

Abstract
HIV-1 CA protein has gained remarkable attention as a promising therapeutic target for the development of new antivirals, due to its pivotal roles in HIV-1 replication (structural and regulatory). Herein, we report the design and synthesis of three series of benzenesulfonamide-containing phenylalanine derivatives obtained by further structural modifications of PF-74 to aid in the discovery of more potent and drug-like HIV-1 CA inhibitors. Structure-activity relationship studies of these compounds led to the identification of new phenylalanine derivatives with a piperazinone moiety, represented by compound 11l, which exhibited anti-HIV-1NL4–3 activity 5.78-fold better than PF-74. Interestingly, 11l also showed anti-HIV-2ROD activity (EC50 = 31 nM), with almost 120 times increased potency over PF-74. However, due to the higher significance of HIV-1 as compared to HIV-2 for the human population, this manuscript focused on the mechanism of action of our compounds in the context of HIV-1. SPR studies on representative compounds confirmed CA as the binding target. The action stage determination assay demonstrated that these inhibitors exhibited antiviral activities with a dual-stage inhibition profile. The early-stage inhibitory activity of compound 11l was 6.25 times more potent as compared to PF-74, but appears to work via accelerating capsid core assembly rather than stabilization. However, the mechanism by which they exert their antiviral activity in the late-stage appears to be the same as PF-74 with less infectious HIV-1 virions are produced in their presence as judged p24 content studies. MD simulations provided the key rationale for the promising antiviral potency of 11l. Additionally, 11l exhibited modest increase in HLM and human plasma metabolic stabilities as compared to PF-74, as well as moderately improved pharmacokinetic profile, favorable oral bioavailability, and no acute toxicity. These studies provide insights and serves as a starting point for subsequent medicinal chemistry efforts in optimizing these promising HIV inhibitors.
Keywords: HIV-1, CA protein, phenylalanine derivatives, assembly, metabolic stability
Graphical Abstract
1. INTRODUCTION
Acquired immunodeficiency syndrome (AIDS) remains a serious threat to public health worldwide, of which the primary etiological agent is the human immunodeficiency virus type 1 (HIV-1).1 Combination antiretroviral therapy (cART) has been the major advancement in clinical treatment over the past decades and has transformed HIV/AIDS into a manageable chronic disease.2 Nevertheless, the long-term application of cART regimens has been plagued by several risks, such as the emergence of resistant HIV-1 mutants, drug-drug interactions, serious toxicity, and adverse side effects.3 The current cART is also limited by complicated lifestyles, treatment fatigue, socioeconomic conditions, etc. Antivirals with novel mechanism of action against underexploited HIV-1 targets are highly desirable to complement and supplant existing cART combinations, enabling persistent efficacy, especially against resistant mutants within current AIDS therapy. One such novel target is the HIV-1 capsid (CA) protein.4
HIV-1 CA assembles into an asymmetric fullerene-shaped cone within the virion, formed by 1000–1500 copies of the CA monomeric protein. This cone-shaped lattice consists of approximately 250 hexamers and 12 pentamers.5,6 The CA protomer can be divided into an N-terminal domain (NTD, residues 1 to 145) and a C-terminal domain (CTD, residues 150 to 231), connected by a flexible linker.7 The HIV-1 CA protein is crucial in both early- and late-stage events of the viral replication cycle, which are essential for virus infectivity.8,9
It has been reported that CA is able to interact with several host cell factors including CypA (cyclophilin A), NUP153 (nucleoporin 153 kDa), CPSF6 (cleavage and polyadenylation specific factor 6), TNPO3 (transportin-3), and RanBP2 (also known as NUP358), to name a few.5 These interactions are crucial for infectivity by mediating the entry of the viral pre-integration complex into the nucleus and facilitate viral escape from innate immune surveillance.10,11 Mutational analysis has revealed that abnormal morphologies or altered stability of the HIV-1 CA are detrimental for viral infectivity.12–15 Therefore, HIV-1 CA has been considered an attractive but underexploited target for clinical drug research.16
A growing number of structurally diverse compounds and peptides targeting the HIV-1 CA have been identified, such as CAI, CAP-1, BI-2, PF-74, GS-CA1 and GS-6207 (Figure 1),17–27 which can inhibit viral replication at early-stage events, late-stage events, or at both stages. As the first small molecule co-crystallized with the HIV-1 CA protein, PF-74 has received significant attention and has been widely studied.22 However, the relatively moderate anti-HIV-1 activity, the poor drug-like profile, and especially the low metabolic stability,28 emphasize the urgency and necessity for further optimization of PF-74. Most recently, GS-CA1 and GS-6207 (an analog of GS-CA1) (Figure 1) with picomolar antiviral activities have been described by Gilead Sciences.25,26 Analyses of their structures showed that both compounds contain a polyphenyl core and a linker region similar to PF-74 (Figure 1, in magenta). Both compounds display greater potency than currently approved antivirals in vitro and have the potential for long-acting inhibitors.25,26 At present, GS-6207 is being administered orally in phase II clinical trials.29 This verifies that the modification of PF-74 is a very promising direction and strategy to find potent CA inhibitors in order to expand current treatment options. Therefore, in our work, we have also selected PF-74 for further structural optimization due to its established mode of action and the availability of multiple crystal structures of its complex with HIV-1 CA protein.
Figure 1.
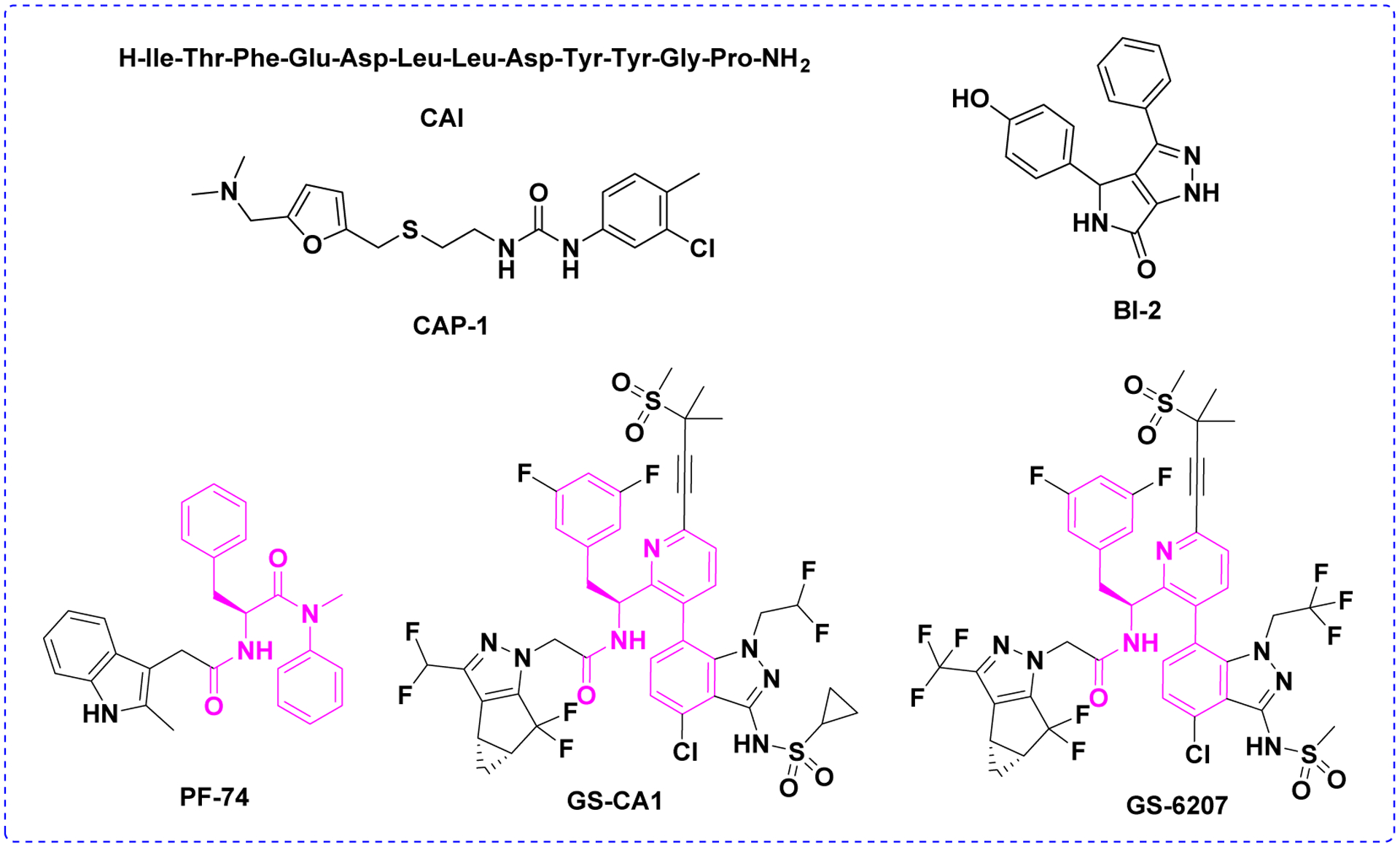
Chemical structures of reported representative HIV-1 CA inhibitors. Polyphenyl core moieties in structures of PF74, GS-CA1 and GS-6207 compounds were shown in magenta.
Analysis of crystal structures of PF-74 in complex with native CA revealed that the indole moiety of PF-74 only occupies a small portion of the interprotomer binding pocket (Figure 2), forming a hydrogen-bond with Gln63 of the NTD (of one protomer), also interacting with Arg173 within the CTD of the adjacent protomer to form a cation-pi interaction. The availability of space in this critical interprotomer pocket allows for further modification of PF-74 to take advantage of additional contacts to improve potency in subsequent analogues. Introducing a methoxy at the para position of the aniline has been shown to be beneficial for antiviral activities,23,30 therefore, in this study, we maintained the methoxy-bearing (in cyan) aniline substituent. Further, we explored the indole moiety with diversely substituted benzenesulfonamide (in blue), aiming to form additional interactions (ideally forming additional hydrogen-bonds) with surrounding key residues (Figure 3) to enhance binding affinity and drug-like properties. Therefore, in the process of scaffold evolution, we initially replaced the methylindole with a benzenesulfonamide group (Series I) and then cyclized it to obtain a benzothiadiazine ring (Series II). Finally, we used bioisosterism and scaffold hopping strategies to obtain a 4-(phenylsulfonyl) piperazinone (Series III).
Figure 2.
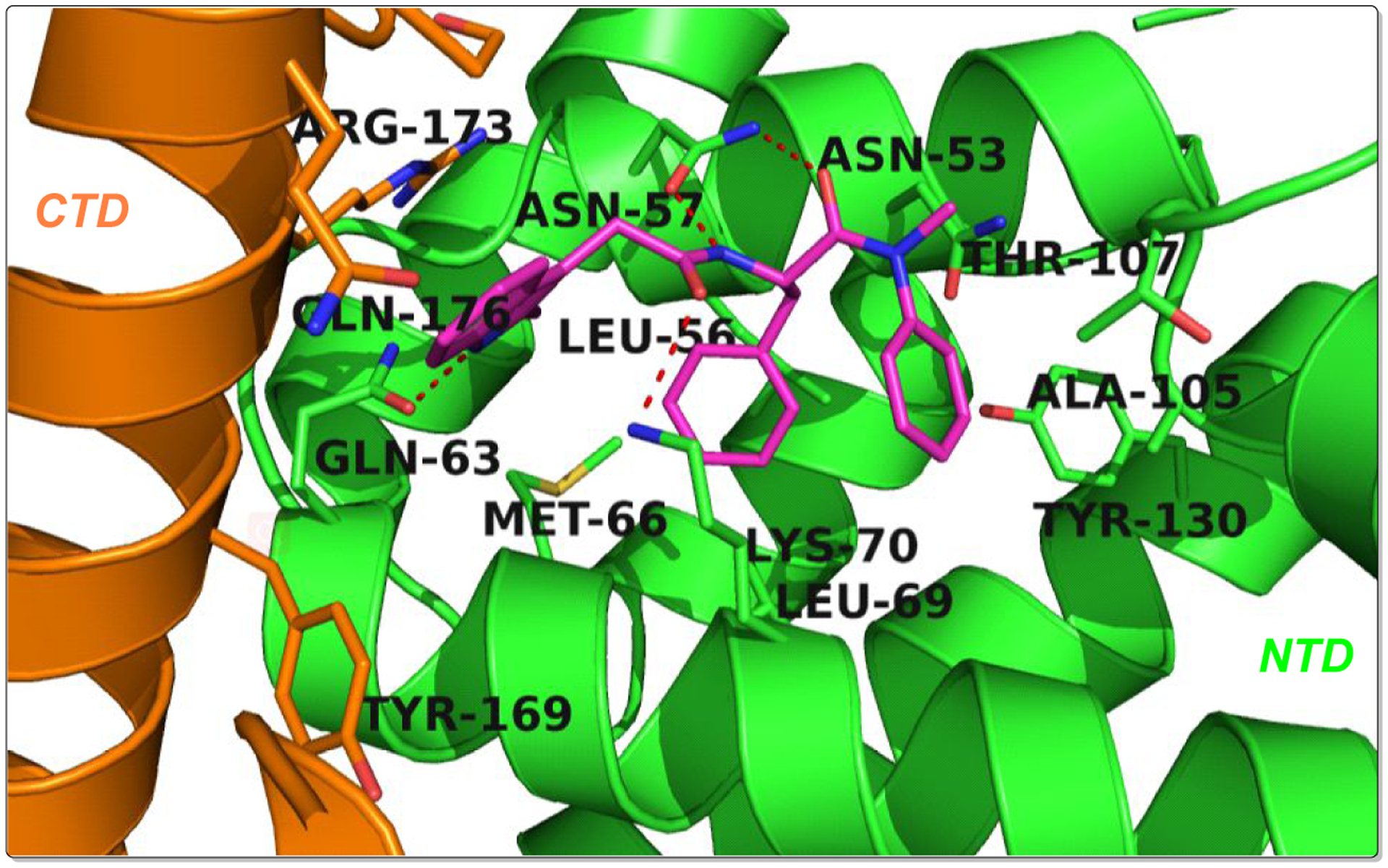
The illustration of the co-crystal structure of PF-74/CA hexamer (PF-74 in magenta, PDB ID: 5HGL) was generated using PyMOL (www.pymol.org). Red dashed lines indicate hydrogen-bond interactions.
Figure 3.
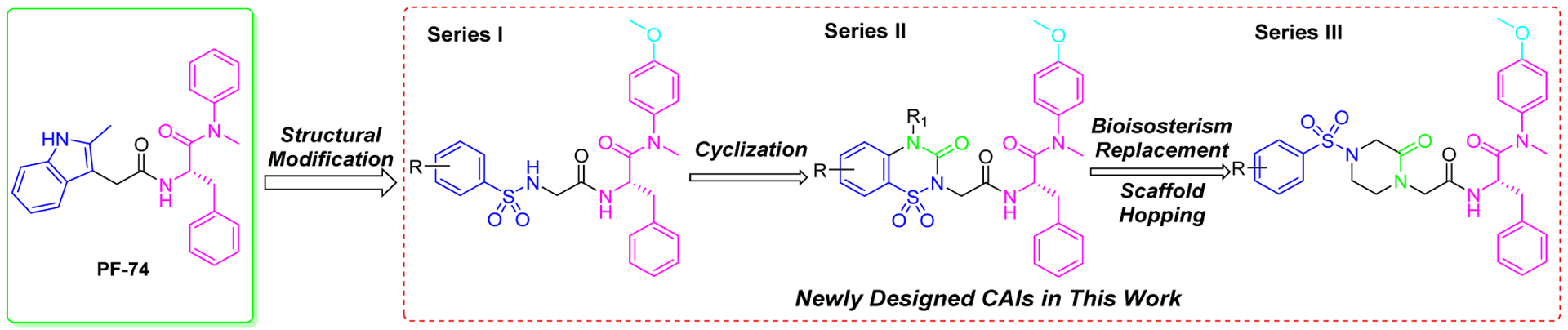
Design pipeline of novel phenylalanine derivatives as HIV-1 CA inhibitors.
Herein we report the design, synthesis, and biological evaluation of three series of phenylalanine derivatives with benzenesulfonamide terminal moieties as HIV-1 CA inhibitors. All synthesized compounds were screened for their antiviral activity in TZM-bl cells and investigated for preliminary structure-activity relationships (SARs). Also, surface plasmon resonance (SPR) direct interaction assays, action stage determination, p24 quantification, CA assembly, molecular dynamics (MD) simulation, metabolic stability, pharmacokinetic profile, and acute toxicity studies were also performed to support the pharmacological characterization of newly synthesized compounds.
2. CHEMISTRY
Starting from commercially available (tert-butoxycarbonyl)-L-phenylalanine (1), the target compounds in Series I were prepared via a concise and well-established synthetic route as outlined in Scheme 1. Treating of 1 with 4-methoxy-N-methylaniline and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBop) in N,N-diisopropylethylamine (DIEA) and dichloromethane to give 2, followed by removal of tert-butyloxycarbonyl (Boc) protection to afford the free amine 3. Acylation of 3 by reacting with Boc-glycine in dichloromethane solution leads to intermediate 4, followed by removal of Boc protection to produce the free amine 5. Finally, 5 reacted with corresponding substituted benzenesulfonyl chloride by acylation reaction to obtain the desired compounds 6a-6n. The other target compounds 6o-6q were obtained by a hydrogenation reduction of the nitro group of 6l-6n.
Scheme 1. Preparation of 6a-6qa.
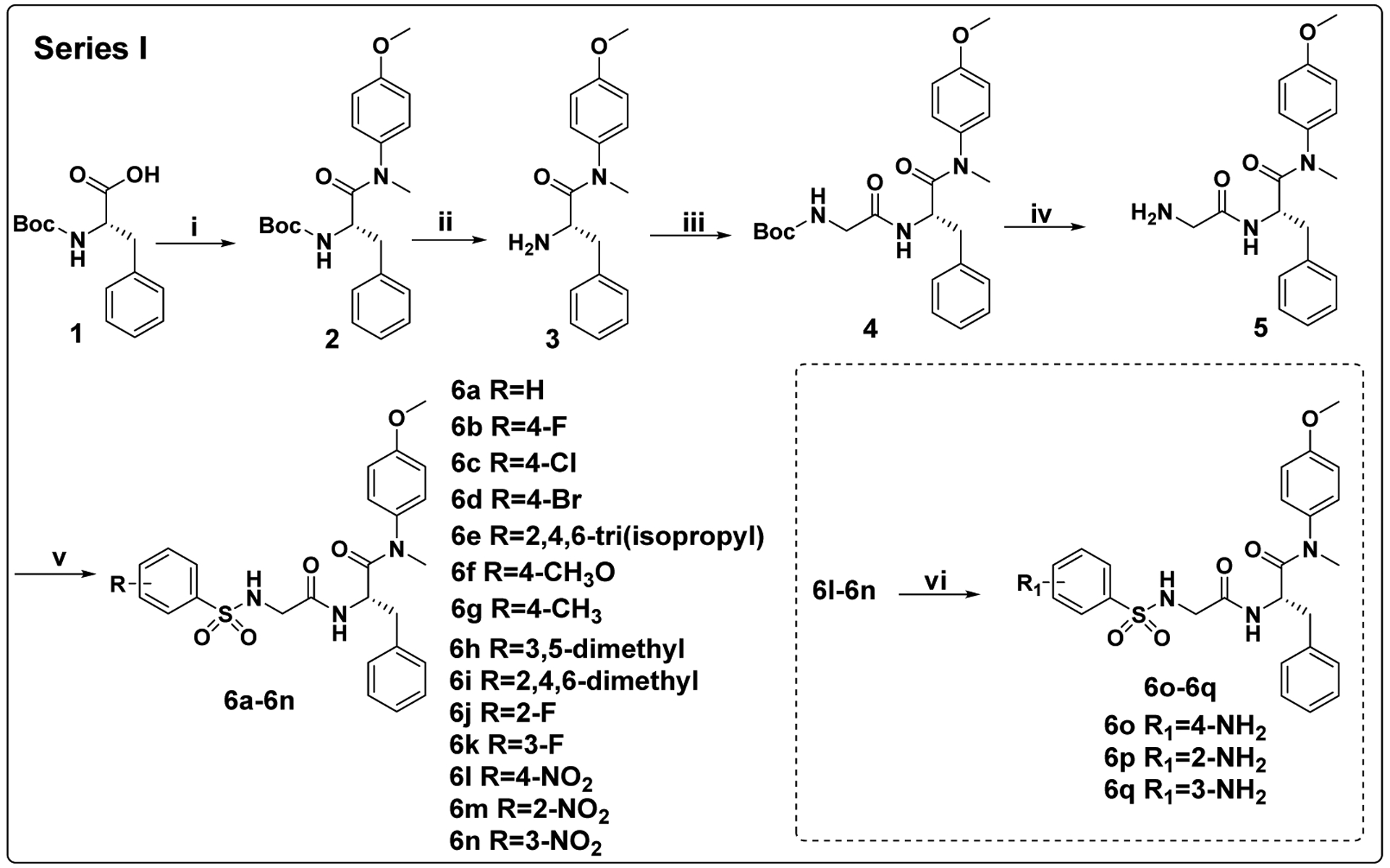
aReagents and conditions: (i) 4-methoxy-N-methylaniline, PyBop, DIEA, dichloromethane, 0°C to r.t.; (ii) trifluoroacetic acid, dichloromethane, r.t.; (iii) Boc-glycine, HATU, DIEA, dichloromethane, 0°C to r.t.; (iv) trifluoroacetic acid, dichloromethane, r.t.; (v) corresponding substituted benzenesulfonyl chloride, TEA, dichloromethane, 0°C to r.t.; (vi) H2, 10% Pd●C, dichloromethane, methanol, r.t..
As shown in Scheme 2, the target compounds in Series II were initiated from intermediate 3. The acylation of 3 with bromoacetic acid in dichloromethane solution leads to the key intermediate 7. Finally, 7 was reacted with corresponding 7-substituted 2H-benzo[e][1,2,4]thiadiazin-3(4H)-one 1,1-dioxide (R1=H, CH3, CH3O; methyl and methoxy are electron-donating groups) under the presented condition by nucleophilic substitution (SN2) reaction to obtain mono-substituted derivatives 8a-8c. However, 7 was reacted with corresponding 7-substituted 2H-benzo[e][1,2,4]thiadiazin-3(4H)-one 1,1-dioxide (R2=F, Cl, Br; all are electron-withdrawing groups) to get di-substituted derivatives 8d-8f, this is because the electron-withdrawing group (R2=halogen) at the 7-position increases the acidity of the NH at the 4-position, so that hydrogen can be captured by the base, and the exposed nitrogen atom participates in the second SN2 reaction.
Scheme 2. Preparation of 8a-8fa.
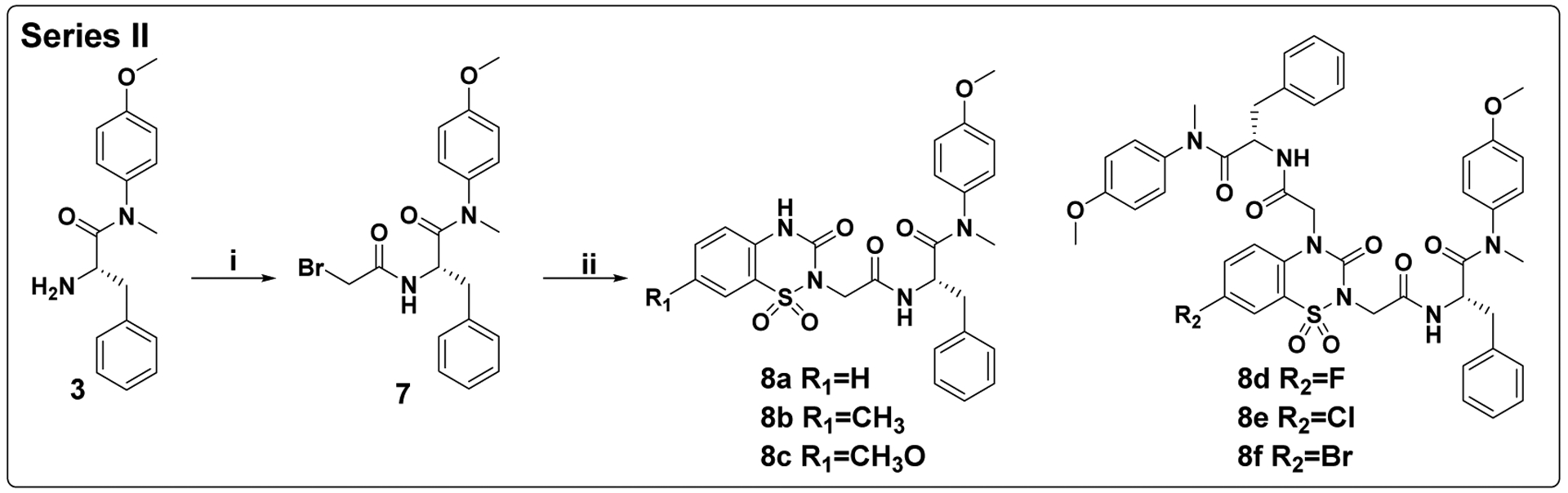
aReagents and conditions: (i) bromoacetic acid, HATU, DIEA, dichloromethane, 0°C to r.t.; (ii) corresponding 7-substituted 2H-benzo[e][1,2,4]thiadiazin-3(4H)-one 1,1-dioxide, Na2CO3, DMF, 40°C.
The target compounds in Series III were prepared via a concise synthetic route, as outlined in Scheme 3. Intermediate 7 was selected as the starting material and reacted with 1-Boc-3-oxopiperazine by nucleophilic substitution (SN2) reaction to produce intermediate 9, followed by removing the Boc protection to afford the free amine 10. Finally, 10 reacted with corresponding substituted benzenesulfonyl chloride by acylation reaction to obtain the desired compounds 11a-11k. The other target compounds 11l-11n were prepared by a hydrogenation reduction of the nitro group of 11i-11k.
Scheme 3. Preparation of 11a-11na.
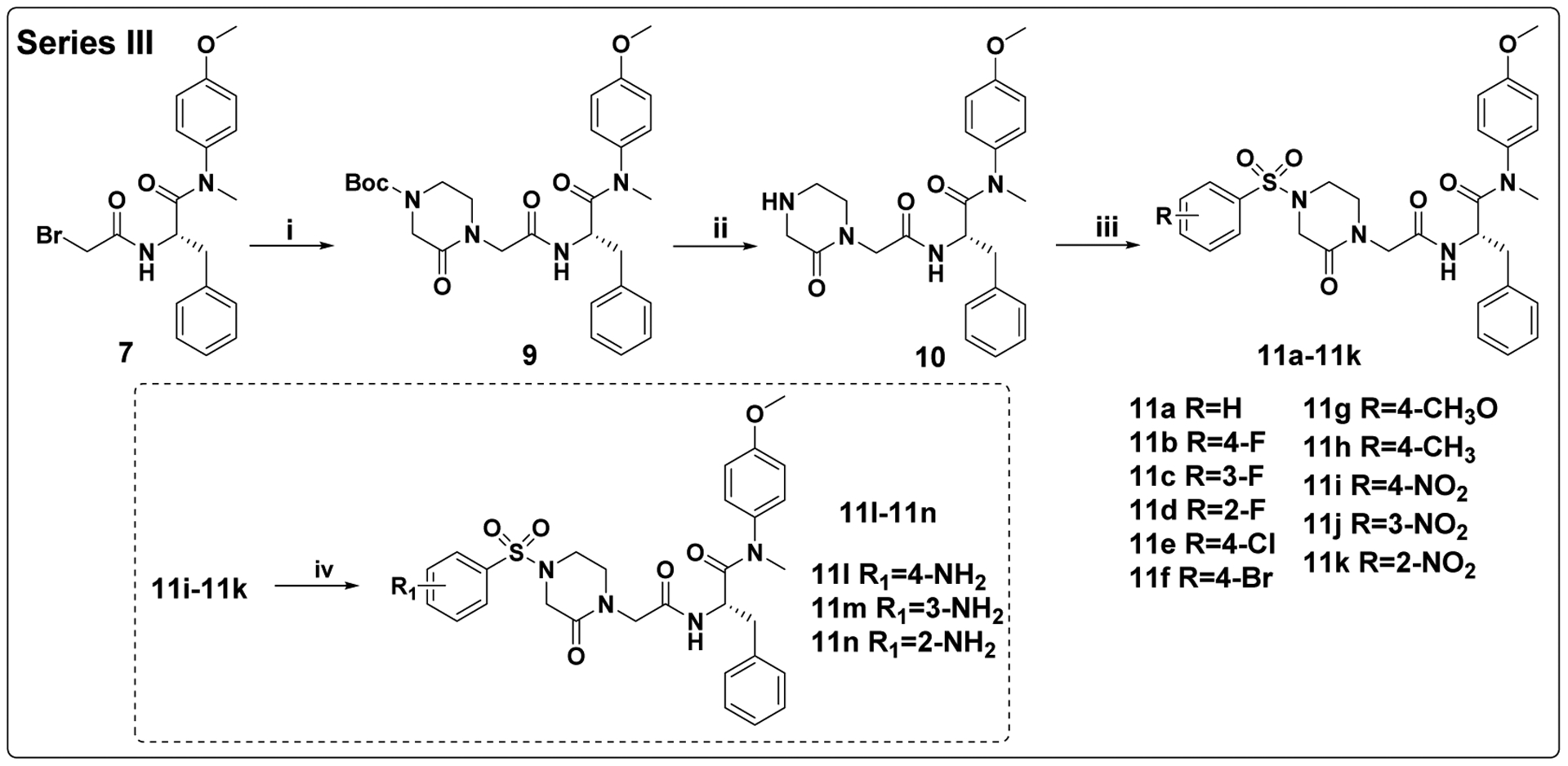
aReagents and conditions: (i) 1-Boc-3-oxopiperazine, K2CO3, DMF, 55°C; (ii) trifluoroacetic acid, dichloromethane, r.t.; (iii) corresponding substituted benzenesulfonyl chloride, TEA, dichloromethane, 0°C to r.t.; (iv) H2, 10% Pd●C, dichloromethane, methanol, r.t..
3. RESULTS AND DISCUSSION
3.1. In Vitro Anti-HIV Assays and SARs Analysis
All the synthesized molecules were tested for their antiviral activity and cytotoxicity using TZM-bl cells fully infected by the HIV-1 NL4–3 virus. EC50 (as measured by a luciferase gene expression assay31) and CC50, as well as selectivity index (SI, the ratio of CC50/EC50) values for compounds in Series I, II and III, are shown in Tables 1, 2 and 3, respectively. PF-74 was utilized as the control drug in this assay.
Table 1.
Anti-HIV-1 activity and cytotoxicity of Series I in TZM-bl cells infected with the HIV-1 NL4–3 virus.
 | ||||
|---|---|---|---|---|
| Compounds | R | EC50a(μM) | CC50b(μM) | SIc |
| 6a | H | 6.23±1.20 | >41.53 | >6.67 |
| 6b | 4-F | 6.81±1.48 | >40.04 | >5.88 |
| 6c | 4-Cl | 9.88±3.30 | >38.76 | >3.92 |
| 6d | 4-Br | 8.21±2.50 | >35.69 | >4.35 |
| 6e | 2,4,6-tri(isopropyl) | 7.90±3.13 | >32.91 | >4.17 |
| 6f | 4-CH3O | 10.36±3.71 | >39.09 | >3.77 |
| 6g | 4-CH3 | 6.26±2.22 | >40.36 | >6.45 |
| 6h | 3,5-dimethyl | 9.22±3.53 | >39.25 | >4.26 |
| 6i | 2,4,6-trimethyl | 10.69±3.06 | >38.19 | >3.57 |
| 6j | 2-F | 10.81±2.20 | >40.04 | >3.70 |
| 6k | 3-F | 5.61±1.54 | >40.04 | >7.14 |
| 6l | 4-NO2 | >37.98 | >37.98 | - |
| 6m | 2-NO2 | 8.74±2.47 | >37.98 | >4.35 |
| 6n | 3-NO2 | 8.93±1.82 | >37.98 | >4.26 |
| 6o | 4-NH2 | 6.65±2.42 | >40.28 | >6.06 |
| 6p | 2-NH2 | 9.06±2.62 | >40.28 | >4.44 |
| 6q | 3-NH2 | 8.26±2.82 | >40.28 | >4.88 |
| PF-74 | - | 0.52±0.18 | >47.00 | >90.91 |
EC50: the concentration of the compound required to achieve 50% protection of TZM-bl cells against HIV-1-induced cytopathic effect, determined in at least triplicate against HIV-1 in TZM-bl cells.
CC50: the concentration of the compound required to reduce the viability of uninfected cells by 50%, determined in at least triplicate against HIV-1 in TZM-bl cells; values were averaged from at least three independent experiments.
SI: selectivity index, the ratio of CC50/EC50.
Table 2.
Anti-HIV-1 activity and cytotoxicity of Series II in TZM-bl cells infected with the HIV-1 NL4–3 virus.
 | |||||
|---|---|---|---|---|---|
| Compounds | R1 | R2 | EC50a(μM) | CC50b(μM) | SIc |
| 8a | H | - | 2.11±0.48 | >38.27 | >18.18 |
| 8b | CH3 | - | 5.96±2.05 | >37.27 | >6.25 |
| 8c | CH3O | - | 4.89±1.01 | >36.19 | >7.41 |
| 8d | - | F | 3.82±0.82 | >23.12 | >6.06 |
| 8e | - | Cl | 4.77±0.99 | >22.69 | >4.76 |
| 8f | - | Br | 5.62±1.84 | >21.60 | >3.85 |
| PF-74 | - | - | 0.52±0.18 | >47.00 | >90.91 |
EC50: the concentration of the compound required to achieve 50% protection of TZM-bl cells against HIV-1-induced cytopathic effect, determined in at least triplicate against HIV-1 in TZM-bl cells.
CC50: the concentration of the compound required to reduce the viability of uninfected cells by 50%, determined in at least triplicate against HIV-1 in TZM-bl cells; values were averaged from at least three independent experiments.
SI: selectivity index, the ratio of CC50/EC50.
Table 3.
Anti-HIV-1 activity and cytotoxicity of the Series III in TZM-bl cells infected with the HIV-1 NL4–3 virus.
 | ||||
|---|---|---|---|---|
| Compounds | R | EC50a(μM) | CC50b(μM) | SIc |
| 11a | H | 0.37±0.12 | >35.42 | >95.22 |
| 11b | 4-F | 1.06±0.26 | 25.74±2.23 | 24.20 |
| 11c | 3-F | 0.45±0.15 | >34.33 | >76.96 |
| 11d | 2-F | 0.48±0.09 | >34.33 | >71.36 |
| 11e | 4-Cl | 1.05±0.37 | >33.38 | >31.73 |
| 11f | 4-Br | 0.36±0.11 | >31.08 | >87.05 |
| 11g | 4-CH3O | 0.64±0.13 | >33.63 | >52.63 |
| 11h | 4-CH3 | 0.50±0.23 | >34.56 | >68.98 |
| 11i | 4-NO2 | 0.30±0.09 | >32.81 | >111.21 |
| 11j | 3-NO2 | 0.67±0.23 | >32.81 | >48.75 |
| 11k | 2-NO2 | 1.00±0.38 | >32.81 | >32.81 |
| 11l | 4-NH2 | 0.09±0.03 | >34.50 | >383.36 |
| 11m | 3-NH2 | 0.33±0.08 | >34.50 | >105.19 |
| 11n | 2-NH2 | 0.54±0.12 | >34.50 | >64.49 |
| PF-74 | - | 0.52±0.18 | >47.00 | >90.91 |
EC50: the concentration of the compound required to achieve 50% protection of TZM-bl cells against HIV-1-induced cytopathic effect, determined in at least triplicate against HIV-1 in TZM-bl cells.
CC50: the concentration of the compound required to reduce the viability of uninfected cells by 50%, determined in at least triplicate against HIV-1 in TZM-bl cells; values were averaged from at least three independent experiments.
SI: selectivity index, the ratio of CC50/EC50.
In general, it can be observed from Tables 1, 2 and 3 that all of the newly synthesized compounds exhibited moderate to excellent activities (except 6l, EC50 > 37.98 μM) against HIV-1 NL4–3 virus with EC50 values ranging from 90 nM (11l) to 10.81 μM (6j).
As shown in Table 1, among the para-substituted benzene derivatives (6a, 6b, 6c, 6d, 6f, 6g, 6l, 6o), taking the unsubstituted compound 6a (EC50 = 6.23 μM) as reference, substitution on the benzene ring (6a) with 4-F (6b, EC50 = 6.81 μM), 4-NH2 (6o, EC50 = 6.65 μM), 4-CH3 (6g, EC50 = 6.26 μM) resulted in no major changes in antiviral activity. While substitution on the benzene ring (6a) with 4-Br (6d, EC50 = 8.21 μM), 4-Cl (6c, EC50 = 9.88 μM), 4-CH3O (6f, EC50 = 10.36 μM) groups showed reduced antiviral activity (1.3, 1.6 and 1.7-fold, respectively). Besides, substitution on the benzene ring (6a) with 4-NO2 (6l, EC50 > 37.98 μM) resulted in almost complete loss of activity. However, the shift of the nitro group from C-4 (6l) to C-2 (6m) or C-3 (6n) position, displayed anti-HIV-1 activity with an EC50 value of 8.74 or 8.93 μM, reveals that the position of the substituent is significant for activity. From the meta-substituted benzene derivatives (6k, 6q, 6n), the order of potency was as follows: 3-F (6k, EC50 = 5.61 μM) > 3-NH2 (6q, EC50 = 8.26 μM) ≥ 3-NO2 (6n, EC50 = 8.93 μM). 6k was also the best active compound in Series I, indicating that fluorine is very favorable for the antiviral activity. Interestingly, among the ortho-substituted derivatives, the order is just the opposite: 2-NO2 (6m, EC50 value of 8.74 μM) ≥ 2-NH2 (6p, EC50 = 9.06 μM) > 2-F (6j, EC50 = 10.81 μM). Among the tri-substituted derivatives (6e, 6i), the antiviral activity of 6e (EC50 = 7.90 μM) was 1.4 times better than that of 6i (EC50 = 10.69 μM), indicating that the volume of the substituent affects the activity, and the larger group seems to be more favorable. However, when comparing the activities of 6g, 6h and 6i, it was found that the addition of methyl groups seems to be unfavorable to activity.
As shown in Table 2, among the compounds in Series II (8a-8f), the activity of thiadiazine ring mono-substituted derivatives (8a-8c, EC50 value ranging from 2.11 μM to 5.96 μM) was almost equivalent to that of di-substituted derivatives (8d-8f, EC50 value ranging from 3.82 μM to 5.62 μM). Among the thiadiazine-bearing derivatives (8a-8c), taking the least substituted compound 8a as reference, it was also the most active compound, the replacement of the 7-H on the thiadiazine ring (8a) with 7-CH3 (8b, EC50 = 5.96 μM) and 7-CH3O (8c, EC50 = 4.89 μM), resulted in 2.8 and 2.3-fold decrease in antiviral activity, respectively, indicating aliphatic (or electron-donating) groups were not beneficial for the potency. Among the di-substituted derivatives (8d-8f, EC50 value of ranging from 3.82 μM to 5.62 μM), the activity is positively correlated with the electronegativity of substituents of 7-position: F (8d, EC50 = 3.82 μM) > Cl (8e, EC50 = 4.77 μM) > Br (8f, EC50 = 5.62 μM), indicating that the electron-withdrawing group is beneficial to activity. As demonstrated, the antiviral activity of thiadiazine derivatives (Series II) was much better than that of Series I and molecular dynamics (MD) simulation studies on 8a and 6k provided plausible clues for this conclusion (see Supporting Information).
As shown in Table 3, through iterative structural optimization, the activity of Series III was further enhanced, indicating that the introduction of piperazinone for Series II via the scaffold hopping and bioisosterism strategies is favorable to the antiviral potency. Most of the compounds in this series exhibited sub-micromolar levels of antiviral activities, of which eight compounds exceeded PF-74 (EC50 = 0.52 μM), especially the most active 11l (EC50 = 90 nM) is nearly 6 times more potent than PF-74. Taking the unsubstituted benzene compound 11a (EC50 = 0.37 μM) as reference: (a) among the para-substituted benzene derivatives, substitution with 4-NH2 (11l, the most potent among all three series), 4-NO2 (11i, EC50 = 0.30 μM), 4-Br (11f, EC50 = 0.36 μM) increased the antiviral activity. However the introduction of other halogen atoms (4-F (11b) and 4-Cl (11e)) decreased the antiviral activity, and the incorporation of aliphatic groups (4-CH3 (11h) and 4-CH3O (11g)) were also not conducive to the inhibition; (b) among the meta-substituted benzene derivatives, the hydrogen-bond donor 3-NH2 (11m) was advantageous for activity, while favorable hydrogen bond acceptors such as 3-F (11c) and 3-nitro (11j) groups caused a significant decrease in activity; (c) all of the ortho-substituted benzene derivatives displayed decreased activity. Interestingly, the activity order of hydrogen-bond donor (NH2) is the same as the hydrogen-bond receptor (NO2): para- > meta- > ortho-. In conclusion, the results showed that para-substitutions were more beneficial to antiviral activity.
Furthermore, we selected representative compounds (6k, 8a, 11a, 11i, and 11l) with the best NL4–3 inhibitory activities in each series and tested their antiviral activities against HIV-1IIIB and HIV-2ROD in MT-4 cells, respectively. The results are shown in Table 4. All selected compounds displayed inhibitory effects on HIV-1 and HIV-2, and are more inclined to inhibit HIV-2, in contrast to PF-74. In addition, it is clear that as the structure of the compound was further optimized, the antiviral activity steadily increased against both isolates, which is consistent with their inhibitory profile on HIV-1NL4–3. It is particularly noteworthy that, in addition to the improved antiviral activity against HIV-1IIIB compared to PF-74, the piperazinone analogs 11a and 11l also have significant antiviral activities against HIV-2ROD, with almost 120 times improvement over PF-74. Also, 11l has much higher safety as compared to PF-74, as indicated by their CC50 and SI values.
Table 4.
Antiviral activity against HIV-1 (IIIB) and HIV-2 (ROD) replicating in MT-4 cells. Corresponding cytotoxicity, and selectivity indices are also given.
| Compounds | EC50a(μM) | CC50b(μM) | SIc | ||
|---|---|---|---|---|---|
| IIIb | ROD | IIIB | ROD | ||
| 6k | 24.48±8.73 | 20.06±4.44 | 136.28±5.26 | 5.57 | 6.79 |
| 8a | 6.07±2.93 | 2.72±1.09 | 129.72±4.88 | 21.37 | 47.69 |
| 11a | 0.71±0.37 | 0.032±0.009 | 108.54±17.00 | 152.87 | 3391.88 |
| 11i | 4.90±1.87 | 0.16±0.05 | 8.89±1.94 | 1.81 | 55.56 |
| 11l | 0.21±0.03 | 0.031±0.012 | 178.81±1.21 | 851.48 | 5768.06 |
| PF-74 | 0.87±0.31 | 3.69±0.59 | 145.18±38.05 | 166.87 | 39.34 |
EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytotoxicity, as determined by the MTT method.
CC50: concentration required to reduce the viability of mock-infected cell cultures by 50%, as determined by the MTT method.
SI: selectivity index, the ratio of CC50/EC50.
Overall, compounds containing a benzenesulfonamide exhibited moderate to excellent antiviral activities, of which 11l displayed the most promising antiviral (HIV-1 and HIV-2) potency. Therefore, 11l will undoubtedly be a useful lead for further structural optimization. Considering the higher infectivity and lethality of HIV-1 worldwide, as compared to HIV-2, we chose to further explore the anti-HIV-1 mechanism of these compounds.
3.2. Compounds from Each Series Interact with the HIV-1 CA Protein as Determined by Surface Plasmon Resonance (SPR)
Due to the potent anti-HIV-1 activity exhibited by most of the derivatives, we then sought to determine whether these compounds bind to the HIV-1 CA protein directly. The most active compounds in their respective series (seven in total), were chosen to determine their binding affinities with monomeric and hexameric CA constructs using an SPR-based method as previously reported32,33 with PF-74 as an in-line control.
As shown in Table 5, PF-74 binds the CA hexamer with the highest affinity among the tested compounds, with an equilibrium dissociation constant (KD) value of 93 nM. As expected, PF-74 binds with a much lower affinity to the CA monomer (KD = 2.80 μM, ratio=30.11). The binding affinities of the tested compounds (except for 8d) are higher to the hexamer than the monomer (indicated by a ratio > 1), comparable to PF-74, indicating that a hexameric CA arrangement is essential for antiviral activity of our compounds. Although the binding affinity of 8d for the monomer is much higher compared to 8a (their affinities to the hexameric CA were almost the same), the antiviral activity has not been improved, confirming that the antiviral activity of our compounds is due to their interaction with the hexameric CA. The binding affinities of 6a, 6k, 8a, 8d as well as PF-74 to CA hexamer were in good agreement with the trend of their cell-based antiviral activities. However, the antiviral activities of 11i, 11l and 11m were improved with respect to PF-74, while in SPR assay, their binding affinities to the CA hexamer were much lower compared to PF-74. This may be due to the steric effects of the disulphide-contrained hexameric CA construct used for the binding assay or that Series III may exert antiviral effects through multiple CA-directed mechanisms of action.8,9
Table 5.
SPR results of representative compounds and PF-74 binding to monomeric and hexameric CA constructs.
| Compound | KDa (μM) | ratiob | |
|---|---|---|---|
| Monomer | Hexamer | ||
| 6a | 15.50±1.76 | 11.80±0.30 | 1.31 |
| 6k | 22.10±12.00 | 7.99±0.08 | 2.77 |
| 8a | 6.63±1.46 | 1.19±0.05 | 5.57 |
| 8d | 0.20±0.07 | 1.30±0.22 | 0.15 |
| 11i | 11.50±5.92 | 4.45±0.32 | 2.58 |
| 11l | 13.60±7.86 | 2.63±0.14 | 5.17 |
| 11m | 10.00±4.38 | 3.02±0.13 | 3.31 |
| PF-74 | 2.80±1.40 | 0.093±0.005 | 30.11 |
All values represent the average response from at least 3 replicates. Error bars represent standard deviation.
KDMonomer/KDHexamer ratio.
In conclusion, these results demonstrated that our new inhibitors appear to exert their antiviral activity by direct binding to the HIV-1 CA protein.
3.3. The New Compounds Series Exhibit A Dual Stage Inhibition Profile
Having characterized their target engagement, we then sought to determine the mechanism of action of these newly synthesized compounds. Given that CA plays important roles in both early- and late-stage events, we decided to determine which stage(s) of the HIV-1 lifecycle can be blocked by these compounds. We selected the most active compounds in each series for testing (6k, 8a and 11l), with PF-74 as a control. The experimental method is the same as previously reported.23,28
As shown in Table 6, according to the ratio results, all tested compounds except for 6k (ratio = 0.51), prefer to act on the early stage of HIV-1 replication (ratio > 1). For example, 8a (ratio = 1.26) had a slightly higher inhibition of the early stage compared to the late stage. 11l, the compound with the most potent antiviral activity (EC50 = 90 nM), had the highest IC50Late/IC50Early ratio (26.79), which was significantly higher than PF-74 (ratio = 4.11). 11l also showed the best inhibitory activity in the early- and late-stages (IC50 = 8.96 nM and 0.24 μM, respectively), with the early stage inhibition being 6.25 times more potent than the lead PF-74 (IC50 = 56 nM), which is consistent with their proportion in the antiviral activity (EC50 value: 90 nM vs 520 nM).
Table 6.
Antiviral activity in early-, late-stages of representative compounds using HIV-1 Env-pseudotyped virus.
| Compound | IC50a (μM) | Ratiob | |
|---|---|---|---|
| Early Stage | Late Stage | ||
| 6k | 8.22 ± 0.99 | 4.20 ± 2.51 | 0.51 |
| 8a | 1.94 ± 0.48 | 2.44 ± 1.52 | 1.26 |
| 11l | 8.96 ± 0.43nM | 0.24 ± 0.11 | 26.79 |
| PF-74 | 56 ± 17 nM | 0.23 ± 0.17 | 4.11 |
the concentration of the compound required to achieve 50% infection of HIV-1 Env-pseudotyped virus in U87.CD4.CCR5 target cells. SD of 3 parallel tests is indicated.
IC50Late/IC50Early ratio.
As demonstrated, all of the new compounds exhibited a dual-stage inhibition profile, which is in accordance with the parental compound PF-74.22 Specifically, in both early- and late-stages, the IC50 values of the tested compounds, including PF-74, were consistent with their antiviral activities. Additionally, in both action stage assays, the IC50 values of the tested compounds (except for 11l) were also consistent with their affinity trends for the CA hexamer.
3.4. New Compounds Do Not Reduce the Amount of Virus Produced in the Late Stages
Encouraged by the potent inhibition of these inhibitors at both early- and late-stages of HIV-1 lifecycle, we sought to determine their effect on the CA/p24 content. We quantified the amount of virus (CA/p24) produced in the presence of 10 μM of compound (6k, 8a and 11l). CA/p24 was captured on an anti-p24 antibody coated ELISA plate, also calorimetrically quantified (with a secondary antibody) and compared to a virus produced in the presence of PF-74.
As depicted in Figure 4, the p24 content of the produced virus in the presence of 6k, 8a, 11l showed only a slight increase (about 0–20% compared to the DMSO control). However, PF-74 increased the CA/p24 content of the produced virus by around 40%. The observed changes are not significant enough to deduce a possible mechanism of action of these new compounds.
Figure 4.
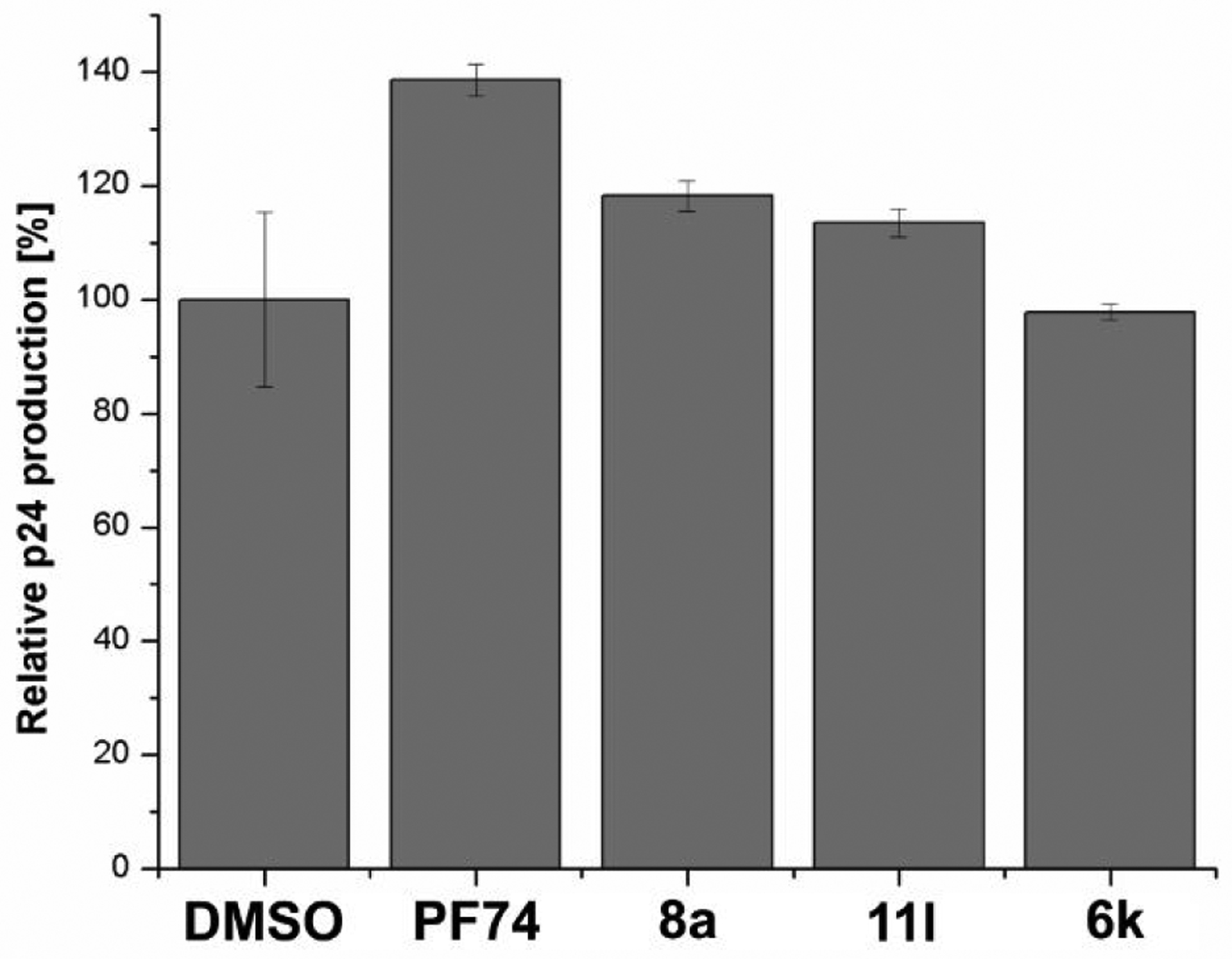
The effect of compounds 6k, 8a, 11l, and PF-74 on viral production. Standard deviation represents 2 replicates performed in duplicate. Performed with NL4–3 Env pseudo-typed HIV-1 virus at 10 μM compound concentration.
3.5. The New Compounds Series Reduce the Assembly of HIV-1 In Vitro and Do Not Inhibit Reverse Transcriptase (RT)
Considering that this new class of compounds does not affect p24 levels, we believe that these inhibitors may disturb CA–CA interactions and its core stability to produce less infectious virus with irregular CA by affecting the rate of CA assembly. To access the effects on HIV-1 CA assembly, we evaluated 6k, 8a, 11l as well as PF-74 in an in vitro CA assembly assay utilized by Blair et al..22
As depicted in Figure 5, PF-74 significantly accelerated the rate of assembly of HIV-1 CA under the in vitro conditions as compared to the DMSO control. By contrast, our compounds dramatically reduce the assembly of HIV-1 CA in the same assay (as compared to both PF-74 and DMSO).
Figure 5.
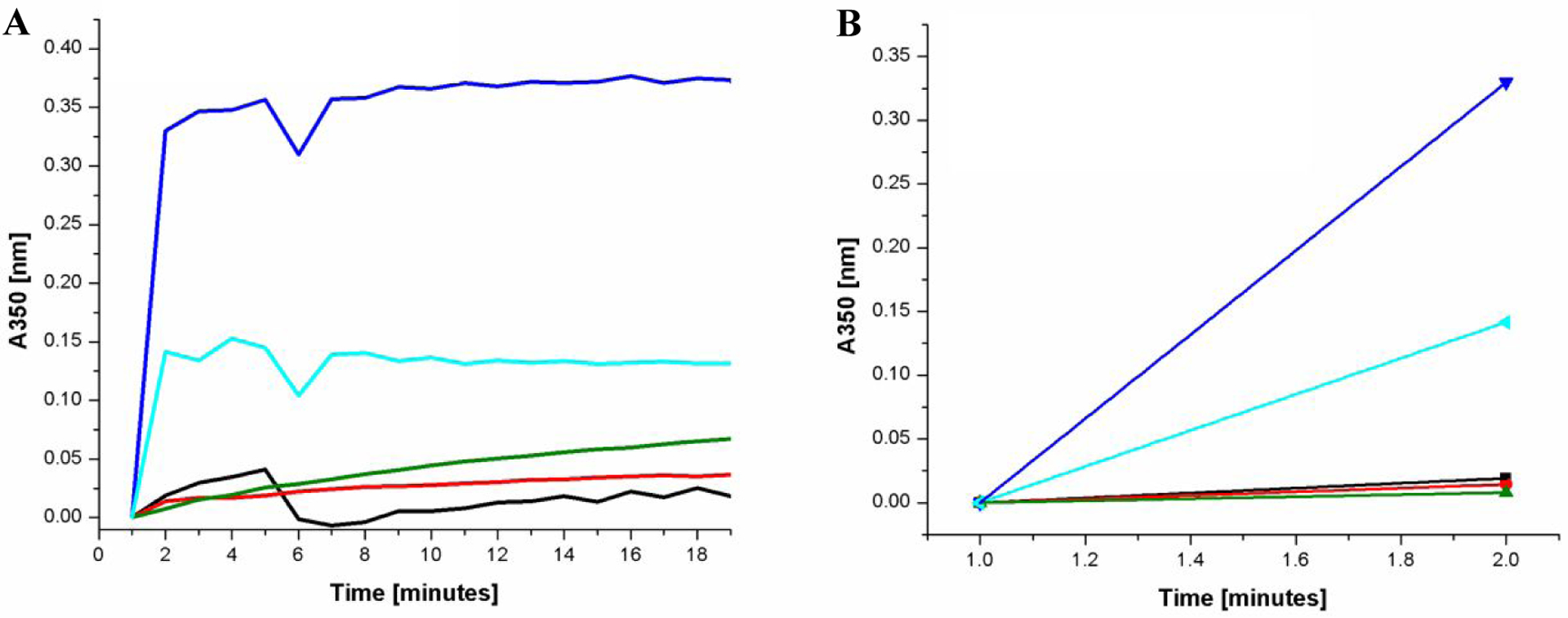
The effect of compounds 6k (green line), 8a (black line), 11l (red line), and PF-74 (blue line) on the NL4–3 capsid assembly in vitro at 3M NaCl. Apo Capsid NL4–3 as control (cyan line). (A) Capsid assembly was monitored by an increase in turbidity using a spectrophotometer at 350 nm over 19 minutes. Capsid was used at a final concentration of 30 μM, and compounds 6k, 8a, 11l and PF-74 at a final concentration of 50 μM. (B) Slope/velocity quantification of capsid assembly during the first 2 minutes. Experiments were performed in triplicate. (AU) Absorption unit.
Based on the results of the p24 content analysis and the CA assembly assay on the representative compounds, we can postulate potential mechanisms of action for these compounds, based upon the CA assay and previous in vitro correlations.28 We propose that in the early stage, the interaction of the compounds with the incoming HIV-1 CA accelerates the disassembly of the core. We have seen this correlation between reduced assembly and accelerated uncoating of the core in another PF-74 derived series.28 The essential but poorly understood process of CA-mediated uncoating is tightly associated with reverse transcription34 To exclude any effect of compound 11l on reverse transcription, we tested the direct inhibitory effect of compound 11l and TMC278 (also known as Rilpivirine) as a positive control on purified recombinant RT itself using an RT assay kit (Roche Diagnostics, IN, USA).
As depicted in Figure 6, it is evident that while the control RT inhibitor TMC278 inhibits RT (51±11% as compared to the DMSO control at 0.73 nM), while 11l does not to any appreciable level (95±15% as compared to the DMSO control at 0.2 μM). Therefore, the early-stage inhibition that we see in the antiviral assay is most likely due to direct action on the capsid core and not by simple inhibition of RT.
Figure 6.
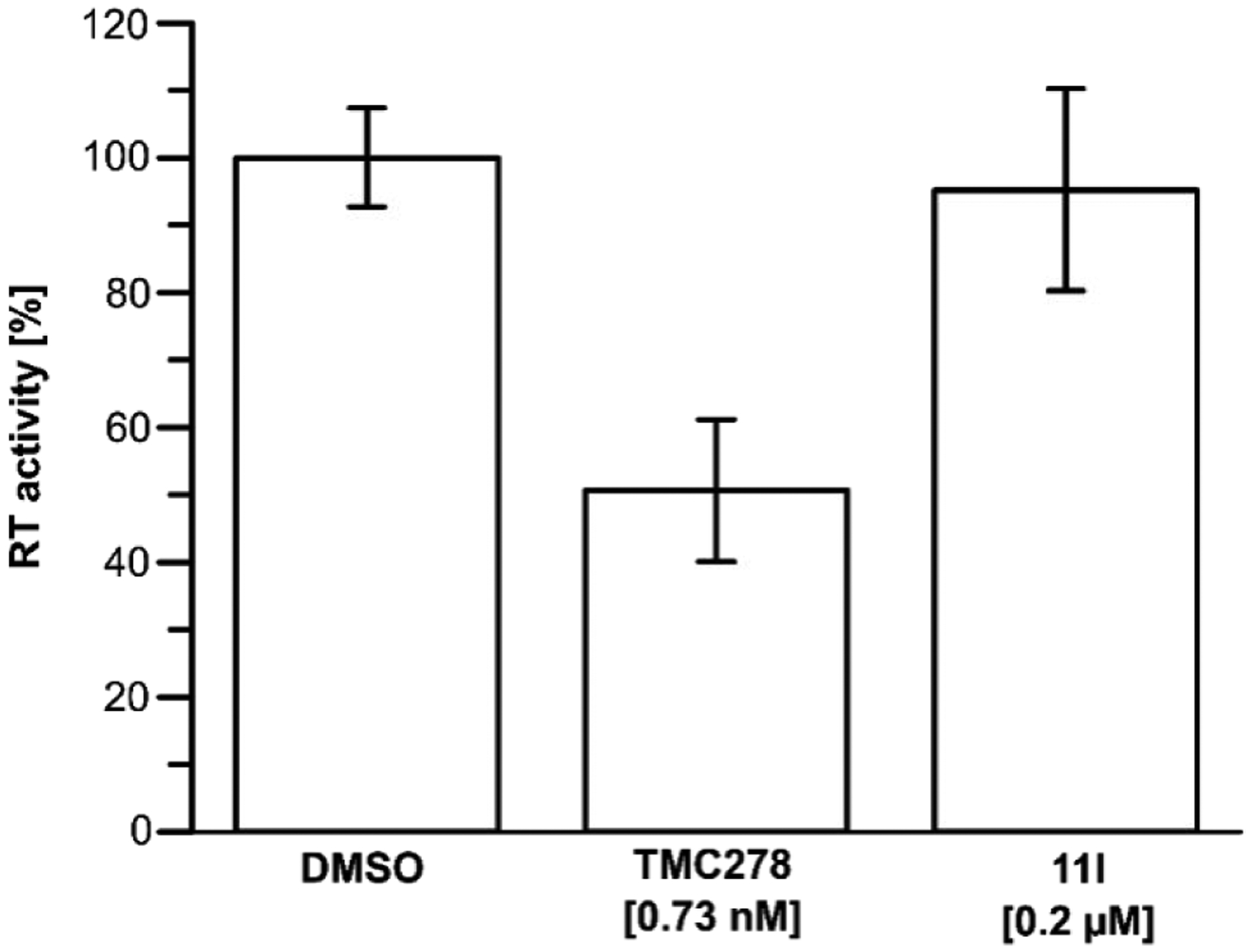
Effect of compound 11l on RT activity in vitro. Compound 11l (0.2 μM) does not affect RT activity as compared to the DMSO control (1%). TMC278/Rilpivirine was used as a positive control at 0.73 nM (IC50). The assay was performed using a calorimetric RT assay developed by Roche. The experiment was performed in triplicate with error bars depicting the standard deviation.
The potential mechanism of action in the late stages is somewhat murkier. Compounds that reduce the assembly of HIV-1 CA in the assembly assay typically reduce the amount of virus produced. However, we do not see this trend. As such, we can only postulate that the reduction in assembly is not sufficient enough to reduce the amount of viral production over the time-frame of the assay and that these compounds most likely produce a virus that has altered morphology that is not compatible with maturation to infectious particles. This mode of action is also exhibited by PF-74.22 Further mechanism of action studies outside the scope of this work are warranted to clarify these postulated mechanisms.
3.6. Molecular Dynamics (MD) Simulations Study
For a better interpretation of SAR of the best CA inhibitor of the series, 11l was simulated for 1 μs to model its binding to the HIV-1 CA monomer using the software Autodock 4.2.6 with default settings.23
As shown in Figure 7A and B, the root mean square deviation (RMSD) and root mean squares fluctuation (RMSF) patterns show that the protein structure is present in different conformational states, which could be accompanied by different binding modes of 11l. To investigate the binding of 11l to the HIV-1 CA monomer and its conformational existence, its RMSD (heavy atoms) was calculated in reference to the first frame of the MD simulation and plotted in Figure 7C. It is clear from the figure that 11l prefers certain conformation during the MD simulation, which shows stable binding to the protein binding site.
Figure 7.
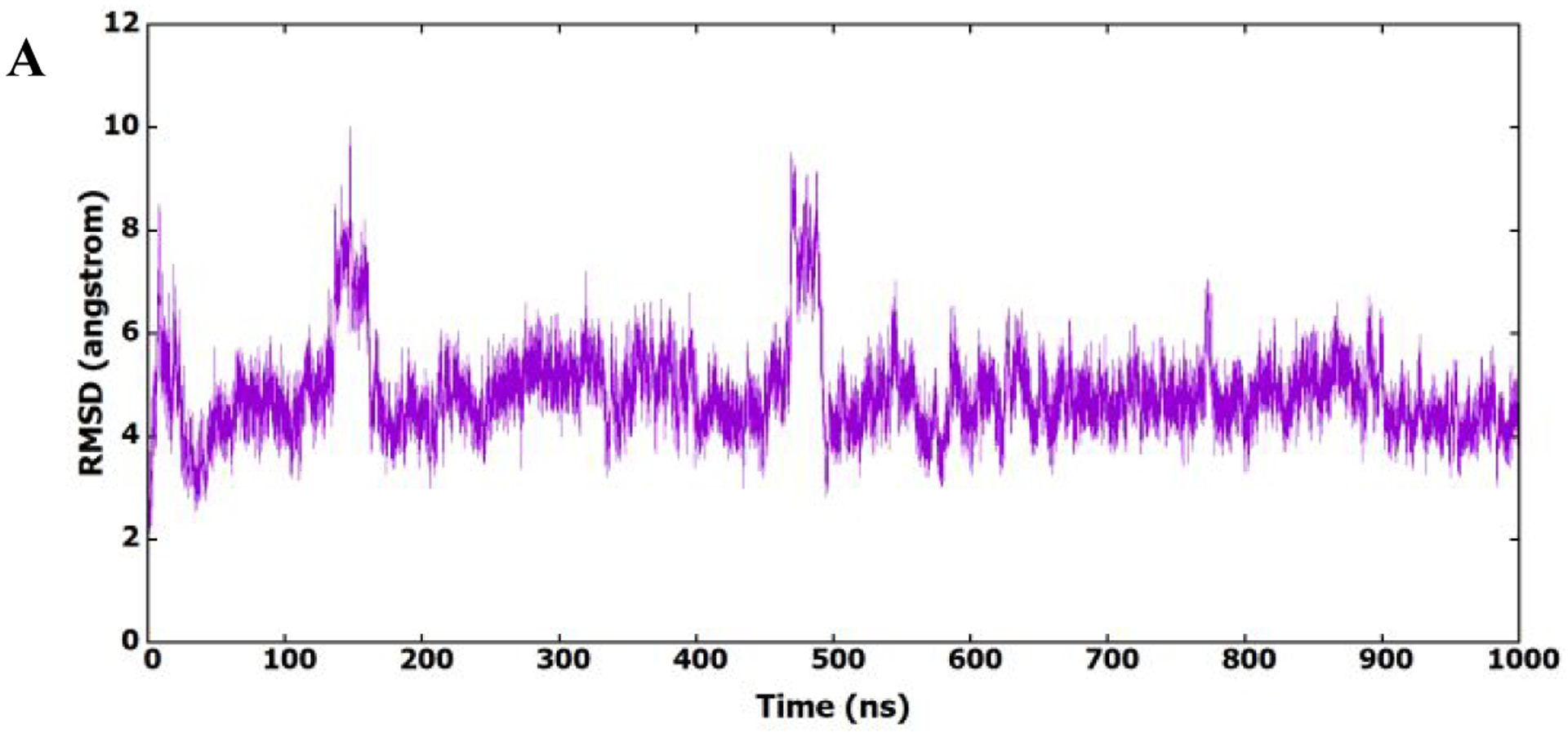
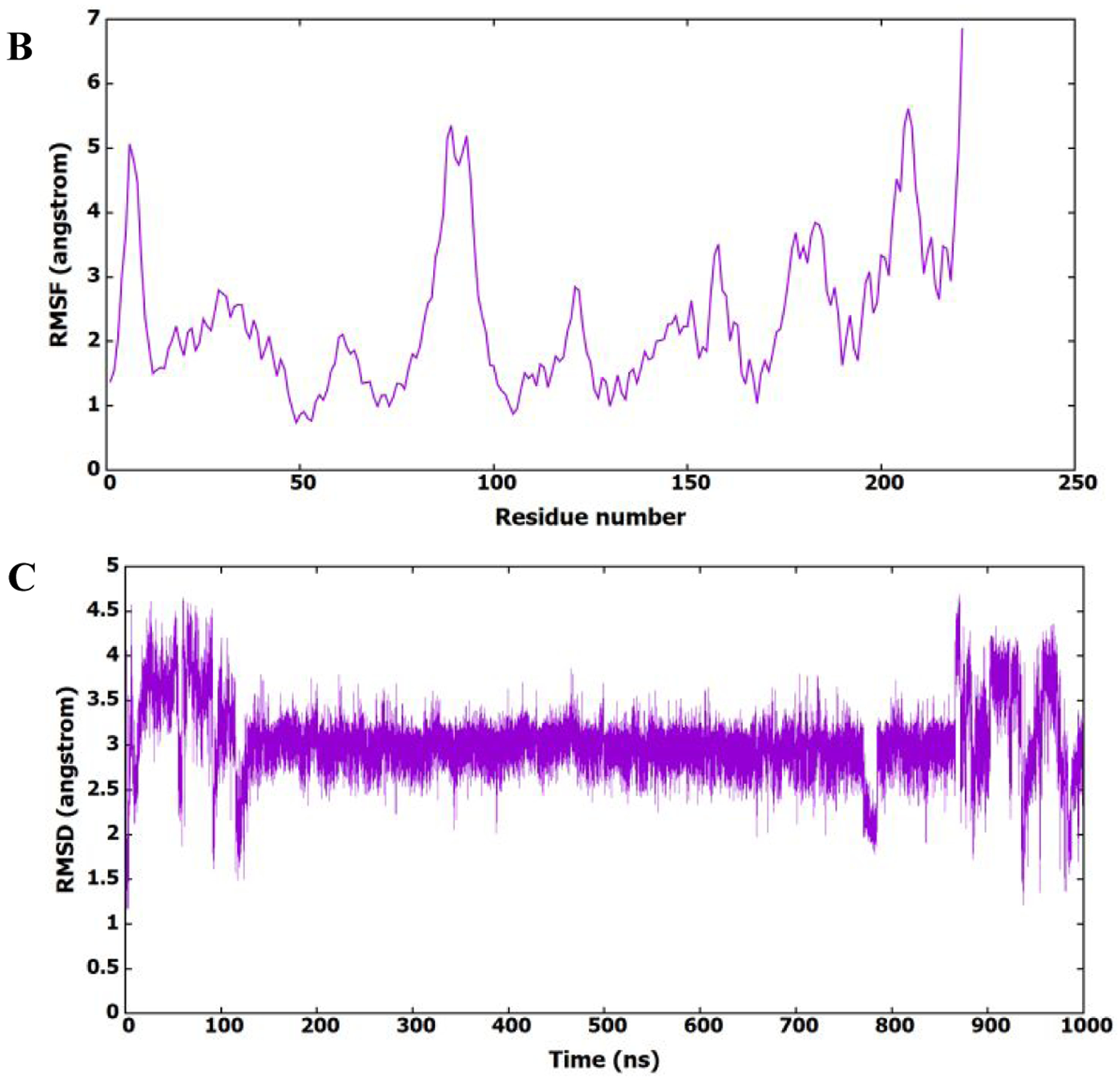
(A) RMSD (heavy atoms) of amino acids of CA HIV-1 monomer in reference to the first frame of the MD simulation. (B) RMSF of the backbone Cα atoms for amino acids of CA HIV-1 monomer. (C) RMSD (heavy atoms) of the bound 11l in reference to the docked conformer.
Results of RMSD and RMSF demonstrated that there is a preferred interaction between 11l and HIV-1 CA monomer, thus the entire trajectory has been clustered based on 11l (no fit). Figure 8A shows a representative structure of the predominant cluster (92.0%): an expanded view for 11l binding to the active site. The binding mode of 11l to the HIV-1 CA monomer is similar to that of PF-74 (the prototype inhibitor), where the core scaffold is oriented to the inside of the active site and the substituent is oriented to the outside of the active site. Furthermore, the binding of 11l is stable, which explains its high activity.
Figure 8.
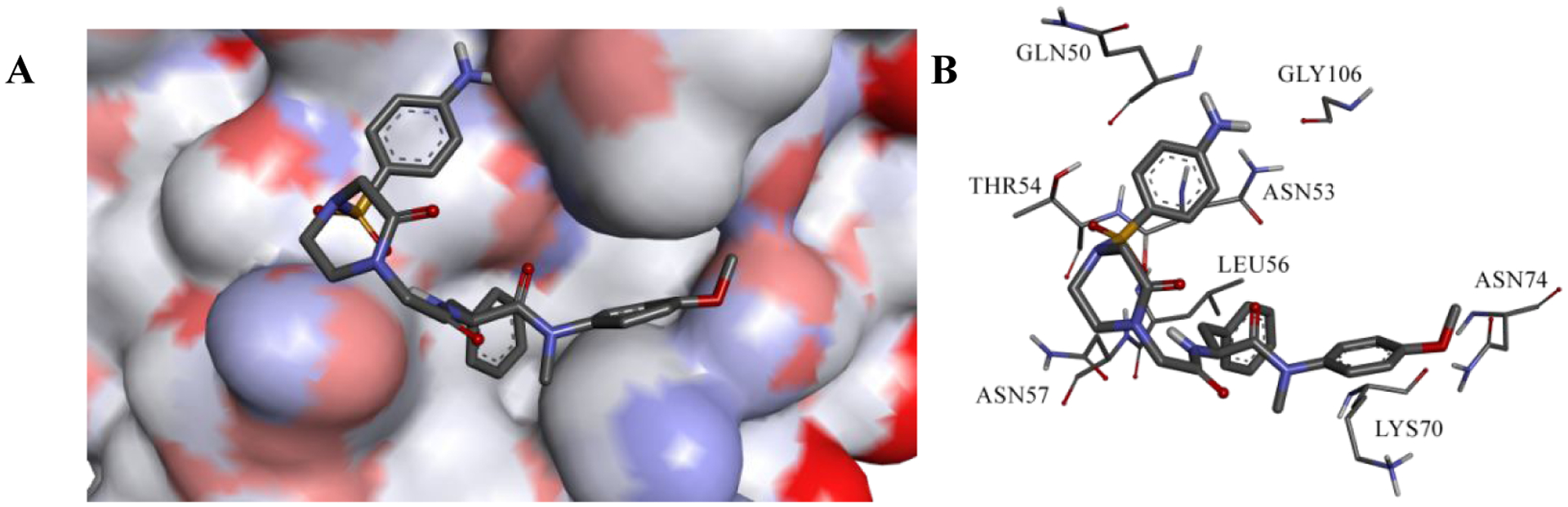
(A) Expanded view of the representative structures of the first clusters. (B) Binding interactions of 11l in the most populated cluster.
Interaction of 11l with HIV-1 CA monomer fixed it to the open conformation as shown by the 5HGL X-ray structure, which could fix the hexamer to one conformation and disables its function.23,35 Figure 8B shows the interactions between the most predominant conformer of 11l and the HIV-1 CA monomer. The phenyl ring of the core region of 11l forms hydrophobic interactions with Lys70, similar to PF-74, and ion-induced dipole interactions with the positively charged nitrogen atom of Lys70. Furthermore, 11l forms aliphatic-aromatic hydrophobic interactions with Leu56 in the same manner as the PF-74 interaction. It also forms a hydrogen bond with Asn74 through its methoxy group of the core region, and a hydrogen bond with Asn57 at the substituent region through its oxopiperazine α-proton.36 Interestingly, this class of inhibitors has an aminophenylsulphonyl moiety, which accessed a new binding region in the HIV-1 CA monomer. Sulphone oxygen atoms are involved in hydrogen bonding to Thr54 NH backbone proton. Also, the protons of the amino group of the aminophenyl are involved in hydrogen bonding with Gly106 and Gln50, which add further interactions and strengthen the binding. Furthermore, the oxygen atoms of the sulphonyl moiety interact by hydrogen bonding with the backbone NH of Thr54. Table 7 emphasizes the frequency of hydrogen bonding represented in Figure 8B, especially the new binding region. The aminophenyl moiety has a high frequency of hydrogen bonding with Gly106 and Gln50 through its amino group, which shows extensive involvement of hydrogen bonding. Results of MD simulation analysis provides potential explanations for the high activity of the newly synthesized inhibitor 11l.
Table 7.
Hydrogen bond analysis and their corresponding frequencies for the whole 1 μs MD trajectory.
| Residues involved | Frequency | |
|---|---|---|
| Gly106 | 11l-N7 | 50.6 |
| Gln50 | 11l-N7 | 31.4 |
| Asn74 | 11l -O40 | 3.5 |
| 11l-O9 | 11l -H21 | 56.4 |
| 11l-O17 | 11l -H21 | 3.8 |
3.7. Plasma and Human Liver Microsome (HLM) Stability as Compared to PF-74
A significant barrier to the use of PF-74 in the clinic, in addition to its potency, its unfavorable drug-like parameters, especially its metabolic stability.28 Therefore, we next examined the stability of the most active compound 11l and lead PF-74 first in human plasma. Propantheline bromide was tested for comparison. As shown in Figure 9, 88.5% of 11l remained intact after incubation for 120 min at 37 °C, and its in vitro half-life was 844.6 min. In contrast, PF-74 was readily metabolized (remaining amounts of 69.0 % at 120 min and t1/2 = 221.1 min). In conclusion, 11l is relatively stable in human plasma, which was considerably more stable than propantheline bromide (0.0% intact after 120 min).
Figure 9.
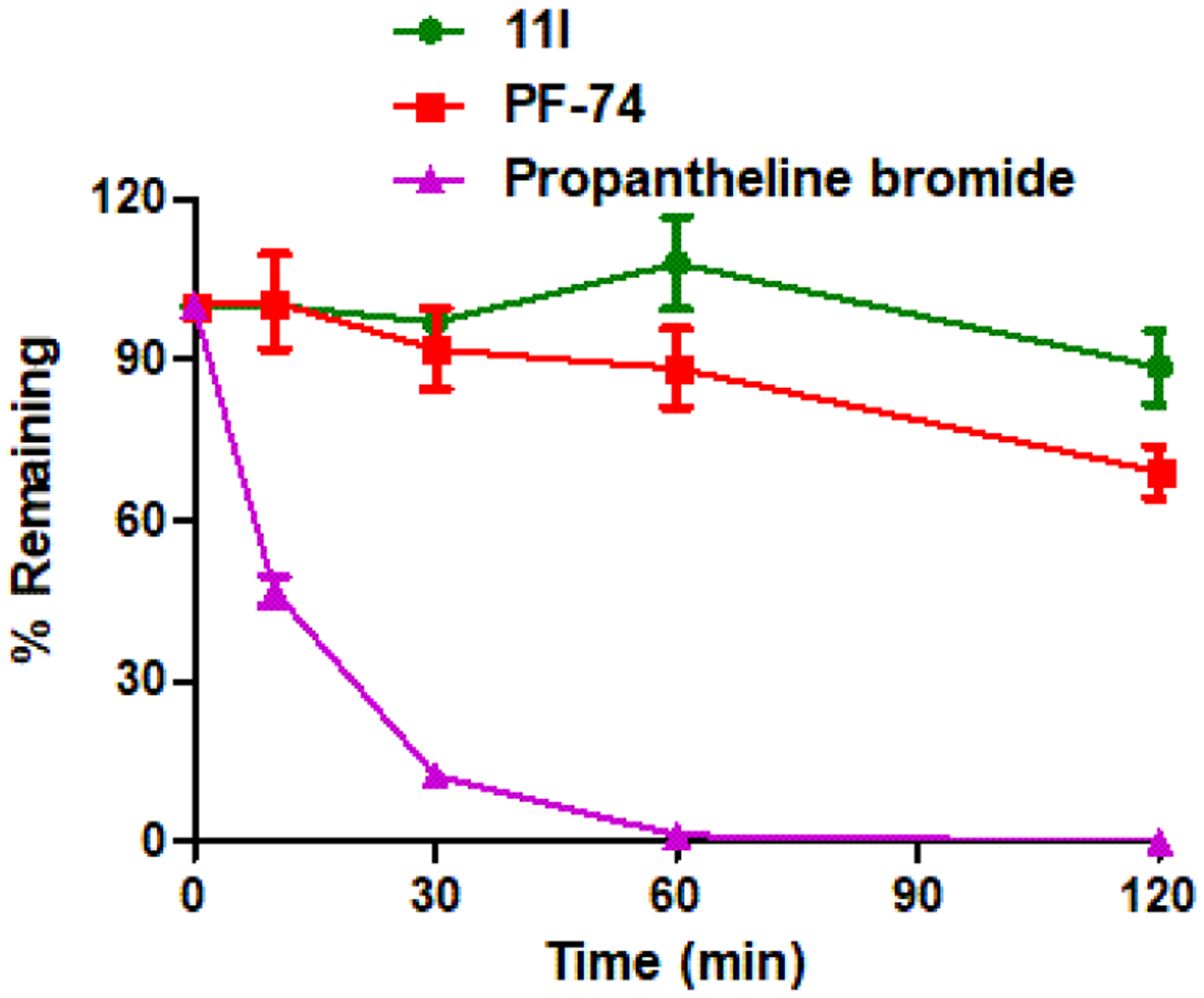
Result summary of human plasma stability assay. Experiments were performed in triplicate. % remaining = 100 × (PAR at appointed incubation time/PAR at time T0). PAR is the peak area ratio of a test compound to the internal standard. Accuracy should be within 80–120% of the indicated value.
We then examined the metabolic stability of 11l and lead PF-74 in a human liver microsomes (HLM) assay. Testosterone, diclofenac, and propranolol were used as controls. As shown in Table 8, PF-74 was rapidly metabolized with a half-life of 1.3 min, while t1/2 of 11l was 4.1 min. CLint(liver) of 11l was also reduced to 1/3 of that of PF-74, with values of 307.8 and 972.3 mL/min/kg, respectively, it meets the basic requirements of ADMET. Based on the results of human plasma and liver microsomes stability assays, the stability of 11l was relatively modest and not much improved compared with the parental PF-74, thus the metabolic stability is an important focus for further compound optimization.
Table 8.
Metabolic Stability Assay in Human Liver Microsomes
| Sample | HLM (Final concentration of 0.5 mg protein/mL) | |||||
|---|---|---|---|---|---|---|
| R2a | T1/2b (min) | CLint(mic)c (μL/min/mg) | CLint(liver)d (mL/min/kg) | Remaining (T=60min) | Remaining (NCFe=60min) | |
| 11l | 0.9863 | 4.1 | 342.0 | 307.8 | 0.0% | 93.4% |
| PF-74 | 0.9761 | 1.3 | 1080.3 | 972.3 | 0.3% | 93.1% |
| Testosterone | 0.9973 | 14.6 | 95.2 | 85.7 | 6.1% | 84.0% |
| Diclofenac | 0.9972 | 14.8 | 93.6 | 84.3 | 5.9% | 94.8% |
| Propafenone | 0.9722 | 6.9 | 201.4 | 181.2 | 0.3% | 100.0% |
R2 is the correlation coefficient of the linear regression for determination of the kinetic constant (see raw data worksheet in the Supporting Information).
T1/2 is half-life, and CLint(mic) is the intrinsic clearance.
CLint(mic) = (0.693/half-life)/mg microsome protein per mL.
CLint(liver) = CLint(mic) × mg microsomal protein/g liver weight × g liver weight/kg body weight.
NCF: no cofactor. No NADPH regenerating system was added to the NCF sample (replaced by buffer) during the 60 min incubation. If the remaining amount is less than 60%, then non-NADPH dependent reaction occurs.
3.8. In Vivo Pharmacokinetics Study and Safety Assessment
The in vivo pharmacokinetic (PK) profile of compound 11l was examined in Sprague-Dawley rats (Table 9 and Figure 10). After a single 2 mg/kg iv dose of 11l, the mean clearance rate (CL) and half-time (t1/2) was 4.06 L h−1 kg−1 and 1.0 h. When administered at 20 mg/kg orally, compound 11l was rapidly absorbed with a Tmax of 0.25 h, a moderate half-life of 1.2 h, and a mean residence time (MRT) of 1.8 h. 11l exhibited a favorable oral bioavailability of 23.0 % in this assay.
Table 9.
Pharmacokinetic Profile of 11la
| Parameter | Unit | 11l (po)b | 11l (iv)c | ||||
|---|---|---|---|---|---|---|---|
| Mean | ± | SD | Mean | ± | SD | ||
| T1/2 | (h) | 1.2 | ± | 0.4 | 1.0 | ± | 0.3 |
| Tmax | (h) | 0.25 | ± | 0.0 | 0.083 | ± | 0.0 |
| Cmax | (ng/mL) | 83.8 | ± | 37.9 | 1280.8 | ± | 64.9 |
| AUC0~t | (h*ng/mL) | 116.3 | ± | 51.0 | 505.6 | ± | 110.0 |
| AUC0~∞ | (h*ng/mL) | 120.6 | ± | 50.9 | 511.5 | ± | 109.4 |
| V | (mL/kg) | NA | ± | NA | 5752.4 | ± | 1292.1 |
| CL | (mL/h/kg) | NA | ± | NA | 4062.3 | ± | 884.6 |
| MRT0~∞ | (h) | 1.8 | ± | 0.8 | 0.7 | ± | 0.3 |
| F | (%) | 23.0 | |||||
PK parameters (mean ± SD, n = 5).
Dosed orally at 20 mg/kg.
Dosed intravenously at 2 mg/kg.
Figure 10.
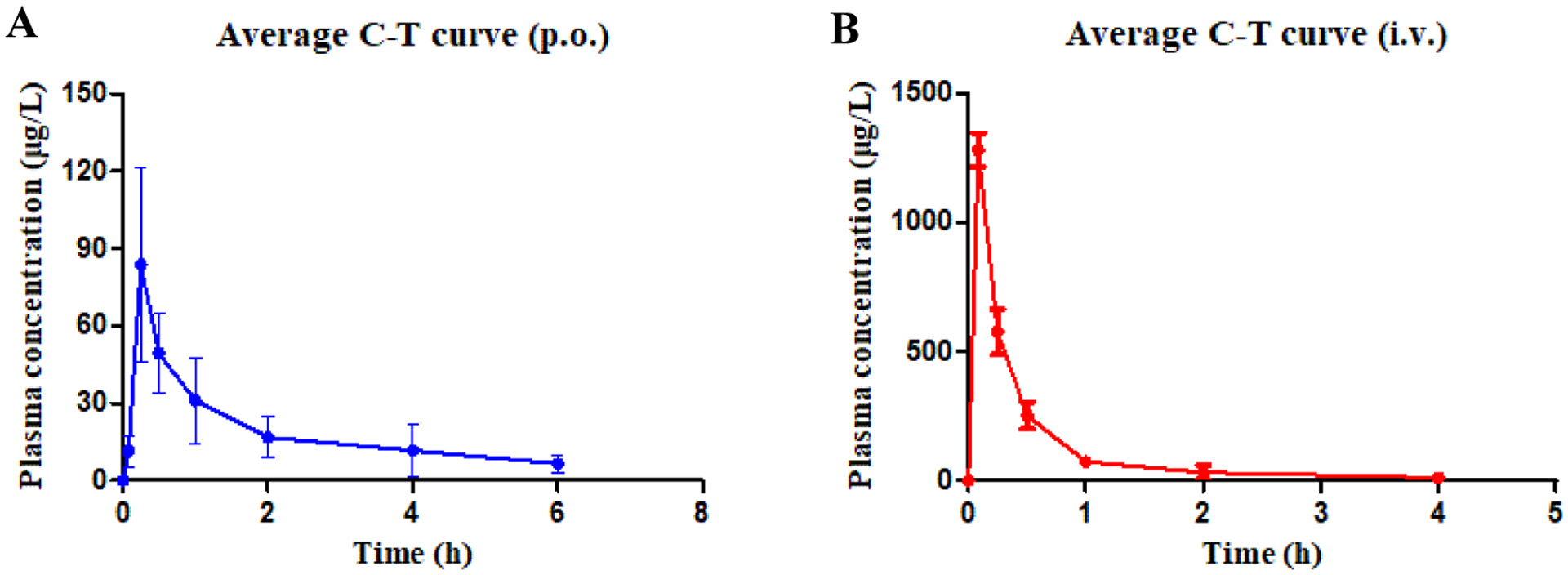
Plasma concentration–time profiles in rats following oral administration (11l, 20 mg/kg, A) and intravenous administration (11l, 2 mg/kg, B).
A single-dose acute toxicity of 11l was studied in Kunming mice (Figure 11). No death was observed after the intragastric administration of 11l with a dose of 1000 mg/kg, and there were no abnormal behaviors (lethargy, clonic convulsion, anorexia, and ruffled fur, etc.). Also, the body weights of all groups increased gradually over the subsequent of one week. Therefore, 11l was well-tolerated at a dose of 1000 mg·kg−1 with no acute toxicity.
Figure 11.
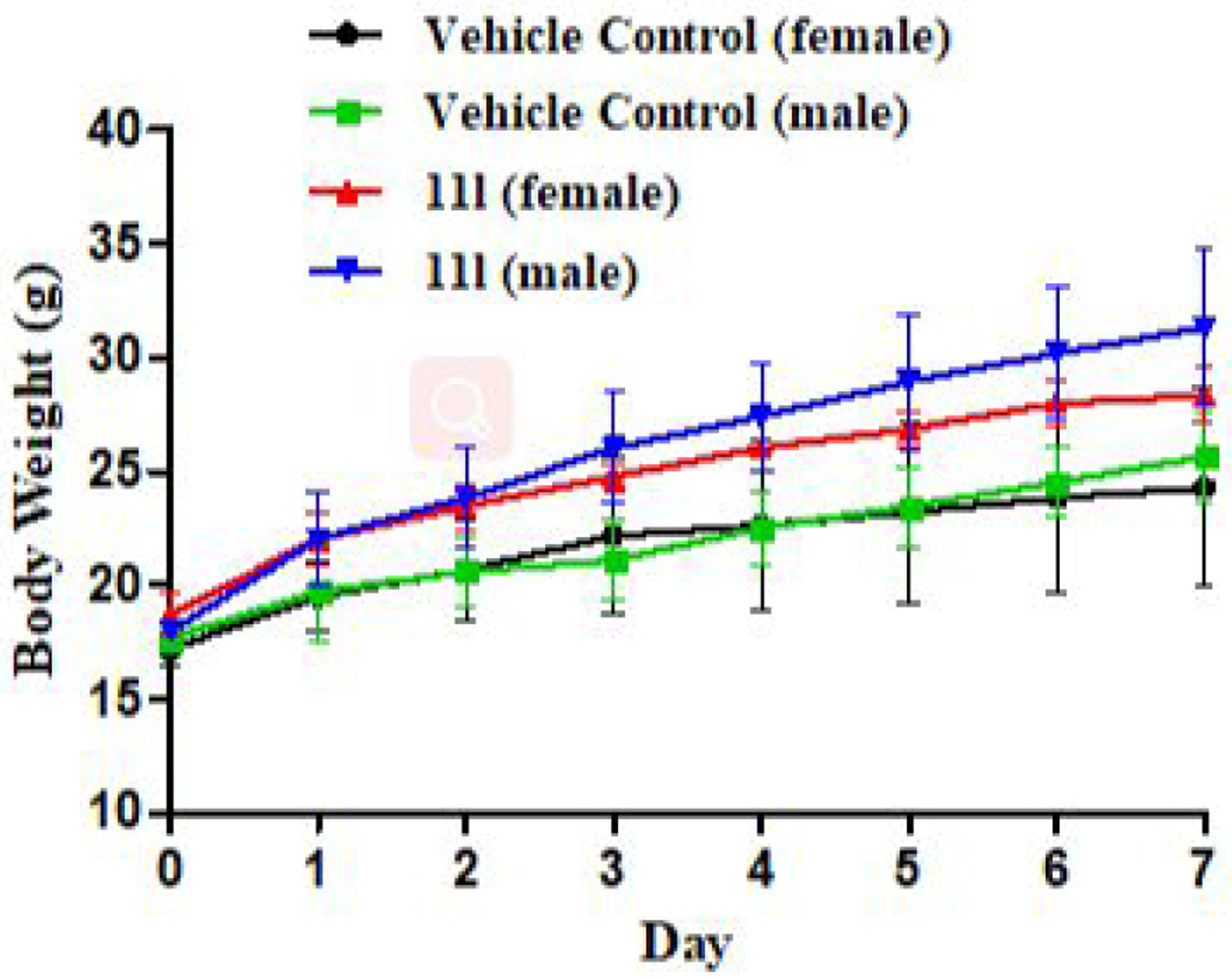
Bodyweight of all mice in four groups (g)-time (day).
4. CONCLUSION
In summary, 37 novel benzenesulfonamide derivatives were designed and synthesized by structural modification of the hit compound PF-74, producing more potent HIV-1 CA inhibitors.
The in vitro anti-HIV-1 assay results showed that all the derivatives (except for 6l) exhibited moderate to excellent antiviral activities against HIV-1 NL4–3 strain. Among the active compounds, the analogues in Series III with benzenesulfonamide substitutions on the piperazin-2-one motif exhibited greater antiviral activities (HIV-1 NL4–3 strain) than those with other structural motifs. The most potent analogue, 11l (EC50 = 90 nM), was 5.78-fold more potent than lead PF-74 (EC50 = 520 nM), with a prominent safety index > 383.36, which verified the rationality of the drug design. In addition, we found that 11a and 11l have antiviral activity against HIV-1IIIB and HIV-2ROD, and are particularly significant against HIV-2, with EC50 values of 32 and 31 nM, respectively. Since there are fewer studies on HIV-2, the discovery of these compounds is of great significance, and it can provide other researchers with valuable lead compounds to conduct their in-depth mechanisms of anti-HIV-2 activities outside the scope of this work. Subject to conditions, this paper only explored the HIV-1 mechanism of action of the compounds. The SPR results confirmed the binding target and demonstrated that these inhibitors could form direct and tight interactions with monomeric and hexameric CA protein constructs. The action stage determination assay also demonstrated that these inhibitors exhibited antiviral effects with a dual-stage inhibition profile. 11l, has the best inhibitory activity in the early- and late-stages (IC50 = 8.96 nM and 0.24 μM, respectively), with the early stage inhibition being 6.25 times more potent than the lead PF-74 (IC50 = 56 nM), thus demonstrating the highest IC50Late/IC50Early ratio (26.79). Skewing the action of a dual-stage inhibitor towards the early stages is attractive as it would preferentially prevent new cell infections, while maintaining a reduction of the infectivity of viruses produced by already infected cells. Based on the results of the p24 content and CA assembly studies, we postulate that the proposed mechanisms of action of these inhibitors differ from that of the parental PF-74. We believe that in the late stages, these compounds work by altering HIV-1 CA assembly, which in turn produces less infectious viral particles, whereas in the early stages, we believe that the compounds work by selectively acting on HIV-1 CA to accelerate uncoating such that it is not compatible with reverse transcription. These proposed modes of action require further investigation outside the scope of the current work. Moreover, MD simulations provided key clues for the outstanding antiviral activity of 11l. Further study on 11l demonstrated modest human plasma and liver microsomes stabilities. In addition, 11l exhibited acceptable in vivo PK profiles and favorable drug-like oral bioavailability in rats. No acute toxicity of 11l was shown at a single dose of 1000 mg·kg−1 in healthy Kunming mice.
Overall, based on the above in vitro and in vivo results, we believe that 11l demonstrated the utility of using bioisosterism replacement and scaffold hopping strategies for improvement of both potency and drug-like parameters and 11l may be a promising lead for further optimization.
5. EXPERIMENTAL SECTION
5.1. Chemistry
1H NMR and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer using solvents as indicated (DMSO-d6, CH3OH-d4). Chemical shifts were reported in δ values (ppm) with tetramethylsilane (TMS) as the internal reference, and J values were reported in hertz (Hz). Melting points (mp) were determined on a micromelting point apparatus and were uncorrected. TLC was performed on Silica Gel GF254 for TLC (Merck) and spots were visualized by iodine vapor or by irradiation with UV light (λ = 254 nm). Flash column chromatography was performed on column packed with Silica Gel60 (200–300 mesh). Thin layer chromatography was performed on pre-coated HUANGHAI_HSGF254, 0.15–0.2 mm TLC-plates. Solvents were of reagent grade and were purified and dried by standard methods when necessary.
Concentration of the reaction solutions involved the use of rotary evaporator at reduced pressure. The solvents of dichloromethane, TEA and methanol etc. were obtained from Sinopharm Chemical Reagent Co., Ltd (SCRC), which were of AR grade. The key reactants including 4-methoxy-N-methylaniline, N-(tert-butoxycarbonyl)-L-phenylalanine, Boc-glycine etc. were purchased from Bide Pharmatech Co. Ltd. The purity of representative final compounds was checked by HPLC and was >95%.
5.1.1. Tert-butyl (S)-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (2).
A solution of (tert-butoxycarbonyl)-L-phenylalanine (1, 2.90 g, 10.93 mmol, 1.5 eq.) in 20 mL dichloromethane was added PyBop (5.69 g, 10.93 mmol, 1.5 eq.) at 0°C, and the mixture stirred for 0.5 h. Subsequently, DIEA (3.61mL, 21.87 mmol, 3 eq.) and 4-methoxy-N-methylaniline (1.0 g, 7.29 mmol, 1 eq.) were added to the mixture and then stirred at room temperature for another 6 h (monitored by TLC). The resulting mixture was evaporated under reduced pressure and the residue was initially washed by 1N HCl and extracted with ethyl acetate (3 × 20 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude product, which was purified by flash column chromatography to afford intermediate 2 as yellow oil with a yield of 88%. 1H NMR (400 MHz, DMSO-d6) δ 7.22 (d, J = 8.3 Hz, 2H, Ph-H), 7.20 – 7.11 (m, 3H, Ph-H), 7.09 (d, J = 8.2 Hz, 1H, NH), 7.03 (d, J = 8.6 Hz, 2H, Ph-H), 6.79 (d, J = 7.3 Hz, 2H, Ph-H), 4.27 – 4.06 (m, 1H, CH), 3.81 (s, 3H, OCH3), 3.13 (s, 3H, NCH3), 2.75 (dd, J = 13.4, 3.8 Hz, 1H, PhCH), 2.61 (dd, J = 13.3, 10.3 Hz, 1H, PhCH), 1.30 (s, 9H, C(CH3)3). 13C NMR (100 MHz, DMSO-d6) δ 172.22 (C=O), 158.98, 155.75 (C=O), 138.53, 136.12, 129.28, 128.47, 126.70, 115.21, 78.33, 55.94, 53.55, 37.86, 37.07, 28.65. ESI-MS: m/z 385.4 (M+1)+, 407.5 (M+23)+. C22H28N2O4 [384.5].
5.1.2. (S)-2-amino-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (3).
Trifluoroacetic acid (3.86 mL, 52.02 mmol, 5.0 eq.) was added dropwise to intermediate 2 (4.0 g, 10.40 mmol, 1.0 eq.) in 30 mL dichloromethane and stirred at room temperature for 1 h. Then, the resulting mixture solution was alkalized to pH ~7 with saturated sodium bicarbonate solution, and then extracted with dichloromethane (40 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude product 3 as yellow oil with the yield of 80%. 1H NMR (400 MHz, DMSO-d6) δ 7.29 – 7.13 (m, 3H, Ph-H), 7.03 – 6.75 (m, 6H, Ph-H), 3.77 (s, 3H, OCH3), 3.44 – 3.35 (m, 1H, CH), 3.06 (s, 3H, NCH3), 2.75 (dd, J = 12.8, 6.7 Hz, 1H, PhCH), 2.45 (dd, J = 12.9, 7.1 Hz, 1H, PhCH), 1.87 (s, 2H, NH2). 13C NMR (100 MHz, DMSO-d6) δ 174.89 (C=O), 158.75, 139.00, 136.35, 129.51, 128.93, 128.47, 126.55, 115.04, 55.85, 53.35, 42.19, 37.45. ESI-MS: m/z 285.05 (M+1)+. C17H20N2O2 [284.36].
5.1.3. Tert-butyl (S)-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino) −2-oxoethyl)carbamate (4).
Boc-glycine (1.62 g, 9.24 mmol, 1.2 eq.) and 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) (4.39 g, 11.15 mmol, 1.5 eq.) were mixed in 20 mL dichloromethane and stirred in an ice bath for 1 h. Then, the intermediate 3 (2.19 g, 7.70 mmol, 1 eq.) and DIEA (2.55 mL, 15.40 mmol, 2 eq.) were added to the above solution slowly at 0°C. The reaction system was then stirred at room temperature for additional 6 h. The reaction was filtered, and concentrated under reduced pressure to afford corresponding crude product, which was purified by flash column chromatography to provide compound 4 as white solid with the yield of 80%, mp: 79–80°C. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 7.8 Hz, 1H, NH), 7.26 – 7.15 (m, 3H, Ph-H), 7.14 – 7.01 (m, 2H, BocNH+Ph-H), 6.96 (d, J = 8.8 Hz, 2H, Ph-H), 6.93 – 6.79 (m, 3H, Ph-H), 4.44 (q, J = 8.0 Hz, 1H, CH), 3.79 (s, 3H, OCH3), 3.53 (dd, J = 16.9, 6.1 Hz, 1H, BocNCH), 3.45 (dd, J = 16.8, 6.0 Hz, 1H, BocNCH), 3.09 (s, 3H, NCH3), 2.84 (dd, J = 13.3, 5.2 Hz, 1H, PhCH), 2.63 (dd, J = 13.3, 8.7 Hz, 1H, PhCH), 1.37 (s, 9H, C(CH3)3). 13C NMR (100 MHz, DMSO-d6) δ 171.46 (C=O), 169.31 (C=O), 158.97, 156.11 (C=O), 137.84, 135.91, 129.36, 129.10, 128.60, 126.89, 115.07, 78.42, 55.87, 51.63, 43.19, 37.99, 37.76, 28.66. ESI-MS: m/z 442.00 (M+1)+, 464.01 (M+23)+, 480.09 (M+39)+. C24H31N3O5 [441.53].
5.1.4. (S)-2-(2-aminoacetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (5).
Trifluoroacetic acid (5 mL) was added dropwise to intermediate 4 (2.7 g, 6.11 mmol) in 20 mL dichloromethane and stirred at room temperature for 12 h. Then, the resulting mixture solution was alkalized to pH ~7 with saturated sodium bicarbonate solution, and then extracted with dichloromethane (30 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude product 5 as yellow oil with the yield of 73%. ESI-MS: m/z 342.3 (M+1)+, 364.4 (M+23)+. C19H23N3O3 [341.4].
5.1.5. General procedure for the synthesis of 6a-6n.
Under ice bath, the key intermediate 5 (1 eq.), corresponding substituted benzenesulfonyl chloride (1 eq.), TEA (1 eq.) were dissolved in the solution of dichloromethane (10 mL). The resulting mixture was then stirred at room temperature (monitored by TLC). Then the reaction mixture was extracted with dichloromethane (30 mL), and the combined organic phase was washed with saturated NaCl solution (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the corresponding crude product, which was purified by recrystallization or preparation thin layer chromatography to afford product 6a-6n.
5.1.5.1. (S)-N-(4-methoxyphenyl)-N-methyl-3-phenyl-2-(2-(phenylsulfonamido)acetamid)propanamide (6a).
White solid, yield: 47%. mp: 69–70°C. 1H NMR (400 MHz, DMSO-d6) δ 8.20 (d, J = 7.9 Hz, 1H, NH), 7.87 (t, J = 6.1 Hz, 1H, SO2NH), 7.76 (d, J = 7.3 Hz, 2H, Ph-H), 7.63 (t, J = 7.3 Hz, 1H, Ph-H), 7.56 (t, J = 7.4 Hz, 2H, Ph-H), 7.24 – 7.14 (m, 3H, Ph-H), 7.02 – 6.89 (m, 4H, Ph-H), 6.89 – 6.81 (m, 2H, Ph-H), 4.36 (td, J = 8.1, 5.9 Hz, 1H, CH), 3.79 (s, 3H, OCH3), 3.49 – 3.36 (m, 2H, SO2NCH2), 3.08 (s, 3H, NCH3), 2.81 (dd, J = 13.4, 5.7 Hz, 1H, PhCH), 2.56 (dd, J = 13.4, 8.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.15 (C=O), 167.24 (C=O), 158.95, 140.74, 137.62, 135.80, 132.84, 129.51, 129.36, 129.05, 128.62, 127.01, 126.95, 115.03, 55.87, 51.52, 45.22, 38.05, 37.75. HRMS: m/z 482.1748 (M+1)+, 985.3245 (2M+23)+. C25H27N3O5S [481.1671].
5.1.5.2. (S)-2-(2-((4-fluorophenyl)sulfonamido)acetamido)-N-(4-methoxyphenyl)-N-meth yl-3-phenylpropanamide (6b).
White solid, yield: 46%. mp: 75–76°C. 1H NMR (400 MHz, DMSO-d6) δ 8.24 (d, J = 7.9 Hz, 1H, NH), 7.92 (t, J = 5.4 Hz, 1H, SO2NH), 7.80 (dd, J = 8.7, 5.3 Hz, 2H, Ph-H), 7.36 (t, J = 8.8 Hz, 2H, Ph-H), 7.25 – 7.15 (m, 3H, Ph-H), 7.01 – 6.88 (m, 4H, Ph-H), 6.88 – 6.80 (m, 2H, Ph-H), 4.32 (td, J = 8.1, 6.0 Hz, 1H, CH), 3.79 (s, 3H, OCH3), 3.49 (dd, J = 16.4, 4.7 Hz, 1H, SO2NCH), 3.39 (dd, J = 16.9, 4.8 Hz, 1H, SO2NCH), 3.08 (s, 3H, NCH3), 2.81 (dd, J = 13.4, 5.6 Hz, 1H, PhCH), 2.55 (dd, J = 13.4, 8.7 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.14 (C=O), 167.18 (C=O), 164.53 (d, 1JCF = 248.8 Hz), 158.96, 137.64, 137.22 (d, 4JCF = 3.0 Hz), 135.80, 130.13, 130.03, 129.35, 129.01, 128.62, 126.95, 116.64, 116.41, 115.00, 55.85, 51.56, 45.15, 38.02, 37.71. HRMS: m/z 500.1651 (M+1)+, 1021.3082 (2M+23)+. C25H26FN3O5S [499.1577].
5.1.5.3. (S)-2-(2-((4-chlorophenyl)sulfonamido)acetamido)-N-(4-methoxyphenyl)-N-meth yl-3-phenylpropanamide (6c).
White solid, yield: 32%. mp: 152–153°C. 1H NMR (400 MHz, DMSO-d6) δ 8.25 (d, J = 7.9 Hz, 1H, NH), 8.00 (s, 1H, SO2NH), 7.73 (d, J = 8.5 Hz, 2H, Ph-H), 7.59 (d, J = 8.6 Hz, 2H, Ph-H), 7.26 – 7.15 (m, 3H, Ph-H), 7.06 – 6.89 (m, 4H, Ph-H), 6.84 (dd, J = 6.9, 2.0 Hz, 2H, Ph-H), 4.31 (td, J = 8.1, 5.9 Hz, 1H, CH), 3.79 (s, 3H, OCH3), 3.51 (d, J = 16.7 Hz, 1H, SO2NCH), 3.40 (d, J = 16.8 Hz, 1H, SO2NCH), 3.08 (s, 3H, NCH3), 2.80 (dd, J = 13.4, 5.7 Hz, 1H, PhCH), 2.61 – 2.52 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.15 (C=O), 167.13 (C=O), 158.96, 139.77, 137.64, 137.62, 135.79, 129.51, 129.35, 129.03, 129.01, 128.62, 126.95, 115.00, 55.86, 51.54, 45.12, 38.03, 37.73. HRMS: m/z 516.1351 (M+1)+, 1053.2422 (2M+23)+. C25H26ClN3O5S [515.1282].
5.1.5.4. (S)-2-(2-((4-bromophenyl)sulfonamido)acetamido)-N-(4-methoxyphenyl)-N-meth yl-3-phenylpropanamide (6d).
White solid, yield: 35%. mp: 167–168°C. 1H NMR (400 MHz, DMSO-d6) δ 8.25 (d, J = 7.9 Hz, 1H, NH), 8.00 (s, 1H, SO2NH), 7.74 (d, J = 8.5 Hz, 2H, Ph-H), 7.66 (d, J = 8.5 Hz, 2H, Ph-H), 7.26 – 7.14 (m, 3H, Ph-H), 7.01 – 6.89 (m, 4H, Ph-H), 6.87 – 6.79 (m, 2H, Ph-H), 4.32 (td, J = 8.1, 6.2 Hz, 1H, CH), 3.79 (s, 3H, OCH3), 3.58 – 3.46 (m, 1H, SO2NCH), 3.40 (dd, J = 17.0, 3.6 Hz, 1H, SO2NCH), 3.09 (s, 3H, NCH3), 2.80 (dd, J = 13.4, 5.7 Hz, 1H, PhCH), 2.60 – 2.52 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.16 (C=O), 167.12 (C=O), 158.96, 140.20, 137.64, 135.78, 132.45, 129.35, 129.13, 129.01, 128.62, 126.95, 126.58, 115.00, 55.87, 51.53, 45.12, 38.05, 37.74. HRMS: m/z 560.0851 (M+1)+, 562.0832 (M+3)+. C25H26BrN3O5S [559.0777].
5.1.5.5. (S)-N-(4-methoxyphenyl)-N-methyl-3-phenyl-2-(2-((2,4,6-triisopropylphenyl)sulf onamido)acetamido)propanamide (6e).
White solid, yield: 38%. mp: 179–180°C. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 8.1 Hz, 1H, NH), 7.53 (t, J = 5.9 Hz, 1H, SO2NH), 7.23 (s, 2H, Ph-H), 7.20 – 7.11 (m, 3H, Ph-H), 6.97 – 6.80 (m, 6H, Ph-H), 4.40 (q, J = 7.8 Hz, 1H, CH), 4.04 (hept, J = 6.2 Hz, 2H, (CH3)2CH×2), 3.78 (s, 3H, OCH3), 3.49 – 3.42 (m, 2H, SO2NCH2), 3.05 (s, 3H, NCH3), 2.90 (p, J = 6.8 Hz, 1H, (CH3)2CH), 2.80 (dd, J = 13.3, 6.1 Hz, 1H, PhCH), 2.55 (d, J = 8.2 Hz, 1H, PhCH), 1.23 – 1.17 (m, 18H, C(CH3)2×3). 13C NMR (100 MHz, DMSO-d6) δ 171.09 (C=O), 167.34 (C=O), 158.91, 152.41, 150.05, 137.52, 135.74, 133.67, 129.37, 129.00, 128.58, 126.93, 123.95, 114.99, 55.84, 51.45, 46.06, 38.27, 37.71, 33.78, 29.44, 25.23, 25.20, 23.92, 23.86. HRMS: m/z 608.3157 (M+1)+, 1237.6070 (2M+23)+. C34H45N3O5S [607.3080].
5.1.5.6. (S)-N-(4-methoxyphenyl)-2-(2-((4-methoxyphenyl)sulfonamido)acetamido)-N-me thyl-3-phenylpropanamide (6f).
White solid, yield: 63%. mp: 76–77°C. 1H NMR (400 MHz, DMSO-d6) δ 8.17 (d, J = 7.9 Hz, 1H, NH), 7.76 – 7.63 (m, 3H, SO2NH+Ph-H×2), 7.27 – 7.16 (m, 3H, Ph-H), 7.05 (d, J = 8.8 Hz, 2H, Ph-H), 7.01 – 6.89 (m, 4H, Ph-H), 6.89 – 6.81 (m, 2H, Ph-H), 4.35 (td, J = 8.0, 5.9 Hz, 1H, CH), 3.80 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.46 – 3.35 (m, 2H, SO2NCH2), 3.08 (s, 3H, NCH3), 2.81 (dd, J = 13.4, 5.7 Hz, 1H, PhCH), 2.55 (dd, J = 13.6, 8.7 Hz, 1H, PhCH).13C NMR (100 MHz, DMSO-d6) δ 171.15 (C=O), 167.31 (C=O), 162.59, 158.95, 137.62, 135.81, 132.34, 129.36, 129.25, 129.02, 128.62, 126.95, 115.01, 114.61, 56.06, 55.86, 51.52, 45.26, 38.09, 37.73. HRMS: m/z 512.1850 (M+1)+, 1045.3434 (2M+23)+. C26H29N3O6S [511.1777].
5.1.5.7. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-((4-methylphenyl)sulfonamido)acetamid o)-3-phenylpropanamide (6g).
White solid, yield: 78%. mp: 66–67°C. 1H NMR (400 MHz, DMSO-d6) δ 8.23 (d, J = 7.9 Hz, 1H, NH), 7.81 (t, J = 6.0 Hz, 1H, SO2NH), 7.69 (d, J = 8.1 Hz, 2H, Ph-H), 7.39 (d, J = 8.1 Hz, 2H, Ph-H), 7.33 – 7.20 (m, 3H, Ph-H), 7.10 – 6.94 (m, 4H, Ph-H), 6.94 – 6.85 (m, 2H, Ph-H), 4.40 (td, J = 8.0, 6.1 Hz, 1H, CH), 3.84 (s, 3H, OCH3), 3.51 – 3.41 (m, 2H, SO2NCH2), 3.13 (s, 3H, NCH3), 2.86 (dd, J = 13.4, 5.7 Hz, 1H, PhCH), 2.65 – 2.57 (m, 1H, PhCH), 2.41 (s, 3H, PhCH3). 13C NMR (100 MHz, DMSO-d6) δ 171.16 (C=O), 167.26 (C=O), 158.95, 143.05, 137.84, 137.62, 135.81, 129.92, 129.35, 129.02, 128.62, 127.10, 126.95, 115.02, 55.87, 51.52, 45.24, 38.08, 37.74, 21.42. HRMS: m/z 496.1898 (M+1)+, 1013.3557 (2M+23)+. C26H29N3O5S [495.1828].
5.1.5.8. (S)-2-(2-((3,5-dimethylphenyl)sulfonamido)acetamido)-N-(4-methoxyphenyl)-N- methyl-3-phenylpropanamide (6h).
White solid, yield: 57%. mp: 156–157°C. 1H NMR (400 MHz, DMSO-d6) δ 8.19 (d, J = 7.9 Hz, 1H, NH), 7.79 (t, J = 6.1 Hz, 1H, SO2NH), 7.49 – 7.41 (m, 2H, Ph-H), 7.32 (s, 1H, Ph-H), 7.29 – 7.19 (m, 3H, Ph-H), 7.07 – 6.94 (m, 4H, Ph-H), 6.94 – 6.84 (m, 2H, Ph-H), 4.44 (q, J = 8.0 Hz, 1H, CH), 3.84 (s, 3H, OCH3), 3.53 – 3.40 (m, 2H, SO2NCH2), 3.13 (s, 3H, NCH3), 2.86 (dd, J = 13.4, 5.8 Hz, 1H, PhCH), 2.68 – 2.58 (m, 1H, PhCH), 2.39 (s, 6H, PhCH3×2). 13C NMR (100 MHz, DMSO-d6) δ 171.13 (C=O), 167.32 (C=O), 158.96, 140.52, 139.02, 137.58, 135.80, 134.18, 129.35, 129.03, 128.63, 126.96, 124.53, 115.02, 55.87, 51.50, 45.26, 38.10, 37.73, 21.23. HRMS: m/z 510.2059 (M+1)+, 1041.3879 (2M+23)+. C27H31N3O5S [509.1984].
5.1.5.9. (S)-N-(4-methoxyphenyl)-N-methyl-3-phenyl-2-(2-((2,4,6-trimethylphenyl)sulfon amido)acetamido)propanamide (6i).
White solid, yield: 34%. mp: 64–65°C. 1H NMR (400 MHz, DMSO-d6) δ 8.14 (d, J = 8.0 Hz, 1H, NH), 7.63 (t, J = 6.1 Hz, 1H, SO2NH), 7.31 – 7.20 (m, 3H, Ph-H), 7.02 (s, 2H, Ph-H), 6.98 – 6.83 (m, 6H, Ph-H), 4.34 (q, J = 7.7 Hz, 1H, CH), 3.83 (s, 3H, OCH3), 3.48 – 3.42 (m, 2H, SO2NCH2), 3.10 (s, 3H, NCH3), 2.81 (dd, J = 13.3, 6.2 Hz, 1H, PhCH), 2.57 (s, 6H, PhCH3×2), 2.50 (dd, J = 13.3, 8.0 Hz, 1H, PhCH), 2.25 (s, 3H, PhCH3). 13C NMR (100 MHz, DMSO-d6) δ 171.06 (C=O), 167.34 (C=O), 158.90, 141.76, 138.93, 137.54, 135.76, 134.70, 131.96, 129.35, 128.95, 128.61, 126.93, 114.98, 55.85, 51.37, 44.49, 38.25, 37.70, 23.05, 20.83. HRMS: m/z 524.2215 (M+1)+, 1069.4181 (2M+23)+. C28H33N3O5S [523.2141].
5.1.5.10. (S)-2-(2-((2-fluorophenyl)sulfonamido)acetamido)-N-(4-methoxyphenyl)-N-meth yl-3-phenylpropanamide (6j).
White solid, yield: 63%. mp: 126–127°C. 1H NMR (400 MHz, DMSO-d6) δ 8.26 (d, J = 7.9 Hz, 1H, NH), 8.14 (s, 1H, SO2NH), 7.84 – 7.67 (m, 2H, Ph-H), 7.51 – 7.40 (m, 1H, Ph-H), 7.37 (t, J = 7.6 Hz, 1H, Ph-H), 7.30 – 7.20 (m, 3H, Ph-H), 6.97 (s, 4H, Ph-H), 6.94 – 6.84 (m, 2H, Ph-H), 4.39 (q, J = 7.9 Hz, 1H, CH), 3.84 (s, 3H, OCH3), 3.61 (q, J = 16.9 Hz, 2H, SO2NCH2), 3.12 (s, 3H, NCH3), 2.85 (dd, J = 13.4, 5.9 Hz, 1H, PhCH), 2.67 – 2.58 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.12 (C=O), 167.32 (C=O), 158.92, 158.77 (d, 1JCF = 251.9 Hz), 137.61, 135.80, 135.45 (d, 3JCF = 8.7 Hz), 129.81, 129.36, 129.02, 128.62, 126.94, 125.02 (d, 4JCF = 3.4 Hz), 117.50 (d, 2JCF = 21.0 Hz), 115.01, 55.87, 51.51, 45.03, 38.10, 37.73. HRMS: m/z 500.1645 (M+1)+, 1021.3026 (2M+23)+. C25H26FN3O5S [499.1577].
5.1.5.11. (S)-2-(2-((3-fluorophenyl)sulfonamido)acetamido)-N-(4-methoxyphenyl)-N-meth yl-3-phenylpropanamide (6k).
White solid, yield: 57%. mp: 114–115°C. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (d, J = 7.9 Hz, 1H, NH), 8.09 (s, 1H, SO2NH), 7.72 – 7.59 (m, 3H, Ph-H), 7.58 – 7.50 (m, 1H, Ph-H), 7.30 – 7.18 (m, 3H, Ph-H), 7.07 – 6.94 (m, 4H, Ph-H), 6.94 – 6.85 (m, 2H, Ph-H), 4.40 (q, J = 8.0 Hz, 1H, CH), 3.84 (s, 3H, OCH3), 3.56 (d, J = 16.5 Hz, 1H, SO2NCH), 3.46 (d, J = 16.6 Hz, 1H, SO2NCH), 3.13 (s, 3H, NCH3), 2.86 (dd, J = 13.4, 5.8 Hz, 1H, PhCH), 2.61 (dd, J = 13.2, 8.2 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.12 (C=O), 167.16 (C=O), 162.11 (d, 1JCF = 246.3 Hz), 158.95, 143.01 (d, 3JCF = 6.7 Hz), 137.60, 135.80, 131.83 (d, 3JCF = 7.9 Hz), 129.34, 129.03, 128.62, 126.95, 123.27, 123.24, 119.94 (d, 2JCF = 21.1 Hz), 115.00, 114.12 (d, 2JCF = 24.2 Hz), 55.86, 51.52, 45.15, 38.05, 37.73. HRMS: m/z 500.1650 (M+1)+, 1021.3056 (2M+23)+. C25H26FN3O5S [499.1577].
5.1.5.12. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-((4-nitrophenyl)sulfonamido)acetamido) −3-phenylpropanamide (6l).
White solid, yield: 39%. mp: 226–227°C. 1H NMR (400 MHz, DMSO-d6) δ 8.33 (d, J = 8.7 Hz, 4H, NH+SO2NH+Ph-H×2), 7.95 (d, J = 8.8 Hz, 2H, Ph-H), 7.32 – 7.13 (m, 3H, Ph-H), 7.01 – 6.86 (m, 4H, Ph-H), 6.86 – 6.75 (m, 2H, Ph-H), 4.34 – 4.19 (m, 1H, CH), 3.78 (s, 3H, OCH3), 3.61 (d, J = 16.4 Hz, 1H, SO2NCH), 3.49 (d, J = 16.6 Hz, 1H, SO2NCH), 3.06 (s, 3H, NCH3), 2.80 (dd, J = 13.4, 5.5 Hz, 1H, PhCH), 2.62 – 2.53 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.17 (C=O), 167.07 (C=O), 158.96, 149.81, 146.66, 137.67, 135.76, 129.32, 128.97, 128.66, 128.62, 126.95, 124.65, 114.92, 55.83, 51.67, 45.06, 37.91, 37.67. HRMS: m/z 527.1596 (M+1)+, 1075.2943 (2M+23)+. C25H26N4O7S [526.1522].
5.1.5.13. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-((2-nitrophenyl)sulfonamido)acetamido) −3-phenylpropanamide (6m).
White solid, yield: 58%. mp: 111–112°C. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (d, J = 7.9 Hz, 1H, NH), 8.15 (s, 1H, SO2NH), 8.03 – 7.92 (m, 2H, Ph-H), 7.89 – 7.76 (m, 2H, Ph-H), 7.24 – 7.14 (m, 3H, Ph-H), 7.02 – 6.88 (m, 4H, Ph-H), 6.88 – 6.79 (m, 2H, Ph-H), 4.35 (q, J = 8.1 Hz, 1H, CH), 3.79 (s, 3H, OCH3), 3.66 (d, J = 16.7 Hz, 1H, SO2NCH), 3.56 (d, J = 16.8 Hz, 1H, SO2NCH), 3.07 (s, 3H, NCH3), 2.81 (dd, J = 13.4, 5.7 Hz, 1H, PhCH), 2.60 – 2.53 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.11 (C=O), 167.16 (C=O), 158.94, 147.88, 137.61, 135.78, 134.33, 133.56, 133.07, 130.15, 129.35, 129.02, 128.62, 126.95, 124.92, 115.02, 55.87, 51.61, 45.29, 38.06, 37.75. HRMS: m/z 527.1593 (M+1)+, 1075.2935 (2M+23)+. C25H26N4O7S [526.1522].
5.1.5.14. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-((3-nitrophenyl)sulfonamido)acetamido) −3-phenylpropanamide (6n).
White solid, yield: 46%. mp: 80–81°C. 1H NMR (400 MHz, DMSO-d6) δ 8.59 – 8.50 (m, 1H, Ph-H), 8.50 – 8.43 (m, 1H, Ph-H), 8.37 – 8.26 (m, 2H, Ph-H), 8.13 (d, J = 7.9 Hz, 1H, NH), 7.83 (t, J = 8.0 Hz, 1H, SO2NH), 7.25 – 7.12 (m, 3H, Ph-H), 6.97 – 6.86 (m, 4H, Ph-H), 6.86 – 6.74 (m, 2H, Ph-H), 4.25 (td, J = 8.1, 5.9 Hz, 1H, CH), 3.78 (s, 3H, OCH3), 3.60 (d, J = 16.6 Hz, 1H, SO2NCH), 3.48 (d, J = 16.8 Hz, 1H, SO2NCH), 3.04 (s, 3H, NCH3), 2.78 (dd, J = 13.4, 5.6 Hz, 1H, PhCH), 2.58 – 2.52 (m, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.06 (C=O), 167.07 (C=O), 158.93, 148.11, 142.64, 137.59, 135.77, 133.23, 131.36, 129.30, 128.99, 128.61, 127.35, 126.95, 122.09, 114.93, 55.85, 51.59, 45.00, 37.91, 37.66. HRMS: m/z 527.1591 (M+1)+, 1075.2938 (2M+23)+. C25H26N4O7S [526.1522].
5.1.6. General procedure for the synthesis of compounds (6o-6q).
(6l−6n; 0.15–0.19 mmol) were dissolved in methanol (5 mL) and dichloromethane (5 mL) and the solution degassed and stirred at room temperature under H2 over 10% Pd●C (10% w/w, 2h). The mixture was filtered and concentrated, and the resulting residue were purified by recrystallization or preparation thin layer chromatography to provide the target compounds (6o-6q).
5.1.6.1. (S)-2-(2-((4-aminophenyl)sulfonamido)acetamido)-N-(4-methoxyphenyl)-N-meth yl-3-phenylpropanamide (6o).
Yellow solid, yield: 94%. mp: 90–91°C. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 8.0 Hz, 1H, NH), 7.39 (d, J = 8.6 Hz, 2H, Ph-H), 7.29 (t, J = 6.2 Hz, 1H, SO2NH), 7.24 – 7.14 (m, 3H, Ph-H), 7.03 – 6.91 (m, 4H, Ph-H), 6.90 – 6.79 (m, 2H, Ph-H), 6.59 (d, J = 8.7 Hz, 2H, Ph-H), 5.96 (s, 2H, NH2), 4.41 (td, J = 8.0, 6.0 Hz, 1H, CH), 3.78 (s, 3H, OCH3), 3.34 – 3.27 (m, 1H, SO2NCH), 3.22 (dd, J = 16.4, 6.3 Hz, 1H, SO2NCH), 3.09 (s, 3H, NCH3), 2.82 (dd, J = 13.3, 5.8 Hz, 1H, PhCH), 2.57 (dd, J = 13.4, 8.3 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.11 (C=O), 167.53 (C=O), 158.95, 153.09, 137.59, 135.79, 129.39, 129.06, 128.62, 126.94, 125.27, 115.05, 113.02, 55.86, 51.40, 45.38, 38.18, 37.75. HRMS: m/z 497.1848 (M+1)+, 1015.3501 (2M+23)+. C25H28N4O5S [496.1780].
5.1.6.2. (S)-2-(2-((2-aminophenyl)sulfonamido)acetamido)-N-(4-methoxyphenyl)-N-meth yl-3-phenylpropanamide (6p).
White solid, yield: 49%. mp: 78–79°C. 1H NMR (400 MHz, DMSO-d6) δ 8.17 (d, J = 7.9 Hz, 1H, NH), 7.68 (t, J = 6.0 Hz, 1H, SO2NH), 7.51 – 7.42 (m, 1H, Ph-H), 7.30 – 7.23 (m, 1H, Ph-H), 7.23 – 7.14 (m, 3H, Ph-H), 7.05 – 6.89 (m, 4H, Ph-H), 6.89 – 6.83 (m, 2H, Ph-H), 6.80 (d, J = 8.1 Hz, 1H, Ph-H), 6.59 (t, J = 7.3 Hz, 1H, Ph-H), 5.95 (s, 2H, NH2), 4.39 (td, J = 8.0, 6.0 Hz, 1H, CH), 3.79 (s, 3H, OCH3), 3.34 – 3.25 (m, 2H, SO2NCH2), 3.08 (s, 3H, NCH3), 2.82 (dd, J = 13.4, 5.8 Hz, 1H, PhCH), 2.56 (dd, J = 13.4, 8.4 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.12 (C=O), 167.34 (C=O), 158.95, 146.79, 137.61, 135.81, 134.05, 129.58, 129.36, 129.05, 128.63, 126.95, 119.66, 117.40, 115.42, 115.04, 55.87, 51.55, 44.77, 38.09, 37.75. HRMS: m/z 497.1855 (M+1)+, 1015.3449 (2M+23)+. C25H28N4O5S [496.1780].
5.1.6.3. (S)-2-(2-((3-aminophenyl)sulfonamido)acetamido)-N-(4-methoxyphenyl)-N-meth yl-3-phenylpropanamide (6q).
White solid, yield: 73%. mp: 74–75°C. 1H NMR (400 MHz, DMSO-d6) δ 8.14 (d, J = 7.9 Hz, 1H, NH), 7.63 (t, J = 6.1 Hz, 1H, SO2NH), 7.27 – 7.11 (m, 4H, Ph-H), 7.08 – 6.90 (m, 5H, Ph-H), 6.90 – 6.81 (m, 3H, Ph-H), 6.79 – 6.71 (m, 1H, Ph-H), 5.58 (s, 2H, NH2), 4.41 (q, J = 7.9 Hz, 1H, CH), 3.79 (s, 3H, OCH3), 3.35 – 3.23 (m, 2H, SO2NCH2), 3.08 (s, 3H, NCH3), 2.82 (dd, J = 13.3, 5.7 Hz, 1H, PhCH), 2.58 (dd, J = 13.4, 8.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.17 (C=O), 167.40 (C=O), 158.96, 149.79, 140.98, 137.61, 135.80, 129.96, 129.38, 129.07, 128.63, 126.95, 117.69, 115.05, 113.64, 111.55, 55.87, 51.53, 45.37, 38.08, 37.76. HRMS: m/z 497.1855 (M+1)+. C25H28N4O5S [496.1780].
5.1.7. (S)-2-(2-bromoacetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamid e (7).
Bromoacetic acid (117 mg, 0.84 mmol, 1.2 eq.) and HATU (401 mg, 1.06 mmol, 1.5 eq.) were mixed in 15 mL dichloromethane and stirred in an ice bath for 1 h. Then, the intermediate 3 (200 mg, 0.70 mmol, 1 eq.) and DIEA (232 μL, 1.41 mmol, 2 eq.) were added to the above solution slowly at 0 °C. The reaction system was then stirred at room temperature for additional 6 h. The reaction was filtered, and concentrated under reduced pressure to afford corresponding crude product 7 as white oil with the yield of 68%. ESI-MS: m/z 405.4 (M+1)+. C19H21BrN2O3 [404.1].
5.1.8. General procedure for the synthesis of 8a-8f.
Under ice bath, the key intermediate 7 (1 eq.), corresponding 7-substituted 2H-benzo[e][1,2,4]thiadiazin-3(4H)-one 1,1-dioxide (1 eq.), Na2CO3 (1 eq.) were dissolved in the solution of DMF (5 mL). The resulting mixture was then stirred at 40°C (monitored by TLC). Then the reaction mixture was extracted with ethyl acetate (30 mL), and the combined organic phase was washed with saturated NaCl solution (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the corresponding crude target product, which was purified by recrystallization or preparation thin layer chromatography to afford product 8a-8f.
5.1.8.1. (S)-2-(2-(1,1-dioxido-3-oxo-3,4-dihydro-2H-benzo[e][1,2,4]thiadiazin-2-yl)acetam ido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (8a).
White solid, yield: 19%. mp: 197–198°C. 1H NMR (400 MHz, DMSO-d6) δ 11.42 (s, 1H, PhNH), 8.61 (d, J = 8.0 Hz, 1H, NH), 7.83 (d, J = 7.9 Hz, 1H, Ph-H), 7.73 (t, J = 7.8 Hz, 1H, Ph-H), 7.39 – 7.27 (m, 2H, Ph-H), 7.19 (d, J = 6.5 Hz, 3H, Ph-H), 7.01 (d, J = 7.6 Hz, 2H, Ph-H), 6.91 (d, J = 8.9 Hz, 2H, Ph-H), 6.89 – 6.79 (m, 2H, Ph-H), 4.46 – 4.22 (m, 3H, CH+NCH2), 3.75 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.85 (dd, J = 13.4, 5.4 Hz, 1H, PhCH), 2.63 (dd, J = 13.4, 8.6 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.11 (C=O), 165.54 (C=O), 158.93, 149.91 (C=O), 137.66, 135.76, 135.23, 134.96, 129.32, 129.01, 128.62, 126.92, 123.86, 122.69, 122.41, 117.63, 115.10, 55.82, 51.87, 42.02, 38.01, 37.75. HRMS: m/z 523.1644 (M+1)+. C26H26N4O6S [522.1573].
5.1.8.2. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(7-methyl-1,1-dioxido-3-oxo-3,4-dihydro −2H-benzo[e][1,2,4]thiadiazin-2-yl)acetamido)-3-phenylpropanamide (8b).
Yellow solid, yield: 36%. mp: 126–127°C.1H NMR (400 MHz, DMSO-d6) δ 11.29 (s, 1H, PhNH), 8.58 (d, J = 7.9 Hz, 1H, NH), 7.64 (s, 1H, Ph-H), 7.54 (d, J = 8.3 Hz, 1H, Ph-H), 7.19 (d, J = 8.1 Hz, 4H, Ph-H), 7.01 (d, J = 7.7 Hz, 2H, Ph-H), 6.91 (d, J = 8.9 Hz, 2H, Ph-H), 6.89 – 6.84 (m, 2H, Ph-H), 4.46 – 4.25 (m, 3H, CH+NCH2), 3.75 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.85 (dd, J = 13.4, 5.5 Hz, 1H, PhCH), 2.63 (dd, J = 13.4, 8.5 Hz, 1H, PhCH), 2.36 (s, 3H, PhCH3). 13C NMR (100 MHz, DMSO-d6) δ 171.12 (C=O), 165.60 (C=O), 158.93, 149.94 (C=O), 137.67, 135.91, 135.77, 133.62, 132.62, 129.32, 129.01, 128.61, 126.91, 122.54, 121.83, 117.56, 115.09, 55.80, 51.87, 41.96, 38.01, 37.75, 20.50. HRMS: m/z 537.1802 (M+1)+, 1095.3346 (2M+23)+. C27H28N4O6S [536.1730].
5.1.8.3. (S)-2-(2-(7-methoxy-1,1-dioxido-3-oxo-3,4-dihydro-2H-benzo[e][1,2,4]thiadiazin- 2-yl)acetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (8c).
White solid, yield: 12%. mp: 114–115°C. 1H NMR (400 MHz, DMSO-d6) δ 11.22 (s, 1H, PhNH), 8.58 (d, J = 7.9 Hz, 1H, NH), 7.34 (dd, J = 8.9, 2.7 Hz, 1H, Ph-H), 7.28 (d, J = 2.6 Hz, 1H, Ph-H), 7.24 (d, J = 8.9 Hz, 1H, Ph-H), 7.22 – 7.15 (m, 3H, Ph-H), 7.02 (d, J = 7.7 Hz, 2H, Ph-H), 6.92 (d, J = 8.9 Hz, 2H, Ph-H), 6.89 – 6.84 (m, 2H, Ph-H), 4.47 – 4.23 (m, 3H, CH+NCH2), 3.83 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 3.09 (s, 3H, NCH3), 2.86 (dd, J = 13.4, 5.4 Hz, 1H, PhCH), 2.63 (dd, J = 13.4, 8.6 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.13 (C=O), 165.63 (C=O), 158.94, 155.39, 149.81 (C=O), 137.68, 135.77, 129.33, 129.02, 128.62, 128.48, 126.92, 123.24, 122.98, 119.45, 115.10, 104.99, 56.50, 55.81, 51.89, 42.03, 38.00, 37.75. HRMS: m/z 553.1755 (M+1)+, 1127.3273 (2M+23)+. C27H28N4O7S [552.1679].
5.1.8.4. (2S,2’S)-2,2’-((2,2’-(7-fluoro-1,1-dioxido-3-oxo-2H-benzo[e][1,2,4]thiadiazine-2,4(3H)-diyl)bis(acetyl))bis(azanediyl))bis(N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide) (8d).
Yellow solid, yield: 39%. mp: 119–120°C. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (d, J = 8.3 Hz, 1H, NH), 8.61 (d, J = 8.0 Hz, 1H, NH), 7.84 (dd, J = 7.0, 2.9 Hz, 1H, Ph-H), 7.54 (td, J = 8.9, 2.8 Hz, 1H, Ph-H), 7.42 – 6.70 (m, 18H, Ph-H), 4.73 – 4.18 (m, 6H, CH×2+NCH2×2), 3.75 (s, 6H, OCH3×2), 3.11 (s, 3H, NCH3), 3.08 (s, 3H, NCH3), 2.87 (ddd, J = 22.2, 13.4, 5.4 Hz, 2H, PhCH×2), 2.72 – 2.57 (m, 2H, PhCH×2). 13C NMR (100 MHz, DMSO-d6) δ 171.14, 171.05, 166.04, 165.32, 158.98, 158.92, 156.58, 150.42, 137.73, 137.59, 135.79, 135.74, 133.33, 133.31, 129.39, 129.34, 129.05, 128.99, 128.67, 128.62, 126.94, 126.92, 126.03, 125.95, 122.17, 121.95, 120.21, 115.16, 115.11, 109.48, 109.21, 55.83, 55.80 , 51.80, 51.74, 48.07, 43.97, 38.22, 38.12, 37.76. HRMS: m/z 865.3021 (M+1)+. C45H45FN6O9S [864.2953].
5.1.8.5. (2S,2’S)-2,2’-((2,2’-(7-chloro-1,1-dioxido-3-oxo-2H-benzo[e][1,2,4]thiadiazine-2,4(3H)-diyl)bis(acetyl))bis(azanediyl))bis(N-(4-methoxyphenyl)-N-methyl-3-phenylp ropanamide) (8e).
White solid, yield: 11%. mp: 115–116°C. 1H NMR (400 MHz, DMSO-d6) δ 8.71 (d, J = 8.2 Hz, 1H, NH), 8.66 – 8.55 (m, 1H, NH), 7.96 (d, J = 2.3 Hz, 1H, Ph-H), 7.69 (dd, J = 9.0, 2.2 Hz, 1H, Ph-H), 7.30 – 7.14 (m, 6H, Ph-H), 7.06 (d, J = 8.1 Hz, 2H, Ph-H), 7.04 – 6.97 (m, 2H, Ph-H), 6.93 (t, J = 8.0 Hz, 5H, Ph-H), 6.90 – 6.84 (m, 4H, Ph-H), 4.74 – 4.24 (m, 6H, CH×2+NCH2×2), 3.75 (s, 6H, OCH3×2), 3.11 (s, 3H, NCH3), 3.09 (s, 3H, NCH3), 2.88 (ddd, J = 19.8, 13.4, 5.4 Hz, 2H, PhCH×2), 2.74 – 2.57 (m, 2H, PhCH×2). 13C NMR (100 MHz, DMSO-d6) δ 171.12, 171.04, 165.94, 165.44, 165.28, 158.98, 158.93, 150.40, 137.72, 137.58,135.78, 135.73, 135.58, 135.22, 134.53, 129.39, 129.34, 129.04, 128.99, 128.67, 128.62, 128.44, 127.65, 126.91, 126.24, 121.79, 119.85, 115.15, 115.11, 55.82, 55.81, 51.80, 51.74, 47.93, 44.06, 38.21, 38.13, 37.76. HRMS: m/z 881.2727 (M+1)+, 883.2799 (M+3)+, 903.2544 (M+23)+. C45H45ClN6O9S [880.2657].
5.1.8.6. (2S,2’S)-2,2’-((2,2’-(7-bromo-1,1-dioxido-3-oxo-2H-benzo[e][1,2,4]thiadiazine-2,4 (3H)-diyl)bis(acetyl))bis(azanediyl))bis(N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide) (8f).
Yellow solid, yield: 7%. mp: 120–121°C. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (d, J = 8.2 Hz, 1H, NH), 8.61 (d, J = 8.0 Hz, 1H, NH), 8.04 (d, J = 2.1 Hz, 1H, Ph-H), 7.85 – 7.76 (m, 1H, Ph-H), 7.31 – 6.79 (m, 19H, Ph-H), 4.72 – 4.18 (m, 6H, CH×2+NCH2×2), 3.75 (s, 6H, OCH3×2), 3.09 (d, J = 8.7 Hz, 6H, NCH3×2), 2.87 (ddd, J = 20.0, 13.4, 5.5 Hz, 2H, PhCH×2), 2.72 – 2.55 (m, 2H, PhCH×2). 13C NMR (100 MHz, DMSO-d6) δ 171.11, 171.04, 165.92, 165.27, 158.98, 158.93, 150.39, 137.72, 137.58, 137.37, 135.95, 135.78, 135.73, 131.70, 129.39, 129.33, 129.10, 129.04, 128.99, 128.67, 128.62, 126.91, 126.49, 124.44, 120.05, 115.88, 115.15, 115.12, 55.82, 55.81, 51.80, 51.75, 47.88, 38.20, 38.14 , 37.76. HRMS: m/z 925.2222 (M+1)+, 927.2210 (M+3)+. C45H45BrN6O9S [924.2152].
5.1.9. Tert-butyl (S)-4-(2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amin o)-2-oxoethyl)-3-oxopiperazine-1-carboxylate (9).
Under ice bath, the key intermediate 7 (700 mg, 1.73 mmol, 1 eq.), 1-Boc-3-oxopiperazine (415 mg, 2.07 mmol, 1.2 eq.), K2CO3 (477 mg, 3.45 mmol, 2 eq.) were dissolved in the solution of DMF (6 mL). The resulting mixture was then stirred at 40°C (monitored by TLC). Then the reaction mixture was extracted with ethyl acetate (20 mL), and the combined organic phase was washed with saturated NaCl solution (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the corresponding crude product 9 as white solid with the yield of 40%. mp: 138–139°C. ESI-MS: m/z 523.09 (M-1)−. C28H36N4O6 [524.62].
5.1.10. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(2-oxopiperazin-1-yl)acetamido)-3-phen ylpropanamide (10).
Trifluoroacetic acid (2 mL) was added dropwise to intermediate 9 (158 mg, 0.30 mmol) in 5 mL dichloromethane and stirred at room temperature for 4 h. Then, the resulting mixture solution was alkalized to pH ~7 with saturated sodium bicarbonate solution and then extracted with dichloromethane (20 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude product 10 as yellow oil with the yield of 63%. ESI-MS: m/z 425.4 (M+1)+, 447.4 (M+23)+. C23H28N4O4 [424.5].
5.1.11. General procedure for the synthesis of 11a-11k.
Under ice bath, the key intermediate 10 (1 eq.), corresponding substituted benzenesulfonyl chloride (1.5 eq.), TEA (2 eq.) were dissolved in the solution of dichloromethane (10 mL). The resulting mixture was then stirred at room temperature (monitored by TLC). Then the reaction mixture was extracted with dichloromethane (20 mL), and the combined organic phase was washed with saturated NaCl solution (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the corresponding crude product, which was purified by recrystallization or preparation thin layer chromatography to afford product 11a-11k.
5.1.11.1. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(2-oxo-4-(phenylsulfonyl)piperazin-1-yl) acetamido)-3-phenylpropanamide (11a).
White solid, yield: 49%. mp: 84–85°C. 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 7.9 Hz, 1H, NH), 7.87 – 7.73 (m, 3H, Ph-H), 7.68 (t, J = 7.5 Hz, 2H, Ph-H), 7.22 – 7.14 (m, 3H, Ph-H), 7.11 (d, J = 8.1 Hz, 2H, Ph-H), 6.97 (d, J = 8.8 Hz, 2H, Ph-H), 6.87 – 6.77 (m, 2H, Ph-H), 4.43 (td, J = 8.8, 5.1 Hz, 1H, CH), 3.88 – 3.80 (m, 2H, piperazine-CH2), 3.78 (s, 3H, OCH3), 3.55 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.49 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.25 – 3.11 (m, 4H, piperazine-CH2×2), 3.09 (s, 3H, NCH3), 2.83 (dd, J = 13.5, 4.8 Hz, 1H, PhCH), 2.62 (dd, J = 13.4, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.36 (C=O), 167.32 (C=O), 163.62 (C=O), 159.00, 137.90, 135.99, 134.83, 134.18, 130.12, 129.28, 129.12, 128.59, 128.12, 126.88, 115.13, 55.89, 51.82, 48.99, 48.41, 46.95, 43.20, 37.77, 37.68. HRMS: m/z 565.2112 (M+1)+, 1151.3958 (2M+23)+. C29H32N4O6S [564.2043].
5.1.11.2. (S)-2-(2-(4-((4-fluorophenyl)sulfonyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-meth oxyphenyl)-N-methyl-3-phenylpropanamide (11b).
White solid, yield: 35%. mp: 199–200°C. 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 7.9 Hz, 1H, NH), 7.90 (dd, J = 8.6, 5.2 Hz, 2H, Ph-H), 7.52 (t, J = 8.7 Hz, 2H, Ph-H), 7.24 – 7.14 (m, 3H, Ph-H), 7.11 (d, J = 8.0 Hz, 2H, Ph-H), 6.97 (d, J = 8.7 Hz, 2H, Ph-H), 6.90 – 6.75 (m, 2H, Ph-H), 4.43 (td, J = 8.7, 5.1 Hz, 1H, CH), 3.88 (d, J = 16.5 Hz, 1H, piperazine-CH), 3.85 (d, J = 16.4 Hz, 1H, piperazine-CH), 3.79 (s, 3H, OCH3), 3.58 (d, J = 16.3 Hz, 1H, piperazine-CH), 3.52 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.28 – 3.18 (m, 2H, piperazine-CH2), 3.16 (m, 2H, piperazine-CH2), 3.10 (s, 3H, NCH3), 2.84 (dd, J = 13.4, 4.7 Hz, 1H, PhCH), 2.62 (dd, J = 13.4, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.37 (C=O), 167.32 (C=O), 165.41 (d, 1JCF = 250.9 Hz), 163.62 (C=O), 159.00, 137.90, 135.89, 131.35 (2×C), 131.25, 129.28 (2×C), 129.12 (2×C), 128.59 (2×C), 126.88, 117.45, 117.22, 115.12 (2×C), 55.88, 51.82, 48.93, 48.40, 46.91, 43.13, 37.76, 37.69. HRMS: m/z 583.2020 (M+1)+, 1187.3740 (2M+23)+. C29H31FN4O6S [582.1948].
5.1.11.3. (S)-2-(2-(4-((3-fluorophenyl)sulfonyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-meth oxyphenyl)-N-methyl-3-phenylpropanamide (11c).
White solid, yield: 41%. mp: 182–183°C. 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 7.9 Hz, 1H, NH), 7.81 – 7.57 (m, 4H, Ph-H), 7.24 – 7.14 (m, 3H, Ph-H), 7.11 (d, J = 7.9 Hz, 2H, Ph-H), 6.97 (d, J = 8.7 Hz, 2H, Ph-H), 6.82 (d, J = 6.6 Hz, 2H, Ph-H), 4.43 (td, J = 8.7, 5.3 Hz, 1H, CH), 3.88 (d, J = 16.6 Hz, 1H, piperazine-CH), 3.83 (d, J = 16.8 Hz, 1H, piperazine-CH), 3.79 (s, 3H, OCH3), 3.62 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.56 (d, J = 16.3 Hz, 1H, piperazine-CH), 3.29 – 3.21 (m, 2H, piperazine-CH2), 3.20 – 3.12 (m, 2H, piperazine-CH2), 3.10 (s, 3H, NCH3), 2.84 (dd, J = 13.5, 4.7 Hz, 1H, PhCH), 2.62 (dd, J = 13.4, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.37 (C=O), 167.33 (C=O), 163.60 (C=O), 162.47 (d, 1JCF = 247.6 Hz), 159.00, 137.90, 137.12 (d, 3JCF = 6.8 Hz), 135.90, 132.48 (d, 3JCF = 7.9 Hz), 129.28, 129.13, 128.59, 126.88, 124.41 (d, 4JCF = 3.0 Hz), 121.35 (d, 2JCF = 21.0 Hz), 115.34, 115.13, 55.88, 51.83, 48.89, 48.42, 46.95, 43.17, 37.76, 37.68. HRMS: m/z 583.2017 (M+1)+, 1187.3844 (M+23)+. C29H31FN4O6S [582.1948].
5.1.11.4. (S)-2-(2-(4-((2-fluorophenyl)sulfonyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-meth oxyphenyl)-N-methyl-3-phenylpropanamide (11d).
White solid, yield: 44%. mp: 170–171°C. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (d, J = 7.9 Hz, 1H, NH), 7.91 – 7.75 (m, 2H, Ph-H), 7.60 – 7.50 (m, 1H, Ph-H), 7.47 (t, J = 7.6 Hz, 1H, Ph-H), 7.23 – 7.15 (m, 3H, Ph-H), 7.12 (d, J = 7.8 Hz, 2H, Ph-H), 6.98 (d, J = 8.7 Hz, 2H, Ph-H), 6.88 – 6.80 (m, 2H, Ph-H), 4.45 (td, J = 8.7, 5.2 Hz, 1H, CH), 3.91 (d, J = 16.5 Hz, 1H, piperazine-CH), 3.86 (d, J = 16.6 Hz, 1H, piperazine-CH), 3.80 (s, 3H, OCH3), 3.73 (d, J = 16.3 Hz, 1H, piperazine-CH), 3.70 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.38 (t, J = 4.9 Hz, 2H, piperazine-CH2), 3.24 – 3.13 (m, 2H, piperazine-CH2), 3.11 (s, 3H, NCH3), 2.85 (dd, J = 13.5, 4.8 Hz, 1H, PhCH), 2.63 (dd, J = 13.4, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.37 (C=O), 167.34 (C=O), 163.69 (C=O), 159.00, 158.82 (d, 1JCF = 252.5 Hz), 137.91, 136.92 (d, 3JCF = 8.7 Hz), 135.91, 131.44, 129.28, 129.13, 128.59, 126.88, 125.81 (d, 4JCF = 3.5 Hz), 124.15 (d, 2JCF = 14.5 Hz), 118.24 (d, 2JCF = 21.5 Hz), 115.14, 55.89, 51.83, 48.47, 48.43, 47.11, 42.79, 37.77, 37.70. HRMS: m/z 583.2017 (M+1)+, 1187.3790 (2M+23)+. C29H31FN4O6S [582.1948].
5.1.11.5. (S)-2-(2-(4-((4-chlorophenyl)sulfonyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-meth oxyphenyl)-N-methyl-3-phenylpropanamide (11e).
White solid, yield: 44%. mp: 203–204°C. 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 8.0 Hz, 1H, NH), 7.83 (d, J = 8.6 Hz, 2H, Ph-H), 7.74 (d, J = 8.6 Hz, 2H, Ph-H), 7.25 – 7.14 (m, 3H, Ph-H), 7.11 (d, J = 8.0 Hz, 2H, Ph-H), 6.97 (d, J = 8.8 Hz, 2H, Ph-H), 6.88 – 6.74 (m, 2H, Ph-H), 4.44 (td, J = 8.7, 5.2 Hz, 1H, CH), 3.88 (d, J = 16.5 Hz, 1H, piperazine-CH), 3.83 (d, J = 16.6 Hz, 1H, piperazine-CH), 3.79 (s, 3H, OCH3), 3.59 (d, J = 16.2 Hz, 1H, piperazineCH), 3.53 (d, J = 16.2 Hz, 1H, piperazineCH), 3.28 – 3.18 (m, 2H, piperazine-CH2), 3.18 – 3.12 (m, 2H, piperazine-CH2), 3.10 (s, 3H, NCH3), 2.84 (dd, J = 13.4, 4.8 Hz, 1H, PhCH), 2.62 (dd, J = 13.4, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.37 (C=O), 167.32 (C=O), 163.59 (C=O), 159.00, 139.16, 137.90, 135.89, 133.91, 130.25, 130.05, 129.28, 129.12, 128.59, 126.88, 115.13, 55.88, 51.81, 48.87, 48.43, 46.94, 43.09, 37.77, 37.69. HRMS: m/z 599.1725 (M+1)+, 1219.3204 (2M+23)+. C29H31ClN4O6S [598.1653].
5.1.11.6. (S)-2-(2-(4-((4-bromophenyl)sulfonyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-meth oxyphenyl)-N-methyl-3-phenylpropanamide (11f).
White solid, yield: 31%. mp: 204–205°C. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.0 Hz, 1H, NH), 7.94 (d, J = 8.5 Hz, 2H, Ph-H), 7.80 (d, J = 8.5 Hz, 2H, Ph-H), 7.30 – 7.20 (m, 3H, Ph-H), 7.17 (d, J = 8.0 Hz, 2H, Ph-H), 7.03 (d, J = 8.8 Hz, 2H, Ph-H), 6.94 – 6.83 (m, 2H, Ph-H), 4.50 (td, J = 8.7, 5.2 Hz, 1H, CH), 3.94 (d, J = 16.5 Hz, 1H, piperazine-CH), 3.89 (d, J = 16.7 Hz, 1H, piperazine-CH), 3.84 (s, 3H, OCH3), 3.65 (d, J = 16.2 Hz, 1H, piperazineCH), 3.59 (d, J = 16.2 Hz, 1H, piperazineCH), 3.33 – 3.24 (m, 2H, piperazine-CH2), 3.24 – 3.19 (m, 2H, piperazine-CH2), 3.15 (s, 3H, NCH3), 2.90 (dd, J = 13.5, 4.8 Hz, 1H, PhCH), 2.68 (dd, J = 13.4, 9.4 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.37 (C=O), 167.32 (C=O), 163.59 (C=O), 159.00, 137.90, 135.89, 134.30, 133.19, 130.10, 129.28, 129.13, 128.59, 128.25, 126.88, 115.13, 55.89, 51.82, 48.86, 48.43, 46.95, 43.08, 37.77, 37.68. HRMS: m/z 643.1224 (M+1)+, 645.1202 (M+3)+. C29H31BrN4O6S [642.1148].
5.1.11.7. (S)-N-(4-methoxyphenyl)-2-(2-(4-((4-methoxyphenyl)sulfonyl)-2-oxopiperazin-1- yl)acetamido)-N-methyl-3-phenylpropanamide (11g).
White solid, yield: 40%. mp: 199–200°C. 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 8.0 Hz, 1H, NH), 7.75 (d, J = 8.8 Hz, 2H, Ph-H), 7.25 – 7.14 (m, 5H, Ph-H), 7.11 (d, J = 7.8 Hz, 2H, Ph-H), 6.98 (d, J = 8.8 Hz, 2H, Ph-H), 6.88 – 6.78 (m, 2H, Ph-H), 4.44 (td, J = 8.7, 5.1 Hz, 1H, CH), 3.95 – 3.83 (m, 5H, PhOCH3+piperazine-CH2), 3.79 (s, 3H, OCH3), 3.51 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.45 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.22 – 3.15 (m, 2H, piperazine-CH2), 3.15 – 3.12 (m, 2H, piperazine-CH2), 3.10 (s, 3H, NCH3), 2.84 (dd, J = 13.5, 4.8 Hz, 1H, PhCH), 2.63 (dd, J = 13.4, 9.4 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.36 (C=O), 167.33 (C=O), 163.71, 163.62 (C=O), 159.00, 137.90, 135.89, 130.47, 129.28, 129.12, 128.59, 126.87, 126.09, 115.26, 115.13, 56.25, 55.88, 51.81, 49.08, 48.39, 46.93, 43.23, 37.77, 37.70. HRMS: m/z 595.2226 (M+1)+, 1211.4210 (2M+23)+. C30H34N4O7S [594.2148].
5.1.11.8. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(2-oxo-4-tosylpiperazin-1-yl)acetamido)- 3-phenylpropanamide (11h).
White solid, yield: 65%. mp: 206–207°C. 1H NMR (400 MHz, DMSO-d6) δ 8.35 (d, J = 8.0 Hz, 1H, NH), 7.70 (d, J = 8.1 Hz, 2H, Ph-H), 7.48 (d, J = 8.1 Hz, 2H, Ph-H), 7.21 – 7.14 (m, 3H, Ph-H), 7.10 (d, J = 7.9 Hz, 2H, Ph-H), 6.97 (d, J = 8.8 Hz, 2H, Ph-H), 6.87 – 6.76 (m, 2H, Ph-H), 4.44 (td, J = 8.7, 5.1 Hz, 1H, CH), 3.87 (d, J = 16.5 Hz, 1H, piperazine-CH), 3.82 (d, J = 17.7 Hz, 1H, piperazine-CH), 3.78 (s, 3H, OCH3), 3.52 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.46 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.21 – 3.12 (m, 4H, piperazine-CH2×2), 3.09 (s, 3H, NCH3), 2.83 (dd, J = 13.5, 4.9 Hz, 1H, PhCH), 2.62 (dd, J = 13.4, 9.4 Hz, 1H, PhCH), 2.42 (s, 3H, PhCH3). 13C NMR (100 MHz, DMSO-d6) δ 171.35 (C=O), 167.32 (C=O), 163.65 (C=O), 158.99, 144.72, 137.90, 135.89, 131.80, 130.55, 129.28, 129.12, 128.58, 128.20, 126.87, 115.13, 55.88, 51.80, 49.01, 48.41, 46.94, 43.21, 37.76, 37.70, 21.53. HRMS: m/z 579.2275 (M+1)+, 1179.4327 (2M+23)+. C30H34N4O6S [578.2199].
5.1.11.9. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(4-((4-nitrophenyl)sulfonyl)-2-oxopipera zin-1-yl)acetamido)-3-phenylpropanamide (11i).
White solid, yield: 68%. mp: 212–213°C. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 8.7 Hz, 2H, Ph-H), 8.38 (d, J = 7.9 Hz, 1H, NH), 8.10 (d, J = 8.7 Hz, 2H, Ph-H), 7.23 – 7.16 (m, 3H, Ph-H), 7.10 (d, J = 7.9 Hz, 2H, Ph-H), 6.97 (d, J = 8.7 Hz, 2H, Ph-H), 6.88 – 6.79 (m, 2H, Ph-H), 4.44 (td, J = 8.6, 5.3 Hz, 1H, CH), 3.90 (d, J = 16.4 Hz, 1H, piperazine-CH), 3.83 (d, J = 16.0 Hz, 1H, piperazine-CH), 3.79 (s, 3H, OCH3), 3.68 (d, J = 16.5 Hz, 1H, piperazineCH), 3.63 (d, J = 16.4 Hz, 1H, piperazineCH), 3.34 – 3.26 (m, 2H, piperazine-CH2), 3.18 (t, J = 5.0 Hz, 2H, piperazine-CH2), 3.10 (s, 3H, NCH3), 2.84 (dd, J = 13.5, 4.8 Hz, 1H, PhCH), 2.62 (dd, J = 13.4, 9.4 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.34 (C=O), 167.29 (C=O), 163.44 (C=O), 158.99, 150.75, 140.99, 137.89, 135.88, 129.71, 129.28, 129.10, 128.59, 126.88, 125.32, 115.12, 55.88, 51.81, 48.69, 48.40, 46.92, 43.06, 37.76, 37.72. HRMS: m/z 610.1961 (M+1)+, 632.1739 (M+23)+. C29H31N5O8S [609.1893].
5.1.11.10. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(4-((3-nitrophenyl)sulfonyl)-2-oxopipera zin-1-yl)acetamido)-3-phenylpropanamide (11j).
White solid, yield: 71%. mp: 159–160°C. 1H NMR (400 MHz, DMSO-d6) δ 8.62 – 8.53 (m, 1H, Ph-H), 8.50 – 8.42 (m, 1H, Ph-H), 8.39 (d, J = 7.9 Hz, 1H, NH), 8.26 (d, J = 7.9 Hz, 1H, Ph-H), 7.97 (t, J = 8.0 Hz, 1H, Ph-H), 7.26 – 7.14 (m, 3H, Ph-H), 7.11 (d, J = 7.9 Hz, 2H, Ph-H), 6.97 (d, J = 8.8 Hz, 2H, Ph-H), 6.87 – 6.76 (m, 2H, Ph-H), 4.43 (td, J = 8.7, 5.3 Hz, 1H, CH), 3.90 (d, J = 16.4 Hz, 1H, piperazine-CH), 3.83 (d, J = 16.7 Hz, 1H, piperazine-CH), 3.79 (s, 3H, OCH3), 3.70 (d, J = 16.3 Hz, 1H, piperazine-CH), 3.64 (d, J = 16.4 Hz, 1H, piperazine-CH), 3.34 – 3.26 (m, 2H, piperazine-CH2), 3.19 (t, J = 5.1 Hz, 2H, piperazine-CH2), 3.10 (s, 3H, NCH3), 2.84 (dd, J = 13.5, 4.9 Hz, 1H, PhCH), 2.62 (dd, J = 13.4, 9.4 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.35 (C=O), 167.30 (C=O), 163.48 (C=O), 158.99, 148.74, 137.89, 136.83, 135.89, 134.01, 132.15, 129.28, 129.11, 128.63, 128.59, 126.88, 122.86, 115.12, 55.89, 51.82, 48.74, 48.36, 46.92, 43.11, 37.76, 37.71. HRMS: m/z 610.1970 (M+1)+, 1241.3681 (2M+23)+. C29H31N5O8S [609.1893].
5.1.11.11. (S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(4-((2-nitrophenyl)sulfonyl)-2-oxopipera zin-1-yl)acetamido)-3-phenylpropanamide (11k).
White solid, yield: 59%. mp: 160–161°C. 1H NMR (400 MHz, DMSO-d6) δ 8.40 (d, J = 8.0 Hz, 1H, NH), 8.09 (d, J = 7.6 Hz, 1H, Ph-H), 8.04 (d, J = 7.7 Hz, 1H, Ph-H), 7.95 (t, J = 7.3 Hz, 1H, Ph-H), 7.88 (t, J = 7.4 Hz, 1H, Ph-H), 7.23 – 7.15 (m, 3H, Ph-H), 7.12 (d, J = 8.1 Hz, 2H, Ph-H), 6.98 (d, J = 8.8 Hz, 2H, Ph-H), 6.84 (d, J = 7.1 Hz, 2H, Ph-H), 4.46 (td, J = 8.7, 5.2 Hz, 1H, CH), 3.94 (d, J = 16.5 Hz, 1H, piperazine-CH), 3.88 (d, J = 16.6 Hz, 1H, piperazine-CH), 3.85 (s, 2H, piperazine-CH2), 3.79 (s, 3H, OCH3), 3.49 (t, J = 5.2 Hz, 2H, piperazine-CH2), 3.20 (t, J = 5.1 Hz, 2H, piperazine-CH2), 3.11 (s, 3H, NCH3), 2.86 (dd, J = 13.5, 4.8 Hz, 1H, PhCH), 2.64 (dd, J = 13.4, 9.4 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.36 (C=O), 167.34 (C=O), 163.60 (C=O), 159.00, 148.34, 137.91, 135.90, 135.65, 133.08, 131.07, 129.29, 129.21, 129.13, 128.60, 126.89, 124.88, 115.14, 55.89, 51.83, 48.54, 48.47, 47.26, 43.00, 37.77, 37.73. HRMS: m/z 610.1965 (M+1)+, 1241.3695 (2M+23)+. C29H31N5O8S [609.1893].
5.1.12. General procedure for the synthesis of compounds 11l-11n.
(11i-11k, 150 mg, 0.246 mmol), 10% Pd●C (10% w/w, 20 mg) were dissolved in methanol (5 mL) and dichloromethane (5 mL) and the solution degassed and stirred at room temperature for 2h under H2. The mixture was filtered and concentrated, and the resulting residue were purified by recrystallization or preparation thin layer chromatography to provide the target compounds (11l-11n).
5.1.12.1. (S)-2-(2-(4-((4-aminophenyl)sulfonyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-meth oxyphenyl)-N-methyl-3-phenylpropanamide (11l).
White solid, yield: 61%. mp: 128–129°C. 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 7.9 Hz, 1H, NH), 7.42 (d, J = 8.6 Hz, 2H, Ph-H), 7.30 – 7.15 (m, 3H, Ph-H), 7.12 (d, J = 7.9 Hz, 2H, Ph-H), 6.98 (d, J = 8.8 Hz, 2H, Ph-H), 6.88 – 6.79 (m, 2H, Ph-H), 6.68 (d, J = 8.7 Hz, 2H, Ph-H), 6.20 (s, 2H, NH2), 4.44 (td, J = 8.8, 5.2 Hz, 1H, CH), 3.86 (s, 2H, piperazine-CH2), 3.79 (s, 3H, OCH3), 3.46 – 3.36 (m, 2H, piperazineCH2), 3.23 – 3.12 (m, 2H, piperazine-CH2), 3.10 (s, 3H, NCH3), 3.07 – 2.95 (m, 2H, piperazine-CH2), 2.84 (dd, J = 13.5, 4.8 Hz, 1H, PhCH), 2.63 (dd, J = 13.4, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.38 (C=O), 167.38 (C=O), 163.98 (C=O), 158.99, 154.17, 137.91, 135.90, 130.31, 129.29, 129.14, 128.58, 126.88, 118.49, 115.14, 113.30, 55.88, 51.83, 49.30, 48.38, 46.94, 43.32, 37.77, 37.66. HRMS: m/z 580.2228 (M+1)+, 1181.4192 (2M+23)+. C29H33N5O6S [579.2152].
5.1.12.2. (S)-2-(2-(4-((3-aminophenyl)sulfonyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-meth oxyphenyl)-N-methyl-3-phenylpropanamide (11m).
White solid, yield: 71%. mp: 106–107°C. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (d, J = 8.0 Hz, 1H, NH), 7.28 (t, J = 7.9 Hz, 1H, Ph-H), 7.24 – 7.15 (m, 3H, Ph-H), 7.12 (d, J = 7.7 Hz, 2H, Ph-H), 7.02 – 6.93 (m, 3H, Ph-H), 6.93 – 6.79 (m, 4H, Ph-H), 5.71 (s, 2H, NH2), 4.44 (td, J = 8.8, 5.2 Hz, 1H, CH), 3.86 (s, 2H, piperazine-CH2), 3.79 (s, 3H, OCH3), 3.50 (d, J = 16.2 Hz, 1H, piperazine-CH), 3.45 (d, J = 16.3 Hz, 1H, piperazine-CH), 3.23 – 3.11 (m, 4H, piperazine-CH2×2), 3.10 (s, 3H, NCH3), 2.84 (dd, J = 13.5, 4.7 Hz, 1H, PhCH), 2.63 (dd, J = 13.4, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.37 (C=O), 167.35 (C=O), 163.72 (C=O), 159.00, 150.26, 137.91, 135.90, 134.84, 130.43, 129.28, 129.14, 128.59, 126.88, 118.83, 115.14, 114.54, 112.15, 55.88, 51.83, 49.15, 48.44, 47.00, 43.31, 37.77, 37.66. HRMS: m/z 580.2220 (M+1)+, 1181.4181 (2M+23)+. C29H33N5O6S [579.2152].
5.1.12.3. (S)-2-(2-(4-((2-aminophenyl)sulfonyl)-2-oxopiperazin-1-yl)acetamido)-N-(4-meth oxyphenyl)-N-methyl-3-phenylpropanamide (11n).
White solid, yield: 80%. mp: 95–96°C. 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 8.0 Hz, 1H, NH), 7.44 (d, J = 7.9 Hz, 1H, Ph-H), 7.36 (t, J = 7.6 Hz, 1H, Ph-H), 7.22 – 7.15 (m, 3H, Ph-H), 7.12 (d, J = 7.7 Hz, 2H, Ph-H), 6.98 (d, J = 8.8 Hz, 2H, Ph-H), 6.90 (d, J = 8.3 Hz, 1H, Ph-H), 6.87 – 6.79 (m, 2H, Ph-H), 6.68 (t, J = 7.5 Hz, 1H, Ph-H), 6.14 (s, 2H, NH2), 4.45 (td, J = 8.7, 5.2 Hz, 1H, CH), 3.86 (s, 2H, piperazine-CH2), 3.79 (s, 3H, OCH3), 3.61 (d, J = 16.3 Hz, 1H, piperazine-CH), 3.56 (d, J = 16.3 Hz, 1H, piperazine-CH), 3.29 – 3.20 (m, 2H, piperazine-CH2), 3.20 – 3.13 (m, 2H, piperazine-CH2), 3.10 (s, 3H, NCH3), 2.84 (dd, J = 13.5, 4.8 Hz, 1H, PhCH), 2.64 (dd, J = 13.4, 9.5 Hz, 1H, PhCH). 13C NMR (100 MHz, DMSO-d6) δ 171.37 (C=O), 167.36 (C=O), 163.85 (C=O), 159.00, 148.26, 137.91, 135.90, 135.17, 130.39, 129.28, 129.13, 128.58, 126.88, 118.02, 115.92, 115.14, 114.39, 55.89, 51.83, 48.91, 48.42, 46.94, 43.14, 37.77, 37.68. HRMS: m/z 580.2225 (M+1)+, 1181.4212 (2M+23)+. C29H33N5O6S [579.2152].
5.2. In Vitro Anti-HIV-1 Assay in TZM-bl Cells and Cytotoxicity Assay31
Inhibition of HIV-1 infection was measured as a reduction in the level of luciferase gene expression after a single round of virus infection of TZM-bl cells as described previously. Briefly, 800 TCID50 of virus (NL4–3) was used to infect TZM-bl cells in the presence of various concentrations of compounds. One day after infection, the culture medium was removed from each well, and 100 μL of Bright Glo reagent (Promega, San Luis Obispo, CA) was added to the cells to measure luminescence using a Victor 2 luminometer. The effective concentration (EC50) against HIV-1 strains was defined as the concentration that caused a 50% decrease in luciferase activity (relative light units) compared to that of virus control wells.
A CytoTox-Glo™ cytotoxicity assay (Promega) was used to determine the cytotoxicity of the synthesized compounds. Parallel to the antiviral assays, TZM-bl cells were cultured in the presence of various concentrations of the compounds for 1 day. The percent of viable cells was determined by following the protocol provided by the manufacturer. The 50% cytotoxic concentration (CC50) was defined as the concentration that caused a 50% reduction in cell viability.
5.3. In Vitro Anti-HIV Assay in MT-4 Cells
Evaluation of the antiviral activity of the compounds against HIV in MT-4 cells was performed using the MTT assay as described below. Stock solutions (10 × final concentration) of test compounds were added in 25 μL volumes to two series of triplicate wells so as to allow simultaneous evaluation of their effects on mock- and HIV-infected cells at the beginning of each experiment. Serial 5-fold dilutions of test compounds were made directly in flat-bottomed 96-well microtiter trays using a Biomek 3000 robot (Beckman instruments, Fullerton, CA). Untreated HIV- and mock-infected cell samples were included as controls. HIV stock (50 μL) at 100–300 CCID50 (50 % cell culture infectious doses) or culture medium was added to either the infected or mock-infected wells of the microtiter tray. Mock-infected cells were used to evaluate the effects of test compound on uninfected cells in order to assess the cytotoxicity of the test compounds. Exponentially growing MT-4 cells were centrifuged for 5 minutes at 220 g and the supernatant was discarded. The MT-4 cells were resuspended at 6 × 105 cells/mL and 50 μL volumes were transferred to the microtiter tray wells. Five days after infection, the viability of mock-and HIV-infected cells was examined spectrophotometrically using the MTT assay. The MTT assay is based on the reduction of yellow colored 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Acros Organics) by mitochondrial dehydrogenase activity in metabolically active cells to a blue-purple formazan that can be measured spectrophotometrically. The absorbances were read in an eight-channel computer-controlled photometer (Infinite M1000, Tecan), at two wavelengths (540 and 690 nm). All data were calculated using the median absorbance value of three wells. The 50% cytotoxic concentration (CC50) was defined as the concentration of the test compound that reduced the absorbance (OD540) of the mock-infected control sample by 50%. The concentration achieving 50% protection against the cytopathic effect of the virus in infected cells was defined as the 50% effective concentration (EC50).
5.4. Binding to CA Proteins Analysis via Surface Plasmon Resonance (SPR)
All binding assays were performed on a ProteOn XPR36 SPR Protein Interaction Array System (Bio-Rad Laboratories, Hercules, CA). The instrument temperature was maintained at 25°C for all kinetic analyses. ProteOn GLH sensor chips were preconditioned with two short pulses each (10 seconds) of 50 mM NaOH, 100 mM HCl, and 0.5% sodium dodecyl sulfide. Then the system was equilibrated with PBS-T buffer (20 mM sodium phosphate, 150 mM NaCl, and 0.005% polysorbate 20, pH 7.4). The surface of a GLH sensorchip was activated with a 1:100 dilution of a 1:1 mixture of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (0.2 M) and sulfo-N-hydroxysuccinimide (0.05 M). Immediately after chip activation, the HIV-1NL4–3 capsid protein constructs, purified as in Xu et al.,37 were prepared at a concentration of 100 μg/ml in 10 mM sodium acetate, pH 5.0 and injected across ligand flow channels for 5 min at a flow rate of 30 μl/min. Then, after unreacted protein had been washed out, excess active ester groups on the sensor surface were capped by a 5 minute injection of 1 M ethanolamine HCl (pH 8.0) at a flow rate of 5 μl/min. A reference surface was similarly created by immobilizing a non-specific protein (IgG b12 anti-HIV-1 gp120; was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Anti-HIV-1 gp120 Monoclonal (IgG1 b12) from Dr. Dennis Burton and Carlos Barbas) and was used as a background to correct non-specific binding.
To prepare a compound for direct binding analysis, compound stock solutions, along with 100% DMSO, and totaling 30 μL was made to a final volume of 1 ml by addition of sample preparation buffer (PBS, pH 7.4). The preparation of analyte in this manner ensured that the concentration of DMSO was matched with that of running buffer with 3% DMSO. Serial dilutions were prepared in the running buffer (PBS, 3% DMSO, 0.005% polysorbate 20, pH 7.4) and injected at a flow rate of 100 μl/min, for a 1 minute association phase, followed by up to a 5 minutes dissociation phase using the “one shot kinetics” capability of the Proteon instrument.38 Data were analyzed using the ProteOn Manager Software version 3.0 (Bio-Rad). The responses from the reference flow cell were subtracted to account for the nonspecific binding and injection artifacts. The equilibrium dissociation constant (KD) for the interactions, and derived from a minimum of three experiments, were calculated in ProteOn Manager Version 3.1.0.6 (Bio-Rad, Hercules, CA), using the equilibrium analysis function.
5.5. Action Stage Determination of 11l
Please refer to our previously published research for methodology.23
5.6. ELISA-based Quantification of Capsid (p24) Content28
An ELISA plate was coated with 50 ng of mouse anti-p24 (Abcam, ab9071) per well for 2 hours at room temperature, blocked with 3% BSA for 2 hours at room temperature and washed with PBST buffer (0.1% Tween-20 in PBS). Pseudovirus stocks were lysed with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO) at 37°C for 1 hour and added to the plate overnight at 4°C. Simultaneously, p24 protein was added for the generation of a standard curve. Following the overnight incubation, the plate was washed with PBST buffer, and 1:5000 dilution of rabbit anti-p24 (Abcam, ab63913) was added for 2 hours at room temperature. After washing the unbound rabbit anti-p24 off the plate with PBST buffer, goat anti-rabbit-HRP at a 1:5000 dilution was added for 1 hour at room temperature. The plate was then extensively washed with PBST buffer. Subsequently, a solution of 0.4 mg/ml o-phenylenediamine in a phosphate-citrate buffer with sodium perborate (Sigma-Aldrich) was added and incubated in the dark for 30 minutes. Optical densities were then obtained at 450 nm in a Multiskan™ GO Microplate Spectrophotometer (Thermo Scientific).
5.7. In Vitro Capsid Assembly Assay28
Briefly, 1.0 μl of concentrated compound (10 mM) in 100% DMSO was added to a 74-μl aqueous solution (the solution was made by mixing 2 ml of 5 M NaCl with 1 ml of 200 mM NaH2PO4, pH 8.0). To initiate the assembly reaction, 25 μl of purified capsid protein (120 μM) was added. An identical reaction mixture was prepared, omitting the compound (i.e., just DMSO as vehicle control). Readings were taken at 350 nm every one minute for 19 minutes. Capsid was used at a final concentration of 30 μM. PF-74 (positive control) and compounds at 50 μM (3% DMSO final).
5.8. Recombinant HIV-1 RT Inhibitory Assay
The Reverse Transcriptase Assay was performed using the colorimetric assay developed by Roche (Reverse Transcriptase Assay, Roche Cat. No. 11468120910) (149). 4 ng recombinant HIV-1 RT in 20 μL lysis buffer (50 mM Tris, 80 mM potassium chloride, 2.5 mM DTT, 0.75 mM EDTA and 0.5% Triton X-100, pH 7.8) was incubated with 20 μL of 200 nM inhibitor (11l) or 0.73 nM of TMC278 (TMC278 was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Rilpivirine (Cat # 12147) from Tibotec Pharmaceuticals, Inc.), and 20 μL reaction mixture (containing template/primer hybrid, digoxigenin-labeled nucleotides and biotin labeled nucleotides) at 37°C for 1 hour. After the incubation, the reaction mixtures were transferred to microplate wells coated with streptavidin and incubated at 37°C for 1 hour. 200 μL of sheep anti-digoxigenin-peroxidase working solution was added per well, and incubated at 37°C for 1 hour. The entire solution was removed and rinsed five times with 250 μL of washing buffer per well. 200 μL of 2,2’-azino-bis(3-ethylbenzothiazoline-6- sulphonic acid) (ABTS) substrate solution was added per well, and the reaction mixture was incubated for another 15 minutes until color development and measured in a Multiskan™ GO Microplate Spectrophotometer (Thermo Scientific) at 405 nM. Inhibitory activity of compound 11l was calculated as percent inhibition as compared to a DMSO (1%) sample.
5.9. Molecular Dynamics Simulation
To keep consistency for MD simulation of different series of CA HIV-1 monomer inhibitors, we used the same procedure for docking, MD simulation and its analysis, please refer to our previously published research for methodology.23
5.10. Human Plasma Stability Assay
The pooled frozen plasma was thawed in a water bath at 37℃ before the experiment. Plasma was centrifuged at 4000 rpm for 5 min and the clots were removed if any. The pH will be adjusted to 7.4 ± 0.1 if required. Preparation of compounds: 1 mM intermediate solution was prepared by diluting 10 μL of the stock solution with 90 μL DMSO; 1 mM intermediate of positive control Propantheline was prepared by diluting 10 μL of the stock solution with 90 μL ultrapure water. 100 μM dosing solution was prepared by diluting 20 μL of the intermediate solution (1 mM) with 180 μL 45% MeOH/H2O. 98 μL of blank plasma was spiked with 2 μL of dosing solution (100 μM) to achieve 2 μM of the final concentration in triplicate and samples were incubated at 37℃ in a water bath. At each time point (0,10, 30, 60 and 120 min), 400 μL of stop solution (200 ng/mL tolbutamide and 200 ng/mL Labetalol in 50% ACN/MeOH) was added to precipitate protein and mixed thoroughly. Centrifuged sample plates at 4,000 rpm for 10 min. An aliquot of supernatant (50 μL) was transferred from each well and mixed with 100 μL ultra pure water. The samples were shaken at 800 rpm for about 10 min before submitting to LC-MS/MS analysis.
5.11. Metabolic Stability in Human Liver Microsomes
Details of the analytical method and raw data are given in the Supporting Information.
5.12. Pharmacokinetics Assay
The oral bioavailability of 11l was examined in male Sprague-Dawley rats, which were randomly divided into 2 groups to receive intravenous (2 mg·kg−1) or oral administration (20 mg·kg−1). The animals were kept in an air-conditioned rat room and used to determine the kinetic profiles. Solutions of 11l were prepared in a mixture of polyethylene glycol (PEG) 400/normal saline/DMSO (40/57/3, v/v/v) before the experiment. Blood samples (200 μL each time) from the intravenous or oral administration group were collected from the sinus jugularis into heparinized centrifugation tubes at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, and 12 h after dosing. Plasma samples were obtained by centrifugation at 8000 rpm, 8 °C for 5 min and immediately frozen (−80 °C). LC–MS analysis was used to determine the concentration of 11l in plasma. Standard curves for 11l in blood were generated by the addition of various concentrations together with an internal standard to blank plasma. Details of the analytical method are given in the Supporting Information. Pharmacokinetic parameters were calculated using DAS2.0 software. The research protocol complied strictly with the institutional guidelines of the Animal Care and Use Committee at the New Drug Evaluation Center of Shandong Academy of Pharmaceutical Sciences.
5.13. Acute Toxicity Experiment
Kunming mice (17–20 g) were purchased from the animal experimental center of Shandong University. The research protocol complied strictly with the institutional guidelines of the Animal Care and Use Committee at Shandong University. 11l was suspended in a mixture of polyethylene glycol (PEG) 400/normal saline (70/30, v/v) up to 100 mg/mL and administered intragastrically by gavage to mice that had been fasted for 12 h. Dosages of 1000 mg·kg−1 were administered to 10 mice per group (5 males, 5 females). Death, body weight, tremor, convulsion, body jerks, hypoactivity, hunched posture, and piloerection were monitored.
Supplementary Material
ACKNOWLEDGEMENTS
Financial support from the National Natural Science Foundation of China (NSFC Nos. 81573347, 81973181), Key Project of NSFC for International Cooperation (No. 81420108027), Young Scholars Program of Shandong University (YSPSDU, No. 2016WLJH32), Key Research and Development Project of Shandong Province (Nos. 2017CXGC1401, 2019JZZY021011), NIH grants R01GM125396 (transitioning to R01AI150491) (Cocklin, PI) and T32-MH079785 are gratefully acknowledged.
ABBREVIATIONS USED
- AIDS
acquired immunodeficiency syndrome
- Boc
tert-butyloxycarbonyl
- CA
capsid
- CTD
C-terminal domain
- CC50
50% cytotoxicity concentration
- 13C NMR
carbon nuclear magnetic resonance
- DIEA
N,N-diisopropylethylamine
- EC50
effective concentration causing 50% inhibition of viral cytopathogenicity
- HRMS
highresolution mass spectrum
- HPLC
high performance liquid chromatography
- cART
combination antiretroviral therapy
- HIV
human immunodeficiency virus
- 1H NMR
proton nuclear magnetic resonance
- MD
molecular dynamics
- NTD
N-terminal domain
- PK
pharmacokinetic
- RMSD
root-mean-square displacement
- RMSF
root-mean-square fluctuation
- SPR
surface plasmon resonance
- SAR
structure–activity relationship
- SI
selectivity index
- TEA
triethylamine
- t1/2
half-time
Footnotes
Publisher's Disclaimer: This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies.
The Supporting Information is available free of charge on the ACS Publications website.
Molecular dynamics simulation on 8a and 6k, metabolic stability in human liver microsomes, analytical method for pharmacokinetics assay, references, and HRMS, 1H-NMR and 13C-NMR spectra for representative target compounds.
Molecular formula strings.
The authors declare that all experimental work complied with the institutional guidelines on animal studies (care and use of laboratory animals).
The authors declare no competing financial interest.
REFERENCES
- [1].De Clercq E Antivirals: past, present and future. Biochem. Pharmacol 2013, 85, 727–744. [DOI] [PubMed] [Google Scholar]
- [2].Zhang J; Crumpacker C Eradication of HIV and cure of AIDS, now and how? Front Immunol. 2013, 4, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhan P; Pannecouque C; De Clercq E; Liu X Anti-HIV drug discovery and development: current innovations and future trends. J. Med. Chem 2016, 59, 2849–2878. [DOI] [PubMed] [Google Scholar]
- [4].Campbell EM; Hope TJ HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol. 2015, 13, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhou J; Price AJ; Halambage UD; James LC; Aiken C HIV-1 resistance to the capsid-targeting inhibitor PF74 results in altered dependence on host factors required for virus nuclear entry. J Virol. 2015, 89, 9068–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen B HIV capsid assembly, mechanism, and structure. Biochemistry. 2016, 55, 2539–2552. [DOI] [PubMed] [Google Scholar]
- [7].Gres AT; Kirby KA; KewalRamani VN; Tanner JJ; Pornillos O; Sarafianos SG Structural virology. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science. 2015, 349, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Le Sage V; Mouland AJ; Valiente-Echeverría F Roles of HIV-1 capsid in viral replication and immune evasion. Virus Res. 2014, 193, 116–129. [DOI] [PubMed] [Google Scholar]
- [9].Perrier M; Bertine M; Le Hingrat Q; Joly V; Visseaux B; Collin G; Landman R; Yazdanpanah Y; Descamps D; Charpentier C Prevalence of gag mutations associated with in vitro resistance to capsid inhibitor GS-CA1 in HIV-1 antiretroviral-naive patients, J Antimicrob. Chemother 2017, 72, 2954–2955. [DOI] [PubMed] [Google Scholar]
- [10].Rasaiyaah J; Tan CP; Fletcher AJ; Price AJ; Blondeau C; Hilditch L; Jacques DA; Selwood DL; James LC; Noursadeghi M; Towers GJ. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013, 503, 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lahaye X; Satoh T; Gentili M; Cerboni S; Conrad C; Hurbain I; El Marjou A; Lacabaratz C; Lelievre JD; Manel N The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013, 39, 1132–1142. [DOI] [PubMed] [Google Scholar]
- [12].Jiang JY; Ablan SD; Derebail S; Hercik K; Soheilian F; Thomas JA; Tang SX; Hewlett I; Nagashima K; Gorelick RJ; Freed EO; Levin JG The interdomain linker region of HIV-1 capsid protein is a critical determinant of proper core assembly and stability. Virology. 2011, 421, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].von Schwedler UK; Stray KM; Garrus JE; Sundquist WI Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol 2003, 77, 5439–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Douglas CC; Thomas D; Lanman J; Prevelige PE Investigation of N-terminal domain charged residues on the assembly and stability of HIV-1 CA. Biochemistry. 2004, 43, 10435–10441. [DOI] [PubMed] [Google Scholar]
- [15].Ganser-Pornillos BK; von Schwedler UK; Stray KM; Aiken C; Sundquist WI Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol 2004, 78, 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang W; Zhou J; Halambage UD; Jurado KA; Jamin AV; Wang Y; Engelman AN; Aiken C Inhibition of HIV-1 maturation via small-molecule targeting of the amino-terminal domain in the viral capsid Protein. J Virol. 2017, 91, pii: e02155–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tang C; Loeliger E; Kinde I; Kyere S; Mayo K; Barklis E; Sun Y; Huang M; Summers MF. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol. 2003, 327, 1013–1020. [DOI] [PubMed] [Google Scholar]
- [18].Kelly BN; Kyere S; Kinde I; Tang C; Howard BR; Robinson H; Sundquist WI; Summers MF; Hill CP Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J Mol Biol. 2007, 373, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sticht J; Humbert M; Findlow S.; Bodem J; Muller B; Dietrich U; Werner J; Krausslich HG A peptide inhibitor of HIV-1 assembly in vitro. Nat Struct Mol Biol. 2005, 12, 671–677. [DOI] [PubMed] [Google Scholar]
- [20].Zhang H; Zhao Q; Bhattacharya S; Waheed AA; Tong X; Hong A; Heck S; Curreli F; Goger M; Cowburn D; Freed EO; Debnath AK A cell-penetrating helical peptide as a potential HIV-1 inhibitor. J Mol Biol. 2008, 378, 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lamorte L; Titolo S; Lemke CT; Goudreau N; Mercier JF; Wardrop E; Shah VB; von Schwedler UK; Langelier C; Banik SS; Aiken C; Sundquist WI; Mason SW. Discovery of novel small molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob Agents Chemother. 2013, 57, 4622–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Blair WS; Pickford C; Irving SL; Brown DG; Anderson M; Bazin R; Cao J; Ciaramella G; Isaacson J; Jackson L; Hunt R; Kjerrstrom A; Nieman JA; Patick AK; Perros M; Scott AD; Whitby K; Wu H; Butler SL HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010, 6: e1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wu G; Zalloum WA; Meuser ME; Jing L; Kang D; Chen CH; Tian Y; Zhang F; Cocklin S; Lee KH; Liu X; Zhan P Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library. Eur J Med Chem. 2018, 158, 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jiang XY; Wu GC; Zalloum WA; Meuser ME; Dick A; Sun L; Chen CH; Kang DW; Jing LL; Jia RF; Cocklin S; Lee KH; Liu XY; Zhan P Discovery of novel 1,4-disubstituted 1,2,3-triazole phenylalanine derivatives as HIV-1 capsid inhibitors. RSC. Adv 2019, 9, 28961–28986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tse WC; Link JO; Mulato A Discovery of novel potent HIV capsid inhibitors with long- acting potential. Conference on Retroviruses and Opportunistic Infections. Seattle, Washington; 2017. [Google Scholar]
- [26].Yant SR; Mulato A; Stepan G, Villasenor AG; Jin D; Margot NA; Ahmadyar S; Ram RR; Somoza JR; Singer E; Wong M; Xu Y; Link JO; Cihlar T; Tse WC. GS-6207, a potent and selective first-in-class long-acting HIV-1 capsid inhibitor. Conference on Retroviruses and Opportunistic Infections, Seattle, Washington; 2019. [Google Scholar]
- [27].Thenin-Houssier S; Valente ST HIV-1 Capsid Inhibitors as Antiretroviral Agents. Curr HIV Res. 2016, 14, 270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xu JP; Francis AC; Meuser ME; Mankowski M; Ptak RG; Rashad AA; Melikyan GB; Cocklin S Exploring modifications of an HIV-1 capsid inhibitor: design, synthesis, and mechanism of action. J. Drug Des. Res 2018, 5, pii: 1070. [PMC free article] [PubMed] [Google Scholar]
- [29].Rana AI; Castillo-Mancilla JR; Tashima KT; Landovitz RL Advances in long-acting agents for the treatment of HIV infection. Drugs. 2020, doi: 10.1007/s40265-020-01284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bondy SS; Chou CH; Link JO; Tse W, C. Antiviral Agents WO2015130966A1. [Google Scholar]
- [31].Liu N; Wei L; Huang L; Yu F; Zheng W; Qin B; Zhu DQ; Morris-Natschke SL; Jiang S; Chen CH; Lee KH; Xie L Novel HIV-1 non-nucleoside reverse transcriptase inhibitor agents: Optimization of diarylanilines with high potency against wild-type and rilpivirine-resistant E138K mutant virus. J. Med. Chem 2016, 59, 3689–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kortagere S; Madani N; Mankowski MK; Schön A; Zentner I; Swaminathan G; Princiotto A; Anthony K; Oza A; Sierra LJ; Passic SR; Wang X; Jones DM; Stavale E; Krebs FC; Martín-García J; Freire E; Ptak RG; Sodroski J; Cocklin S; Smith AB 3rd. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J. Virol 2012, 86, 8472–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kortagere S; Xu JP; Mankowski MK; Ptak RG; Cocklin S Structure-activity relationships of a novel capsid targeted inhibitor of HIV-1 replication. J. Chem. Inf. Model 2014, 54, 3080–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ambrose Z; Aiken C HIV-1 uncoating: connection to nuclear entry and regulation by host proteins. Virology. 2014, 454, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jacques DA; Mcewan WA; Hilditch L; Price AJ; Towers GJ; James LC HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature. 2016, 536, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sarma B; Thakuria R; Nath NK; Nangia A Crystal structures of mirtazapine molecular salts. CrystEngComm. 2011, 13, 3232–3240. [Google Scholar]
- [37].Xu JP; Branson JD; Lawrence R; Cocklin S Identification of a small molecule HIV-1 inhibitor that targets the capsid hexamer. Bioorg. Med. Chem. Lett 2016, 26, 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bravman T; Bronner V; Lavie K; Notcovich A; Papalia GA; Myszka DG Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal. Biochem 2006, 358, 281–288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.