and the [Cu2(NH3)4O2]2+ complexes are almost completely reduced. The formation of N2 indicates that the NH3-SCR cycle involves a reaction of the [Cu2(NH3)4O2]2+ complex with NO. The CuI species formed in this reaction is probably a Z[CuI(NH3)], which is attached to the zeolite framework. A minor part (<10%) of the Cu remains in a CuII state, due to a lack of reactive NH3 in the catalyst. Addition of NH3 at this stage leads to the restoration of the linear [Cu(NH3)2]+ complexes with N2 evolution, further confirming the role of the reaction of NO with the [Cu2(NH3)4O2]2+ complexes in the NH3-SCR reaction cycle.
Acknowledgments
M.C. would like to express his gratitude to his scientific mentor, Prof. P. Ugliengo, for his permission to carry out research and build up collaborations, independently and with freedom. T.S. would like to deeply acknowledge Prof. Enrico Tronconi for his encouragement in pursuing this collaboration independently and in total freedom, as well as for the enlightening discussions and feedbacks.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c06270.
Experimental details on spectroscopic experiments; computational details; EXAFS fitting methods and results; wavelet transform (WT) analysis of EXAFS spectra, methods and additional results; additional in situ spectroscopy results; mass spectrometry results (PDF)
Author Present Address
# C.N.: Department of Chemistry, Center for Materials Science and Nanotechnology, University of Oslo, Sem Sælands vei 26, 0371 Oslo, Norway
Author Present Address
∇ T.S.: European Commission Joint Research Centre, 21027 Ispra, Italy
Author Contributions
¶ C.N. and T.S. contributed equally to this article.
The authors declare no competing financial interest.
Supplementary Material
References
- Beale A. M.; Gao F.; Lezcano-Gonzalez I.; Peden C. H. F.; Szanyi J. Recent advances in automotive catalysis for NOx emission control by small-pore microporous materials. Chem. Soc. Rev. 2015, 44 (20), 7371–7405. 10.1039/C5CS00108K. [DOI] [PubMed] [Google Scholar]
- Borfecchia E.; Beato P.; Svelle S.; Olsbye U.; Lamberti C.; Bordiga S. Bordiga Cu-CHA – a model system for applied selective redox catalysis. Chem. Soc. Rev. 2018, 47, 8097–8133. 10.1039/C8CS00373D. [DOI] [PubMed] [Google Scholar]
- Paolucci C.; Verma A. A.; Bates S. A.; Kispersky V. F.; Miller J. T.; Gounder R.; Delgass W. N.; Ribeiro F. H.; Schneider W. F. Isolation of the Copper Redox Steps in the Standard Selective Catalytic Reduction on Cu-SSZ-13. Angew. Chem., Int. Ed. 2014, 53 (44), 11828–11833. 10.1002/anie.201407030. [DOI] [PubMed] [Google Scholar]
- Gao F.; Mei D.; Wang Y.; Szanyi J.; Peden C. H. F. Selective Catalytic Reduction over Cu/SSZ-13: Linking Homo- and Heterogeneous Catalysis. J. Am. Chem. Soc. 2017, 139, 4935–4942. 10.1021/jacs.7b01128. [DOI] [PubMed] [Google Scholar]
- Greenaway A. G.; Marberger A.; Thetford A.; Lezcano-Gonzalez I.; Agote-Aran M.; Nachtegaal M.; Ferri D.; Krocher O.; Catlow C. R. A.; Beale A. M. Detection of key transient Cu intermediates in SSZ-13 during NH3-SCR deNO(x) by modulation excitation IR spectroscopy. Chem. Sci. 2020, 11 (2), 447–455. 10.1039/C9SC04905C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens T. V. W.; Falsig H.; Lundegaard L. F.; Vennestrom P. N. R.; Rasmussen S. B.; Moses P. G.; Giordanino F.; Borfecchia E.; Lomachenko K. A.; Lamberti C.; Bordiga S.; Godiksen A.; Mossin S.; Beato P. A Consistent Reaction Scheme for the Selective Catalytic Reduction of Nitrogen Oxides with Ammonia. ACS Catal. 2015, 5 (5), 2832–2845. 10.1021/cs501673g. [DOI] [Google Scholar]
- Paolucci C.; Parekh A. A.; Khurana I.; Di Iorio J. R.; Li H.; Caballero J. D. A.; Shih A. J.; Anggara T.; Delgass W. N.; Miller J. T.; Ribeiro F. H.; Gounder R.; Schneider W. F. Catalysis in a Cage: Condition-Dependent Speciation and Dynamics of Exchanged Cu Cations in SSZ-13 Zeolites. J. Am. Chem. Soc. 2016, 138 (18), 6028–6048. 10.1021/jacs.6b02651. [DOI] [PubMed] [Google Scholar]
- Hammershøi P. S.; Negri C.; Berlier G.; Bordiga S.; Beato P.; Janssens T. V. W. Temperature-programmed reduction with NO as a characterization of active Cu in Cu-CHA catalysts for NH3-SCR. Catal. Sci. Technol. 2019, 9 (10), 2608–2619. 10.1039/C9CY00358D. [DOI] [Google Scholar]
- Lezcano-Gonzalez I.; Wragg D. S.; Slawinski W. A.; Hemelsoet K.; Van Yperen-De Deyne A.; Waroquier M.; Van Speybroeck V.; Beale A. M. Determination of the Nature of the Cu Coordination Complexes Formed in the Presence of NO and NH3 within SSZ-13. J. Phys. Chem. C 2015, 119 (43), 24393–24403. 10.1021/acs.jpcc.5b06875. [DOI] [Google Scholar]
- Lomachenko K. A.; Borfecchia E.; Negri C.; Berlier G.; Lamberti C.; Beato P.; Falsig H.; Bordiga S. The Cu-CHA deNO(x) Catalyst in Action: Temperature-Dependent NH3-Assisted Selective Catalytic Reduction Monitored by Operando XAS and XES. J. Am. Chem. Soc. 2016, 138 (37), 12025–12028. 10.1021/jacs.6b06809. [DOI] [PubMed] [Google Scholar]
- Chen L.; Jansson J.; Skoglundh M.; Grönbeck H. Mechanism for Solid-State Ion Exchange of Cu+ into Zeolites. J. Phys. Chem. C 2016, 120 (51), 29182–29189. 10.1021/acs.jpcc.6b09553. [DOI] [Google Scholar]
- Jones C. B.; Khurana I.; Krishna S. H.; Shih A. J.; Delgass W. N.; Miller J. T.; Ribeiro F. H.; Schneider W. F.; Gounder R. Effects of dioxygen pressure on rates of NOx selective catalytic reduction with NH3 on Cu-CHA zeolites. J. Catal. 2020, 389, 140–149. 10.1016/j.jcat.2020.05.022. [DOI] [Google Scholar]
- Chen L.; Janssens T. V. W.; Vennestrøm P. N. R.; Jansson J.; Skoglundh M.; Grönbeck H. A Complete Multisite Reaction Mechanism for Low-Temperature NH3-SCR over Cu-CHA. ACS Catal. 2020, 10 (10), 5646–5656. 10.1021/acscatal.0c00440. [DOI] [Google Scholar]
- Selleri T.; Ruggeri M. P.; Nova I.; Tronconi E. The Low Temperature Interaction of NO + O-2 with a Commercial Cu-CHA Catalyst: A Chemical Trapping Study. Top. Catal. 2016, 59 (8–9), 678–685. 10.1007/s11244-016-0543-9. [DOI] [Google Scholar]
- Falsig H.; Vennestrom P. N. R.; Moses P. G.; Janssens T. V. W. Activation of Oxygen and NO in NH3-SCR over Cu-CHA Catalysts Evaluated by Density Functional Theory. Top. Catal. 2016, 59 (10–12), 861–865. 10.1007/s11244-016-0560-8. [DOI] [Google Scholar]
- Chen L.; Falsig H.; Janssens T. V. W.; Gronbeck H. Activation of oxygen on (NH3-Cu-NH3)(+) in NH3-SCR over Cu-CHA. J. Catal. 2018, 358, 179–186. 10.1016/j.jcat.2017.12.009. [DOI] [Google Scholar]
- Paolucci C.; Khurana I.; Parekh A. A.; Li S. C.; Shih A. J.; Li H.; Di Iorio J. R.; Albarracin-Caballero J. D.; Yezerets A.; Miller J. T.; Delgass W. N.; Ribeiro F. H.; Schneider W. F.; Gounder R. Dynamic multinuclear sites formed by mobilized copper ions in NOx selective catalytic reduction. Science 2017, 357 (6354), 898–903. 10.1126/science.aan5630. [DOI] [PubMed] [Google Scholar]
- Gao F.; Walter E. D.; Kollar M.; Wang Y.; Szanyi J.; Peden C. H. F. Understanding ammonia selective catalytic reduction kinetics over Cu/SSZ-13 from motion of the Cu ions. J. Catal. 2014, 319 (0), 1–14. 10.1016/j.jcat.2014.08.010. [DOI] [Google Scholar]
- Borfecchia E.; Negri C.; Lomachenko K. A.; Lamberti C.; Janssens T. V. W.; Berlier G. Temperature-dependent dynamics of NH3-derived Cu species in the Cu-CHA SCR catalyst. React. Chem. Eng. 2019, 4 (6), 1067–1080. 10.1039/C8RE00322J. [DOI] [Google Scholar]
- Tyrsted C.; Borfecchia E.; Berlier G.; Lomachenko K. A.; Lamberti C.; Bordiga S.; Vennestrom P. N. R.; Janssens T. V. W.; Falsig H.; Beato P.; Puig-Molina A. Nitrate-nitrite equilibrium in the reaction of NO with a Cu-CHA catalyst for NH3-SCR. Catal. Sci. Technol. 2016, 6 (23), 8314–8324. 10.1039/C6CY01820C. [DOI] [Google Scholar]
- Chen L.; Falsig H.; Janssens T. V. W.; Jansson J.; Skoglundh M.; Gronbeck H. Effect of Al-distribution on oxygen activation over Cu-CHA. Catal. Sci. Technol. 2018, 8 (8), 2131–2136. 10.1039/C8CY00083B. [DOI] [Google Scholar]
- Chen L.; Janssens T. V. W.; Gronbeck H. A comparative test of different density functionals for calculations of NH3-SCR over Cu-Chabazite. Phys. Chem. Chem. Phys. 2019, 21 (21), 10923–10930. 10.1039/C9CP01576K. [DOI] [PubMed] [Google Scholar]
- Cramer C. J.; Kinal A.; Wloch M.; Piecuch P.; Gagliardi L. Theoretical characterization of end-on and side-on peroxide coordination in ligated Cu2O2 models. J. Phys. Chem. A 2006, 110 (40), 11557–11568. 10.1021/jp064232h. [DOI] [PubMed] [Google Scholar]
- Cramer C. J.; Wloch M.; Piecuch P.; Puzzarini C.; Gagliardi L. Theoretical models on the Cu2O2 torture track: Mechanistic implications for oxytyrosinase and small-molecule analogues. J. Phys. Chem. A 2006, 110 (5), 1991–2004. 10.1021/jp056791e. [DOI] [PubMed] [Google Scholar]
- Borfecchia E.; Lomachenko K. A.; Giordanino F.; Falsig H.; Beato P.; Soldatov A. V.; Bordiga S.; Lamberti C. Revisiting the nature of Cu sites in the activated Cu-SSZ-13 catalyst for SCR reaction. Chem. Sci. 2015, 6 (1), 548–563. 10.1039/C4SC02907K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri C.; Signorile M.; Porcaro N. G.; Borfecchia E.; Berlier G.; Janssens T. V. W.; Bordiga S. Dynamic Cu-II/Cu-I speciation in Cu-CHA catalysts by in situ Diffuse Reflectance UV-vis-NIR spectroscopy. Appl. Catal., A 2019, 578, 1–9. 10.1016/j.apcata.2019.03.018. [DOI] [Google Scholar]
- Kieger S.; Delahay G.; Coq B.; Neveu B. Selective catalytic reduction of nitric oxide by ammonia over Cu-FAU catalysts in oxygen-rich atmosphere. J. Catal. 1999, 183 (2), 267–280. 10.1006/jcat.1999.2398. [DOI] [Google Scholar]
- Mathon O.; Beteva A.; Borrel J.; Bugnazet D.; Gatla S.; Hino R.; Kantor I.; Mairs T.; Munoz M.; Pasternak S.; Perrin F.; Pascarelli S. The time-resolved and extreme conditions XAS (TEXAS) facility at the European Synchrotron Radiation Facility: the general-purpose EXAFS bending-magnet beamline BM23. J. Synchrotron Radiat. 2015, 22, 1548–1554. 10.1107/S1600577515017786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas D. K.; Borfecchia E.; Dyballa M.; Pankin I. A.; Lomachenko K. A.; Martini A.; Signorile M.; Teketel S.; Arstad B.; Berlier G.; Lamberti C.; Bordiga S.; Olsbye U.; Lillerud K. P.; Svelle S.; Beato P. Methane to Methanol: Structure Activity Relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139 (42), 14961–14975. 10.1021/jacs.7b06472. [DOI] [PubMed] [Google Scholar]
- Pankin I. A.; Martini A.; Lomachenko K. A.; Soldatov A. V.; Bordiga S.; Borfecchia E. Identifying Cu-oxo species in Cu-zeolites by XAS: A theoretical survey by DFT-assisted XANES simulation and EXAFS wavelet transform. Catal. Today 2020, 345, 125–135. 10.1016/j.cattod.2019.09.032. [DOI] [Google Scholar]
- Vilella L.; Studt F. The Stability of Copper Oxo Species in Zeolite Frameworks. Eur. J. Inorg. Chem. 2016, 2016 (10), 1514–1520. 10.1002/ejic.201501270. [DOI] [Google Scholar]
- Negri C.; Borfecchia E.; Cutini M.; Lomachenko K. A.; Janssens T. V. W.; Berlier G.; Bordiga S. Evidence of Mixed-Ligand Complexes in Cu-CHA by Reaction of Cu Nitrates with NO/NH3 at Low Temperature. ChemCatChem 2019, 11 (16), 3828–3838. 10.1002/cctc.201900590. [DOI] [Google Scholar]
- Giordanino F.; Vennestrom P. N. R.; Lundegaard L. F.; Stappen F. N.; Mossin S.; Beato P.; Bordiga S.; Lamberti C. Characterization of Cu-exchanged SSZ-13: a comparative FTIR, UV-Vis, and EPR study with Cu-ZSM-5 and Cu-beta with similar Si/Al and Cu/Al ratios. Dalton T. 2013, 42 (35), 12741–12761. 10.1039/c3dt50732g. [DOI] [PubMed] [Google Scholar]
- Li H.; Paolucci C.; Khurana I.; Wilcox L.; Goltl F.; Albarracin-Caballero J. D.; Shih A. J.; Ribeiro F. H.; Gounder R.; Schneider W. F. Consequences of exchange-site heterogeneity and dynamics on the UV-visible spectrum of Cu-exchanged SSZ-13. Chem. Sci. 2019, 10 (8), 2373–2384. 10.1039/C8SC05056B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipek B.; Wulfers M. J.; Kim H.; Goltl F.; Hermans I.; Smith J. P.; Booksh K. S.; Brown C. M.; Lobo R. F. Formation of Cu2O2 (2+) and Cu2O (2+) toward C-H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39. ACS Catal. 2017, 7 (7), 4291–4303. 10.1021/acscatal.6b03005. [DOI] [Google Scholar]
- Wulfers M. J.; Teketel S.; Ipek B.; Lobo R. F. Conversion of methane to methanol on copper-containing small-pore zeolites and zeotypes. Chem. Commun. 2015, 51 (21), 4447–4450. 10.1039/C4CC09645B. [DOI] [PubMed] [Google Scholar]
- Mahadevan V.; Hou Z. G.; Cole A. P.; Root D. E.; Lal T. K.; Solomon E. I.; Stack T. D. P. Irreversible reduction of dioxygen by simple peralkylated diamine-copper(I) complexes: Characterization and thermal stability of a Cu-2(mu-O)(2) (2+) core. J. Am. Chem. Soc. 1997, 119 (49), 11996–11997. 10.1021/ja972946n. [DOI] [Google Scholar]
- Park G. Y.; Qayyum M. F.; Woertink J.; Hodgson K. O.; Hedman B.; Sarjeant A. A. N.; Solomon E. I.; Karlin K. D. Geometric and Electronic Structure of {Cu(MeAN)}(2)(mu-eta(2):eta(2)(O-2(2-))) (2+) with an Unusually Long O-O Bond: O-O Bond Weakening vs Activation for Reductive Cleavage. J. Am. Chem. Soc. 2012, 134 (20), 8513–8524. 10.1021/ja300674m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutsail G. E.; Gagnon N. L.; Spaeth A. D.; Tolman W. B.; DeBeer S. Valence-to-Core X-ray Emission Spectroscopy as a Probe of O-O Bond Activation in Cu2O2 Complexes. Angew. Chem., Int. Ed. 2019, 58 (27), 9114–9119. 10.1002/anie.201903749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I.; Chen P.; Metz M.; Lee S. K.; Palmer A. E. Oxygen binding, activation, and reduction to water by copper proteins. Angew. Chem., Int. Ed. 2001, 40 (24), 4570–4590. . [DOI] [PubMed] [Google Scholar]
- Baldwin M. J.; Ross P. K.; Pate J. E.; Tyeklar Z.; Karlin K. D.; Solomon E. I. Spectroscopic and Theoretical-Studies of an End-on Peroxide-Bridged Coupled Binuclear Copper(II) Model Complex of Relevance to the Active-Sites in Hemocyanin and Tyrosinase. J. Am. Chem. Soc. 1991, 113 (23), 8671–8679. 10.1021/ja00023a014. [DOI] [Google Scholar]
- Lam B. M. T.; Halfen J. A.; Young V. G.; Hagadorn J. R.; Holland P. L.; Lledos A.; Cucurull-Sanchez L.; Novoa J. J.; Alvarez S.; Tolman W. B. Ligand macrocycle structural effects on copper-dioxygen reactivity. Inorg. Chem. 2000, 39 (18), 4059–4072. 10.1021/ic000248p. [DOI] [PubMed] [Google Scholar]
- Martini A.; Pankin I. A.; Marsicano A.; Lomachenko K. A.; Borfecchia E. Wavelet analysis of a Cu-oxo zeolite EXAFS simulated spectrum. Radiat. Phys. Chem. 2020, 175, 108333. 10.1016/j.radphyschem.2019.05.023. [DOI] [Google Scholar]
- Timoshenko J.; Kuzmin A. Wavelet data analysis of EXAFS spectra. Comput. Phys. Commun. 2009, 180 (6), 920–925. 10.1016/j.cpc.2008.12.020. [DOI] [Google Scholar]
- Funke H.; Chukalina M.; Scheinost A. C. A new FEFF-based wavelet for EXAFS data analysis. J. Synchrotron Radiat. 2007, 14, 426–432. 10.1107/S0909049507031901. [DOI] [PubMed] [Google Scholar]
- Funke H.; Scheinost A. C.; Chukalina M. Wavelet analysis of extended x-ray absorption fine structure data. Phys. Rev. B: Condens. Matter Mater. Phys. 2005, 71 (9), 094110. 10.1103/PhysRevB.71.094110. [DOI] [Google Scholar]
- Sushkevich V. L.; Safonova O. V.; Palagin D.; Newton M. A.; van Bokhoven J. A. Structure of copper sites in zeolites examined by Fourier and wavelet transform analysis of EXAFS. Chem. Sci. 2020, 11 (20), 5299–5312. 10.1039/D0SC01472A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolucci C.; Di Iorio J. R.; Ribeiro F. H.; Gounder R.; Schneider W. F.. Catalysis Science of NOx Selective Catalytic Reduction With Ammonia Over Cu-SSZ-13 and Cu-SAPO-34. In Advances in Catalysis, Vol 59; Song C., Ed.; Elsevier, 2016; 59, pp 1–107. [Google Scholar]
- Trevani L. N.; Roberts J. C.; Tremaine P. R. Copper(II)-ammonia complexation equilibria in aqueous solutions at temperatures from 30 to 250 degrees C by visible spectroscopy. J. Solution Chem. 2001, 30 (7), 585–622. 10.1023/A:1010453412802. [DOI] [Google Scholar]
- Colombo M.; Koltsakis G.; Nova I.; Tronconi E. Modelling the ammonia adsorption-desorption process over an Fe-zeolite catalyst for SCR automotive applications. Catal. Today 2012, 188 (1), 42–52. 10.1016/j.cattod.2011.09.002. [DOI] [Google Scholar]
- Selleri T.; Gramigni F.; Nova I.; Tronconi E. NO oxidation on Fe- and Cu-zeolites mixed with BaO/Al2O3: Free oxidation regime and relevance for the NH3-SCR chemistry at low temperature. Appl. Catal., B 2018, 225, 324–331. 10.1016/j.apcatb.2017.11.068. [DOI] [Google Scholar]
- Marberger A.; Petrov A. W.; Steiger P.; Elsener M.; Kröcher O.; Nachtegaal M.; Ferri D. Time-resolved copper speciation during selective catalytic reduction of NO on Cu-SSZ-13. Nat. Catal. 2018, 1 (3), 221–227. 10.1038/s41929-018-0032-6. [DOI] [Google Scholar]
- Lezcano-Gonzalez I.; Deka U.; Arstad B.; Van Yperen-De Deyne A.; Hemelsoet K.; Waroquier M.; Van Speybroeck V.; Weckhuysen B. M.; Beale A. M. Determining the storage, availability and reactivity of NH3 within Cu-Chabazite-based Ammonia Selective Catalytic Reduction systems. Phys. Chem. Chem. Phys. 2014, 16 (4), 1639–1650. 10.1039/C3CP54132K. [DOI] [PubMed] [Google Scholar]
- Godiksen A.; Stappen F. N.; Vennestrom P. N. R.; Giordanino F.; Rasmussen S. B.; Lundegaard L. F.; Mossin S. Coordination Environment of Copper Sites in Cu-CHA Zeolite Investigated by Electron Paramagnetic Resonance. J. Phys. Chem. C 2014, 118 (40), 23126–23138. 10.1021/jp5065616. [DOI] [Google Scholar]
- Solomon E. I. Spectroscopic methods in bioinorganic chemistry: Blue to green to red copper sites. Inorg. Chem. 2006, 45 (20), 8012–8025. 10.1021/ic060450d. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Heppner D. E.; Johnston E. M.; Ginsbach J. W.; Cirera J.; Qayyum M.; Kieber-Emmons M. T.; Kjaergaard C. H.; Hadt R. G.; Tian L. Copper Active Sites in Biology. Chem. Rev. 2014, 114 (7), 3659–3853. 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.