

Abstract
A growing number of in vitro hepatic models exist to study human genetics, liver biology, disease modeling and drug development and range from 2D hepatocytes to 3D multi-cellular tissues that are derived from human stem cells. However, stem cell-based models generally suffer from batch-, clone- and donor-dependent variability, hindering broader usage in diverse biomedical applications. To circumvent this challenge, we herein describe a reproducible protocol to generate human liver organoids in 20-25 days derived from human pluripotent stem cells (PSCs). These organoids are intra-luminally polarized to form canalicular structures and are comprised of mainly hepatic epithelial cells, co-differentiated with stellate-like and hepatic macrophage-like cells that enables hepatic inflammatory disease modeling in vitro. These multi-lineage liver organoids express hepatocyte genes, secrete albumin and have vital metabolic functions. This protocol utilizes PSC derived 3D human liver organoids as a renewable, reproducible and personalized cell source, thus facilitating disease modeling and mechanistic studies with a future goal of developing novel therapeutics against currently intractable diseases.
Keywords: pluripotent stem cells, human liver organoids, steatohepatitis, self-organization
1. Introduction
The liver is the largest internal organ in the human body and is involved in a wide variety of processes including synthesis of many plasma proteins, maintaining constant blood glucose levels, metabolism, drug detoxification, and enzyme production. Blood from the entire gastrointestinal tract is delivered through the portal vein to the liver where it is then filtered before being passed to the rest of the body (Freitas-Lopes, Mafra, David, Carvalho-Gontijo, & Menezes, 2017). Many of these physiological processes are primarily performed by hepatocytes which make up >80% of the liver mass and 60% of its cellular composition, whereas the rest of the liver is composed of supporting cell types including biliary epithelial cells or cholangiocytes and non-parenchymal hepatic stellate cells, immune cells such as macrophages, and liver endothelial cells (Stanger, 2015). These non-parenchymal cells also play distinct roles in the liver including production of coagulation Factor VIII by the sinusoidal endothelium, and immune surveillance by macrophages and other immune cells (Freitas-Lopes et al., 2017; Heinz & Braspenning, 2015). Further, the progression of certain liver diseases such as non-alcoholic fatty liver disease (NAFLD) are dependent on activation of these supporting cell types, which become inflammatory and fibrotic (Higashi, Friedman, & Hoshida, 2017; Kazankov et al., 2019; Li, He, Li, Liu, & Gong, 2017).
In the liver, exogenous compounds including drugs are metabolized through a multifaceted process for removal from the body. Following transport of compounds into hepatocytes, specific enzymes are responsible for metabolizing drugs for excretion/secretion (Almazroo, Miah, & Venkataramanan, 2017). For example, the cytochrome p450 (Cyp450) enzymes found in the smooth ER and mitochondria of hepatocytes convert lipophilic chemicals to hydrophilic products allowing them to be excreted (Almazroo et al., 2017). Exposure to certain drugs can result in hepatocyte toxicity, wherein oxidative stress, production of reactive metabolites, and induction of mitochondrial dysfunction can result in immune cell activation, causing inflammation (Mosedale & Watkins, 2017; Shehu, Ma, & Venkataramanan, 2017; Yuan & Kaplowitz, 2013). In order to expedite the inefficient drug development process, preclinical identification and removal of potentially hepatotoxic compounds is crucial before entering clinical trials.
The current gold standard for in vitro assessment of drug metabolism and hepatoxicity are primary human hepatocytes (PHH) grown in 2D; however, ex vivo PHH rapidly dedifferentiate and lose hepatic functionality in short-term culture (Heslop et al., 2017; Zhou, Shen, & Lauschke, 2019). Moreover, the lack of a renewable source of PHH, donor scarcity, and high inter-individual differences in drug metabolism make studying drug induced liver injury (DILI) challenging. Some DILI is due to interactions between hepatocytes and resident immune cells and therefore cannot be modeled in PHH (Bale et al., 2014). Furthermore, these 2D PHH lack the cellular complexity and 3D architecture of the liver and thus may not accurately predict drug responses and reflect disease mechanisms (Zhou et al., 2019). 3D models utilizing multiple cell types such as primary hepatocytes and primary non-parenchymal cells such as macrophage and stellate cells co-cultured into spheroids offer promise of more physiologically relevant studies. It is reported that these 3D spheroid models maintain metabolic activity and have increased sensitivity and specificity for cytotoxicity to recognize known human hepatotoxicants compared to PHH (Proctor et al., 2017). However, these models are limited by tissue availability, and genetic studies cannot be performed.
To overcome these challenges, self-organizing, 3D hepatic organoid systems derived from biopsied liver tissue or PSCs have been developed that more closely mimic the in vivo liver microenvironment (Fiorotto et al., 2018; L.-J. Wu et al., 2019; Zhou et al., 2019). Several of these models can be maintained for months or years yet still retain hepatic functions after differentiation (Akbari et al., 2019; Huch et al., 2015; Mun et al., 2019). To date, biopsy-derived hepatic organoids are predominantly comprised of epithelial cell types: hepatocytes and/or cholangiocytes (Akbari et al., 2019; Broutier et al., 2017; Huch et al., 2015; Mun et al., 2019; Wang et al., 2019; F. Wu et al., 2019), whereas hepatic models derived from PSCs offer the possibility of creating multi-lineage liver organoids from the same cell line. The protocol described herein utilizes either embryonic stem cells (ESC) or induced PSCs (iPSCs) as a renewable, reproducible and scalable source of 3D organoids that contain both hepatocytes and supportive cells relevant for disease modeling, genetic studies, and drug screening.
2. Applications of liver organoids:
2.1. Genetic and developmental studies
Human iPSCs, which can be made from any donor’s cells, for example, from cells taken during a simple blood-draw, offer a near unlimited cellular source which is critical for generating a large number of organoids for high-throughput screening. Organoids can be derived from patients with recognized disease or with confirmed genetic mutations and compared to healthy matched controls to study personalized medicine across a genetically diverse set of humans (Ouchi et al., 2019). Additionally, single base changes can be efficiently introduced to existing iPSCs using the CRISPR/Cas system to create isogenic pairs of mutant and control iPSCs, providing a platform for unequivocal analysis of the effects of a disease causing mutation or a specific polymorphism on drug metabolism (Ben Jehuda, Shemer, & Binah, 2018). Further, guiding pluripotent stem cells into different lineages utilizes the process of directed differentiation, which recapitulates many aspects of ‘development in a dish’ thus informing fundamental understanding of human development. Directed differentiation has successfully resulted in methods to guide the step-wise differentiation of pluripotent stem cells into many different digestive organoid systems (Broda, McCracken, & Wells, 2019; McCracken, Howell, Wells, & Spence, 2011). These experimental organoid model systems, including liver organoids as described here, thus serve as tractable biological systems ex vivo, to mechanistically interrogate complex human organogenesis in a way that is otherwise not possible.
2.2. Toxicity and efficacy studies
In the liver organoid model, hepatocytes share anatomical characteristics with those cells found in vivo. For example, induced hepatocytes have discrete cellular polarity as Type IV collagen is expressed on the outer surface of the cells and Zonula occludens-1 (ZO-1) is expressed only on the apical cell surface of the intraluminal lining of the organoids, and using transmission electron microscopy we have demonstrated that liver organoid hepatocytes contain microvilli directed toward the canalicular structure of the organoid (Shinozawa, et al, submitted). Induced hepatocytes express many hepatic genes (such as HNF4-a, ASGR1, A1At, TDO2, TTR) and become functional in multiple ways including secretion of the serum protein albumin, as well as producing and transporting bile acids (Ouchi et al., 2019) (Shinozawa et al). Liver organoids can model drug toxicity in a high-throughput manner based on metabolic functionalities of the hepatocytes, including their Cyp450 enzyme expression and inducibility, and their ability to model mitochondrial dysfunction after known toxic drug treatment (Shinozawa et al).
The pathology of many liver diseases is complex, but several liver diseases including NAFLD/non-alcoholic steatohepatitis (NASH), alcoholic steatohepatitis (ASH), and chronic liver hepatitis have strong inflammatory components that involve activation of resident immune cells and stellate cells (Higashi et al., 2017; Kazankov et al., 2019; Li et al., 2017). In NAFLD the accumulation of lipids in fatty hepatocytes can activate macrophages to produce pro-inflammatory cytokines through a variety of mechanisms (Kazankov et al., 2019; Li et al., 2017). These pro-inflammatory mediators can recruit other immune cells, damage the epithelial cells and activate stellate cells to produce excessive extracellular matrix proteins. These accumulating matrix proteins can result in measurable tissue stiffness, and ultimately fibrosis. Understanding the interplay between hepatocytes, immune and stellate cells is critical for studying relevant liver diseases. The liver organoids contain mainly hepatocyte cells, along with macrophage-like and stellate-like cells which is a more physiologically relevant model for studying pathological diseases involving inflammation and fibrosis (Ouchi et al., 2019). In fact, these organoids are able to model NAFLD and display corresponding tissue stiffness and inflammation (Ouchi et al., 2019). These organoid-based pathological models are a powerful tool that mirrors many aspects of the patients’ phenotype at a personalized level and can be ultimately utilized for the selection of efficacious compounds in preclinical testing.
3. Overview of differentiation protocol
This protocol will describe the step-wise differentiation of liver organoids from iPSC into definitive endoderm (DE) followed by early foregut stage and finally to functional liver organoids through the addition of cytokines and small molecules (see Fig. 1) and this protocol will discuss specific markers that can be used to benchmark each stage of differentiation. Although an endoderm differentiation method is utilized, a consistent percentage of these cells will co-differentiate into mesoderm cells. This co-differentiation is key to generating multi-lineage liver organoids. After the foregut stage, clusters of cells are embedded into Matrigel where multi-lineage organoids form. Also described is a protocol for whole mount staining, an essential methodology for characterization of the organoids, and fatty acid treatment and live imaging of liver organoids.
Figure 1.
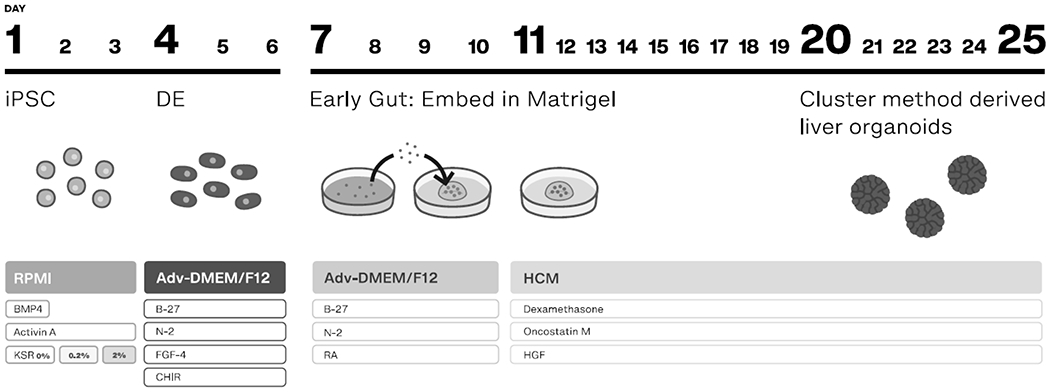
Schematic of liver organoid formation. This method follows a differentiation sequence that goes through DE and then an early gut stage. Clusters or clumps of cells are then embedded into Matrigel. The protocol ends with treatment of complete HCM and liver organoids at day 20-25.
4. Step-by-step methods
4.1. Coating plates for stem cell culture and organoid differentiation
It is necessary to pre-coat plates with a functional extracellular matrix for iPSCs to attach, proliferate, and differentiate. The Laminin protein is the main component of Matrigel, a commonly used cell culture matrix. Laminin iMatrix-511 is a truncated, functional form of Laminin that is xeno-free, is optimal for single cell passaging, promotes adhesion and continued proliferation (Miyazaki et al., 2012). The coated plates can be wrapped in parafilm and kept for up to two weeks at 4oC.
iPSCs are routinely passaged and maintained using a single cell passaging protocol and grown on Laminin with mTeSR1 media. Maintenance of the cell lines involves using Accutase to produce a single cell suspension, counting cells to the desired cell concentration, and adding fresh mTeSR1 with Y-27632. This single cell passaging method still produces colonies, and depending on cell line and passaging concentration needs to be passaged every 4-7 days.
4.1.2. Materials and reagents
Laminin iMatrix-511 Silk, stock 0.5 mg/ml (892021, Nacalai USA).
Sterile Dulbecco's phosphate-buffered saline, no calcium, no magnesium (DPBS-/-) (14190, Gibco).
Cell culture plates. BD Multi-well Flat-Bottom Plates (353046, 353047, Corning).
4.1.3. Protocol
The Laminin dilution used to coat the plate is based on the surface area of the well, and the final concentration of Laminin diluted in dPBS-/- should be 0.5 ug/cm2. For example, for one well of a 6 well plate (9.5 cm2), add 9.5 uL (4.75 ug) into 990 μL dPBS-/-, and add this solution to the well. Swirl the solution in the well so the entire surface of the well is coated with solution.
Place the plate in an incubator for 1 hour at 37oC, 3 hours at room temperature (RT), or overnight at 4oC. Although the plate can be stored for up to 2 weeks at 4oC, 1 mL additional dPBS-/- should be added per well of a 6 well plate and parafilm wrapped around the outside of the plate to ensure that the wells do not become dry.
4.2. Differentiation of stem cells into liver organoids
This protocol will reproducibly produce multi-lineage liver organoids between 20-25 days after the initial plating of the cells. The stages of DE (McCracken et al., 2011) and early gut both have reproducible and defined percentages of contributing endoderm and mesoderm populations. On day 7, the monolayer undergoes morphogenetic movements that give rise to floating and attached 3D structures, called spheroids (see Fig. 2b). For the initial cell plating, we utilize Accutase and a single-cell seeding method that starts with a known cell number and results in more reproducible differentiation.
Figure 2.
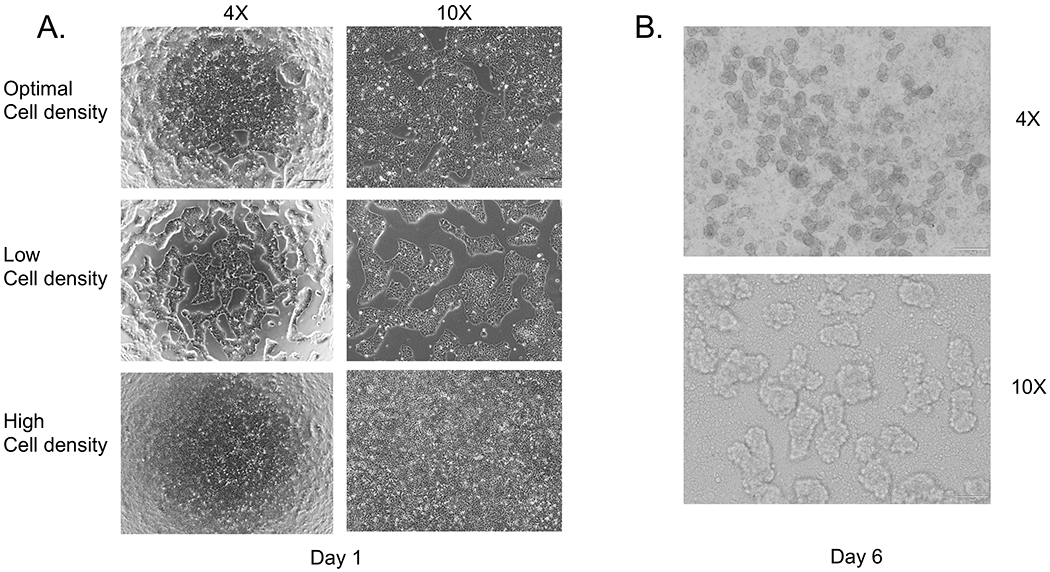
Successful co-differentiation is critically dependent on the starting cell density. A. top shows an optimal cell density of about 90% confluence, the middle panel shows a low cell density, and the lower panel is overly confluent. B. On day 6-7 spheroids will form as 3D structures attached to the monolayer and floating in the media.
4.2.1. Materials and reagents
iPSC lines, 72.3 (CCHMC Pluripotent Stem Cell Facility, (McCracken et al., 2014)), TkDA3-4 (N. Takayama et al., 2010), and 1383D6 (K. Takayama et al., 2017) are all robust, proliferative lines. This protocol has also been performed with the ES cell line WA01 (H1) from WiCell.
mTeSR1 (85850, Stem Cell Technologies) optionally supplemented with 1X Penicillin/Streptomycin (Pen/Strep) (15140, Thermo-fisher).
10 μM Y27632, Rock inhibitor (1254, Tocris). Y27632 is used to increase cell viability during single cell passaging.
Accutase, (AT104, Innovative Cell Technologies). This is a gentle enzyme mixture used for single cell passaging.
Wash media: DMEM/F12 (11330, Gibco) supplemented with 1X Pen/Strep.
RPMI Base: RPMI 1640, (11875, Gibco) supplemented with 1X Pen/Strep and 25 mM Hepes, (15630, Gibco).
Advanced DMEM Base: Advanced DMEM/F12, (12634, Gibco) supplemented with 1X B27 (17504, Gibco), 1X N2 (17502, Gibco), 10 mM Hepes, 1X Glutamax (35050, Thermo), 1X Gentamycin/Amphotericin solution (R01510, Gibco).
Complete Hepatocyte Culture Media (HCM): Lonza HCM bullet kit (CC-3198, Lonza), prepared as per manufacturer’s instructions, except no EGF, supplemented with 100 nM Dexamethasone (D4902, Sigma), 20 ng/ml recombinant human oncostatin M (300-10, Peprotech), and 10 ng/ml recombinant human hepatocyte growth factor (HGF) (100-39, Peprotech).
Day 1 Media: RPMI Base supplemented with 50 ng/ml recombinant human bone morphogenetic protein-4 (314-BP, R&D Technologies), and 100 ng/ml recombinant human Activin A (338-AC, R&D Technologies).
Day 2 Media: RPMI Base supplemented with 100 ng/ml Activin A and 0.2% Knockout serum replacement (KSR) (A3181502, Gibco).
Day 3 Media: RPMI Base supplemented with 100 ng/ml Activin A and 2% KSR.
Day 4-6 Media: Advanced DMEM Base supplemented with 500 ng/ml recombinant human fibroblast growth factor-4 (FGF-4), (100-31, Peprotech), and 3 μM CHIR99021, (4423, R&D Technologies).
Retinoic Acid (RA) Media: Advanced DMEM Base supplemented with 2 μM RA (R2625, Sigma). After preparing, keep the media in the dark or wrapped in foil.
Matrigel, (356237, Corning).
VWR multi-well plates (10062-892, 10062-896, VWR). Using these plates ensures that during the embedding step the Matrigel forms a proper domed bead shape in the well and does not spread.
Corning ultra-low attachment multi well plates (3471, 3473, Corning). These plates ensure that the organoids are kept in a floating culture in the Matrigel, and do not attach to the bottom of the wells, stabilizing the organoids’ spheroid shapes and function.
100 μm cell strainers, (352360, Corning).
Note: The Day 1, 2, 3, 4-6, and RA media are optimally made fresh each day, but can be kept for up to one week at 4oC.
4.2.2. Liver organoid protocol day 1-6, 2D culture
Day -1.
It is critical to start with high quality iPSCs between the passage numbers of 20-60. The cells should be 70-80% confluent to start and show no signs of differentiation. Before beginning, warm the mTeSR1, Accutase, and wash media to RT.
Aspirate mTeSR1 from the iPSCs and add 1 mL Accutase/ well to a 6 well plate or 0.25 mL/well to a 24 well plate. Place the plate into a 37oC, 5% CO2 incubator for 3-5 minutes, checking on the cells in the incubator to confirm that the cells have not detached from the plate. The cells are ready when they are easily detached by gentle pipetting, so it is not necessary to incubate the cells with Accutase until they detach. Add 5 mL wash media to a 15 mL conical tube.
Remove the plate from the incubator, and release the cells from the bottom of the well by gently pipetting. Add this cell suspension in Accutase to the 5 mL wash media in the conical tube. Gently pipet up and down twice to rinse the cells in the wash buffer.
Measure the viable cells with trypan blue exclusion cell counting and calculate the number of viable cells/mL in the cell suspension.
Spin down the cell suspension at 300 x g, RT for 3 minutes.
For each cell line, the optimal cell density will need to be experimentally determined to achieve a 90% cell density on day 1. For TkDA, 1383D6 and 72-3, 1.3 x 106 cells/well for a 6 well plate or 3 x 105 cells/well for a 24 well plate is ideal. This density will ensure that the DE is a flat confluent monolayer and all areas of the well are more evenly differentiated. For the purposes of this protocol, detailed instructions for a 6 well plate will be described below.
Aspirate the wash media supernatant from the cell pellet, and add the appropriate amount of mTeSR1 supplemented with 10 μM Y27632 to generate a 1 x 106 cells/mL suspension. Y27632 is critical to ensure cell survival of the single cell suspension. Add the appropriate amount of cell suspension (i.e. 1mL/well of a 6 well plate) to Laminin coated wells, and gently shake the plate side to side three times, and forward and backward three times to ensure an even cell distribution. For example, for TkDA, add 1 mL of the 1 x 106 cells/mL suspension per well.
Add 1 mL of additional mTeSR1 supplemented with Y27632 per well (2 mL total per well). Place the plate back in the incubator for 24 hours.
Day 0.
Warm mTeSR1 to RT. Confirm that the plated cells are healthy and are growing at a uniformly high density. Aspirate mTeSR1 with Y27632, and add mTeSR1 without Y27632, 2 mL/well for a 6 well plate. Place the plate back in the incubator for 24 hours.
Day 1.
On this day, the cells should have reached ~85-90% confluence (see Fig. 2a). It is imperative that the cell confluence is as close to this as possible. Too low or too high a cell density can result in overgrowth of mesoderm, cell death after Activin A treatment, and/or incomplete differentiation. Before beginning differentiation, titrate the cell number for each line to ensure 85-90% confluence on day 1. It is possible to wait an additional 24 hours until this confluency is reached, and in this case, aspirate the mTeSR1 and add 2 mL fresh mTESR1 to each well.
Warm Day 1 media to 37oC. Activin A treatment will result in cell death, so pre-warming the media to 37oC on this day helps to increase cell survival. Aspirate mTeSR1 media. Add 2 mL/well for a 6 well plate of Day 1 media. Place the plate back in the incubator for 24 hours.
Day 2.
After one day of treatment with ‘Day 1 Media’ there will be cell death and floating cells in the media. However, there should still be a layer of cells attached to the plate after treatment. View cells under an inverted microscope to determine if cells are still attached to the tissue culture plate. If there are few or no cells attached do not continue with the differentiation protocol and restart the protocol, otherwise proceed.
Warm Day 2 media to 37oC. Aspirate Day 1 media and replace with 2 mL/well for a 6 well plate of Day 2 media. Place back in the incubator for 24 hours.
Day 3.
Warm Day 3 media to 37oC. The cells will have started to recover on this day and have started proliferating rapidly. Aspirate Day 2 media and replace with 2 mL/well for a 6 well plate of Day 3 media. Place back in the incubator for 24 hours.
**On day 4, the endoderm should be confluent after differentiation. If cells are too dense or too sparse, survival and differentiation efficiency will be lower.
Checkpoint: At this stage of the protocol, immunofluorescence can be performed for Sox17 and FoxA2 which should co-stain the DE, and 85-90% of cells should be Sox17 and FoxA2 double positive (McCracken et al., 2011). If 85-90% of cells are not double positive, do not continue with the differentiation protocol and restart the protocol.
Days 4-6.
For days 4-6, warm Day 4-6 media to 37oC. Each day aspirate the media and replace with 2 mL/well for a 6 well plate of the prewarmed Day 4-6 media. Due to the amount of cells in each well, the media will turn yellow each day. Confirm the presence of a 3-dimensional structures forming from the monolayer of cells, including attached and floating spheroids (see Fig. 2b).
4.2.3. Liver organoid protocol day 7-25, 3D culture
After the 2D stage, we have found both inter-line and inter-batch variation in the amount of spheroids produced, as well as growth of mesoderm. Due to this we have an optional step after day 18 to remove excessive mesenchymal growth using a cell strainer.
Day 7 - Embedding into Matrigel
Checkpoint: At this stage immunofluorescence can be performed for the following markers with the expected staining frequency: FoxA2 (~85%+) and FoxF1 (~10-15%+), and CDX2 (>90%+).
Mechanical Dissociation
Warm wash media and RA media to RT. In this step, the entire attached cell layer as well as any floating spheroids will be collected by mechanically dislodging the cell layer through forceful pipetting. Do not aspirate the media, rather using the media already in each well, forcefully pipet the media up and down repeatedly with a P1000 tip to delaminate the cells in the well from the plate. Since the cells are growing densely they will often come off in small sheets, they can be pipetted up and down and broken into smaller clusters. Collect 1 well of a 6 well plate into a 15 mL conical tube. Rinse each well of a 6 well plate with 1 mL wash media and add this to the conical tube. At this point, there may still be large clusters of cells in the conical tube, and using a P1000, pipet up and down gently 5-10 times to break up any large cell clusters.
Centrifuge for 3 min at 300 x g, at RT.
Completely aspirate the media from the cell pellet, so that it does not interfere with the Matrigel.
Remove the Matrigel from 4oC, and keep it on ice in the biosafety cabinet. Using a P1000 pipet, add 1000 µL of Matrigel to the cell pellet in the 15 mL conical tube from 1 well of a 6 well plate and gently and slowly pipet the cell pellet up and down several times to ensure an even distribution of cells. When pipetting sample up and down to mix, be careful to avoid introducing air bubbles into the Matrigel-cell suspension. Keep this tube on ice.
For plating the cells in Matrigel, always use VWR plates. To each well of a 24 well plate, add 75 µL of the cell/cluster suspension to the center of the well, forming a dome-shape. Optionally, different volumes of Matrigel drops are also successful for generating liver organoids, for example, plate 6 drops of 50 μL Matrigel in one well of a 6 well plate.
Gently place the plate in a 37oC incubator for 5 minutes to solidify the Matrigel.
Add 0.5 mL of RA media to each well of a 24 well plate and place back in the incubator. At this stage, there will be different sized clusters of cells within the Matrigel drop (see Fig. 3a) ranging from 25-100 μm.
On Day 9, 48 hours later, warm RA media to RT. Gently aspirate the RA media from each well, taking care to not disturb the Matrigel drops and add 0.5 mL fresh RA media to each well.
On Day 11, switch to complete HCM which is a hepatocyte specification media. Small (<100 μm organoids can be seen starting to grow at this stage). Warm complete HCM to RT. Aspirate the RA media and add 0.5 mL of complete HCM to each well of a 24 well plate.
Every 3 days exchange the media by aspirating the media in the well and adding 0.5 mL fresh complete HCM.
Figure 3.
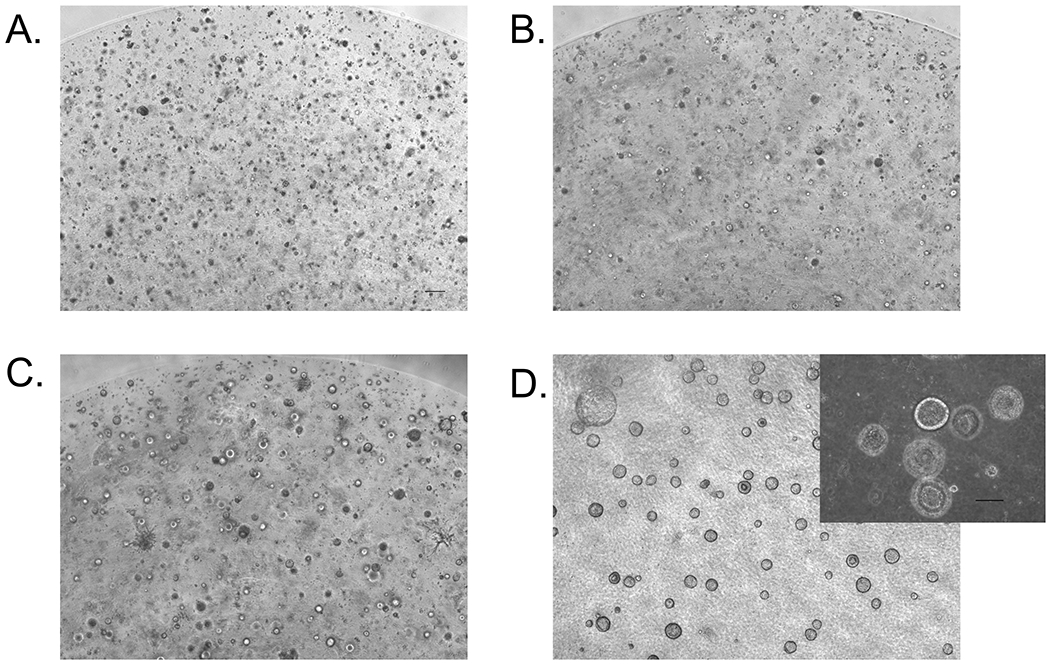
Visualization of liver organoid formation morphology in Matrigel over time. A) Day 8, B) Day 12, C) Day 15, D) Day 21.
Day 17-18 Passage Day:
On this day, many spherical organoids with diameters less than 100 µm will be visible throughout the entire Matrigel drop (see Fig. 3c). Depending on the cell line, there will also be varying amounts of mesenchymal-like cell types: some cells will be attached to the bottom of the plate, and some will be growing directly in the Matrigel. Optional: To remove these mesenchymal-like larger aggregates of cells, passage the organoid suspension through a 100 µm cell strainer as described below. In cases where organoids will be individually sorted and analyzed, separating out the mesenchyme is recommended. Also, the amount of mesenchyme can depend on cell line and differentiation batch and will need to be experimentally determined.
Warm the complete HCM to RT. Aspirate the HCM, and add 0.5 mL of complete HCM with 10% Matrigel (read optional step 2) added to each well of the 24 well plate. Tilt the plate towards you, and using a P1000, pipet the media into the pipet tip and expel the liquid towards each Matrigel drop. The drops should easily dislodge from the bottom of the plate into the media. To dissociate the Matrigel into solution, pipet gently up and down 5-10 times using the P1000 or until no pieces of Matrigel are visible.
Optional: Place a 100 µm cell strainer over a 50 mL conical tube. Using a P1000, pipet the media and organoid solution through the strainer. If using this optional step, add the 10% Matrigel after using the strainer.
Transition organoids to floating culture in low attachment plates: Add the media and 10% Matrigel solution from one well of the VWR 24 well plate, to one corresponding well of a 24 well ultra-low attachment plate. In this “floating” culture, the liver organoids are still supported by the Matrigel. Place the plate back in the incubator.
Optional: The organoids can be left in the Matrigel drop for a longer period of time, until day 20-25 depending on what the downstream applications are. For more high-throughput applications that require individual, distinct organoids in each well, passaging on day 17-18 results in a high number of single organoids that can be separated into 96 or 384 well plates for assays.
Day 21
The organoids can be used starting on this day for functional assays or staining.
Warm complete HCM to RT. Confirm the organoids are healthy and growing in size (see Fig. 3d). No aspiration is needed; add 0.5 mL complete HCM with 10% Matrigel to each well of the 24 well plate. Place the plate back in the incubator.
Day 23
On this day the media can be changed to fresh complete HCM or the organoids can be used for downstream applications. For media change, transfer the organoids and media from one well of a 24 well plate to a 15 mL conical tube. Spin the tube at 300 x g, RT for 3 minutes.
Aspirate the media, leaving the cell pellet and layer of Matrigel. Add 0.5 mL complete HCM with 10% Matrigel and gently pipet up and down. Transfer the organoids to a new ultra-low attachment plate.
Checkpoint: To check for hepatic markers, albumin can be assayed by ELISA and hepatic genes by qRT-PCR.
4.3. Whole mount staining of liver organoids
Whole mount imaging of liver organoids allows visualization of the entire structure at once using 3-D reconstructed Z-stacks on a confocal microscope, as well as to examine individual Z-stacks. This enables localization of individual proteins within organoids, and identification of different cell types within the organoid, which includes hepatocytes, stellate-like cells, and macrophage-like cells. The cell polarity of the hepatocytes can be examined using antibodies specific for the outer surface (type IV collagen) and for the intraluminal lining (ZO-1) or the canalicular region. The tissue morphology of the organoids is preserved needing no specimen preparation except for fixing and permeabilization of the organoids.
4.3.1. Materials and Reagents
Day 20-25 liver organoids.
4% Paraformaldehyde (PFA) (J19943K2, Thermo Scientific).
8 well glass bottom plate slide (80827, Ibidi).
DPBS -/-
DAPI mounting medium (F6057, Sigma).
10% BSA Solution (SRE0036, Sigma).
1% Triton X-100 (T8787, Sigma)/ DPBS -/- solution.
E-cadherin Polyclonal Goat Antibody, (AF648, R&D Systems), located on the cell surface of epithelial cells and is a marker for hepatocytes (and cholangiocytes).
ZO-1 Mouse Monoclonal Antibody, (BD610966, BD Biosciences), a tight junction associated protein. This antibody will stain the intraluminal lining and is seen in the inner surface of the hepatocytes, and demonstrates a distinct polarity of the liver organoids.
Hepatocyte Nuclear Factor 4- (HNF4-α) Rabbit Monoclonal Antibody, (Ab200142, Abcam), a transcription factor regulating several hepatic genes.
Donkey anti-Rabbit 555 Secondary Antibody (A32794, Thermo Scientific).
Donkey anti-Goat 488 Secondary Antibody (A11055, Thermo Scientific).
Donkey anti-Mouse 647 Secondary Antibody (A31571, Thermo Scientific).
Mini-centrifuge
Rocker
4.3.2. Protocol
Optional: Before starting the staining, pre-coat the tubes and pipet tips with 1% BSA/DPBS -/- (or use glass pipets) to help reduce the loss of liver organoids that adhere to the sides of the tubes and tips.
Collect 1 well of organoids from a 24 well plate into a 15 mL conical tube. If processing an entire 24 well plate of organoids, use 24 15 mL conical tubes. Add 6 mL DPBS -/- per tube and gently pipet up and down to wash the Matrigel from the organoids.
Spin down the conical tube at 300 x g for 3 min. There will be a pellet of organoids, a small layer of Matrigel above the organoids, and the media will be layered on top. Aspirate the media/DPBS -/- down to about midway in the Matrigel layer. Since this protocol involves many wash steps, it is imperative to gently aspirate the washes to not remove too many of the small floating organoids.
Wash a second time with 6 mL DPBS -/- and spin down 300 x g for 3 min.
After aspirating DPBS -/- add 1 mL 4% PFA per conical tube and pipet gently up and down to make an even suspension of organoids in solution. Leave at RT for 2 hours, rocking gently.
The PFA will need to be diluted before washing or the organoids will not settle after centrifuging. Add 10 mL DPBS -/- per tube and gently pipet up and down.
Spin down at 400 x g for 3 minutes. Remove the PFA solution and dispose of properly.
Prepare the permeabilization buffer (0.5% Triton X/DPBS -/-): For 10 mL: 5 mL DPBS -/-, 5 mL 1% Triton X solution. Add 1 mL permeabilization buffer per tube and transfer the organoids in solution to a 1.7 mL centrifuge tube, organoids from one 15 mL conical tube to one 1.7 mL centrifuge tube.
Gently rock for 15-20 minutes. Perform a quick spin down in a microcentrifuge tube, aspirate the buffer and repeat this step with permeabilization buffer twice more.
Prepare blocking buffer (1% BSA/0.5 Triton X/DPBS -/-): For 10 mL: 1 mL 10 % BSA, 5 mL 1% Triton X solution, 4 mL DPBS -/-. After aspirating the permeabilization buffer add 1 mL blocking buffer and leave for 1 hour at RT on the rocker.
Prepare the primary antibody mixture: 1:500 dilution of anti- E-cadherin, and 1:200 dilution of anti-ZO-1, and anti-HNF4-α in blocking buffer. Perform a quick spin down of the permeabilization buffer and aspirate it off. Add the primary antibody mixture, and incubate overnight at 4 oC on a rocker.
Aspirate the primary antibody mixture, and perform 3 washes with 1 mL 0.2% Triton X/DPBS -/- for 30 minutes each on the rocker, spinning down and aspirating the wash buffer each time.
Prepare the secondary antibody mixture: 1:200 dilution of Donkey anti-Rabbit 555, Donkey anti-Goat 488, and Donkey anti-Mouse 647 in blocking buffer. Add this mixture to the tube with the organoids and leave at RT for 2-4 hours on the rocker or overnight at 4oC.
Wash 3 times with 1 mL DPBS -/- for 10 minutes each on the rocker, spinning down and aspirating the PBS each time.
Add 50-100 μL of DPBS -/- to each tube of organoids and transfer to one well of an IBIDI glass slide. Add one drop of DAPI mounting medium/well. Gently tilt the slide side to side to mix the DAPI mixture.
Image using a confocal microscope.
4.4. Fatty acid treatment of liver organoids
Steatosis, or excessive fatty acid accumulation in hepatocytes, is a significant health concern and associated with liver inflammation, and in some cases, progression to cirrhosis. Currently, no effective therapies are available, in part due to the lack of robust human models. To model steatosis in organoids a treatment is used with a BSA conjugated form of sodium oleate (oleic acid sodium salt), a fatty acid found in many animal and vegetable fats and oils. Oleic acid (OA) is a mono-unsaturated omega-9 fatty acid. The organoids are treated for 3 days with fatty acid and a BODIPY stain is used to visualize accumulated fatty acid inside hepatocytes. This protocol is designed as a starting point for studying inflammation and fibrosis in liver organoids. Due to the involvement of macrophages and stellate cells in disease progression, it is feasible to use liver organoids as a model for studying steatosis and associated diseases like NAFLD and fibrosis. For example, as previously published, treatment with OA in liver organoids resulted in hepatocyte ballooning, increases in inflammatory markers (IL-6 and TNF-alpha) and fibrosis markers (Ouchi et al., 2019).
4.4.1. Preparation of sodium oleate/BSA
4.4.2. Materials and reagents
1M Oleic acid (OA) (O1383, Sigma) in 100% ethanol. Oleic acid is a liquid at RT and is soluble in alcohols. Aliquot and store -20oC.
37.5 mM NaOH, (72068, Sigma)
30% Bovine Serum Albumin (BSA) in saline, fatty acid free, (A9205, Sigma) diluted to 5% in DPBS-/-. Using fatty acid free BSA ensures that there are no biological effects from endogenous fatty acids in the BSA.
DPBS-/-
Bead bath
Heat block
4.4.3. Protocol
OA is very insoluble in cell culture media and will form crystals and fat droplets; prepare a sodium oleate solution to aid in solubility. Prepare a 20 mM sodium oleate solution by adding 80 μL 1M OA to 3920 μL 37.5 mM NaOH in a 15 mL conical tube. Mix well by pipetting and vortexing.
Heat this solution at 70oC for 30 minutes in the heat block.
BSA is a carrier for fatty acid in the serum, and so sodium oleate is conjugated to BSA in this step. The final stock concentration of sodium oleate/BSA is 5mM. Add 4 mL of the 20 mM sodium oleate to 12 mL of the 5% BSA/ DPBS-/-. Mix well by pipetting and vortexing.
Heat this solution at 37oC for 1 hour in the bead bath. The fatty acid:BSA ratio is 6:1 (Alsabeeh, Chausse, Kakimoto, Kowaltowski, & Shirihai, 2018). Aliquot and store at -20oC.
Prepare the control solution: Add 4 mL of 37.5 mM NaOH to 12 mL of 5% BSA/DPBS-/- and mix well by pipetting and vortexing. Heat at 37oC for 1 hour. Aliquot and store at -20oC.
4.4.4. Liver organoid treatment and live imaging
4.4.5. Materials and reagents
5 mM sodium oleate/BSA or control NaOH/BSA
Day 20-25 liver organoids
24 well low attachment plates
8 well glass bottom plate slide
complete HCM
3.8 mM BODIPY 493/503 (D3922, Thermofisher Scientific) in DMSO, for imaging lipid droplet accumulation. Aliquot and store at -20oC, protected from light.
500 μM SiR-actin (CY-SC001, Cytoskeleton Inc.) in DMSO, for F actin membrane staining, store at -20oC.
10 mg/ml Hoechst nuclear stain, (33342, Invitrogen).
DPBS-/-
4.4.6. Protocol
Collect one well of liver organoids from a 24 well plate into a 15 mL conical tube, and add 10 mL DPBS-/-. Pipet up and down with a 10 mL pipet to thoroughly wash the Matrigel from the organoids.
Spin down for 1000 x g for 3 min. Aspirate the DPBS-/- and media from the conical tube, leaving the pellet of organoids. Repeat this wash step.
Prepare the 500 μΜ sodium oleate/BSA solution or control solution: Per well of a 24 well plate (500 μL total): 50 μL 5 mM sodium oleate/BSA solution (or control) into 450 μL HCM.
Add the 500 μΜ sodium oleate/BSA or control solution to the pellet of organoids, and pipet up and down gently to break up the pellet.
Use a P1000 to transfer the organoids in solution to one well of a 24 well ultra low attachment plate.
Place back in a 37oC incubator for 3 days. Over the 3 days, the sodium oleate/BSA treated organoids will become darker in color as they accumulate lipid. (See Fig. 5a,c)
For live staining of fatty acid treated organoids, collect the organoids and media into a 15 mL conical tube, add DPBS-/-, and spin down 200 x g for 3 min. Aspirate the media and DPBS-/-.
While this is spinning, prepare the BODIPY/ F-actin/Hoechst stain solution. The BODIPY stain will need to be diluted from 1:1000-5000 and the concentration that has a low background will need to be empirically determined. The SiR-actin stain works well with the organoids at a 1:1000 stain, and Hoechst between 0.1-1 μg/ml. Dilute all into 1 mL HCM and add 500 μL to the 15 mL conical tube containing the organoids.
Gently pipet up and down with a P1000 to break up the pellet. Incubate for 1 hr at 37oC in the incubator.
After 1 hour, add 10 mL DPBS-/- to the conical tube to wash the stains and pipet up down. Spin down for 200 x g for 3 min.
Add 300 μL HCM per tube and pipet up and down with a P1000 to break apart the pellet.
Transfer organoids and media, 150 μL each, to 2 wells of an 8 well glass bottom slide (1 well of a 24 well plate will go into 2 wells of a chamber slide). Keep covered with foil or in the dark at 4oC if the slides won’t be imaged straightaway.
Visualize and image liver organoids on a confocal microscope. See Fig. 5b,d for example images.
Figure. 5.
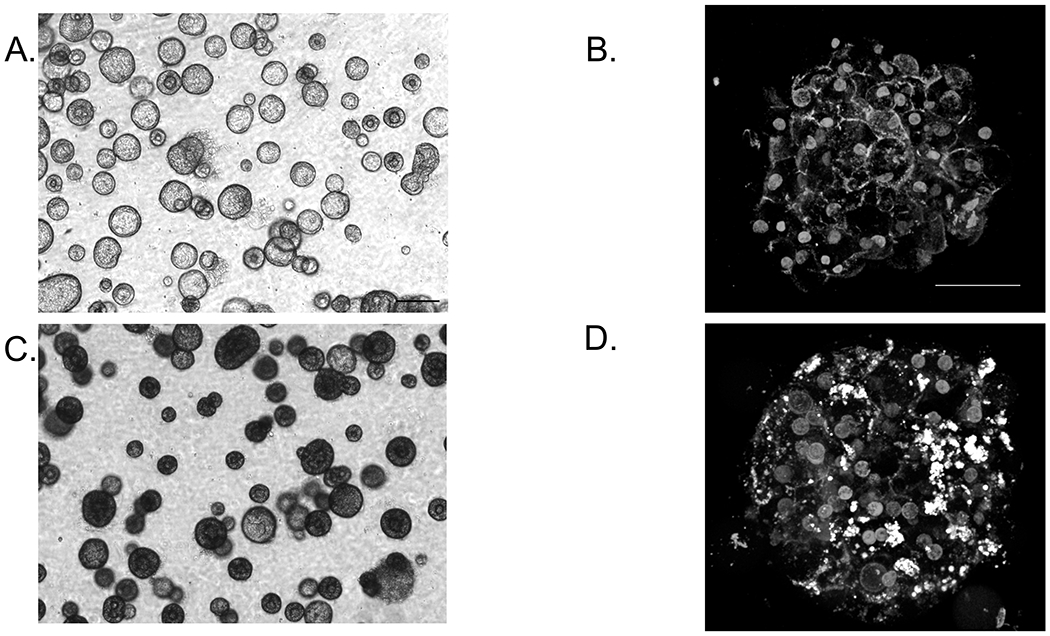
OA treatment of liver organoids, day 23. A,C, are phase contrast images of before and after treatment of liver organoids with OA. B,D are live confocal images before and after treatment with OA showing lipid accumulation within an organoid. F-actin (red) is a membrane stain, DAPI (blue) is a nuclear stain, and BODIPY (green) is a lipid stain.
5. Technical and design considerations
5.1. Room for improvement
Different PSC lines can be used to successfully generate liver organoids, however like other PSC organoid models, liver organoids have inter-line and inter-batch variability in production robustness and mesenchyme growth. Since the starting material on day 7 consists of small clusters of cells, it is difficult to obtain uniform cluster size, leading to some variability in the organoids’ composition at day 20-25. It is possible for some organoids to have more stellate-like and macrophage-like cells than other organoids. Some batches have more mesenchymal growth than others, requiring the use of the cell strainers to remove this extra mesenchyme.
Optimization of cell density at initial plating is crucial for proper and complete differentiation of the PSCs to endoderm and mesoderm. Since this step is critical to proper differentiation it will need to be optimized for each new cell line tested. In addition to the starting density being critical to continue the differentiation, the appearance at day 4, DE stage is also important. The DE should grow as a uniform confluent layer of cells, without having overgrowth of cells on this monolayer.
Successfully differentiating PSC to functionally mature hepatic cells also has its own set of challenges. Despite high levels of genes related to cholesterol metabolism, coagulation and complement factors, the hepatic cells in liver organoids express alpha-fetoprotein (AFP) and have lower levels of Cyp450 genes and activity than primary hepatocytes, indicating that the cells are still immature (Shinozawa, et al). Embryonic stem cells primarily use glycolysis as the main source of energy; however, upon differentiation into the hepatic lineage, mitochondria undergo morphological changes and oxidative phosphorylation becomes increasingly important (Hopkinson et al., 2017). Additional manipulations to induce metabolic switching will aid in developing mature hepatocytes to model the global physiology of human liver.
6. Conclusions
An in vitro human model of the liver is a particularly useful tool because it allow us to better understand diverse biological functions in the body, and to study human disease. It is likely that 3D spheroid and organoid models will be employed alongside current 2D models to gain insight into drug toxicity and disease mechanisms, as each model has distinct advantages and drawbacks. Future studies with this liver organoid model can be used to investigate mechanistic pathways at molecular, cellular and tissue levels that govern human liver development, disease and drug responses.
Figure 4.
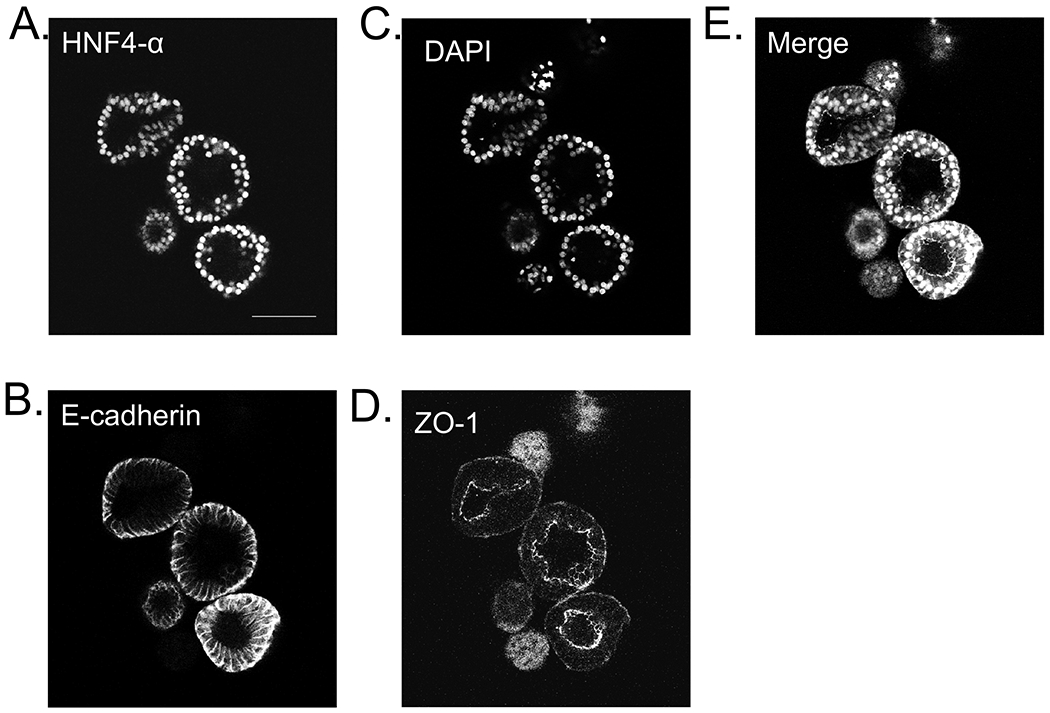
Whole mount staining of liver organoids, day 23. Representative images of A. HNF4-α (red), B. E-cadherin (green), C. DAPI (white), D. ZO-1 (purple), and merge.
Acknowledgements
The authors would like to appreciate communication design items by Ms. Asuka Kodaka for graphics. We would like to also express sincere gratitude to Drs. Yuqi Cai, Kentaro Iwasawa, Masaki Kimura, Christopher N. Mayhew and other Takebe, Wells, Zorn lab members for their support and excellent technical assistance. This work was supported by Cincinnati Children’s Research Foundation grant and PRESTO grant from Japan Science and Technology Agency (JST) to TT. This work was also supported by an NIH grant UG3 DK119982, Cincinnati Center for Autoimmune Liver Disease Fellowship Award, PHS Grant P30 DK078392 (Integrative Morphology Core and Pluripotent Stem Cell and Organoid Core) of the Digestive Disease Research Core Center in Cincinnati, Takeda Science Foundation award, Mitsubishi Foundation award and AMED JP19fk0210037, JP19bm0704025, JP19fk0210060, JP19bm0404045, and JSPS JP18H02800, 19K22416. TT is a New York Stem Cell Foundation – Robertson Investigator.
References:
- Akbari S, Sevinç GG, Ersoy N, Basak O, Kaplan K, Sevinç K, … Erdal E (2019). Robust, Long-Term Culture of Endoderm-Derived Hepatic Organoids for Disease Modeling. Stem Cell Reports. 10.1016/j.stemcr.2019.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almazroo OA, Miah MK, & Venkataramanan R (2017). Drug Metabolism in the Liver. Clinics in Liver Disease, 21(1), 1–20. 10.1016/j.cld.2016.08.001 [DOI] [PubMed] [Google Scholar]
- Alsabeeh N, Chausse B, Kakimoto PA, Kowaltowski AJ, & Shirihai O (2018). Cell culture models of fatty acid overload: Problems and solutions. Biochimica Et Biophysica Acta. Molecular and Cell Biology of Lipids, 1863(2), 143–151. 10.1016/j.bbalip.2017.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale SS, Vernetti L, Senutovitch N, Jindal R, Hegde M, Gough A, … Yarmush ML (2014). In Vitro Platforms for Evaluating Liver Toxicity. Experimental Biology and Medicine (Maywood, N.J.), 239(9), 1180–1191. 10.1177/1535370214531872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Jehuda R, Shemer Y, & Binah O (2018). Genome Editing in Induced Pluripotent Stem Cells using CRISPR/Cas9. Stem Cell Reviews and Reports, 14(3), 323–336. 10.1007/s12015-018-9811-3 [DOI] [PubMed] [Google Scholar]
- Broda TR, McCracken KW, & Wells JM (2019). Generation of human antral and fundic gastric organoids from pluripotent stem cells. Nature Protocols, 14(1), 28. 10.1038/s41596-018-0080-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarró LM, Bradshaw CR, … Huch M (2017). Human primary liver cancer–derived organoid cultures for disease modeling and drug screening. Nature Medicine, 23(12), 1424. 10.1038/nm.4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorotto R, Amenduni M, Mariotti V, Fabris L, Spirli C, & Strazzabosco M (2018). Liver diseases in the dish: IPSC and organoids as a new approach to modeling liver diseases. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 10.1016/j.bbadis.2018.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas-Lopes MA, Mafra K, David BA, Carvalho-Gontijo R, & Menezes GB (2017). Differential Location and Distribution of Hepatic Immune Cells. Cells, 6(4). 10.3390/cells6040048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, & Braspenning J (2015). Measurement of Blood Coagulation Factor Synthesis in Cultures of Human Hepatocytes. Methods in Molecular Biology (Clifton, N.J.), 1250, 309–316. 10.1007/978-1-4939-2074-7_23 [DOI] [PubMed] [Google Scholar]
- Heslop JA, Rowe C, Walsh J, Sison-Young R, Jenkins R, Kamalian L, … Kevin Park B (2017). Mechanistic evaluation of primary human hepatocyte culture using global proteomic analysis reveals a selective dedifferentiation profile. Archives of Toxicology, 91(1), 439–452. 10.1007/s00204-016-1694-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T, Friedman SL, & Hoshida Y (2017). Hepatic stellate cells as key target in liver fibrosis. Advanced Drug Delivery Reviews, 121, 27–42. 10.1016/j.addr.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MMA, … Clevers H (2015). Long-Term Culture of Genome-Stable Bipotent Stem Cells from Adult Human Liver. Cell, 160(1–2), 299–312. 10.1016/j.cell.2014.11.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, … Grønbæk H (2019). The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nature Reviews Gastroenterology & Hepatology, 16(3), 145–159. 10.1038/s41575-018-0082-x [DOI] [PubMed] [Google Scholar]
- Li P, He K, Li J, Liu Z, & Gong J (2017). The role of Kupffer cells in hepatic diseases. Molecular Immunology, 85, 222–229. 10.1016/j.molimm.2017.02.018 [DOI] [PubMed] [Google Scholar]
- McCracken KW, Howell JC, Wells JM, & Spence JR (2011). Generating human intestinal tissue from pluripotent stem cells in vitro. Nature Protocols, 6(12), 1920–1928. 10.1038/nprot.2011.410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Futaki S, Suemori H, Taniguchi Y, Yamada M, Kawasaki M, … Kawase E (2012). Laminin E8 fragments support efficient adhesion and expansion of dissociated human pluripotent stem cells. Nature Communications, 3, 1236. 10.1038/ncomms2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosedale M, & Watkins PB (2017). Drug-Induced Liver Injury: Advances in Mechanistic Understanding that will Inform Risk Management. Clinical Pharmacology and Therapeutics, 101(4), 469–480. 10.1002/cpt.564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun SJ, Ryu J-S, Lee M-O, Son YS, Oh SJ, Cho H-S, … Son MJ (2019). Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. Journal of Hepatology, 0(0). 10.1016/j.jhep.2019.06.030 [DOI] [PubMed] [Google Scholar]
- Ouchi R, Togo S, Kimura M, Shinozawa T, Koido M, Koike H, … Takebe T (2019). Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metabolism, 30(2), 374–384.e6. 10.1016/j.cmet.2019.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehu AI, Ma X, & Venkataramanan R (2017). Mechanisms of Drug-Induced Hepatotoxicity. Clinics in Liver Disease, 21(1), 35–54. 10.1016/j.cld.2016.08.002 [DOI] [PubMed] [Google Scholar]
- Stanger BZ (2015). Cellular Homeostasis and Repair in the Mammalian Liver. Annual Review of Physiology, 77, 179–200. 10.1146/annurev-physiol-021113-170255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama N, Nishimura S, Nakamura S, Shimizu T, Ohnishi R, Endo H, … Eto K (2010). Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. The Journal of Experimental Medicine, 207(13), 2817–2830. 10.1084/jem.20100844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang X, Tan Z, Su Y, Liu J, Chang M, … Wang Y (2019). Human ESC-derived expandable hepatic organoids enable therapeutic liver repopulation and pathophysiological modeling of alcoholic liver injury. Cell Research. 10.1038/s41422-019-0242-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Wu D, Ren Y, Huang Y, Feng B, Zhao N, … Xu A (2019). Generation of hepatobiliary organoids from human induced pluripotent stem cells. Journal of Hepatology, 70(6), 1145–1158. 10.1016/j.jhep.2018.12.028 [DOI] [PubMed] [Google Scholar]
- Wu L-J, Chen Z-Y, Wang Y, Zhao J-G, Xie X-Z, & Chen G (2019). Organoids of liver diseases: From bench to bedside. World Journal of Gastroenterology, 25(16), 1913–1927. 10.3748/wjg.v25.i16.1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, & Kaplowitz N (2013). Mechanisms of Drug Induced Liver Injury. Clinics in Liver Disease, 17(4), 507–518. 10.1016/j.cld.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Shen JX, & Lauschke VM (2019). Comprehensive Evaluation of Organotypic and Microphysiological Liver Models for Prediction of Drug-Induced Liver Injury. Frontiers in Pharmacology, 10, 1093. 10.3389/fphar.2019.01093 [DOI] [PMC free article] [PubMed] [Google Scholar]