

Abstract
MUC1 glycopeptides are attractive antigens for anti-cancer vaccine development. One potential drawback in using the native MUC1 glycopeptide for vaccine design is the instability of the O-glycosyl linkage between the glycan and the peptide backbone to glycosidase. To overcome this challenge, a MUC1 glycopeptide mimic has been synthesized with the galactose-galactosamine disaccharide linked with threonine (Thomsen-Friedenreich or Tf antigen) through an unnatural β-glycosyl bond. The resulting MUC1-β-Tf had a much-enhanced stability toward a glycosidase capable of cleaving the glycan from the corresponding MUC1 glycopeptide with the natural α-Tf linkage. The MUC1-β-Tf was subsequently conjugated with a powerful carrier bacteriophage Qβ. The conjugate induced high levels of IgG antibodies in clinically relevant human MUC1 transgenic mice, which cross-recognized not only the natural MUC1-α-Tf glycopeptide but also MUC1 expressing tumor cells, supporting the notion that a simple switch of the stereochemistry of the glycan/peptide linkage can be a strategy for anti-cancer vaccine epitope design for glycopeptides.
Graphical Abstract
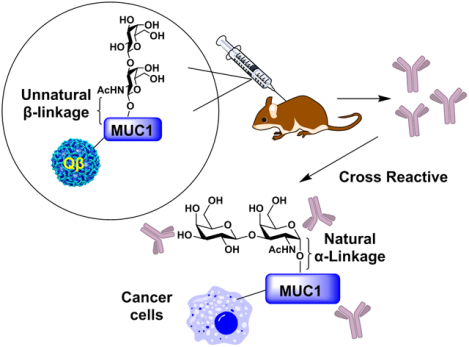
A MUC1 glycopeptide bearing an unnatural β-glycosyl bond between the glycan and the peptide backbone was synthesized. The mimic can induce high levels of IgG antibodies cross-recognizing cancer cells expressing the native MUC1 glycoprotein.
Introduction
Cancer is a leading cause of death in the world. With an estimated ~1.8 million new cases and ~600,000 deaths in the US alone in 2020,1 there is a pressing need for new and better cancer therapeutics. One of the rising modalities is immunotherapy including vaccination.2, 3 For the development of effective anti-cancer vaccines, an attractive class of antigenic targets is tumor associated carbohydrate antigen (TACA), a representative example of which is the mucin-1 antigen (MUC1).4
MUC1 is overexpressed in multiple cancers including breast, lung, pancreatic, colon, prostate, and ovarian cancers, with some cancer cells expressing MUC1 100 times more than normal cells.5, 6 In addition, as a glycoprotein, the structures of MUC1 glycans are distinct between normal and cancerous cells with O-linked glycans attached to cancer associated MUC1 more truncated and less branched.6–8 Multiple clinical studies have shown that patients with high titers of anti-MUC1 IgG antibodies are correlated with better prognosis in multiple cancers.9, 10 For example, non-small cell lung cancer with high MUC1 antibody titers have a 91% one-year survival rate, which compares favorably with the 21% rate for the patients with low levels of MUC1 antibodies.9
Much effort has been devoted to produce MUC1 based constructs that can elicit high levels of anti-MUC1 antibodies to fight cancer.11 Initial attempts focused on the MUC1 peptide as the antigen (Figure 1A).12 However, because MUC1 is an endogenously expressed protein in humans, the majority of B and T cells that strongly recognize MUC1 peptides would undergo cell death during the maturation process, rendering it challenging to elicit a powerful MUC1 based anti-cancer immune response.13 One strategy to overcome this immunotolerance is to target glycosylated MUC1 peptides, as MUC1 glycopeptides bearing Thomsen-nouveau (Tn) antigen (αGalNAc-Ser/Thr), sialyl-Tn (STn, αNeu5Ac-(2,6)-αGalNAc-Ser/Thr) or Thomsen-Friedenreich (Tf) antigen (βGal-(1,3)-αGalNAc-Ser/Thr) have been shown to be more immunogenic than the peptide alone (Figure 1B).14–16 Nevertheless, no MUC1 based vaccines have achieved clinical successes yet requiring continued innovation in development.
Figure 1.
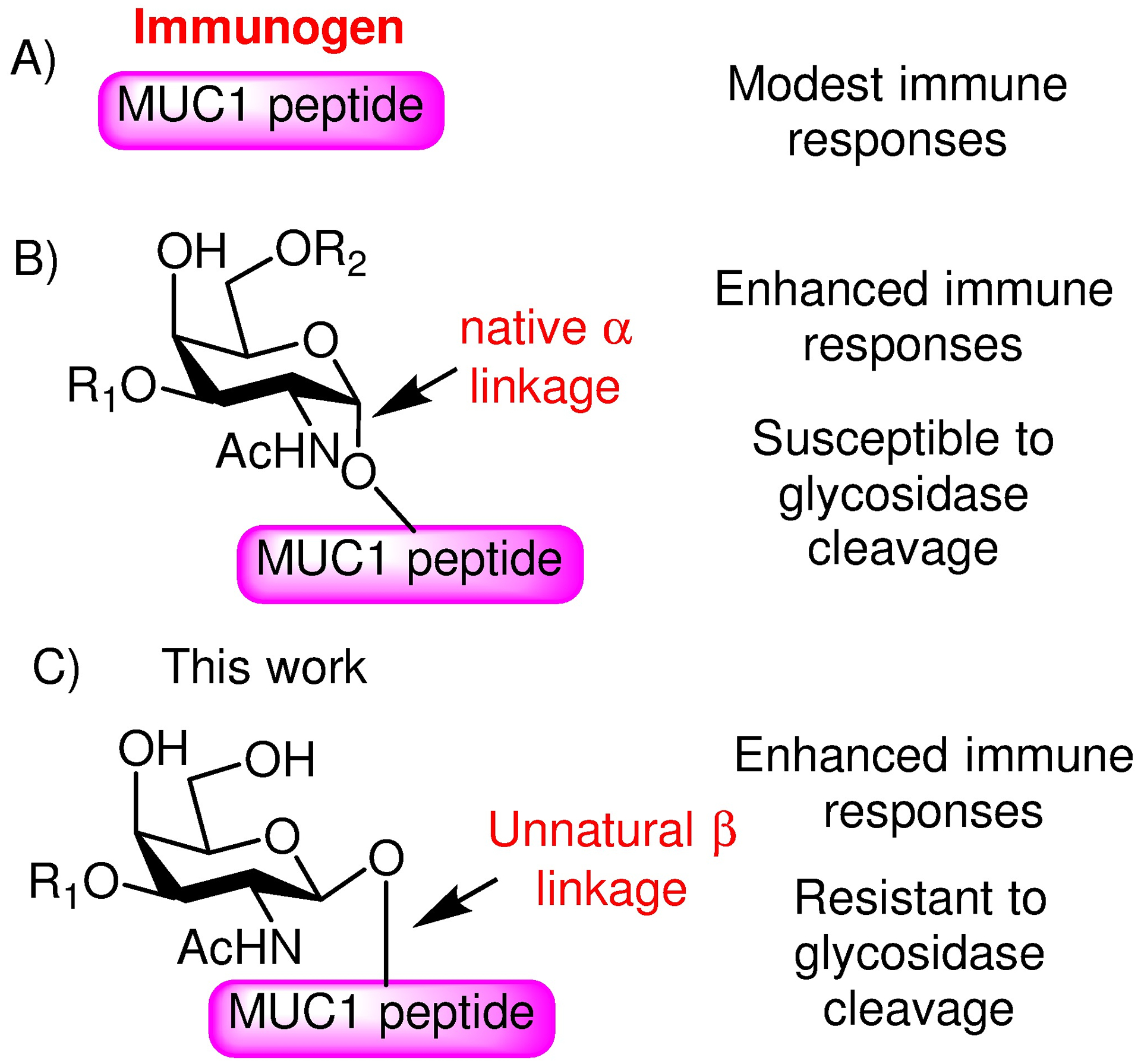
Comparison of various MUC1 immunogens investigated in the literature and this work. A) Early vaccines utilized MUC1 peptides, which induced modest immune responses; B) MUC1 glycopeptides can enhance the immune responses. The native α-linkage between the glycan and the peptide backbone is susceptible to glycosidase cleavage. C) In this work, we investigate a MUC1 glycopeptide mimetic with the unnatural β-glycosyl linkage, which can help enhance the stability toward a glycosidase.
In order to boost the immune response against carbohydrate containing antigens, an interesting direction to pursue is to design mimetics such as C-glycosides,17–19 S-glycosides,20 N-acyl-modified glycosides,21, 22 as well as deoxyfluoroglycosides.23 These analogs are considered more resistant to hydrolysis by glycosidases in the body, thus potentially providing longer immune stimulation upon in vivo administration. For glycopeptides, mimetics have also been designed and synthesized.23–30 Campo and colleagues explored the role of stereochemistry at the glycosidic bond between the carbohydrate and the peptide.29 Using the monosaccharide Tn conjugated to the peptide backbone through the unnatural β-O-linkage, antibodies elicited by the constructs were cross-reactive to the natural epitope. In comparison, Vogel and coworkers prepared Tf analogs with a C-linkage replacing the native β-O glycosidic bond between Gal and GalNAc of the disaccharide and incorporated them into peptides.24 Mouse immunization with these glycopeptide analogs demonstrated that the β-C-linked analog was able to induce IgG antibodies, which bound the β-C-anomer specifically with little recognition of the α-C-anomer. In contrast, the α-C-linked glycopeptide analog did not elicit detectable levels of antibodies at all. No cross-recognition of the O-linked disaccharide bearing glycopeptide was investigated.24 Glycopeptides with the unnatural β-O-linked Tf disaccharide have not been explored for vaccine design to date.
As MUC1-Tf has been shown to be a superior epitope for vaccine development,31 we have become interested in studying MUC1-Tf glycopeptide mimetics to enhance the stability of the antigen. The interstitial fluid of tumors and sera of cancer patients is known to have increased levels of glycosidases.32 As glycosylation of the peptide can significantly improve immune responses,14–16 we focused on the glycosidic linkage between the glycan chain and the peptide backbone. With challenges in synthesizing C-glycosides, we have investigated the glycopeptide bearing the more accessible unnatural β-O-linkage between the Gal-GalNAc disaccharide and the MUC1 peptide backbone to mimic the native MUC1-α-Tf glycopeptide (Figure 1C). The unnatural MUC1-β-Tf glycopeptide was found to have a higher stability toward the glycosidase capable of hydrolyzing the natural α-anomer. Furthermore, antibodies generated against the MUC1-β-Tf glycopeptide in human MUC1 transgenic mice can cross-recognize the native MUC1 glycopeptide on tumor cells, providing a new lead in anti-cancer vaccine development.
Results and Discussion
Synthesis of MUC1-β-Tf glycopeptide
Previously, we have synthesized MUC1 glycopeptide bearing the natural α-linked Tf antigen as potential anti-cancer vaccines.31 During the synthesis of the α-Tf antigen (Scheme 1A), the β-Tf building block 1 was generated via glycosylation of Fmoc protected threonine 2 by disaccharide donor 3, albeit in a low 18% yield.31 In order to obtain sufficient quantities of the β-linked isomer, we redesigned the synthesis. To favor the formation of the β-anomer, the 2-amine of the galactosamine was protected using 2,2,2-trichloroethoxy carbonyl (Troc), which is known to favor the 1,2-trans glycosyl linkage through neighboring group participation.33, 34 Our initial approach to prepare the disaccharide was to perform 1+1 glycosylation between 4 and the Troc bearing 5,35 which gave little of the desired disaccharide 6 (Scheme 1B). Varying the reaction solvent, temperature, and the promoter did not improve the outcome. As similar glycosylations with an analogous N3 bearing acceptor proceeded in good yields,31, 36 it was likely the difficulties encountered in this 1+1 glycosylation was due to the steric hindrance and/or electron withdrawing effects of the Troc moiety. To overcome the difficulty encountered in 1+1 glycosylation, an alternative strategy was pursued by forming the glycosyl linkage between galactosamine and threonine first (Scheme 1C). The 3-OH of the galactosamine was temporarily protected as tert-butyldimethylsilyl (TBS) ether (donor 7), which successfully glycosylated Fmoc protected threonine 2 with subsequent TBS deprotection giving compound 8 in 75% yield for the two steps. The stereochemistry of the newly formed glycosyl linkage in 8 was confirmed to be β by 1H-NMR analysis (3JH1H2 = 7.7 Hz). The extension of the glycan chain was carried out using the per-acetylated Gal donor 4 without the need for elaborate protective groups to glycosylate acceptor 8 producing disaccharide threonine 9 in 86% yield. The divergent outcome in glycosylation of acceptor 5 vs 8 by the thiogalactoside donor 4 was possibly due to the presence of the nucleophilic thiotolyl aglycon in 5 competing with the glycosylation of the free hydroxyl group.37 Protective group manipulation of 9 led to the glycosyl amino acid 11 (Scheme 1C). Compounds 9 and 10 proved difficult to fully dry, although the remaining solvent did not affect the following steps. The acetylation to give 10 did not go to completion. The trace free OH did not affect the remaining steps.
Scheme 1.
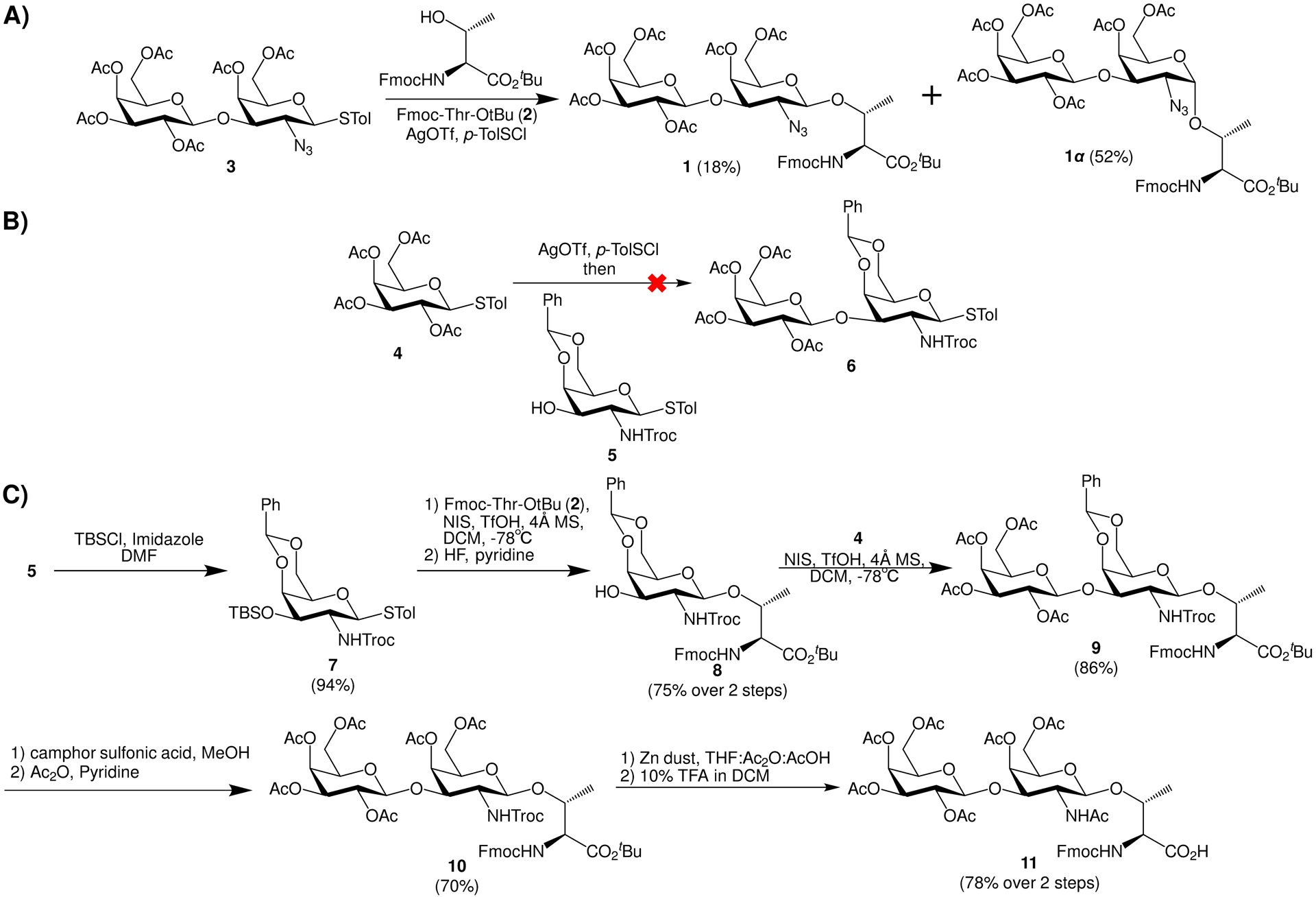
A) The azido bearing disaccharide donor 3 gave a low yield of the β-Tf building block 1. B) 1+1 glycosylation of 4 and 5 failed to give disaccharide 6. C) Successful synthesis of β-Tf amino acid 11.
The assembly of MUC1-β-Tf glycopeptide was performed next by solid phase peptide synthesis using Fmoc chemistry, which started with Wang resins functionalized with 1,4-diaminobutane15 (Scheme 2). The addition of Fmoc-protected amino acids to peptide chains was promoted by (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU)/hydroxybenzotriazole (HOBt). For coupling of the glycosyl amino acid (Glyco-AA) 11, the combination of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU)/1-hydroxy-7-azabenzotriazole (HOAt) was utilized. Upon completion of the synthesis, the glycopeptide was cleaved from the resins and deprotected using 5% (v/v) hydrazine in water. The resulting glycopeptide 12 bearing a free amine at its C-terminus was obtained from HPLC purification in 30–40% yield (Scheme 2), which was characterized by NMR and mass spectrometry.
Scheme 2.
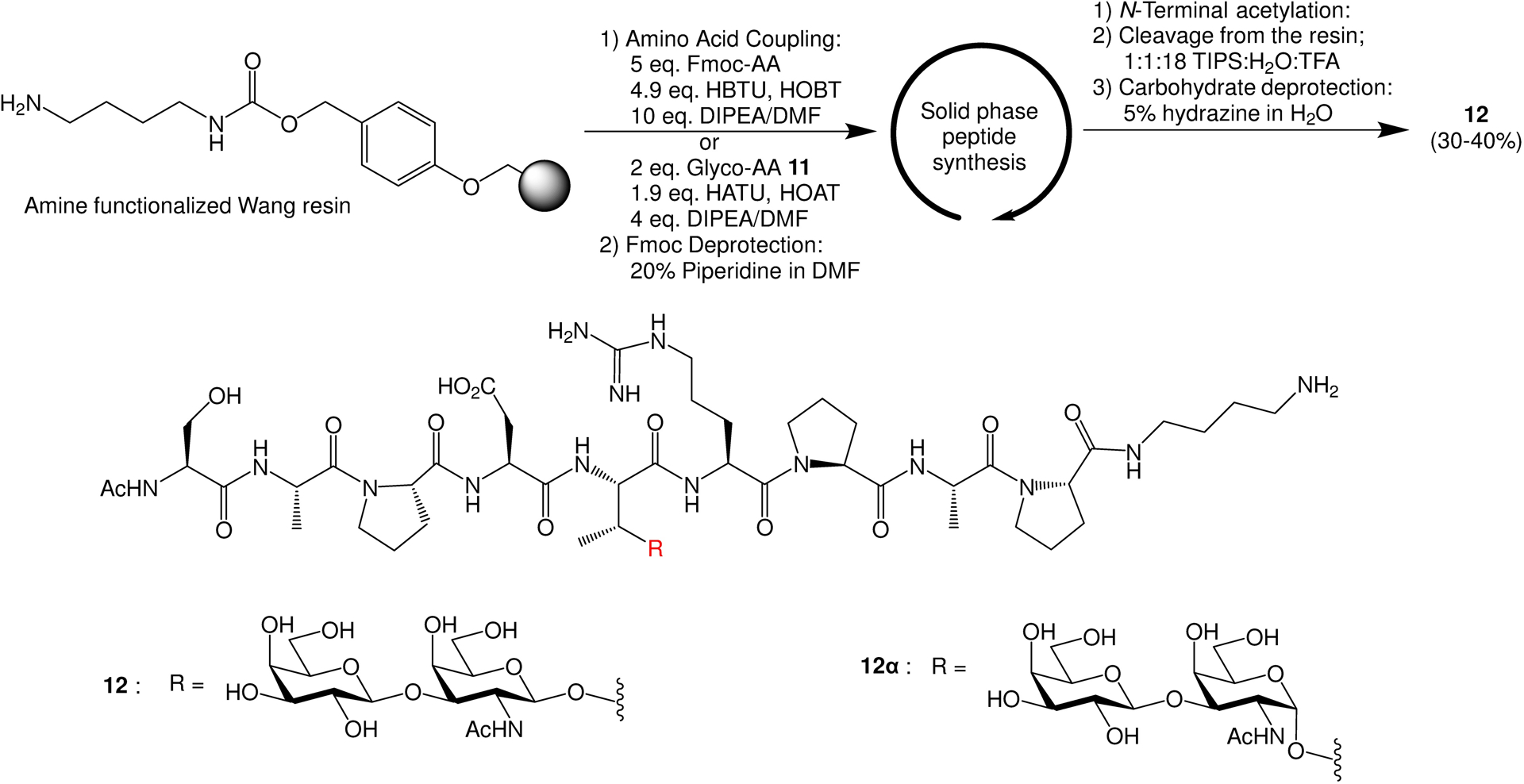
Solid phase synthesis of MUC1 glycopeptide 12 containing the β-linked Tf.
MUC1-β-Tf glycopeptide 12 has enhanced stability to enzymatic degradation
To test the hypothesis that the MUC1-β-Tf glycopeptide 12 is more resistant to glycosidase mediated hydrolysis, we treated glycopeptide 12 with the endo-α-N-acetylgalactosaminidase from Enterococcus faecalis (E. faecalis),38 as E. faecalis is a commensal bacterium found in humans.39 MUC1-β-Tf glycopeptide 12 and the corresponding MUC1-α-Tf glycopeptide (12α)31 were incubated with E. faecalis endo-α-N-acetylgalactosaminidase respectively. Aliquots of the reaction mixture were taken out and subjected to mass spectrometry analysis. After only 20 minutes, a significant amount of the cleavage peptide product for the α-isomer 12α was observed with nearly complete cleavage after 1.75 hours (Supporting Information Figure S1). In contrast, the β-anomer 12 showed no detectable cleavage even after 24 hours. These observations are consistent with the crystal structure of endo-α-N-acetylgalactosaminidase (PDB:6M77),38 where the complex of endo-α-N-acetylgalactosaminidase with β-GalNAc monosaccharide showed the 1-hydroxy group pointing toward the protein presumably hindering the binding of β-linked Tf glycopeptide to the enzyme (Supporting Information Figure S2). Thus, changing the stereochemical linkage of the glycopeptide could enhance its stability toward the endo-α-N-acetylgalactosaminidase in the biological system.
Synthesis of MUC1-β-Tf glycopeptide 12 conjugate with bacteriophage Qβ carrier
As MUC1 glycopeptide itself is a weak immunogen, an immunogenic carrier is necessary to covalently conjugate with the glycopeptide to boost the anti-MUC1 antibody responses. Bacteriophage Qβ has been shown to be a superior carrier for vaccine development.40 In order to conjugate MUC1-β-Tf glycopeptide 12 with Qβ, glycopeptide 12 was derivatized with a bifunctional linker 13,41 and subsequently incubated with Qβ in KPB (0.1M, pH 7.0) at 37℃ (Scheme 3). Mass spectrometry analysis of the resulting conjugate (Supporting Information Figure S3) showed an average of 220 glycopeptides per capsid.
Scheme 3.
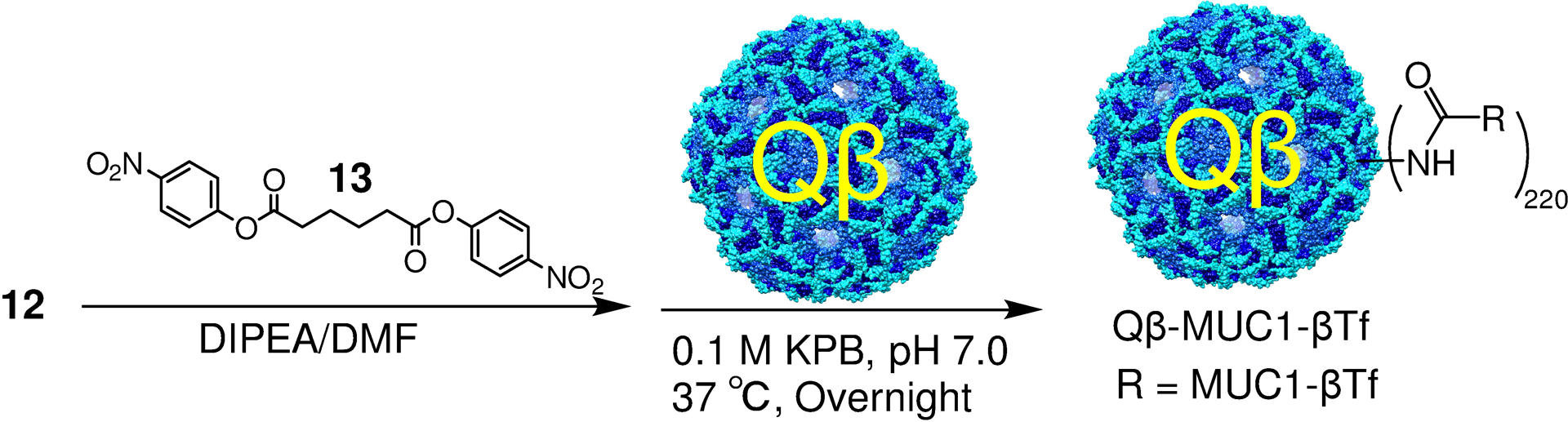
Conjugation of MUC1 glycopeptides to bacteriophage Qβ.
Generation of anti-MUC1 IgG antibodies with Qβ-MUC1-β-Tf conjugate
Immunological evaluation of Qβ-MUC1-β-Tf was performed in human MUC1 transgenic (MUC1.Tg) mice. As mouse mucins have low sequence identities with human MUC1, compared to wild type mice, MUC1.Tg mice are a more suitable model for evaluation of MUC1 based vaccines. MUC1.Tg mice have similar expression levels and patterns of human MUC1 and are immunotolerant toward MUC1 similar to that encountered in humans.42 MUC1.Tg mice were immunized with Qβ-MUC1-β-Tf (8.6 nmol MUC1) and monophosphoryl Lipid A (MPLA from Salmonella enterica serotype Minnesota Re 595, Re mutant) on day 0 with two booster injections two weeks apart. MPLA is an FDA approved adjuvant, which can enhance immune responses through the activation of Toll like receptor-4.43 As a control, mice were immunized with Qβ only following an identical protocol as the Qβ-MUC1-β-Tf group. The post-immune sera were collected on day 35. The levels of anti-MUC1 antibodies in post-immune sera were analyzed with enzyme linked immunosorbent assay (ELISA). To avoid the interference of Qβ carrier for anti-MUC1 antibody analysis, MUC1-β-Tf, MUC1-α-Tf and MUC1 peptide were conjugated to bovine serum albumin (BSA) to generate the corresponding BSA-conjugates (Supporting Information Scheme S1 and Figure S4).
The sera from immunized MUC1.Tg mice showed strong anti-MUC1-β-Tf IgG responses and the mean IgG titer was 2,200,000 ELISA units (Figure 2A), more than 1,000 folds higher than control mice receiving Qβ only. The high IgG antibody titers suggest the induction of helper T cell activation and antibody isotype switching. The sera gave a similar average IgG titer of 2,500,000 against MUC1-α-Tf indicating the antibodies induced could recognize the MUC1 glycopeptide with the native glycosyl linkage. In contrast, the average antibody titer against the MUC1 peptide was only 310,000, highlighting that the glycan was critical for antibody recognition. The subtypes of IgG antibodies were also determined (Figure 2B). All major IgG subtypes were produced, indicating both Th1 and Th2 types of immune responses were elicited by the Qβ-MUC1-β-Tf conjugate.
Figure 2.
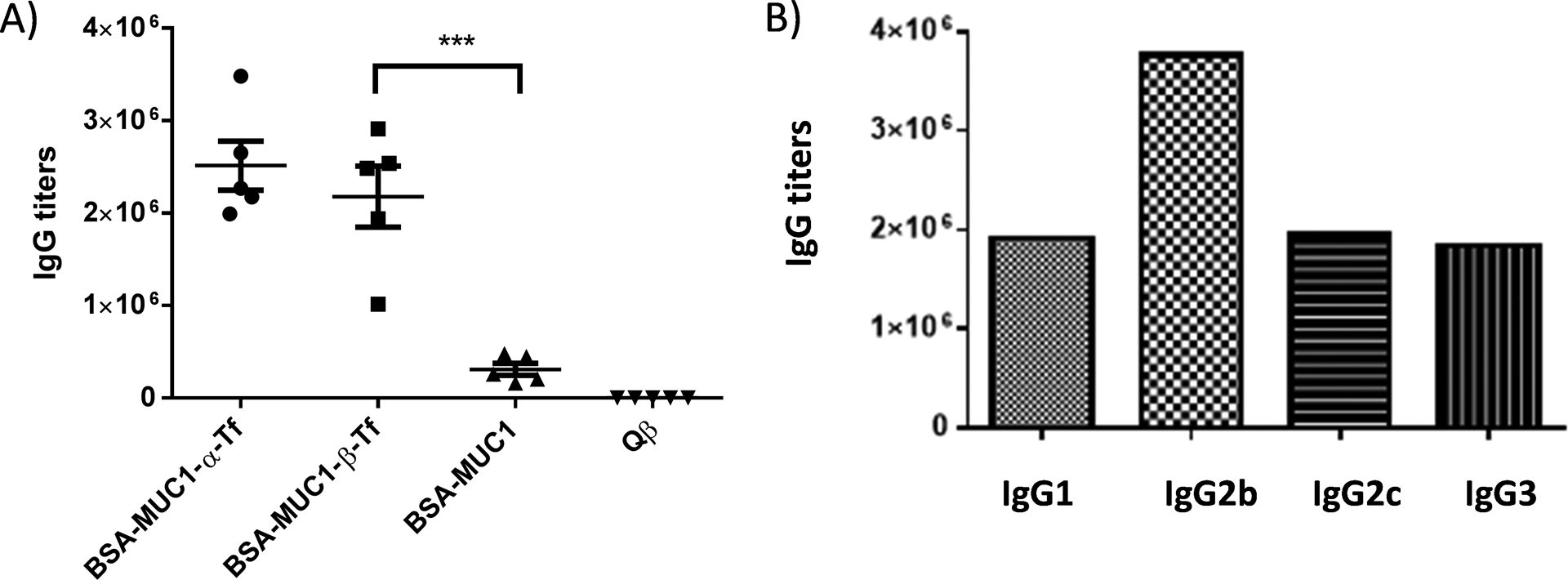
A) Titers of IgG antibodies from MUC1.Tg mice immunized with the Qβ-MUC1-β-Tf conjugate. For determination of anti-MUC1 IgG titers, the ELISA measurements were performed against the corresponding BSA-MUC1-α-Tf, BSA-MUC1-β-Tf, and BSA-MUC1. Each symbol represents one mouse (n = 5 mice for each group). The titer numbers were determined by regression analysis with log10 dilution plotted with optical density and reported as the highest fold of dilution giving the optical absorbance value of 0.1 over those of the pre-immune control sera (OD ~ 0.2). ***p < 0.001. The p values were determined through a two-tailed unpaired Student’s t-test using GraphPad Prism. B) Titers of anti-MUC1-β-Tf IgG subtypes from a representative mouse immunized with the Qβ-MUC1-β-Tf conjugate indicating the generation of all major subtypes of IgG antibodies.
Antibodies Induced by Qβ-MUC1-β-Tf Conjugates Recognized Multiple MUC1 Glycoforms in Glycopeptide Microarray Screening
To better understand the epitope profile of the antibodies induced by Qβ-MUC1-β-Tf, the post-immune sera were subject to MUC1 glycopeptide microarray analysis.44, 45 The microarray contained a total of 72 MUC1 glycopeptide components with the common backbone sequence of PAHGVTSAPDTRPAPGSTAP and α-linked Tn, Tf or cores 1–4 glycans at various serine and threonine locations representing some of the typical MUC1 glycopeptide structures. In addition, other glycopeptides/glycoproteins including mucin-5B (MUC5B) glycopeptides, fetuin, transferrin, mucins from porcine stomach and bovine submaxillary glands were included on the microarray. After incubation of arrays with individual mouse serum, unbound antibodies were removed by thorough washing and fluorescently labeled anti-mouse IgG secondary antibody was used to semi-quantify the amounts of antibodies bound on the microarray through measuring the relative fluorescence intensities.
MUC1 glycopeptides 14–20 bear Tf at various locations of the peptide backbone. Sera from Qβ-MUC1-β-Tf immunized mice recognized well these glycopeptides (Figure 3). The position of the glycan within the peptide backbone moderately affected antibody recognition, which may be due to the influence of the peptide on the conformation of the glycan or the peptide being recognized as part of the epitope. In addition, MUC1 glycopeptides containing other glycans ranging from core 1 to core 4 pentasaccharide at the same threonine as glycopeptide 12 were also bound by the post-immune sera. In contrast, these post-immune sera exhibited little reactivities with various MUC5B glycopeptides 101–103 or glycoproteins 105–111 fetuin, transferrin, ICAM-1, porcine stomach mucin, and bovine submaxillary mucin). Combined with the low binding to MUC1 peptide (Figure 1), these results confirmed the specificity of generated antibodies toward MUC1 glycopeptides suggesting both the glycan and the MUC1 peptide backbone are important for antibody recognition. The antibodies could bind MUC1 glycopeptides with diverse glycosylation patterns (e.g., 22, 29, 36, 50, 64, 71, 78, 85, 92, 98-100 in Figure 2), which is attractive as tumor associated MUC1 can be highly heterogeneous in the glycan structures.46–48
Figure 3.
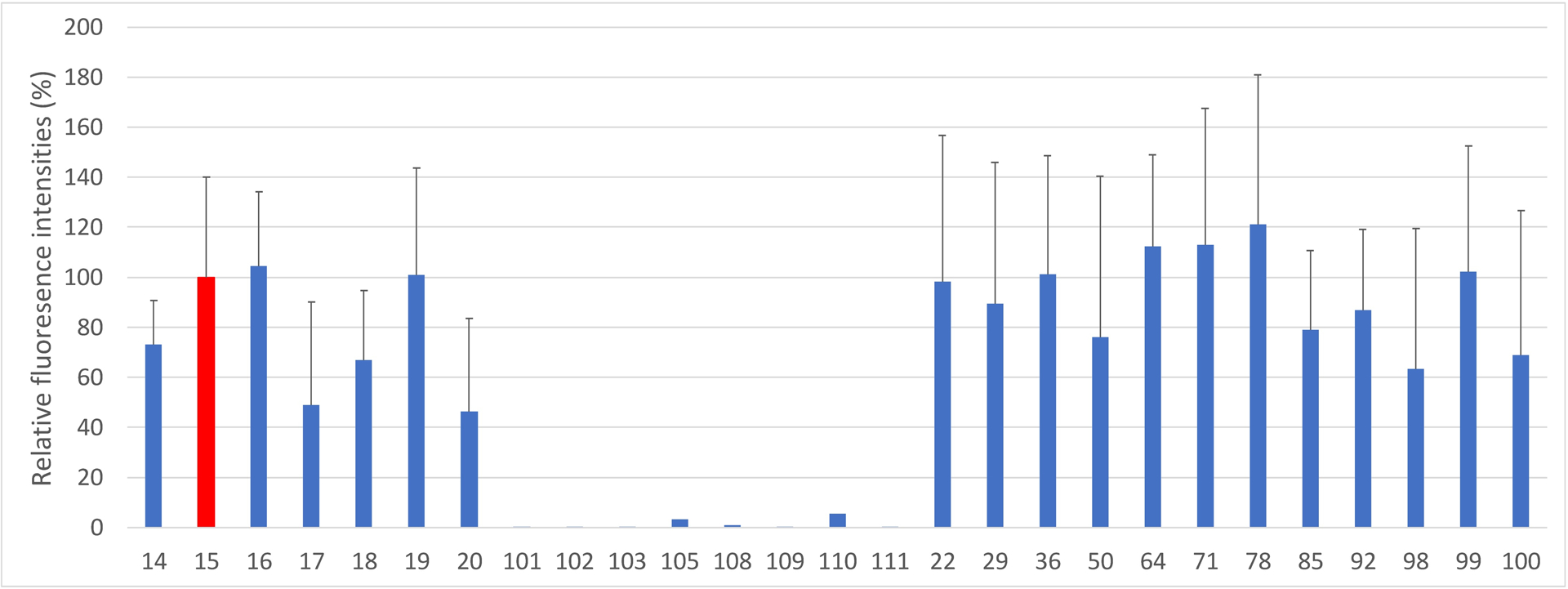
Representative results of MUC1 glycopeptide microarray screening of post immune sera from Qβ-MUC1-β-Tf immunized mice. The anti-sera could recognize MUC1 glycopeptides bearing diverse glycan structures, while exhibiting little recognition to non-MUC1 glycopeptides/glycoproteins. Glycopeptide 14: PAHGVT*SAPDTRPAPGSTA; 15: PAHGVTSAPDT*RPAPGSTA; 16: PAHGVTSAPDTRPAPGST*A; 17: PAHGVT*SAPDT*RPAPGSTA; 18: PAHGVT*SAPDTRPAPGST*A 19: PAHGVTSAPDT*RPAPGST*A; 20: PAHGVT*SAPDT*RPAPGST*A. T* denotes the Tf antigen. Glycopeptides 101–103 are various MUC5B glycopeptides. 105 is Fetuin and 108–111 are ICAM-1, transferrin, porcine stomach mucin, and bovine submaxillary mucin respectively. Glycan structures: glycopeptide 14: Tf; 22: C1Tf1 (for abbreviations and structures, see Supporting Information Figure S6); 29: C1Tf2; 36: C2Tf1te; 50: C2Tf2te; 64: Tn; 71: C3Tf1; 78: C3Tf2; 85: C4Tf1; 92: C4Tf2; 98: core 1 PDT; 99: core 3 PDT; 100: core 2 PDT. The error bars represent standard deviation (SD) from five mice with each mouse sample measured five times.
Antibodies Elicited by Qβ-MUC1-β-Tf could Recognize and Kill MUC1-Expressing Cancer Cells
For an effective vaccine, the induced anti-MUC1 antibodies should be capable of recognizing the native antigen in its native environment, i.e., tumor cells. For this purpose, the binding of post-immune sera with the MUC1 expressing tumor cells was evaluated by flow cytometry. When sera from MUC1.Tg mice at day 35 post-immunization were incubated with mouse melanoma cells, B16-MUC1, significant IgG binding to cancer cell was observed (Figure 4A). The abilities of Qβ-MUC1-β-Tf induced IgG antibodies to lyse tumor cells were measured next. B16-MUC1 cells were incubated with post-immune sera and rabbit complement. Subsequently, cancer cell death was determined via a cell viability assay. The sera from Qβ-MUC1-β-Tf immunized mice exhibited significantly higher cytotoxicity toward B16-MUC1 cells compared to the sera from control mice administered with Qβ (Figure 4B). The levels of cytotoxicity of these sera to tumor cells were statistically equivalent to those by sera from MUC1-α-Tf immunized mice.31
Figure 4.
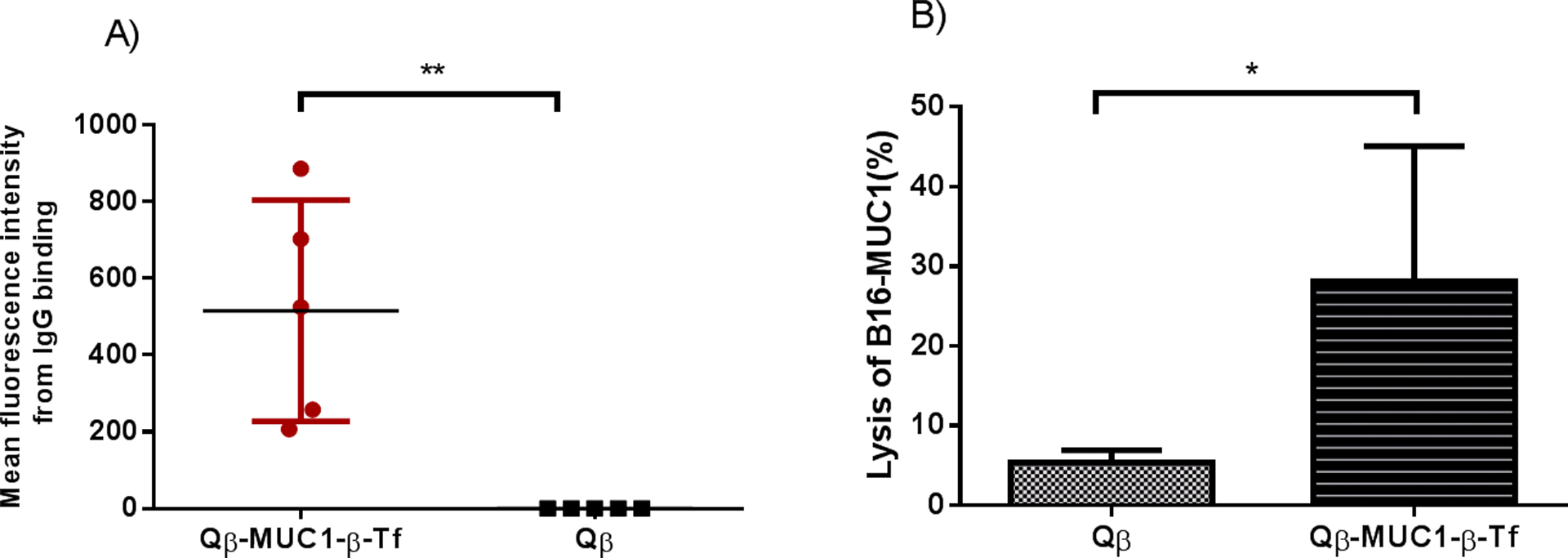
A) Flow cytometry analysis of sera from Qβ-MUC1-β-Tf immunized mice showed significant IgG antibody binding to tumor cells compared with sera from Qβ immunized mice. Binding to B16-MUC1 cells was tested with 1:20 dilution of the corresponding sera. B) Antibodies induced by Qβ-MUC1 conjugates exhibited significant cytotoxicity towards B16-MUC1 cells through complement mediated cytotoxicity (CDC). CDC toward B16-MUC1 cells was determined by an MTS cell viability assay. Each group represents 5 mice. * p < 0.05. **p < 0.01. The p values were determined through a two-tailed unpaired Student’s t-test using GraphPad Prism.
Conclusion:
An efficient synthetic route has been developed to access the β-linked Tf MUC1 as a mimic of the native MUC1-α-Tf glycopeptide. The MUC1-β-Tf has a greatly enhanced stability toward the glycosidase that can efficiently cleave the native α-linkage. The conjugate of MUC1-β-Tf with bacteriophage Qβ carrier induced a strong anti-MUC1 IgG antibody response in immune-tolerant human MUC1 transgenic mice. The antibodies produced could cross-recognize the MUC1-α-Tf glycopeptide as well as a diverse range of tumor associated MUC1 glycoforms. In addition, the antibodies induced by Qβ-MUC1-β-Tf could kill tumor cells via complement mediated cytotoxicity. To the best of our knowledge, this is the first time the use of glycopeptides with an unnatural stereochemistry of the glycosyl bond has been shown to elicit an immune response capable of killing tumor cells. With the ease in synthesis and the enhanced stability toward a glycosidase, glycomimetics with an artificial anomeric stereochemistry could be a direction to pursue in epitope design for anti-cancer vaccines.
Supplementary Material
Acknowledgments
We are grateful to the National Cancer Institute, National Institutes of Health (Grant R01 CA225105), Michigan Economic Development Corporation, and Michigan State University Foundation for financial support of our work.
Footnotes
Supporting Information: The Supporting Information includes detailed experimental procedures, characterization data and spectra, supplementary figures, and supplementary schemes.
References:
- 1.Cancer Facts and Figures, https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html.
- 2.Romero P, Banchereau J, Bhardwaj N, Cockett M, Disis ML, Dranoff G, Gilboa E, Hammond SA, Hershberg R, Korman AJ, Kvistborg P, Melief C, Mellman I, Palucka AK, Redchenko I, Robins H, Sallusto F, Schenkelberg T, Schoenberger S, Sosman J, Türeci Ö, Van den Eynde B, Koff W and Coukos G, Sci. Transl. Med, 2016, 8, 334PS339. [DOI] [PubMed] [Google Scholar]
- 3.Van der Burg SH, Arens R, Ossendorp F, Van Hall T and Melief CJM, Nat. Rev. Cancer, 2016, 16, 219–233. [DOI] [PubMed] [Google Scholar]
- 4.Apostolopoulos V, Hu XF, Pouniotis DS and Xing PX, Curr. Trends Immunol, 2004, 12, 629–639. [Google Scholar]
- 5.Rakha EA, Boyce RW, Abd El-Rehim D, Kurien T, Green AR, Paish EC, Robertson JF and Ellis IO, Modern Pathol, 2005, 18, 1295–1304. [DOI] [PubMed] [Google Scholar]
- 6.Nath S and Mukherjee P, Trends Mol. Med, 2014, 20, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Mensdorff-Pouilly S, Moreno M and Verheijen RHM, Cancers, 2011, 3, 3073–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hattrup CL and Gendler SJ, Annu. Rev. Physiol, 2008, 70, 431–457. [DOI] [PubMed] [Google Scholar]
- 9.Hirasawa Y, Kohno N, Yokoyama A, Kondo K, Hiwada K and Miyake M, Am. J. Respir. Crit. Care Med, 2000, 161, 589–594. [DOI] [PubMed] [Google Scholar]
- 10.Hamanaka Y, Suehiro Y, Fukui M, Shikichi K, Imai K and Hinoda Y, Int. J. Cancer, 2003, 103, 97–100. [DOI] [PubMed] [Google Scholar]
- 11.Hossain MK and Wall KA, Vaccines, 2016, 4, 25 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor-Papadimitriou J, Burchell JM, Graham R and Beatson R, Biochem Soc Trans, 2018, 46, 659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soares MM, Mehta V and Finn OJ, J. Immunol, 2001, 166, 6555–6563. [DOI] [PubMed] [Google Scholar]
- 14.Ryan SO, Turner MS, Gariépy J and Finn OJ, Cancer Res, 2010, 70, 5788–5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Yin Z, McKay C, Pett C, Yu J, Schorlemer M, Gohl T, Sungsuwan S, Ramadan S, Baniel C, Allmon A, D. R., Westerlind U, Finn MG and Huang X, J. Am. Chem. Soc, 2018, 140, 16596–16609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai H, Huang Z-H, Shi L, Sun Z-Y, Zhao Y-F, K. H. and Li Y-M, Angew. Chem. Int. Ed, 2012, 51, 1719–1723. [DOI] [PubMed] [Google Scholar]
- 17.Sadraei SI, Reynolds MR and Trant JF, Adv. Carbohydr. Chem. Biochem, 2017, 74, 137–237. [DOI] [PubMed] [Google Scholar]
- 18.Weïwer M, Huang F, Chen C, Yuan X, Tokuzaki K, Tomiyama H and Linhardt RJ, ACS Symp. Ser, 2008, 990, 216–238. [Google Scholar]
- 19.Peri F, Cipolla L, Recigno M, La Ferla B and Nicotra F, Bioconjugate Chem, 2001, 12, 325–328. [DOI] [PubMed] [Google Scholar]
- 20.Geraci C, Consoli GML, Galante E, Bousquet E, Pappalardo M and Spadaro A, Bioconjugate Chem, 2008, 19, 751–758. [DOI] [PubMed] [Google Scholar]
- 21.Song C, Sun S, Huo C-X, Li Q, Zheng X-J, Tai G, Zhou Y and Ye X-S, Bioorg. Med, 2016, 24, 915–920. [DOI] [PubMed] [Google Scholar]
- 22.Sun S, Zheng X-J, Huo C-X, Song C, Li Q and Ye X-S, ChemMedChem, 2016, 11, 1090–1096. [DOI] [PubMed] [Google Scholar]
- 23.Daum M, Broszeit F and Hoffmann-Röder A, Eur. J. Org. Chem, 2016, 2016, 3709–3720. [Google Scholar]
- 24.Awad L, Madani R, Gillig A, Kolympadi M, Philgren M, Muhs A, Gérard C and Vogel P, Chem. Eur. J, 2012, 18, 8578–8582. [DOI] [PubMed] [Google Scholar]
- 25.Cipolla L, Rescigno M, Leone A, Peri F, La Ferla B and Nicotra F, Bioorg. Med. Chem, 2002, 10, 1639–1646. [DOI] [PubMed] [Google Scholar]
- 26.Rojas-Ocáriz V, Compañón I, Aydillo C, Castro-Lopez J, Jiménez-Barbero J, Hurtado-Guerrero R, Avenoza A, Zurbano MM, Peregrina J. m., Busto JH and Corzana F, J. Org. Chem, 2016, 81, 5929–5941. [DOI] [PubMed] [Google Scholar]
- 27.Bousquet E, Spadaro A, Pappalardo MS, Bernardini R, Romeo R, Panza L and Ronsisvalle G, J. Carbohydr. Chem, 2000, 19, 527–541. [Google Scholar]
- 28.Wagner S, Mersch C and Hoffmann-Röder A, Chem. Eur. J, 2010, 16, 7319–7330. [DOI] [PubMed] [Google Scholar]
- 29.Campo VL, Riul TB, Bortot LO, Martins-Teixeira MB, Marchiori MF, Iaccarino E, Ruvo M, Dias-Baruffi M and Carvalho I, ChemBioChem, 2017, 18, 527–538. [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann-Röder A, Kaiser A, Wagner S, Gaidzik N, Kowalczyk D, Westerlind U, Gerlitzki B, Schmitt E and Kunz H, Angew. Chem. Int. Ed, 2010, 49, 8498–8503. [DOI] [PubMed] [Google Scholar]
- 31.Wu X, McKay C, Pett C, Yu J, Schorlemer M, Ramadan S, Lang S, Behren S, Westerlind U, Finn MG and Huang X, ACS Chem. Biol, 2019, 14, 2176–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernacki RJ, Niedbala MJ and Korytnyk W, Cancer Metastasis Rev, 1985, 4, 81–102. [DOI] [PubMed] [Google Scholar]
- 33.Kusumoto S, Yoshimura H, Imoto M, Shinamoto T and Shiba T, Tetrahedron Lett, 1985, 26, 909–912. [Google Scholar]
- 34.Ellervik E and Magnusson G, Carbohydr. Res, 1996, 280, 251–260. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Zhou L, El-boubbou K, Ye X-S and Huang X, J. Org. Chem, 2007, 72, 6409–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilsson J, Brinkmalm G, Ramadan S, Gilborne L, Noborn F, Blennow K, Wallin A, Svensson J, Abo-Riya MA, Huang X and Larson G, Sci. Rep, 2019, 9, 5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z and Gildersleeve JC, J. Am. Chem. Soc, 2006, 128, 11612–11619. [DOI] [PubMed] [Google Scholar]
- 38.Miyazaki T and Park EY, FEBS Lett, 2020, 594, 2282–2293. [DOI] [PubMed] [Google Scholar]
- 39.Murray BE, Clin. Microbiol. Rev, 1990, 3, 46–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin Z, Comellas-Aragones M, Chowdhury S, Bentley P, Kaczanowska K, BenMohamed L, Gildersleeve JC, Finn MG and Huang X, ACS Chem. Biol, 2013, 8, 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu X, Ling C-C and Bundle DR, Org. Lett, 2004, 6, 4407–4410. [DOI] [PubMed] [Google Scholar]
- 42.Rowse GJ, Tempero RM, VanLith ML, Hollingsworth MA and Gendler SJ, Cancer Res, 1998, 58, 315–321. [PubMed] [Google Scholar]
- 43.Alving CR, Peachman KK, Rao M and Reed SG, Curr. Opin. Immunol, 2012, 24, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pett C, Cai H, Liu J, Palitzsch B, Schorlemer M, Hartmann S, Stergiou N, Lu M, Kunz H, Schmitt E and Westerlind U, Chem. Eur. J, 2017, 23, 3875–3884. [DOI] [PubMed] [Google Scholar]
- 45.Yin Z, Wu X, Kaczanowska K, Sungsuwan S, Comellas-Aragones M, Pett C, Yu J, Baniel C, Westerlind U, Finn MG and Huang X, ACS Chem. Biol, 2018, 13, 1668–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beatty P, Hanisch FG, Stolz DB, Finn OJ and Ciborowski P, Clin. Cancer Res, 2001, 7, 781s–787s. [PubMed] [Google Scholar]
- 47.Storr SJ, Royle L, Chapman CJ, Hamid UM, Robertson JF, Murray A, Dwek RA and Rudd PM, Glycobiology, 2008, 18, 456–462. [DOI] [PubMed] [Google Scholar]
- 48.Hanisch FG, Stadie TR, Deutzmann F and Peter-Katalinic J, Eur. J. Biochem, 1996, 236, 318–327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.