

Abstract
Mass spectrometry (MS) is routinely used to identify, characterize, and quantify biological molecules. For protein analysis, MS‐based workflows can be broadly categorized as top‐down or bottom‐up, depending on whether the proteins are analyzed as intact molecules or first digested into peptides. This article outlines steps for preparing peptide samples for MS as part of a bottom‐up proteomics workflow, providing versatile methods suitable for discovery and targeted analyses in qualitative and quantitative workflows. Resulting samples contain peptides of suitable size for analysis by MS instrumentation generally available to modern research laboratories, including MS coupled to either liquid chromatography (LC) or matrix‐assisted laser desorption/ionization (MALDI) interfaces. This article incorporates recent developments in methodologies and consumables to facilitate sample preparation. The protocols are well‐suited to users without prior experience in proteomics and include methods for universally applicable suspension trap processing and for alternate in‐solution processing to accommodate a range of sample types. Cleanup, quantification, and fractionation procedures are also described. © 2021 The Authors.
Basic Protocol: Preparation of high‐complexity peptide samples for mass spectrometry analysis using S‐Trap™ processing
Alternate Protocol 1: Preparation of low‐ to moderate‐complexity peptide samples for mass spectrometry analysis using in‐solution processing
Alternate Protocol 2: Detergent, polymer, and salt removal from peptide samples before mass spectrometry analysis using SP2 processing
Support Protocol 1: Protein quantification using Pierce 660 nm assay
Support Protocol 2: Peptide quantification using Pierce quantitative fluorometric peptide assay
Support Protocol 3: High‐pH fractionation of complex peptide samples
Keywords: bottom‐up proteomics, detergent removal, mass spectrometry, S‐Trap™
INTRODUCTION
Mass spectrometry (MS) is routinely used to identify, characterize, and quantify biological molecules. In the past 20 years, numerous MS‐based proteomics methods have been developed to determine protein identity, quantity, and interaction partners, and the presence and localization of post‐translational modifications. MS‐based proteomics workflows can be broadly categorized as top‐down or bottom‐up (Kelleher et al., 1999). Top‐down approaches involve the MS analysis of intact proteins to obtain amino acid sequence information in a way that preserves the stoichiometry among post‐translational modifications, proteolytic cleavage products, and products of splicing events. Bottom‐up approaches, on the other hand, involve the enzymatic or chemical digestion of proteins into peptides before MS analysis, and can be used to identify and quantify peptides, which are then used to infer protein identity and quantity. The presence and sites of post‐translational modifications can also be determined. Although top‐down proteomics methods offer the advantage of preserving the relationship between the protein sequence and modifications, bottom‐up strategies remain among the most popular strategies for the analysis of biological samples. The popularity of bottom‐up methods is tied to their versatility and the availability of instrumentation and bioinformatic tools for data analysis (Mayne et al., 2016). Briefly, bottom‐up methods can be used for discovery and targeted analyses in both qualitative and quantitative workflows. In addition, samples resulting from bottom‐up workflows contain peptides of suitable size for analysis by MS instrumentation most commonly available to modern research laboratories. Finally, many open‐source and commercially developed software tools are available to support data analysis for a broad range of experimental designs (Bruce et al., 2013; Chen et al., 2020).
Bottom‐up strategies can be used to identify and quantify thousands of proteins from a single sample, all without prior knowledge of the sample composition or reliance on antibodies. Such workflows typically involve a series of steps to digest the protein into peptides through either enzymatic or chemical digestion, followed by removal of contaminants before analysis by MS (see Fig. 1A for general overview of the workflow). Once high‐quality tandem mass spectrometry (MS/MS) data are recorded for the peptides, modern bioinformatics approaches can be used to determine peptide and inferred protein identity, sites of post‐translational modifications, and, with an appropriate experimental design, relative abundances of peptides among samples (Tsiamis et al., 2019).
Figure 1.
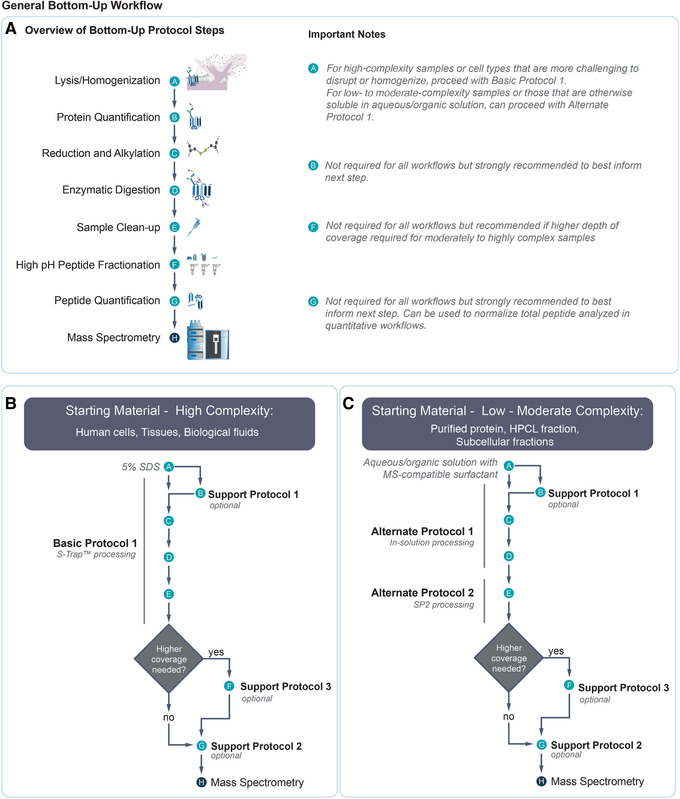
General bottom‐up workflow. (A) Overview of a general bottom‐up proteomics preparation workflow. Relevant notes for various steps are provided at right. (B) Overview of the workflow for processing high‐complexity samples, resulting from a combination of Basic Protocol with Support Protocols 1, 2, and 3. (C) Overview of the workflow for processing low‐ to moderate‐complexity samples, resulting from a combination of Alternate Protocols 1 and 2, together with Support Protocols 1, 2, and 3. For (B) and (C), examples of sample types appropriate for each workflow are provided.
Many different types of samples, including purified proteins, cells, tissues, and biological fluids, can be used as starting material for bottom‐up workflows. Recent developments in sample preparation methodologies and consumables make it easier than ever to prepare high‐quality samples from various types of starting material. Since the publication of our previous article describing the preparation of protein from polyacrylamide gels (Gundry et al., 2009), the field has increasingly come to favor gel‐free approaches because of advantages in time, ease, and minimization of sample loss. Also, whereas previous methods have included protein precipitation as a method to remove detergents and other contaminants from protein samples, the use of suspension trapping allows proteins to be solubilized in harsh detergent and directly digested without precipitation. Finally, although traditional peptide cleanup methods using reverse‐phase chromatography could be used for desalting but not detergent or polymer removal, new approaches that use carboxylate‐coated magnetic particles achieve the needed removal of detergents, polymers, and salts.
Numerous bottom‐up sample preparation strategies are available, and the number of protocols and variations in experimental detail can be overwhelming for users without prior experience in proteomics. This article outlines the basic steps required to prepare samples for MS analysis within the context of a bottom‐up proteomics workflow. The protocols take advantage of new developments in methodologies and consumables to make sample processing easier. Also, guidance for selecting the appropriate method for various types of biological samples is provided. The choice of bottom‐up workflow will depend on the sample complexity and the goals of the experiment. Figure 1A outlines the major steps in a bottom‐up workflow, and Figure 1B and 1C illustrate how the Basic, Alternate, and Support Protocols described here can be mixed and matched together into a seamless workflow tailored to the experimental goals. Sample complexity (i.e., the number of proteins and dynamic range of protein concentration) and the relative ease with which the sample can be solubilized will determine whether a workflow involving rigorous homogenization followed by suspension trapping using an S‐trap™ column (Basic Protocol) or an in‐solution processing workflow (Alternate Protocol 1) should be used. A fast and easy method for detergent and salt removal from peptide mixtures (Alternate Protocol 2) is effective for cleaning samples generated by Alternate Protocol 1 and can also be applied to samples generated by virtually any other bottom‐up workflow described elsewhere. The Basic Protocol, on the other hand, includes sample cleanup as part of its procedure by nature of the S‐trap™ column. Although protein and peptide quantification before MS analysis are not universally required for all bottom‐up experiments, we strongly urge their inclusion when a sufficient amount of sample is available. Accurate protein quantification (Support Protocol 1) is useful for calculating how much enzyme or chemical is required for digestion. It is also important for selecting the column size with suitable capacity in the S‐trap™ processing step (Basic Protocol), as well as for ensuring that sufficient volume is used to solubilize proteins in the in‐solution processing workflow (Alternate Protocol 1). Peptide quantification (Support Protocol 2) is useful for determining whether the peptide sample should be diluted before MS analysis, to avoid overloading the liquid chromatography (LC) column used for separation in LC‐MS analyses. Peptide quantification is essential in quantitative workflows in which equivalent amounts of total peptide are compared to reveal differences in relative abundance of individual peptides. Finally, the peptide mixtures generated by the Basic Protocol or Alternate Protocol 1 may be fractionated by separating peptides according to their hydrophobicity (Support Protocol 3) to enhance coverage of the proteome for moderate‐ to high‐complexity samples, as lower‐abundance proteins may be more difficult to detect in these sample types.
STRATEGIC PLANNING
Protein samples for bottom‐up MS analysis can be prepared from a variety of sources, and the complexity of the sample should be considered when choosing between the Basic Protocol and Alternate Protocol 1. Low‐complexity samples are defined here as purified proteins and mixtures containing up to hundreds of proteins. These types of samples commonly result from affinity purification or reverse‐phase high‐performance liquid chromatography (RP‐HPLC) used to concentrate or purify intact proteins. Moderate‐complexity samples are those with several thousand proteins and include whole cell lysates or subcellular fractions, while high‐complexity samples are those with several thousand proteins from multiple cell types, such as tissue samples. Biological fluids such as plasma are also considered highly complex in this context because they can contain many non‐protein substances (e.g., lipids, small molecules), which can pose challenges to sample preparation, and can exhibit protein concentrations spanning 12 orders of magnitude (Anderson and Anderson, 2002). Alternate Protocol 1 is suitable for low‐ to moderate‐complexity samples in which the proteins of interest are readily soluble in the digestion buffer used in this protocol (i.e., aqueous/organic solution containing MS‐compatible surfactant). Therefore, it is advisable to first perform a solubilization test to ensure that the sample can be solubilized in this buffer. For example, we find that a variety of cell types (e.g., B lymphocytes, HEK293T, cardiac fibroblast, and HeLa cells) can be processed using Alternate Protocol 1. However, cells that contain a high content of insoluble cytoskeletal proteins, such as muscle cells, perform better in the Basic Protocol. Importantly, for cultured cells, no specific culture conditions are required. Rather, the Basic Protocol is generalizable to any culture conditions necessary to address the biological question of interest.
There is no absolute lower or upper limit to the amount of starting material that can be processed by Alternate Protocol 1. Generally, a larger amount of protein present in the starting sample will yield more peptide identifications by MS. However, for the Basic Protocol, each S‐trap™ column has a defined optimal binding capacity. Therefore, it is advisable to determine how much protein is in a sample when planning an experiment, to maximize peptide and inferred protein identifications. For sample‐limited cases, it is advisable to perform protein quantification (Support Protocol 1) on a representative sample (e.g., from a control that is not sample limited) to provide an estimate of the amount of protein that will be available from the experimental sample. If using cells, it is advised that the number of cells to be used for the experiment be counted ahead of time (e.g., via manual or automated counting of trypan‐blue‐excluded cells) to provide a general guide as to the expected amount of total protein present. It is advisable to begin with at least 10 µg of protein to recover enough peptides after processing for successful identification by most modern MS/MS instruments. If protein and peptide quantification (Support Protocols 1 and 2) will be performed, starting Alternate Protocol 1 with 50 µg protein is recommended, so that there is enough material to be used for these quantification assays. The upper limit is defined by solubility and the maximum volume of the tube. If 1.5‐ml microcentrifuge tubes are used, practically this limits the digestion volume to 1.2 ml, which is typically sufficient for at least 2500 µg of protein.
The Basic Protocol and Alternate Protocol 1 are described for trypsin/LysC digestion at pH 8.0. If another enzyme is used, the pH should be adjusted according to the corresponding manufacturer's guidelines.
The Basic Protocol is described for a spin column format but can be adapted to higher‐throughput analyses using the S‐Trap™ spin column plate format following the manufacturer's recommendations for sample loading amounts and solution volumes.
Before starting Alternate Protocol 2, it is advisable to perform Support Protocol 2 to determine the concentration of peptides in the sample so that the mass of total peptide for cleanup can be inferred. The mass of total peptide will be used to determine the volume of particle solution needed to clean the peptide sample. When performing Support Protocol 2, recovery is maximized using 0.2‐0.4 µl of 50 µg/µl particle suspension per microgram of peptide (representing a 10:1 to 20:1 particle:peptide ratio). Over 80% recovery has been observed, but it is advisable to conservatively estimate that 50% of peptides will be recovered, and therefore the amount of peptide desired for MS analysis will determine how much peptide sample to cleanup.
PREPARATION OF HIGH‐COMPLEXITY PEPTIDE SAMPLES FOR MASS SPECTROMETRY ANALYSIS USING S‐TRAP™ PROCESSING
This protocol outlines the steps required to prepare high‐complexity samples for MS analysis by suspension trapping using an S‐Trap™ column (Figs. 1B and 2). This protocol is suitable for processing a wide variety of sample types, and details are provided for processing cells and tissues. By omitting the homogenization and sonication step used for cells and tissues, the workflow can be adopted for plasma, serum, urine, and cerebral spinal fluid, as described elsewhere (Ding et al., 2020; Mindikoglu et al., 2020). The protocol describes how to homogenize the sample in 5% sodium dodecyl sulfate (SDS), then reduce, alkylate, immbolize, and digest the proteins on the S‐Trap™ column. Peptides are then eluted from the column and can be analyzed directly by MS. Alternatively, with samples for which enhanced sequence coverage of lower‐abundance peptides is required, Support Protocol 3 can be optionally used to fractionate the mixture of peptides based on their hydrophobicity (Fig. 1B). Finally, Support Protocol 2 can be used directly after the Basic Protocol or Support Protocol 3 to assess how much of the sample should be used for MS analysis, or to normalize total peptide in the case of quantitative analyses.
Figure 2.
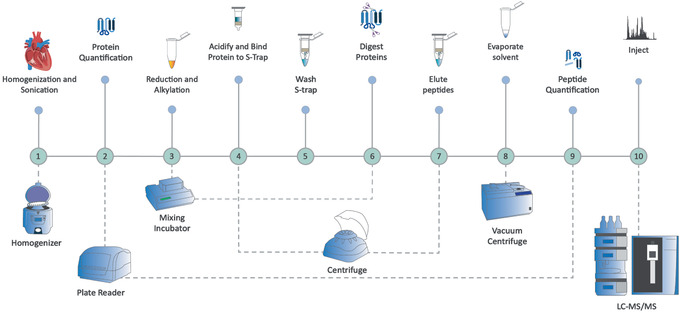
Overview of a bottom‐up proteomics preparation workflow for high to moderate‐complexity samples using S‐Trap™ processing, following the Basic Protocol and Support Protocols 1 and 2. The sample is homogenized and sonicated before protein quantification. Disulfide bonds are reduced, and free thiols are alkylated. The sample is acidified, bound onto the S‐trap™, and then washed free of contaminants. Digestion takes place on the spin column. Peptides are eluted, solvent is evaporated, and peptides are quantified.
NOTE: Use only HPLC‐ and/or LC‐MS‐grade solvents throughout the protocol. All steps are performed at room temperature unless otherwise noted.
Materials
High‐complexity sample (see protocol introduction)
Precellys lysing kit (Precellys, cat. no. P000926‐LYSK0‐A), or other homogenizing bead tubes
Lysis buffer: if using tissue, SDS tissue lysis buffer; if using cells, SDS cell lysis buffer (see recipes)
Benzonase® Nuclease (Sigma Aldrich, cat. no. E1014‐25KU; optional)
100 mM tris(2‐carboxyethyl)phosphine hydrochloride (TCEP; diluted from 1 M TCEP, Sigma, cat. no. C4706)
400 mM acrylamide (Sigma, cat. no. A3553)
12% phosphoric acid (for adjusting pH of solvents as well as S‐Trap™ protocol; see recipe)
S‐Trap™ binding/wash buffer (see recipe)
S‐Trap™ micro columns (≤100 μg, 10 pack, Protifi, cat. no. C02‐micro‐10)
Trypsin/LysC (Promega, cat. no. V5071)
S‐Trap™ digestion buffer: 50 mM TEAB in LC‐MS‐grade water
0.2% FA in LC‐MS‐grade water (see recipe)
0.2% FA in 50% LC‐MS‐grade acetonitrile (see recipe)
0.1% FA in 2% LC‐MS‐grade acetonitrile (see recipe)
50‐ml conical centrifuge tubes (for solution storage; Fisher Scientific, cat. no. 06‐443‐19)
20‐ml disposable glass vials (for solution storage)
Dissection tweezers
Razor blade
Homogenizer (e.g., Bertin Instruments, Precellys 24)
Block sonicator (e.g., Hielscher, UP200St)
Ice bucket with lid (alternatively, use aluminum foil to cover ice bucket)
Refrigerated centrifuge (ideally with swinging bucket, but if none none is available, a fixed‐angle centrifuge can be used)
Variety of positive‐displacement pipets (e.g., P10‐P1000)
Low‐retention pipet tips (e.g., 0.5 µl‐1 ml)
10‐µl glass syringe or other glass pipet device
15‐ml conical tubes
Mixing incubator (e.g., Thermomixer)
Vortexer
pH meter (for recipe preparation only)
2.0‐ml microcentrifuge tubes (Thermo Fisher Scientific, cat. no. 05‐408‐138, for waste collection only)
Microcentrifuge with rotor speed adjustable up to 7000 × g
1.5‐ml low‐retention microcentrifuge tubes (Thermo Fisher Scientific, cat. no. 02‐681‐320)
Vacuum centrifuge (e.g., SpeedVac)
Homogenize and sonicate tissue or cells
-
1a
If using tissue: Dice the flash‐frozen or fresh tissue with razor in a disposable weigh dish. Tare 2‐ml bead beater tube (from Precellys lysing kit) and then add ∼50‐100 mg of tissue to the tube. Record weights.
Dice frozen tissue while still frozen. Keep samples on ice throughout protocol unless stated otherwise to reduce enzymatic digestion.
Process as follows:
Add 9 time the tissue weight (in ml) of SDS tissue lysis buffer to the sample. For example, for 50 mg of tissue, add 450 ml SDS buffer. Cap the tube tightly.
Using the Precellys bead beater 24 or other homogenizer, homogenize the samples twice for 15 s each at 6500 rpm, with a 10‐s pause in between. Immediately place tubes on ice.
-
1b
If using cells: The amount of lysis buffer to use is based on the number of cells. Into the tube containing the pelleted cells, add the appropriate amount of SDS cell lysis buffer and Benzonase to degrade DNA, using the information below as a guide.
| Number of cells | 1‐2 million | 5 million | 10 million | 15‐20 million |
| Lysis buffer | 250 µl | 300 µl | 500 µl | 600‐800 µl |
| Benzonase | 0.5 µl | 0.5 µl | 1 µl | 1.5 µl |
-
2
Suspend the tube adaptor from the block sonicator above an ice bucket, and place sample tube from step 1a or 1b in the tube adaptor. Make sure that both the bottom and top of the tube are embedded in ice. Run sonicator for 10 s on and then 10 s off, at 30% power, three times. Wait 1 min, and repeat the cycle two more times. Keep on ice afterwards.
Sonication is necessary to shear DNA within the sample, which would otherwise clog the S‐Trap™. Optimize the power levels according to your sonicator. Although a probe sonicator may be used, this increases the chance for cross‐contamination, as the probe is reused and requires careful cleaning between uses. The block sonicator used here enables samples in closed tubes to be processed without cross‐contamination.
-
3
Clarify the sample by centrifugation for 10 min at 13,000 × g, 4°C. If there is visible particulate of insoluble material, use a P200 pipet to aspirate at least 100 µl of cell lysis off the top without disturbing particulate at the bottom of the tube, and transfer to a fresh centrifuge tube.
-
4
Perform protein quantification (see Support Protocol 1).
Determining the amount of protein in the sample will provide guidance about the amount of enzyme to use in later steps. This information can also be used to normalize among samples intended for quantitative analyses. As stated in Strategic Planning, for sample‐limited cases, it is advisable to have an estimate of the total amount of protein based on a similar sample type, when possible. If, based on a representative sample, the amount of protein in the experimental sample is <10 µg, it will not be possible to perform protein quantification without using a majority of the sample for this step. However, if starting with 50‐100 mg tissue or 5 million cells as stated above, there will be sufficient material to perform protein quantification.
Reduce disulfide bonds and alkylate free cysteine residues
-
5
If the protein concentration exceeds 2 µg/µl, dilute samples to 2 µg/µl with lysis buffer and transfer 25 µl (i.e., 50 µg of protein) to a fresh tube.
It is possible to use >25 µl, but this will require additional loading steps onto the S‐trap™.
-
6
Reduce disulfides by adding 2.5 µl of 100 mM TCEP (to 10 mM TCEP final). Cap and vortex to mix.
-
7
Place tube(s) on an incubating mixer for 30 min at 1200 rpm, 37°C.
-
8
Alkylate cysteines by adding 2.75 µl of 400 mM acrylamide (to 40 mM acrylamide final). Cap and vortex to mix.
-
9
Place tube(s) on the incubating mixer for 30 min at 1200 rpm, 37°C.
Immobilize and digest proteins
-
10
Add 3 µl of 12% phosphoric acid. Cap and vortex to mix.
This step is essential as it brings the protein to the appropriate pH for trapping on the S‐Trap™. Sample will be lost if this step is not performed. If starting with more volume than presented in these steps, add at least 10% volume of 12% phosphoric acid.
-
11
Add 165 µl S‐Trap™ binding/wash buffer to the 33 µl of acidified sample from step 10, giving a total volume of ∼198 µl. Cap and vortex to mix. Protein particulate may form initially, but solution should appear translucent after vortexing. Do not centrifuge.
Centrifuging the sample at this step increases the risk of precipitating proteins and increases sample loss.
-
12
Place the S‐Trap™ column in a 2.0‐ml microcentrifuge tube and add the acidified sample mixture to the column.
Column equilibration is not needed. Do not load >200 µl to the spin column. Do not allow the waste volume at the bottom of the microcentrifuge tube to rise above the level of the bottom surface of the S‐Trap™ column.
-
13
Centrifuge the S‐Trap™ column at 4000 × g for 2 min or until all of the sample has passed through the S‐Trap™ column. Discard flowthrough.
-
14
Wash the S‐Trap™ column by adding 150 µl S‐Trap™ binding/wash buffer to each trap. Repeat centrifugation. Discard flowthrough.
If using fixed angle, rotate the S‐Trap™ columns a half turn between the centrifugation steps to avoid asymmetrical packing and washing. Remove wash flowthrough as necessary (i.e., after two centrifuge cycles for 2‐ml waste tubes, but every cycle for smaller tubes) or transfer to additional tubes. Vacuum manifolds that apply pressure (e.g., Promega Vac‐Man®) may also be used.
-
15
Move the S‐Trap™ column to a fresh 1.5‐ml low‐retention microcentrifuge tube for the digestion.
-
16
Add 100 µl digestion buffer to one 20‐µg vial of trypsin/LysC. Add 20 µl of the resulting dilution to the top of the S‐trap™ column.
It is important to verify that there is no air bubble between the digestion solution and the trap bed by placing the tip of the pipet directly on the S‐Trap™ sorbent bed when dispensing. The S‐Trap™ will immediately begin to absorb the solution but may not flow through completely. Any air bubbles may be removed by gently tapping the side of the spin column. This protocol has been optimized for a complex sample with a total protein amount of 50 µg and uses an enzyme:protein ratio of ∼1:12.
-
17
Cap the S‐Trap™ column to limit evaporative loss.
-
18
Proceed with trypsin/LysC digestion by incubating the sample for 2 hr at 37°C without agitation. After incubation, uncap the samples and add 40 µl S‐Trap™ digestion buffer (50 mM TEAB).
-
19
Centrifuge the S‐Trap™ column in the sample collection tube for 2 min at 4000 × g.
The eluted peptides will collect in the sample tube. Do not discard this tube, as you will do three elutions and all of them will be collected in the same tube.
-
20
Elute again into the same tube with 40 µl of 0.2% formic acid in water. Centrifuge again for 2 min at 4000 × g.
-
21
Elute hydrophobic peptides into the same tube with 35 µl of 0.2% formic acid in 50% acetonitrile. Centrifuge again for 2 min at 4000 × g. Discard S‐Trap™ column.
-
22
Dry eluted peptides in a collection tube using a vacuum centrifuge 1‐2 hr at room temperature, checking every half hour. Remove promptly when dry.
These peptides can now be used directly for MS analysis, by diluting to desired concentration with 0.1% FA in 2% LC‐MS‐grade acetonitrile (e.g., 50 μl), but we recommend performing peptide quantification (Support Protocol 2) to provide guidance on how much sample to load onto the MS. Alternatively, if additional depth of coverage is needed, peptides can be further fractionated using Support Protocol 3.
Starting from 50 mg human heart tissue, the Basic Protocol will typically yield sufficient peptide quantity for 30‐40 injections of 1 μg peptide per injection onto a nanospray LC‐MS system, with >4000 unique proteins identified per injection (Fig. 3).
Figure 3.
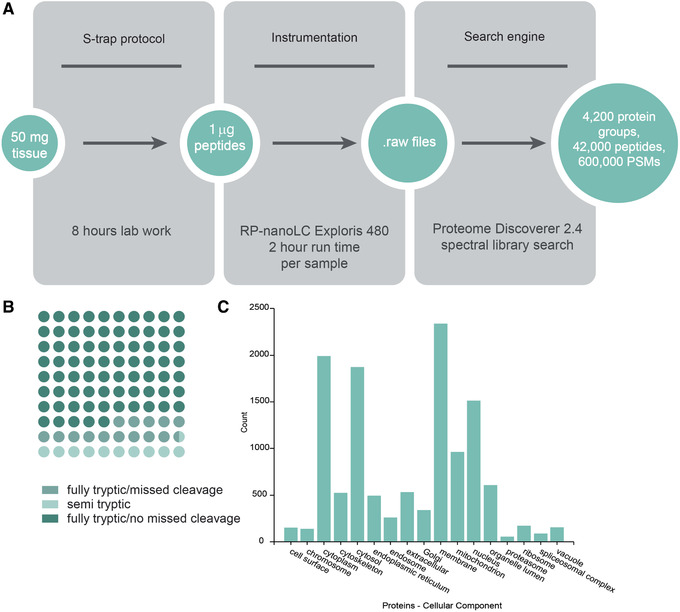
Sample data for Basic Protocol. (A) Protocol workflow: S‐trap™ processing of 50 mg human heart tissue yields sufficient peptide material for 30‐40 injections of 1 μg per injection. This workflow yields >4000 proteins per injection. PSMs, peptide spectrum matches. (B) Dot plot of percentage of peptides identified as fully tryptic, with and without missed cleavages, or semi‐tryptic. (C) Number of proteins annotated among various cellular components based on Gene Ontology, as annotated by ProteomeDiscoverer 2.4.
Alternate Protocol 1. PREPARATION OF LOW‐ TO MODERATE‐COMPLEXITY PEPTIDE SAMPLES FOR MASS SPECTROMETRY ANALYSIS USING IN‐SOLUTION PROCESSING
This protocol outlines the steps required to prepare low‐ to moderate‐complexity samples for MS analysis, and describes how to (1) solubilize proteins from RP‐HPLC fractions or affinity purification fractions, (2) prepare lyophilized protein samples, and (3) prepare a protein sample from cultured and counted cells. Then, steps to reduce disulfide bonds, alkylate free cysteines, and digest the protein enzymatically are presented (Fig. 4). At the end of Alternate Protocol 1, the user should proceed to Alternate Protocol 2 for sample cleanup. Subsequently, Support Protocol 3 can be optionally used to fractionate peptides based on their hydrophobicity in cases where enhanced sequence coverage of lower‐abundance peptides is required. Finally, Support Protocol 2 can be used directly after Alternate Protocol 2 or Support Protocol 3 to provide guidance about how much of the sample should be used for MS analysis, or to normalize total peptide in the case of quantitative analyses (Fig. 1C).
Figure 4.
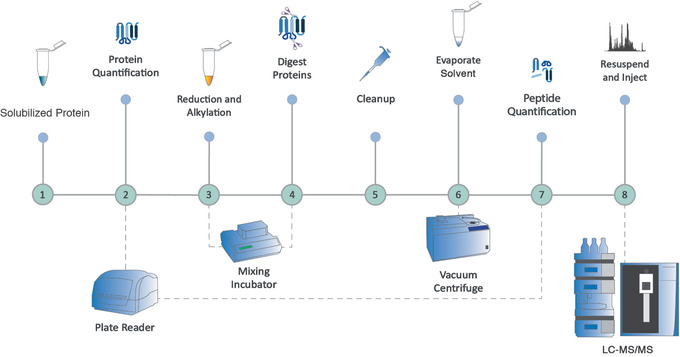
Overview of a bottom‐up proteomics preparation workflow for low‐ to moderate‐complexity samples, using Alternate Protocol 1 followed by Alternate Protocol 2, and also including Support Protocols 1 and 2. First, protein quantity is determined; then disulfide bonds are reduced and free thiols are alkylated. Proteins are digested enzymatically, contaminants are removed, and solvents are evaporated before peptide quantification.
This protocol is suitable for processing a wide range of sample types, including purified proteins in lyophilized form, RP‐HPLC and affinity chromatography fractions containing protein(s) of interest, and whole or fractionated lysates of cells. See Strategic Planning for important notes, but in general 1‐2500 μg protein is the suggested starting amount.
NOTE: Use only HPLC‐ and/or LC‐MS‐grade solvents throughout the protocol. All steps are performed at room temperature unless otherwise noted.
Materials
Sample containing protein of interest (i.e., proteins from RP‐HPLC and affinity chromatography), lyophilized proteins, or cultured cells; see protocol introduction)
100 mM aqueous ammonium bicarbonate (NH4HCO3) in LC‐MS‐grade water, pH 8.5 (see recipe)
Glacial acetic acid (Thermo Fisher Scientific, cat. no. BP1185)
1 N sodium hydroxide (NaOH; diluted from 10 N NaOH, Sigma Aldrich, SX0607N‐6)
Appropriate cell culture medium, if using cell sample
1× phosphate‐buffered saline (PBS; Gibco, cat. no. 10010‐023)
Cell lysis buffer (see recipe)
100 mM tris(2‐carboxyethyl)phosphine hydrochloride (TCEP; Sigma, cat. no. C4706) in LC‐MS‐grade water
400 mM acrylamide (Sigma, cat. no. A3553)
Trypsin/LysC enzyme combination (Promega, cat. no. V5071)
1.0% (v/v) trifluoroacetic (TFA) acid in LC‐MS‐grade water (see recipe)
20‐ml disposable glass vials (for solution storage)
Vacuum centrifuge (e.g., SpeedVac)
Vortexer
Narrow‐range pH paper
Centrifuge
Low‐retention 1.5‐ml microcentrifuge tubes (Thermo Fisher Scientific, cat. no. 02‐681‐320)
10‐µl glass syringe or other glass pipet device
Variety of positive displacement pipets (e.g., P10‐P1000)
Low‐retention pipet tips (e.g., 0.5 µl‐1 ml)
Block sonicator (e.g., Hielscher, UP200St)
Mixing incubator (e.g., Thermomixer)
Laboratory sealing film (e.g., Parafilm)
Prepare the protein sample
-
1a
To prepare a sample from an RP‐HPLC or affinity purification fraction: Dry the sample containing the proteins of interest via vacuum centrifugation. The evaporation should be performed at room temperature to avoid degradation of the sample; the amount of time required will be dependent on the volume and organic content of the sample. Resuspend dried protein in 100 µl of 100 mM NH4HCO3, pH 8.5, with rigorous vortexing for 30 s. Spot an aliquot of the solution (1 µl) on narrow‐range pH paper. Adjust pH to 8.0 with glacial acetic acid or 1 N sodium hydroxide as necessary.
The volume used for resuspension depends on the amount of protein in the original sample. Generally, a solution of 2 µg/µl works well for protein quantification and digestion. A volume of 100 µl would be appropriate for an unknown amount, but further dilution may be necessary depending on solubility (Strategic Planning).
The NH4HCO3 solution should be prepared fresh, as its pH will increase over time at room temperature. It is possible to store the NH4HCO3 solution for several days at 4°C, but the pH should be checked before use. The pH also makes it prone to bacterial growth, which can contaminate samples.
-
1b
To prepare a lyophilized protein sample: Resuspend dried protein (50‐1000 µg; see note on concentration above) in 100 µl of 100 mM NH4HCO3, pH 8.5, with rigorous vortexing for 30 s. Spot a 1‐µl aliquot of the solution on narrow‐range pH paper. Adjust pH to 8.0 with acetic acid or 1 N sodium hydroxide as necessary.
-
1cTo prepare a protein sample from cultured and counted cells:
- Count cells and add a minimum of 1 million cells and a maximum of 20 million cells to each 1.5‐ml tube (e.g., a minimum of 1 million cells in 250 µl and a maximum of 20 million cells in 800 µl).
- Collect cells by centrifugation (5 min at 300 × g, room temperature).
- Aspirate and discard supernatant.
- Add 1 ml of 1× PBS. Gently pipet up and down 3‐5 times using a P1000 pipet until cells are well resuspended.
- Collect cells by centrifugation for 5 min at 300 × g, room temperature.
- Aspirate and discard liquid.
- Add 1 ml of 1× PBS. Gently pipette up and down 3‐5 times using a P1000 pipette until cells are well resuspended.
- Collect cells by centrifugation for 5 min at 300 × g, room temperature.
- Aspirate and discard liquid.
-
Add cell lysis buffer according to cell count shown below. Mix by pipetting up and down 3‐5 times. Keep on ice throughout the protocol.If samples appear to contain a large, insoluble pellet after sonication in step xi, add more volume in this step, up to a maximum of 800 µl.
Number of cells 1‐2 million 5 million 10 million 15‐20 million Lysis buffer 250 µl 300 µl 500 µl 600‐800 µl -
Suspend the tube adaptor from the block sonicator above an ice bucket, and place sample tube in the tube adaptor. Make sure that both the bottom and top of the tube are embedded in ice. Sonicate for 10 s on and then 10 s off, at 30% power, three times. Wait 1 min, and repeat the cycle two more times if ≥5 million cells are digested. Keep on ice afterwards.These parameters are suitable for the Hielscher UP200St block sonicator. Optimize the power levels according to your sonicator. Although a probe sonicator may be used, this increases the chance for cross‐contamination, as the probe is reused and requires careful cleaning between uses. The block sonicator used here enables samples in a closed tube to be processed without cross‐contamination.
-
2
Perform protein quantification (see Support Protocol 1).
Determining the amount of protein in the sample will inform the amount of enzyme to use in later steps. This information can also be used to normalize among samples intended for quantitative analyses. As stated in Strategic Planning, for sample‐limited cases, it is advisable to at least have an estimate of the total amount of protein based on a similar sample type, when possible. If, based on a representative sample, the amount of protein in the experimental sample is <10 µg, it will not be possible to perform protein quantification without using a majority of the sample for this step. However, if sufficient protein is indeed available, protein quantification is strongly advised.
Reduce disulfide bonds and alkylate free cysteine residues
-
3
Reduce disulfides by adding 100 mM TCEP to the sample from step 1 (to 5 mM TCEP final).
Although reduction and alkylation (steps 3‐5) are not required before enzymatic or chemical cleavage of proteins that do not contain cysteines, these steps will typically provide enhanced protein sequence coverage of most proteins as they help unfold the protein and maintain it in an open configuration, thereby facilitating cleavage.
-
4
Cap tubes, and place samples in a mixing incubator at 37°C for 30 min.
Throughout this protocol, agitation can be achieved with end‐over‐end rotation or vortexing.
-
5
Alkylate cysteines by adding 400 mM acrylamide (to 40 mM final). Cap tubes, and place samples in a mixing incubator for 30 min at 37°C.
Digest protein
-
6
Add appropriate amount of trypsin/LysC or other digestion enzyme (e.g., chymotrypsin, trypsin, LysC, or AspN) or chemical (e.g., cyanogen bromide/70% formic acid) based on total amount of protein present. This will vary among enzymes. If using trypsin, add sufficient enzyme for a final trypsin:protein ratio between 1:20 and 1:100 (w/w).
The amount of trypsin needed will vary depending on the amount of protein in the sample. Although longer digestion times may lead to higher sequence coverage, they will also result in increased loss of some peptides, which can affect quantification. Shorter digestion times can benefit workflow speed but may result in lower sequence coverage.
Many commercial vendors provide proteomics‐grade trypsin in convenient aliquots (e.g., 1‐ to 25‐μg vials; such as Promega cat. no. V5083), which eliminate the need to weigh out the enzyme. To minimize trypsin autolysis, which will add extra peaks to the MS spectra, use the minimum amount of trypsin. Typically, a ratio of 1:20 (w/w) is sufficient for complete digestion of 10 μg protein within 4 hr (e.g., 0.5 μg of enzyme). If protein concentration is especially high (e.g., 2500 μg) or the accessibility of trypsin to the cleavage sites is hampered (e.g., due to insolubility), it may be helpful to add another aliquot of trypsin, perform a short digestion (4 hr) with LysC before the addition of trypsin, or use combination enzymes such as the trypsin/LysC recommended here.
The activity of trypsin is enhanced in the presence of acetonitrile (10%‐50% v/v), and thus acetonitrile may be added to aid protein solubilization during tryptic digestion (Waas et al., 2014).
-
7
Use 1 µl of the sample to test the pH using a narrow‐range pH paper. As trypsin activity is greatest at pH 8.0, adjust pH to 8.0 with acetic acid or 1 N sodium hydroxide as necessary.
-
8
Vortex briefly, cap tube, seal tube with Parafilm, and incubate in a mixing incubator at 37°C for 4‐17 hr.
-
9
Proceed to Support Protocol 2 for peptide quantification, which will be used to then inform sample cleanup (Alternate Protocol 2), before MS analysis. Alternatively, if additional depth of coverage is needed, peptides can be further fractionated using Support Protocol 3.
Alternate Protocol 2. REMOVAL OF DETERGENT, POLYMER, AND SALT FROM PEPTIDE SAMPLES BEFORE MASS SPECTROMETRY ANALYSIS USING SP2 PROCESSING
Detergents and salts are common ingredients in sample preparation buffers. Polymers (e.g., polyethylene glycol, PEG) resulting from the manufacturing process of laboratory consumables (e.g., tubes, filters) can also contaminate proteomics samples. Detergents, polymers, and salts will interfere with chromatographic separation and ionization of peptides and must therefore be removed from the samples before MS analysis. Although reverse‐phase methods using filter cartridges or tips are popular for peptide desalting, they are not capable of removing detergents or PEG. The recently described SP2 method uses carboxylate‐modified paramagnetic particles to remove detergents, PEG, and salts from peptide samples (Waas, Pereckas, Jones Lipinski, Ashwood, & Gundry, 2019). This protocol describes how to perform the SP2 method to remove contaminants from peptide samples before MS analysis (Fig. 5). Peptides resulting from step 9 in Alternate Protocol 1, or alternative sample preparation strategies such as on‐filter digestions, can be cleaned by this method.
Figure 5.
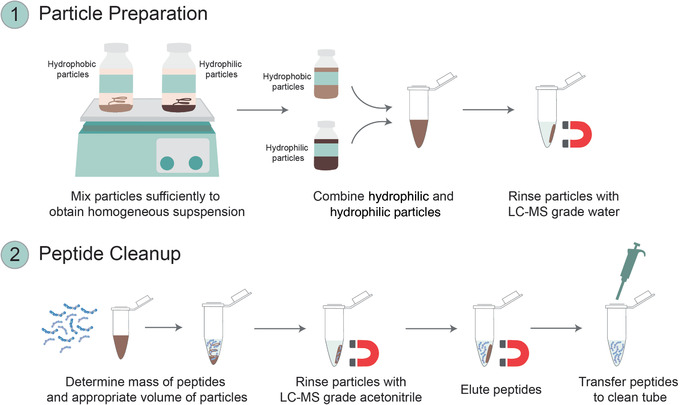
Overview of the SP2 protocol for detergent and salt removal from peptide samples using Alternate Protocol 2. (1) During particle preparation, hydrophobic and hydrophilic Sera‐Mag SpeedBeads carboxylate‐modified particles are combined and washed for subsequent use during peptide cleanup. (2) Particles are then combined with an appropriate mass of peptides, and cleaned peptides are eluted.
NOTE: Use only HPLC‐ and/or LC‐MS‐grade solvents throughout the protocol. All steps are performed at room temperature unless otherwise noted.
Materials
Peptide sample from Alternate Protocol 1, step 9 (with concentration and mass determined beforehand following Support Protocol 2)
Sera‐Mag SpeedBeads Carboxylate‐Modified Particles, 50 µg/µl, hydrophilic (Cytivia, 45152105050250)
Sera‐Mag SpeedBeads Carboxylate‐Modified Particles, 50 µg/µl, hydrophobic (Cytivia, 65152105050250)
Acetonitrile (LC‐MS grade, Thermo Fisher Scientific, cat. no. A955)
Water (LC‐MS grade, Thermo Fisher Scientific, cat. no. W6)
10% formic acid (FA) in LC‐MS water (see recipe)
Elution solution: 2% (v/v) LC‐MS‐grade acetonitrile in LC‐MS‐grade water
20‐ml disposable glass vials (for solution storage)
Orbital shaker
Vortexer
1.5‐ml low‐retention microcentrifuge tubes (Thermo Fisher Scientific, cat. no. 02‐681‐320)
Magnetic rack (e.g., Permagen, cat. no. MSR24)
10‐µl glass syringe or other glass pipet device
Variety of positive‐displacement pipets (e.g., P10‐P1000)
Low‐retention pipet tips (e.g., 0.5 µl‐1 ml)
Gel‐loading tips (200 µl)
Benchtop minicentrifuge
Prepare particles
-
1
Remove both sets (hydrophilic and hydrophobic) of Sera‐Mag SpeedBeads Carboxylate‐Modified particle stocks from 4°C storage.
-
2
Place particle stocks on an orbital shaker for 30 min to allow them to warm to room temperature and to obtain homogeneous suspensions.
-
3
Shake and vortex sufficiently to fully suspend the particles before use.
This may require prolonged and vigorous mixing.
-
4
Combine 100 µl hydrophilic particles with 100 µl hydrophobic particles into a 1.5‐ml low‐retention microcentrifuge tube, and add 800 µl LC‐MS‐grade water.
-
5
Place the microcentrifuge tube on a magnetic rack and let the particles settle for 2 min.
-
6
Carefully remove the supernatant with a 200‐µl gel‐loading tip, and discard supernatant and tip.
-
7
Remove the microcentrifuge tube from the magnetic rack and rinse the particles with 1000 µl LC‐MS‐grade water.
-
8
Mix thoroughly by pipetting up and down 3‐5 times using a P1000 pipet.
-
9
Return the microcentrifuge tube to the magnetic rack and let the particles settle for 2 min.
-
10
Carefully remove the supernatant with a 200 µl gel‐loading tip, and discard supernatant and tip.
-
11
Repeat steps 7‐10 twice more.
-
12
Add 200 µl LC‐MS‐grade water to resuspend the particles at 50 μg/µl, and store at 4°C.
Prepared particles can be stored at 4°C for 6 months. Do not freeze the particles.
Clean up peptides
-
13
Remove prepared particles from step 12 from 4°C storage.
-
14
Vortex prepared particles immediately before use to ensure they are well suspended.
-
15
Obtain peptides resulting from step 9 in Alternate Protocol 1.
-
16
Add prepared particles corresponding to 20 times the mass of peptides to a 1.5‐ml low‐retention microcentrifuge tube containing peptides to be cleaned.
Example: For 15 µg of peptide solution at 1 µg/µl, use 6 µl of particles. 6 µl of particles at 50 μg/µl equals 300 μg of particles, which is 20 times the mass of peptides.
-
17
Flick the microcentrifuge tube containing the peptides and particle suspension 3‐5 times to mix the sample.
-
18Determine the amount of acetonitrile required to bring sample to a final concentration of 95% acetonitrile. The peptides in step 15 contain 20% acetonitrile. Therefore, use the following equation to determine amount of acetonitrile to be added:Example: If digest contains 20% acetonitrile, the volume of particles used is 6 µl, and the volume of sample is 15 µl, add 339 µl acetonitrile:
-
19
Add sufficient 100% acetonitrile to the microcentrifuge tube to obtain a final concentration of 95% acetonitrile.
Example: if digest contains 20% acetonitrile, add 339 µl acetonitrile
-
20
Mix by pipetting up and down 3‐5 times using a P1000 pipet to ensure particles are well dispersed, and then allow the mixture to settle for 2 min.
-
21
Place microcentrifuge tube on magnetic rack for 1 min.
-
22
Remove supernatant with a 200‐µl gel‐loading tip, taking care not to disturb the particles. Discard supernatant and tip.
-
23
Remove microcentrifuge tube from magnetic rack and add 500 µl of 100% acetonitrile to cover the particles completely.
At this step, particles may remain stuck on the side of the tube, so ensure the acetonitrile wash covers the surface area of the particles.
-
24
Repeat steps 20‐22 once more.
-
25
Remove microcentrifuge tube from magnetic rack.
-
26
Determine the elution volume, considering that the percent recovery is maximized when using 9 times the volume of particles.
-
27
Add desired volume of elution solution to particles.
Example: if 6 µl of particles was added in step 16, add 54 µl (9 times the volume of particles).
-
28
Vortex microcentrifuge tube containing particles in elution solution on the side for 30 s, to remove the particles from the tube wall and disperse them.
-
29
To collect any sample that may be left on the wall of the tube, briefly centrifuge the samples for 3 s using a benchtop minicentrifuge (i.e., place tubes in minicentrifuge, turn the centrifuge on, and then immediately turn it off).
-
30
Place microcentrifuge tube on magnetic rack and allow the particles to settle for 1 min.
-
31
Using a 200 µl gel‐loading tip, remove supernatant and transfer to a fresh microcentrifuge tube, taking care not to disturb the particles.
The supernatant contains cleaned peptides.
-
32
Repeat steps 30 and 31 once more.
-
33
Add 10% FA (to 0.1% FA final).
-
34
Centrifuge sample 10 min at 14,000 × g and then transfer the supernatant to a sample vial for MS analysis.
Starting with 5 million B lymphocytes, sample preparation by Alternate Protocol 1 followed by cleanup using Alternate Protocol 2 will typically yield enough peptide for 50 injections of 1 μg peptide per injection onto a nanospray LC‐MS system, with >4000 unique proteins being discovered per injection (Fig. 6).
Figure 6.
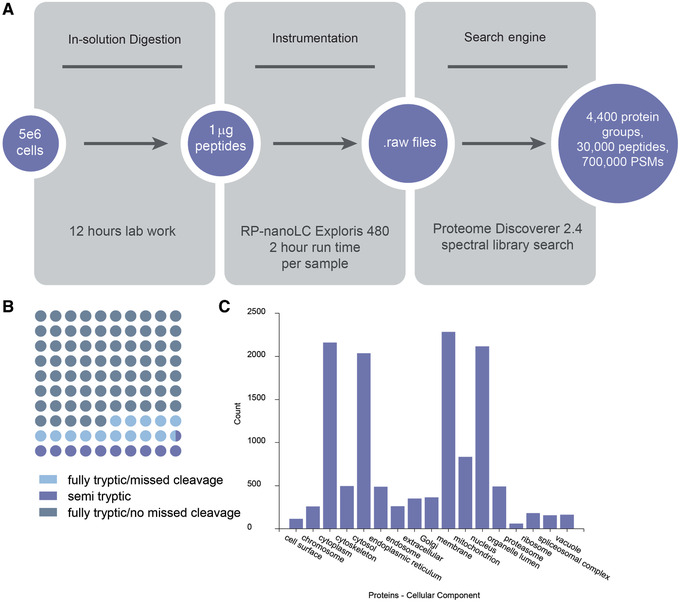
Sample data for Alternate Protocol 1 followed by Alternate Protocol 2. (A) Protocol workflow: In‐solution digestion of 5e6 RMPI 1788 cells followed by SP2 cleanup of the sample yields sufficient peptide material for 50 injections of 1 μg per injection. This workflow yields >4000 proteins per injection. PSMs, peptide spectrum matches. (B) Dot plot of percentage of peptides identified as fully tryptic with and without missed cleavages or semi‐tryptic. (C) Number of proteins annotated among various cellular components based on Gene Ontology, as annotated by ProteomeDiscoverer 2.4.
Support Protocol 1. PROTEIN QUANTIFICATION USING PIERCE™ 660 NM ASSAY
Protein quantification is an important step in many bottom‐up proteomics workflows (including those described here), as it provides information on how much enzyme to use for optimal digestion efficiency and which size (binding capacity) of S‐Trap™ column to use. Several strategies are available for the determination of protein concentration (e.g., Bradford, bicinchoninic acid assay, NanoDrop™, and Qubit™), but they vary according to interferences allowed (e.g., SDS, Tween® 20, Triton), sensitivity, and dynamic range. This protocol describes how to perform protein quantification using the Pierce™ 660 nm Protein Assay, which is suitable for a range of proteomics applications and can be used to quantify samples that contain detergents such as SDS; hence, it is compatible with workflows that require this detergent for enhancing protein solubilization (such as the Basic Protocol).
The assay requires standards: the Pierce Bovine Serum Albumin (BSA) Set, which contains standardized BSA solutions at a specific concentration, from 125 to 2000 µg/ml in 0.9% saline and 0.05% sodium azide, along with two additional standards, of 62.5 and 31.25 µg/ml, which you will need to prepare (see steps 2 and 3 below).
Materials
Sample from Alternate Protocol 1, step 2, or Basic Protocol, step 4
Purified water (18 MΩ)
-
Pierce™ 660 nm Protein Assay Kit (Pierce, cat. no. 22662), containing:
Pierce™ 660 nm Protein Assay Reagent (Pierce, cat. no. 22660)
Pre‐diluted Protein Assay Standards
Ionic Detergent Compatibility Reagent (IDCR, Pierce, cat. no. 22663; if using Pierce™ 660 nm Protein Assay kit for S‐Trap™ samples, the ionic detergent compatibility reagent is necessary for consistent readings.
1.5‐ml low‐retention microcentrifuge tube
96‐well clear flat‐bottom plate (Thermo Fisher Scientific, cat. no. 12‐565‐501)
Multichannel pipet
25‐ml reservoir
Spectrophotometer (e.g., Varioskan LUX Multimode Microplate Reader)
Prepare sample
-
1
Dilute samples before protein quantification. For this, add 5 µl of each sample to two tubes. Then, add 95 µl purified water to one tube and label it DF = 20 (dilution factor 20). Last, add 195 µl to the other tube and label it DF = 40. Mix well.
In our experience, these dilutions typically work well when starting with 5 million B cells for Alternate Protocol 1 or 50 mg heart tissue for Basic Protocol. If the sample is found to be outside the concentration range of the protein standards, dilute to within the range and reanalyze.
-
2
Add 100 µl purified water to a 1.5‐ml low‐retention microcentrifuge tube and then add 100 µl of the 125 µg/ml BSA standard from the Pierce protein assay kit. This will be the 62.5 µg/ml standard.
-
3
Add 100 µl purified water to a 1.5‐ml low‐retention microcentrifuge tube and then add 100 µl of the 62.5 µg/ml standard. This will be the 31.25 µg/ml standard.
-
4
Add 10 µl of each standard, each unknown sample, and the appropriate blank water sample into their respective wells of a 96‐well microplate well.
Standards, blanks, and samples should be added in duplicate.
-
5
Add 20 ml Pierce™ 660 nm Protein Assay Reagent to a 50‐ml conical tube, and add the contents of an Ionic Detergent Compatibility Reagent (IDCR) packet according to the manufacturer's instructions. Cap and mix well to dissolve.
IDCR is an additive to the assay reagent that improves the sensitivity and reproducibility of the assay in the presence of SDS.
-
6
Add 150 µl of the IDCR Protein Assay Reagent to each plate well, for a final well volume of ∼160 µl.
-
7
Mix plate by gently tapping palm against the side to the plate 3 times.
-
8
Incubate plate 5 min in the dark.
Perform measurement and calculations
-
9
Measure absorbance at 660 nm using a spectrophotometer.
-
10
Determine concentration. Depending on the spectrophotometer used, the concentration including the dilution factor may be calculated for you. In this case, the measurement will result in a unit of µg/ml. To convert to total µg/µl, divide the concentration by 1000. To calculate the total micrograms of protein in your sample, multiply that number by the volume remaining in the sample.
Table 1 shows some typical concentration results obtained from various starting samples.
Table 1.
Examples of Protein Quantity Values for Various Cell and Tissue Types Obtained Using the Pierce™ 660 nm Assay
| Sample | Typical protein quantity (µg) |
|---|---|
| 1 million human B cells (RPMI 1788) | 100 |
| 1 million human embryonic kidney cells (HEK) | 200 |
| 1 million human pluripotent stem cells | 300‐400 |
| 50 mg wet weight human heart tissue, ventricle | 1000 |
Support Protocol 2. PEPTIDE QUANTIFICATION USING PIERCE QUANTITATIVE FLUOROMETRIC PEPTIDE ASSAY
Peptide quantification is an important step for evaluating the yield of the processing steps used to generate peptides from proteins and for normalizing the total peptide amount analyzed in quantitative workflows. Peptide quantification can be done immediately before MS to help determine whether the sample should be diluted before analysis. It can also be performed before Alternate Protocol 2 and Support Protocol 3 to provide guidance about the mass of peptides to use for each application. This protocol describes how to perform peptide quantification using the Pierce™ Quantitative Fluorometric Peptide Assay.
Materials
-
Pierce™ Quantitative Fluorometric Peptide Assay (do not premix items), containing:
Fluorometric Peptide Assay Buffer
Fluorometric Peptide Assay Reagent
Peptide Digest Assay Standard (1 mg/ml)
Purified water (18 MΩ)
Sample resulting from Alternate Protocol 1, step 9, or Basic Protocol, step 23
Multichannel pipet
96‐well flat‐bottom black plate (Thermo Fisher Scientific, cat. no. 12‐566‐07)
25‐ml reservoir
Spectrophotometer (e.g., Varioskan LUX Multimode Microplate Reader)
Prepare sample
-
1
Use the information below to prepare a dilution series of the Peptide Digest Assay Standard from the Pierce assay kit in separate tubes. Use purified water as diluent.
The volumes below will be enough for one set of measurements in which each standard is placed into two wells of a 96‐well plate and used as reference for all experimental samples in the plate. If quantifying more samples than can fit into a single 96‐well plate, additional standards should be prepared so that each plate will contain standards in duplicate. Make standards fresh before use.
| Tubes | Volume of diluent, µl | Volume of standard, µl | Final standard concentration, μg/ml |
| A | 0 | 50 of stock | 1000 |
| B | 25 | 25 of Tube A dilution | 500 |
| C | 25 | 25 of Tube B dilution | 250 |
| D | 25 | 25 of Tube C dilution | 125 |
| E | 25 | 25 of Tube D dilution | 62.5 |
| F | 25 | 25 of Tube E dilution | 31.3 |
| G | 25 | 25 of Tube F dilution | 15.3 |
| H | 25 | 25 of Tube G dilution | 7.8 |
| Blank | 20 | 0 | 0 |
-
2
Dilute samples before quantification by adding 5 µl of each sample to two tubes. Add 20 µl purified water to one tube and label it DF = 5. Add 45 µl to the other tube and label it DF = 10. Mix well.
These dilutions typically work well when starting with 5 million B cells for Alternate Protocol 1 or 50 mg heart tissue for Basic Protocol. If the sample is found to be outside the concentration range of the peptide standards, dilute within the range and reanalyze.
-
3
Add 10 µl of each standard, unknown sample, and the appropriate blank sample into a microplate well.
Standards, blanks, and samples should be aliquoted in duplicates.
-
4
Add 70 µl Fluorometric Peptide Assay Buffer to each well.
-
5
Add 20 µl Fluorometric Peptide Assay Reagent to each well.
-
6
Mix plate by gently tapping palm against the side to the plate 3 times.
-
7
Incubate plate for 5 min in the dark.
Perform measurement and calculations
-
8
Measure absorbance using a spectrophotometer with a wavelength excitation at 390 nm and emission of 475 nm.
-
9
Determine concentration. Depending on the spectrophotometer used, the concentration including the dilution factor may be calculated for you. In this case, the measurement will result in a unit of µg/ml. To convert to total µg/µl, divide the concentration by 1000. To calculate the total µg in your sample, multiply that number by the volume remaining in the sample. Dilute samples to desired concentration for MS analysis with with 0.1% FA in 2% LC‐MS‐grade acetonitrile.
Support Protocol 3. HI‐pH FRACTIONATION OF COMPLEX PEPTIDE SAMPLES
This protocol describes how to perform peptide fractionation using high‐pH reverse‐phase peptide fractionation spin columns (Fig. 7). Fractionation of a single peptide mixture by this protocol will generate ten lower‐complexity samples that can be analyzed separately by MS. In doing so, greater depth of proteome coverage can be achieved for moderate‐to high‐complexity samples for which the dynamic range of protein abundances makes it more challenging to detect low‐abundance peptides. This protocol can be used directly after the Basic Protocol, Alternate Protocol 1, or other preparation strategies. The sample is loaded onto a washed and equilibrated spin column, and peptides are eluted with increasing amounts of acetonitrile, at a high pH. Solvent is evaporated and peptides are then quantified before MS analysis.
Figure 7.
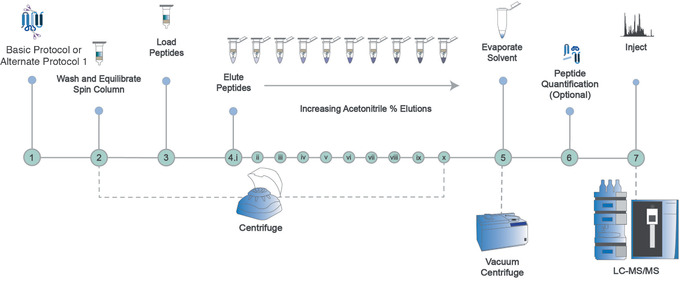
Overview of the protocol for high‐pH fractionation of peptide samples to obtain deeper coverage of the proteome for highly complex samples. After Basic Protocol or Alternate Protocol 1 is performed, the resulting sample is loaded onto a washed and equilibrated spin column, and peptides are eluted with increasing amounts of acetonitrile at a high pH. Solvent is evaporated and peptides are then quantified before MS analysis.
NOTE: Use only HPLC‐ and/or LC‐MS‐grade solvents throughout the protocol. All steps are performed at room temperature unless otherwise noted.
Materials
100% acetonitrile (LC‐MS grade, Fisher Scientific, cat. no. A955)
0.1% triethylamine (TEA; included in kit)
High pH Reversed‐Phase Peptide Fractionation Kit (Pierce, cat. no. 84868)
Sample from Alternate Protocol 1 that has been cleaned using Alternate Protocol 2, step 34, or sample from Basic Protocol, step 23 (maximum 100 µg)
1% TFA in LC‐MS‐grade water (see recipe)
Water (LC‐MS grade, Fisher Scientific, cat. no. W6)
5% (v/v) formic acid (FA) in 95% (v/v) LC‐MS‐grade water (see recipe)
5% (v/v) FA in 95% (v/v) LC‐MS‐grade acetonitrile (see recipe)
0.1% (v/v) FA in 2% (v/v) LC‐MS‐grade acetonitrile in LC‐MS‐grade water (see recipe)
2.0‐μl low‐retention microcentrifuge tubes (Fisher Scientific, cat. no. 02‐681‐321)
Microcentrifuge with rotor speed adjustable up to 7000 × g
Vacuum centrifuge
Prepare the elution solvents
-
1
Prepare solutions in 2.0‐ml low‐retention microcentrifuge tubes according to the information below. This provides sufficient volume for one sample. If fractionating more than one sample, scale accordingly.
| Fraction no. | % acetonitrile | 100% acetonitrile, µl | 0.1% TEA, µl |
| 1 | 5 | 15 | 285 |
| 2 | 7.5 | 22.5 | 277.5 |
| 3 | 10 | 30 | 270 |
| 4 | 12.5 | 37.5 | 262.5 |
| 5 | 15 | 45 | 255 |
| 6 | 17.5 | 52.5 | 247.5 |
| 7 | 20 | 60 | 340 |
| 8 | 50 | 150 | 150 |
| Fraction no. | % acetonitrile | 5% FA in acetonitrile, µl | 5% FA, µl |
| 9 | 15 | 45 | 255 |
| 10 | 50 | 150 | 150 |
Condition the spin columns
Do not exceed recommended centrifugation speeds. All steps can be performed at room temperature unless otherwise noted.
-
2
Remove the protective white tip from the bottom of the high‐pH fractionation spin column and discard.
-
3
Place the column into a 2.0‐ml microcentrifuge tube.
-
4
Centrifuge 2 min at 5000 × g to remove the solution and pack the resin material. Discard the liquid.
-
5
Wash the spin column by removing the top screw cap and adding 300 µl of 100% acetonitrile onto the column. Cap and centrifuge 2 min at 5000 × g. Discard acetonitrile wash.
Replace the cap during each centrifuge step to ensure that the spin column does not dry out, which could affect the final results.
-
6
Repeat step 4 once more.
-
7
Wash the spin column by removing the top screw cap and adding 300 µl of 0.1% TFA solution into the column. Cap and centrifuge 2 min at 5000 × g. Discard 0.1% TFA wash. Repeat step 6 once more.
Fractionate the peptide mixture
-
8
If using peptides from Alternate Protocol 1 that were cleaned with Alternate Protocol 2, dry them in a 1.5‐ml low‐retention microcentrifuge tube for 1‐2 hr using a vacuum centrifuge set at room temperature. Check every 30 min and remove promptly once dry. If using dried peptides from the Basic Protocol, proceed to step 9.
-
9
Dissolve all of the peptide sample in 300 µl of 0.1% TFA solution.
The maximum amount of peptide that can be loaded onto the high‐pH fractionation column is 100 µg. If following Basic Protocol and Alternate Protocol 1 as described above, the total amount of peptide in the resulting samples will be <100 µg; therefore, the entire sample can be loaded onto the fractionation column. If using other than the recommended starting material or amounts described for the Basic Protocol and Alternate Protocol 1, perform peptide quantification after Support Protocol 2 and use a maximum of 100 µg for this step.
-
10
Place the spin column into a fresh 2.0‐ml microcentrifuge tube.
-
11
Add the 300 µl of sample solution from step 9 into the top of the column, cap, and centrifuge 2 min at 3000 × g. Retain eluate as “flowthrough” fraction for troubleshooting only.
As with most loading and washing steps, it is necessary to retain flowthrough solutions in case peptides were not retained on the packing material. If there are no peptides in the samples, this solution can be tested during troubleshooting.
-
12
Place the column into a fresh 2.0‐ml microcentrifuge tube.
-
13
Add 300 µl LC‐MS‐grade water into the top of the column, cap, and centrifuge 1 min at 3000 × g. Retain wash for troubleshooting only.
As with most loading and washing steps, it is necessary to retain flowthrough solutions in case peptides were not retained on the packing material. If there are no peptides in the samples, this solution can be tested during troubleshooting.
-
14
Place the column into a fresh 2.0‐ml microcentrifuge tube labeled “Fraction 1.”
-
15
Add 300 µl of the appropriate elution solution described in step 1 (e.g., 5% acetonitrile, 0.1% TEA) and centrifuge for 2 min at 3000 × g to collect the fraction. Retain eluate as “fraction #1”.
The first elution solution is fraction #1. A total of ten elution solutions (prepared according to the table in step 1) will be added to the same column in ten steps.
-
16
Repeat steps 14 and 15 for the remaining step gradient fractions using the appropriate elution solutions and a fresh microcentrifuge tube each time (i.e., elution solutions for fractions #2‐#10).
-
17
Evaporate eluted peptides in a 2.0‐ml low‐retention microcentrifuge tube using a vacuum centrifuge set at room temperature for 1‐2 hr. promptly once dry. The resulting peptide samples can then be used for MS analysis.
Starting with 50 µg of peptides resulting from applying Alternate Protocol 1 to human ventricle heart tissue, the high‐pH fractionation method in Support Protocol 3 will typically yield sufficient peptide quantity for three injections of 1 μg peptide per injection per fraction onto a nanospray LC‐MS system. In total, >7000 unique protein identifications per set of fractions can typically be made (Fig. 8).
Figure 8.
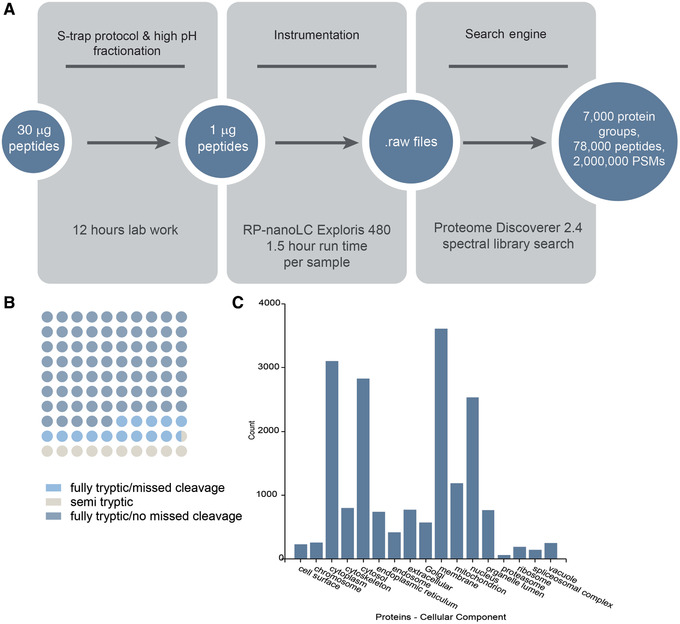
Sample data for Basic Protocol followed by Support Protocol 3. (A) S‐trap™ processing of human heart tissue followed by high‐pH fractionation to increase proteome coverage yielded 7000 proteins. The protocol workflow and results by step are shown. PSMs, peptide spectrum matches. (B) Dot plot of percentage of peptides identified as fully tryptic with and without missed cleavages or semi‐tryptic. (C) Number of proteins annotated among various cellular components based on Gene Ontology.
REAGENTS AND SOLUTIONS
Aqueous ammonium bicarbonate (NH4HCO3), pH 8.5, 250 mM (10 ml)
-
1.
Weigh 198 mg ammonium bicarbonate (Sigma Aldrich, cat. no. 09830) and add to 10 ml LC‐MS‐grade water (Fisher Scientific, cat. no. W6). Mix well.
-
2.
Adjust pH to 8.5 with glacial acetic acid.
Prepare fresh before use if possible. Can also be stored for several days at 4°C, but pH must be checked and solution discarded if pH exceeds >8.5.
Aqueous ammonium bicarbonate (NH4HCO3), pH 8.5, 100 mM (25 ml)
-
1.
Weigh 198 mg ammonium bicarbonate and add to 25 ml LC‐MS‐grade water. Mix well.
-
2.
Adjust pH to 8.5 with glacial acetic acid.
Prepare fresh before use. Can also be stored for several days at 4°C, but pH must be checked and solution discarded if pH exceeds >8.5.
Cell lysis buffer (40% Invitrosol, 100 mM ammonium bicarbonate, 40% acetonitrile)
-
1.
Prepare cell lysis buffer for Alternate Protocol 1 according to the number of cells to be used, as described below. (For example, if using 5 million cells, 300 µl lysis buffer will be needed.)
| Number of cells | 1‐2 million | 5 million | 10 million | 15‐20 million |
| Lysis buffer | 250 µl | 300 µl | 500 µl | 600‐800 µl |
-
2.
To prepare 250 µl lysis buffer, mix 100 µl Invitrosol (from Invitrosol™ LC‐MS Protein Solubilizer Kit; Invitrogen, cat. no. MS10007), 100 µl acetonitrile (LC‐MS grade, Thermo Fisher Scientific, cat. no. A955), and 50 µl of 250 mM ammonium bicarbonate (see recipe).
-
3.
Make buffer fresh before use and keep on ice.
Formic acid in LC‐MS‐grade water, 10% (10 ml)
-
1.
In a glass scintillation vial, add 1.0 ml LC‐MS‐grade 100% formic acid (Fisher Scientific, cat. no. A117) to 9 ml LC‐MS‐grade water using a glass syringe dedicated to acid or other disposable glass pipet (to avoid plastic leaching contamination, do not use plastic).
-
2.
Mix well and store up to 1 month at room temperature.
Formic acid in LC‐MS‐grade water, 5% (5 ml)
-
1.
Add 4.75 ml LC‐MS‐grade water to a glass scintillation vial.
-
2.
Add 0.25 ml LC‐MS‐grade 100% formic acid using a glass syringe dedicated to acid or other disposable glass pipette (to avoid plastic leaching contamination, do not use plastic).
-
3.
Mix well and store up to 2 weeks at room temperature.
Formic acid in LC‐MS‐grade water, 0.2% (5 ml)
-
1.
Add 0.1 ml of 10% formic acid to 4.9 ml LC‐MS‐grade water in a glass scintillation vial.
-
2.
Mix well and store up to 2 weeks at room temperature.
Formic acid in LC‐MS‐grade acetonitrile, 5% (5 ml)
-
1.
Add 4.75 ml LC‐MS‐grade acetonitrile (Fisher Scientific, PN A955) to a glass scintillation vial.
-
2.
Add 0.25 ml of 100% formic acid using a glass syringe dedicated to acid or other disposable glass pipette (to avoid plastic leaching contamination, do not use plastic).
-
3.
Mix well and store up to 2 weeks at room temperature.
Formic acid in 50% LC‐MS‐grade acetonitrile, 0.2% (5 ml)
-
1.
Add 2.5 ml LC‐MS‐grade acetonitrile to 2.4 ml LC‐MS‐grade water in a glass scintillation vial.
-
2.
Add 0.1 ml of 10% formic acid in LC‐MS‐grade water (see recipe).
-
3.
Mix well and store up to 2 weeks at room temperature.
Formic acid in 2% LC‐MS‐grade acetonitrile, 0.1% (5 ml)
-
1.
Add 0.1 ml LC‐MS‐grade acetonitrile to 4.85 ml LC‐MS‐grade water in a glass scintillation vial.
-
2.
Add 0.05 ml of 10% formic acid in LC‐MS‐grade water (see recipe).
-
3.
Mix well and store up to 2 weeks at room temperature.
Phosphoric acid, 12% (10 ml)
-
1.
Add 1.2 ml ACS‐grade 85% phosphoric acid (Thermo Fisher Scientific, cat. no. A260) to 8.8 ml purified water. Mix well.
-
2.
Store up to 3 months at room temperature.
SDS tissue lysis buffer (5% SDS, 50 mM TEAB), pH 7.55 (10 ml)
-
1.
Add 5 ml of 10% SDS buffer to 4.5 ml of purified water and 0.5 ml of 1 M TEAB. Mix well.
-
2.
Adjust pH to 7.55 with 12% phosphoric acid (∼20 µl).
-
3.
Store up to 1 day at room temperature.
SDS cell lysis buffer (5% SDS, 4 mM MgCl2, 50 mM TEAB), pH 7.55 (10 ml)
-
1.
Add 5 ml of 10% SDS buffer, 0.02 ml of 1 M magnesium chloride, and 0.5 ml of 1 M TEAB to 4.5 ml purified water. Mix well.
-
2.
Adjust pH to 7.55 with 12% phosphoric acid (∼20 µl).
-
3.
Store up to 1 day at room temperature.
S‐Trap™ binding/wash buffer (90% methanol, 100 mM TEAB), pH 7.1 (10 ml)
-
1.
Add 90 ml HPLC‐grade methanol to 10 ml of 1 M TEAB and mix well.
-
2.
Adjust pH to 7.1 with 12% phosphoric acid (∼0.1 ml).
-
3.
Store up to 1 month at room temperature.
Trifluoroacetic acid in LC‐MS‐grade water, 10% (10 ml)
-
1.
In a glass scintillation vial, add 1.0 ml LC‐MS‐grade 100% trifluoroacetic acid (Fisher Scientific, PN A116) to 9 ml LC‐MS‐grade water using a glass syringe dedicated to acid or other disposable glass pipet (to avoid plastic leaching contamination, do not use plastic).
-
2.
Mix well and store up to 1 month at room temperature.
Trifluoroacetic acid in LC‐MS‐grade water, 1% (5 ml)
-
1.
Add 4.5 ml LC‐MS‐grade water to a glass scintillation vial.
-
2.
Add 0.5 ml of 10% trifluoroacetic acid (see recipe).
-
3.
Mix well and store up to 2 weeks at room temperature.
COMMENTARY
Background Information
Mass spectrometry (MS) is an essential analytical technique in modern biological and physical science research, where it plays a principal role in the identification, characterization, and quantification of proteins (Aebersold and Mann, 2003, 2016; Biemann, 2014). Its popularity for protein analysis is due to multiple reasons. For instance, unlike gel electrophoresis, which separates molecules according to their relative molecular mass (M r) and can be affected by molecular shape, polarity, temperature, electric field, and solvent, MS is dependent on the intrinsic property of a protein (i.e., its mass). MS measures the mass‐to‐charge ratio of ions and is independent of many of the variables that affect size determination by electrophoresis. In addition, whereas antibody‐based detection methods (e.g., flow cytometry, immunocytochemistry, and western blotting) are dependent on antibody specificity, which is itself dependent on protein isoform, post‐translational modifications, and secondary/tertiary structure, MS can identify and characterize proteins despite these variables. Finally, MS requires no a priori knowledge of proteome composition to identity thousands of proteins from a single sample.
MS‐based methods for proteomics can generally be categorized as top‐down or bottom‐up. Overall, bottom‐up and top‐down approaches are often complementary, as they provide different levels of information and each has its own technical challenges and advantages. Top‐down methods preserve the stoichiometry among post‐translational modifications, proteolytic cleavage products, and products of splicing events, among other features (Kelleher et al., 1999). Despite these advantages, bottom‐up methods remain popular for peptide identification, quantification, identification of interaction partners, and site localization of post‐translational modifications (Abello et al., 2010; Frei et al., 2013; Gillet et al., 2016; Kho et al., 2004; Mayne et al., 2016; Rhee et al., 2013; Roux et al., 2012; Sabarth et al., 2002; Sprung et al., 2005; Wollscheid et al., 2009). The popularity of bottom‐up methods is due in part to their ease of implementation and to the availability of instrumentation and bioinformatic tools suitable for many different experimental workflows (Cox and Mann, 2008; Dorfer et al., 2014; Eng et al., 1994; Eng et al., 2013; Gluck et al., 2013; Kim and Pevzner, 2014; Perkins et al., 1999; Risk et al., 2013; Wenger and Coon, 2013).
Every sample preparation approach is biased and may not be suitable for all proteins. In other words, there is no universal bottom‐up sample preparation approach that allows the simultaneous identification and characterization of high‐ and low‐abundance, soluble and membrane‐bound, unmodified and modified, and secreted and localized proteins. For example, there are >200 known post‐translational modifications, and identifying proteins that carry these modifications requires specialized approaches to enrich specifically for the modification of interest in ways that typically exclude the detection of other protein classes. The protocols described here, for instance, are suited to the identification and quantification of peptides and proteins, but generally not to the identification of post‐translational modifications unless combined with enrichment methods specific to each modification type.
Importantly, all proteomics methods are biased towards identifying proteins that are most soluble or abundant under the experimental conditions used. For example, Alternate Protocol 1 will be biased towards soluble, abundant proteins and will provide limited coverage of plasma membrane proteins compared to specialized approaches that are specifically designed to solubilize and enrich for this class of hydrophobic and low‐abundance proteins (Kim et al., 2011; Shin et al., 2003; Wollscheid et al., 2009). Moreover, Alternate Protocol 1 is unlikely to provide suitable proteome coverage of tissue due to the high molecular heterogeneity, limited solubility of membrane and extracellular matrix proteins, and large dynamic range of protein concentrations present in such samples. When analyzing tissue or cells that are difficult to disrupt (e.g., muscle), the use of harsh detergents coupled with rigorous physical disruption methods is typically necessary to enhance coverage of the proteome. However, standard laboratory detergents (e.g., SDS, Triton™X‐100, Tween®20, CHAPS) are incompatible with MS. Although MS‐compatible surfactants are now commercially available (e.g., Invitrosol™, ProteaseMax™, RapiGest™ SF), they commonly need to be paired with chaotropic agents or organic solvents for maximum solubility of the proteome (Waas et al., 2014). The recent development of suspension trapping sample preparation (Zougman, Selby, & Banks, 2014), Strap or S‐Trap™, enables streamlined processing of samples that have been historically more challenging to work with because it achieves enzymatic digestion of proteins solubilized in SDS in a way that yields MS‐compatible peptides that do not require further cleanup.
It is expected that the facile preparation methods described in this article will be easy to implement, with high success rates in laboratories with little to no experience in proteomics. However, alternative approaches can be considered. For the Basic Protocol, alternatives to the S‐Trap™ method for streamlined protein immobilization, digestion, and cleanup include spin filter‐aided sample preparation (Manza, Stamer, Ham, Codreanu, & Liebler, 2005; Wiśniewski, Zougman, Nagaraj, & Mann, 2009), although another study found that S‐Trap™ provides superior performance (Ludwig, Schroll, & Hummon, 2018). The PreOmics iST Sample preparation kit and Thermo Scientific™ EasyPep™ are also designed for easy use, although we have not directly compared performance with the S‐Trap™. SP3 is another strategy that uses carboxylate‐coated magnetic beads to achieve these three steps, but it may be more challenging to implement for novice users (Moggridge, Sorensen, Morin, & Hughes, 2018; Sielaff et al., 2017; Waas et al., 2019).
Relevant to Alternate Protocol 2, the SP2 protocol will remove many common contaminants from peptide samples, including salts, detergents (e.g., SDS, Tween®20, Triton), and PEG (Fig. 9), which overall improves chromatography and reduces time and costs associated with having to clean LC‐MS systems after contaminants have been introduced. Overall, SP2 cleanup allows higher sample throughput, improves instrument performance, and results in less downtime due to system maintenance compared to other cleanup methods (e.g., C18; Waas et al., 2019).
Figure 9.
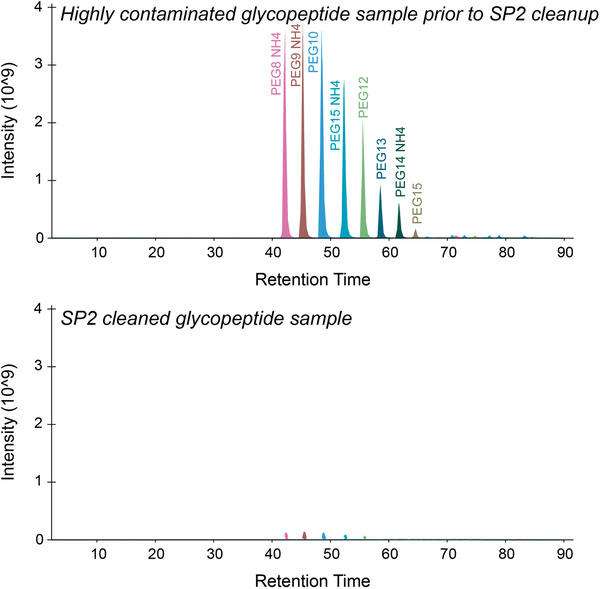
Base peak chromatogram for PEG contaminants before and after SP2 cleanup of a peptide sample. Before SP2 cleanup (Alternate Protocol 2), the sample showed high levels of PEG contaminants (top panel), which were greatly reduced after the cleanup (bottom panel).
Low‐abundance proteins, such as cytokines, play important roles in many physiological processes (Hirano et al., 1990; Kitagawa and Saito, 1988). Furthermore, some biological processes are regulated by subtle changes in abundance or post‐translational modifications of high‐ and low‐abundance proteins (Andelova et al., 2020; Cundiff et al., 2016; Karve and Cheema, 2011). Therefore, the ability to dig deep into the proteome and detect both low‐ and high‐abundance proteins is critical for many study designs. Several strategies are available for protein‐ and peptide‐level separation, which aim to spread the highly complex proteome across multiple fractions, creating multiple lower‐complexity samples from a single high‐complexity sample. Although LC‐MS methods typically include on‐line, low‐pH reverse‐phase chromatography to separate complex peptide mixtures, there is a limit to the amount of separation achieved with typical gradients and column dimensions. For this reason, several multidimensional fractionation methods have been developed to further separate protein and peptide mixtures before MS analysis, including methods that use size exclusion chromatography (SEC) or hydrophilic interaction chromatography (HILIC; Boersema, Divecha, Heck, & Mohammed, 2007; Opiteck, Ramirez, Jorgenson, & Moseley, 1998; Schirmer, Yates, & Gerace, 2003). Variations of these approaches can be performed using HPLC or spin‐column‐based tips and filter tubes. For the off‐line formats of SEC and HILIC, some form of desalting or cleanup is often required before MS analysis. In another off‐line approach that is the basis of Support Protocol 3, high‐pH reverse‐phase fractionation provides separation that is orthogonal to low‐pH reverse‐phased separation and yields peptides in a solution that is directly compatible with MS analysis without requiring any further cleanup or desalting.
Critical Parameters
Generally, the ability to detect multiple peptides per protein contributes to higher‐confidence data regarding the identification and abundance of the protein. As such, the degree to which the protein is digested into peptides is a major contributor to success—or lack thereof—in a bottom‐up proteomics experiment. This is not to say that identifications based on a single peptide sequence are universally unacceptable, but rather that for general proteomics workflows aimed to simultaneously discover and quantify many proteins in a single experiment, a higher number of peptides generated and detected by MS will yield more protein identifications and more quantifiable data. Consequently, a key principle in bottom‐up proteomics workflows is that the enzyme or chemical used for digestion must have access to the cleavage sites within the protein, and the solution must be at an appropriate pH for enzyme or chemical activity. The three‐dimensional conformation of a protein in solution will affect the accessibility of cleavage sites. Therefore, experimental conditions that enhance protein solubilization and unfolding—and thus promote enzymatic activity—will overall lead to maximum efficiency of cleavage (at least 85% of peptides with zero missed cleavages), yield more peptides, and result in better‐quality data. For this reason, users may consider sample‐specific optimization of the steps described in the Basic Protocol and Alternative Protocol 1 to determine which conditions lead to the greatest amount of sequence coverage and the most reproducible detection of peptides of interest. An overview of common optimization steps is provided in Figure 10.
Figure 10.
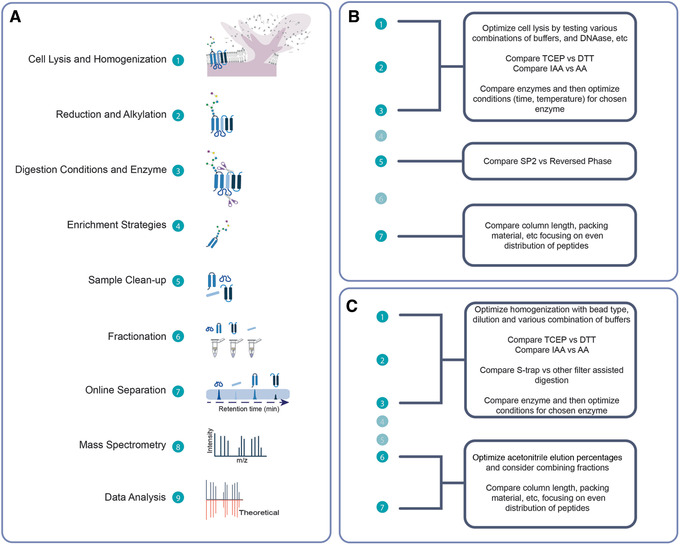
Bottom‐up workflow method optimization. (A) Various steps within a bottom‐up proteomics workflow that can be optimized according to sample type and experimental goals. (B) Example of sample preparation method optimization using Alternate Protocol 1 for in‐solution processing of cells. (C) Example of sample preparation method optimization using Basic Protocol for S‐trap™ processing of tissue.
Reduction and alkylation of thiols to cleave disulfide bonds and prevent their reformation is a strategy that commonly leads to higher protein sequence coverage. Reduction of disulfide bonds can be achieved with either dithiothreitol (DTT) or TCEP. Compared to DTT, which works optimally at pH 7‐9, TCEP is typically more efficient, works in a wider pH range (pH 2‐11), and has no offensive odor. Therefore, our protocols use TCEP. Iodoacetamide (IAA) and acrylamide are both popular choices for alkylating reagents. Although we have found them to be interchangeable for the in‐solution processing of lower‐complexity mixtures, acrylamide provides a slight advantage for reducing missed cleavages and increasing the number of protein identifications for very complex mixtures when using the Basic Protocol (Fig. 11). Importantly, when reduction and alkylation are performed, the alkylation results in modification of the cysteine residue (carbamidomethyl for IAA, propionamide for acrylamide), and this modification must be considered when performing MS data analysis. The concentrations and incubation times for DTT, TCEP, acrylamide, and IAA may vary depending on the amount of protein in the sample. In our experience, the concentrations and reduction/alkylation times provided in these protocols work well for the sample types we use most commonly (e.g., B lymphocytes, HeLa cells, human heart tissue), but concentrations of 5‐10 mM for DTT and TCEP and 10‐50 mM for IAA may also be tested. Incubation times may range from 5 min to 1 hr for the reduction step and from 10 min to 1 hr for the alkylation step. Similarly, reduction can be performed at room temperature, 37°C, or 56°C.
Figure 11.
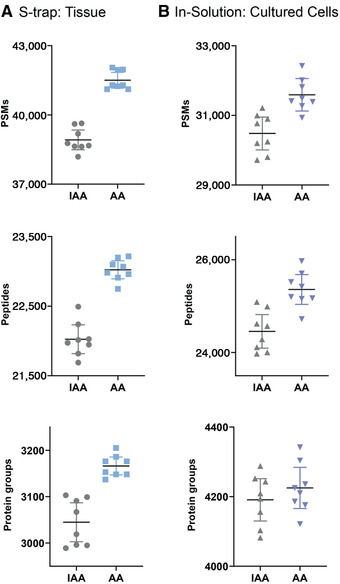
Numbers of protein groups, peptides, and peptide spectrum matches (PSMs) obtained when using iodoacetamide (IAA) or acrylamide (AA) as alkylating reagents. (A) Results of IAA and then AA performed with Basic Protocol S‐trap™ processing of human heart tissue. (B) Results of IAA and AA performed with Alternate Protocol 1 in solution processing of RPMI 1788 cell lysis. Shown are data for 8 technical replicates. Line represents the mean and error bars the 95% confidence interval.
Another key principle is that the enzyme or chemical selected will determine which peptide sequences are available for analysis. Only a fraction of the peptides resulting from digestion will be of a suitable mass‐to‐charge ratio (m/z) for MS/MS analysis. Therefore, any prerequisites for coverage of a specific site or region within a selected protein or proteins must be considered when planning the experimental design to ensure that the appropriate digestion enzyme or chemical is used.
For Alternate Protocol 1, one of the most important factors that can affect the efficiency of a trypsin digestion is pH. Users should be aware that some trypsin products come as lyophilized powder and others come in solution format. Typically, trypsin is dissolved in acidic conditions. Another important consideration is the solubility of the protein and the access of trypsin to the digestion sites within the protein sequence. Standard laboratory detergents (e.g., SDS, CHAPS, Triton, Tween® 20) can suppress trypsin activity. Therefore, for in‐solution processing, the MS‐compatible surfactant Invitrosol (Invitrogen) and acetonitrile are added to increase the effectiveness of the enzymatic digestion (Waas et al., 2014). Alternative MS‐compatible surfactants include Rapigest (Waters), PPS (ProteinDiscovery), MaSDeS (Chang et al., 2015), and Azo (Brown et al., 2019).
For the Basic Protocol, when using tissue as a starting material, the choice of homogenization and subsequent sonication methods are critical for success. We discourage the use of a mortar and pestle, which results in incomplete homogenization. Multisample benchtop homogenizers (e.g., Barocycler, Pressure BioSciences; Bead Beater, MP Biomedical; or BeadBlaster, Benchmark) are available at a range of prices and capabilities, and in our experience, the bead‐based homogenization method works well for disrupting human heart tissue. Equally important, the sonication step is critical for lysing the cells and shearing DNA. We discourage the use of probe sonicators, as using the same item to contact multiple samples can result in cross‐contamination. Thus, we use a block sonicator, which allows individual samples to be sonicated within enclosed microcentrifuge tubes. If homogenization or sonication are not optimized, the S‐trap™ may become clogged. Much as in Alternate Protocol 1, it is important to optimize digestion time and temperature for the sample and enzyme choice in the Basic Protocol by inspecting the sequence coverage and rate of missed cleavages in the search results. For example, using that protocol, we find that a 2‐hr trypsin/LysC digestion works well for heart tissue, but other cell types (e.g., B lymphocytes) are well served by digestion with trypsin for only 1 hr at 47°C. Finally, the maximum volume that can be loaded at one time onto the S‐trap™ micro spin column is 300 µl (25 µl sample + 275 µl digestion buffer and other components). Therefore, although it is possible to begin with protein in a volume >25 µl, this would require using multiple 300‐µl aliquots and performing multiple centrifugation steps to process the entire sample. S‐trap™ columns, however, are available in various sizes—e.g., micro (≤100 µg protein), mini (100‐300 µg protein), and midi (≥300 µg protein)—and if it is desirable to process a larger amount of protein, the Basic Protocol can be adjusted according to the manufacturer's guidelines to facilitate the use of mini or midi S‐trap™ columns.
The magnetic particles used in Alternate Protocol 2 settle during storage and form a mass that can be difficult to dissociate. A homogeneous mixture can be obtained through a combination of shaking (e.g., placing the particle stock on an orbital shaker for 30 min) and vortexing. It is advisable to prepare a stock of particles before sample cleanup and store them at 4°C, for up to 6 months. In our experience, the Sera‐Mag SpeedBeads as well as the original Sera‐Mag versions perform equivalently in terms of recovery and cleaning capacity. Although the SpeedBeads settle within 15 s when placed on a magnetic rack, either type of particle is fully settled against the magnet after 1 min. If using a low‐retention 1.5‐ml microcentrifuge tube, the total volume of sample + diluent + particle can be 75 µl. Maintaining the total volume at ≤50 µl facilitates sample handling, as the addition of sufficient acetonitrile to achieve 95% can be performed in one step. Alternatively, SP2 cleanup with larger volumes can be performed in a 15‐ml tube using a 15‐ml‐tube magnetic rack.
For Support Protocols 1 and 2, accurate pipetting is essential. The user should follow best practices and check the accuracy of their pipets and skill set via weight determination. Standards and blanks should be performed in duplicate, and samples should be processed with two different dilutions in duplicate.
For Support Protocol 3, the percentage of acetonitrile can be changed to spread the distribution of peptides across fractions. Fractions may be combined to reduce MS instrument time needed to analyze the sample if their elution profile indicates they are complementary (e.g., fraction no. 1 would contain mostly hydrophilic peptides, whereas later fractions may not and would not elute at the same time with reverse phase chromatography).
The peptide samples resulting from the Basic Protocol and Alternate Protocol 1 are suitable for MS coupled to either liquid chromatography (LC) or matrix‐assisted laser desorption/ionization (MALDI) interfaces. Based on peptide quantification (measured or estimated), the samples can be used directly or diluted to an appropriate concentration before placing them in the sample vial for MS analysis. If using LC‐MS, we typically dilute samples in aqueous mobile phase (or 2% acetonitrile in aqueous 0.1% FA) and inject ≤1 μg per replicate. However, the appropriate peptide concentration will depend on many factors, including instrumentation, method, column, sample complexity, and experimental goals. For the Basic Protocol and Alternate Protocol 1, the performance of the method will ultimately be assessed based on MS results. Although we report results that are typical of our experience, MS results (e.g., number of peptides and proteins identified) will be directly affected by the condition and type of the column used in the case of LC‐MS analyses, type and capabilities of the MS instrumentation, data acquisition settings, and data analysis details, as detailed previously (Dave et al., 2011; Han et al., 2008; Zhang et al., 2014). Therefore, it is possible that another user could apply the protocols expertly to the same sample types as described here but identify a different number of proteins because the MS instrument is configured slightly differently. Ultimately, results will depend on performance of the instrumentation and software used to analyze the data. Consequently, users are strongly encouraged to consult with the MS instrumentation operator before beginning an experiment, to ensure the experimental design is appropriate and that the peptide amount and sample volume are adequate for the instrumentation that will be used for analysis.
Troubleshooting
In Table 2, we provide a list of common problems associated with each of the protocols described, and their solutions.
Table 2.
Summary of Common Problems Encountered and Their Solutions
| Problem | Solution |
|---|---|
| Basic Protocol | |
| Sample clogged on S‐Trap™ | Centrifuge the sample thoroughly before pipetting, and be sure to shear DNA with sonication or with the addition of MgCl2 and Benzonase. |
| Contaminants in MS data, such as polyethylene glycol or lipids, or high backpressure during sample loading | Perform additional wash steps on S‐trap™ at step 14. For removal of lipids, add three washes of 50:50 (v/v) methanol/chloroform before standard S‐trap™ buffer washes. |
| Percentage of peptides identified with at least one missed cleavage is >15% | Increase the concentration of trypsin if digesting a large amount of protein. |
| Alternate Protocol 1 | |
| Sample contaminated with detergents or polymers, as evidenced by poor LCMS data | Use detergent‐free glassware; be sure to perform SP2 cleanup, as C18 cleanup protocols do not remove PEG. |
| Large number of missed cleavages in database search results | Check that the digestion solution pH is ∼8 after adding trypsin to the protein, and adjust with small amounts of concentrated acid or base as necessary. |
| Alternate Protocol 2 | |
| Low peptide recovery |
During particle preparation, let particles warm to room temperature under constant shaking on an orbital shaker for ≥30 min. Sufficiently suspend particles by shaking and vortexing to yield a homogenous suspension. Particle preparation should be made fresh every 6 months. Ensure that math was performed correctly during peptide quantification assay. Alternatively, repeat peptide quantification assay. To maximize recovery, use 0.2‐0.4 µl of 50 µg/µl particle suspension per microgram of peptide to be cleaned (i.e., a particle to peptide ratio of 10:1‐20:1). |
| PEG or other contaminants in MS data |
Use detergent‐free glassware. Make fresh elution solution. |
| Support Protocols 1 and 2 | |
| No protein in sample, or low protein concentration after lysis | Use ionic detergent compatibility reagent included with the Pierce kit. |
| Variable numbers in duplicate values | Be sure to allow the reagents to come to room temperature. Consider checking pipet technique. |
| No protein in sample, or low protein concentration after tissue lysis | Be sure to use ionic detergent compatibility reagent included in the Pierce kit if there are detergents in the sample. |
| Too much protein for standard range | Dilute sample further with lysis buffer before attempting protein quantification again. |
| Support Protocol 3 | |
| Variable peptide amounts in fractions | Consider changing acetonitrile percentages to optimize spread of peptides. |
| No peptides in any fractions | Check the saved flowthrough and wash tubes for non‐retained peptides. |
Understanding Results
Examples of data we obtain from application of these methods are provided within each protocol. In general, sample‐specific optimization of each step of the workflows illustrated in Figures 1 and 10 will result in increased digestion efficiency, sequence coverage, and proteome coverage. Examining the total number of proteins identified, number of peptides per protein, sequence coverage, enzyme specificity (e.g., full vs. semi‐tryptic peptides), and missed cleavages can inform whether the method requires optimization. High‐pH fractionation should increase the number of identified peptides compared to those from a non‐fractionated sample.
Time Considerations
The protocols described here can be completed as follows.
Basic Protocol: One person can typically prepare a maximum of 18 samples in 8 hr, assuming the centrifuge used can hold this number of microcentrifuge tubes simultaneously.
Alternate Protocol 1: One person can typically prepare a maximum of 18 samples in 12 hr, assuming the centrifuge used can hold this number of microcentrifuge tubes simultaneously.
Alternate Protocol 2: One person can prepare 8‐12 samples in 30 min, assuming the magnetic rack can hold this number of microcentrifuge tubes simultaneously.
Support Protocol 1: One person can typically prepare 6‐8 samples in 30 min and 12 samples in 1 hr.
Support Protocol 2: One person can typically prepare 6‐8 samples in 30 min and 12 samples in 1 hr.
Support Protocol 3: One person can typically prepare 3‐6 samples in 3 hr.
Conflicts of Interest Statement
R.L.G. is on the advisory board of ProtiFi but receives no compensation of any kind for this role.
Author Contributions
Melinda Wojtkiewicz: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing‐original draft, Writing‐review & editing, Linda Berg Luecke: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing‐original draft, Writing‐review & editing, Maia I. Kelly: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing‐original draft, Writing‐review & editing, Rebekah L. Gundry: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing‐review & editing.
Acknowledgments
This work was supported by the National Institutes of Health (R01‐HL134010, R01‐HL126785 to R.L.G.; TL1TR001437, UL1TR001436 to L.B.L), American Heart Association (20PRE35200049 to L.B.L.), and Juvenile Diabetes Research Foundation (2‐SRA‐2019‐829‐S‐B to R.L.G.). L.B.L. is a member of the MCW‐MSTP, which is partially supported by a T32 grant from the National Institute of General Medical Sciences (GM080202). Funding sources were not involved in study design, data collection, interpretation, analysis, or publication.
Wojtkiewicz, M. , Berg Luecke, L. , Kelly, M. I. , & Gundry, R. L. (2021). Facile preparation of peptides for mass spectrometry analysis in bottom‐up proteomics workflows. Current Protocols, 1, e85. doi: 10.1002/cpz1.85
Data Availability Statement
The data that support the findings will be available in MassIVE (https://massive.ucsd.edu; MSV000086814) following an embargo of 1 year from the date of publication to allow for commercialization of research findings.
Literature Cited
- Alagesan, K. , Khilji, S. K. , & Kolarich, D. (2017). It is all about the solvent: On the importance of the mobile phase for ZIC‐HILIC glycopeptide enrichment. Analytical and Bioanalytical Chemistry, 409, 529–538. doi: 10.1007/s00216-016-0051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert, A. J. (1990). Hydrophilic‐interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. Journal of Chromatography, 499, 177–196. doi: 10.1016/S0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- Bache, N. , Geyer, P. E. , Bekker‐Jensen, D. B. , Hoerning, O. , Falkenby, L. , Treit, P. V. , … Mann, M. (2018). A novel LC system embeds analytes in pre‐formed gradients for rapid, ultra‐robust proteomics. Molecular & Cellular Proteomics, 17, 2284–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersema, P. J. , Divecha, N. , Heck, A. J. R. , & Mohammed, S. (2007). Evaluation and optimization of ZIC‐HILIC‐RP as an alternative MudPIT strategy. Journal of Proteome Research, 6, 937–946. doi: 10.1021/pr060589m. [DOI] [PubMed] [Google Scholar]
- Fu, Q. , Kowalski, M. P. , Mastali, M. , Parker, S. J. , Sobhani, K. , van den Broek, I. , … Van Eyk, J. E. (2018). Highly reproducible automated proteomics sample preparation workflow for quantitative mass spectrometry. Journal of Proteome Research, 17, 420–428. doi: 10.1021/acs.jproteome.7b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher, N. L. , Lin, H. Y. , Valaskovic, G. A. , Aaserud, D. J. , Fridriksson, E. K. , & McLafferty, F. W. (1999). Top down versus bottom up protein characterization by tandem high‐resolution mass spectrometry. Journal of the American Chemical Society, 121, 806–812. [Google Scholar]
- Kim, Y. , Elschenbroich, S. , Sharma, P. , Sepiashvili, L. , Gramolini, A. O. , & Kislinger, T. (2011). Use of colloidal silica‐beads for the isolation of cell‐surface proteins for mass spectrometry‐based proteomics. Methods in Molecular Biology, 748, 227–241. doi: 10.1007/978-1-61779-139-0_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig, K. R. , Schroll, M. M. , & Hummon, A. B. (2018). Comparison of in‐solution, FASP, and S‐Trap based digestion methods for bottom‐up proteomic studies. Journal of Proteome Research, 17, 2480–2490. doi: 10.1021/acs.jproteome.8b00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manza, L. L. , Stamer, S. L. , Ham, A.‐J. L. , Codreanu, S. G. , & Liebler, D. C. (2005). Sample preparation and digestion for proteomic analyses using spin filters. Proteomics, 5, 1742–1745. doi: 10.1002/pmic.200401063. [DOI] [PubMed] [Google Scholar]
- Moggridge, S. , Sorensen, P. H. , Morin, G. B. , & Hughes, C. S. (2018). Extending the compatibility of the SP3 paramagnetic bead processing approach for proteomics. Journal of Proteome Research, 17, 1730–1740. doi: 10.1021/acs.jproteome.7b00913. [DOI] [PubMed] [Google Scholar]
- Opiteck, G. J. , Ramirez, S. M. , Jorgenson, J. W. , & Moseley, M. A. (1998). Comprehensive two‐dimensional high‐performance liquid chromatography for the isolation of overexpressed proteins and proteome mapping. Analytical Biochemistry, 258, 349–361. doi: 10.1006/abio.1998.2588. [DOI] [PubMed] [Google Scholar]
- Schirmer, E. C. , Yates, J. R. , & Gerace, L. (2003). MudPIT: A powerful proteomics tool for discovery. Discovery Medicine, 3, 38–39. [PubMed] [Google Scholar]
- Shin, B. K. , Wang, H. , Yim, A. M. , Le Naour, F. , Brichory, F. , Jang, J. H. , … Hanash, S. M. (2003). Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. Journal of Biological Chemistry, 278, 7607–7616. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- Sielaff, M. , Kuharev, J. , Bohn, T. , Hahlbrock, J. , Bopp, T. , Tenzer, S. , & Distler, U. (2017). Evaluation of FASP, SP3, and iST protocols for proteomic sample preparation in the low microgram range. Journal of Proteome Research, 16, 4060–4072. doi: 10.1021/acs.jproteome.7b00433. [DOI] [PubMed] [Google Scholar]
- Waas, M. , Bhattacharya, S. , Chuppa, S. , Wu, X. , Jensen, D. R. , Omasits, U. , … Gundry, R. L. (2014). Combine and conquer: Surfactants, solvents, and chaotropes for robust mass spectrometry based analyses of membrane proteins. Analytical Chemistry, 86, 1551–1559. doi: 10.1021/ac403185a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waas, M. , Pereckas, M. , Jones Lipinski, R. A. , Ashwood, C. , & Gundry, R. L. (2019). SP2: Rapid and automatable contaminant removal from peptide samples for proteomic analyses. Journal of Proteome Research, 18, 1644–1656. doi: 10.1021/acs.jproteome.8b00916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski, J. R. , Zougman, A. , Nagaraj, N. , & Mann, M. (2009). Universal sample preparation method for proteome analysis. Nature Methods, 6, 359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Wollscheid, B. , Bausch‐Fluck, D. , Henderson, C. , O'Brien, R. , Bibel, M. , Schiess, R. , … Watts, J. D. (2009). Mass‐spectrometric identification and relative quantification of N‐linked cell surface glycoproteins. Nature Biotechnology, 27, 378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, J. B. , Keppel, T. R. , Waas, M. , Salmon, A. E. , Buchberger, A. R. , Skumatz, C. M. B. , … Kassem, I. S. (2020). Quantitative proteomic analysis of aqueous humor after rabbit lensectomy reveals differences in coagulation and immunomodulatory proteins. Molecular Omics, 16, 126–137. doi: 10.1039/C9MO00169G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Z. , & Desaire, H. (2015). Carbohydrates on proteins: Site‐specific glycosylation analysis by mass spectrometry. Annual Review of Analytical Chemistry, 8, 463–483. doi: 10.1146/annurev-anchem-071114-040240. [DOI] [PubMed] [Google Scholar]
- Zougman, A. , Selby, P. J. , & Banks, R. E. (2014). Suspension trapping (STrap) sample preparation method for bottom‐up proteomics analysis. Proteomics, 14, 1006–1000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings will be available in MassIVE (https://massive.ucsd.edu; MSV000086814) following an embargo of 1 year from the date of publication to allow for commercialization of research findings.