

Abstract
Ketamine produces rapid and robust antidepressant effects in depressed patients within hours of administration, often when traditional antidepressant compounds have failed to alleviate symptoms. We hypothesized that ketamine would translocate Gαs from lipid rafts to non-raft microdomains, similarly to other antidepressants but with a distinct, abbreviated treatment duration. C6 glioma cells were treated with 10 μM ketamine for 15 min, which translocated Gαs from lipid raft domains to non-raft domains. Other NMDA antagonist did not translocate Gαs from lipid raft to non-raft domains. The ketamine-induced Gαs plasma membrane redistribution allows increased functional coupling of Gαs and adenylyl cyclase to increase intracellular cyclic adenosine monophosphate (cAMP). Moreover, increased intracellular cAMP increased phosphorylation of cAMP response element-binding protein (CREB), which, in turn, increased BDNF expression. The ketamine-induced increase in intracellular cAMP persisted after knocking out the NMDA receptor indicating an NMDA receptor-independent effect. Furthermore, 10 μM of the ketamine metabolite (2R,6R)-hydroxynorketamine (HNK) also induced Gαs redistribution and increased cAMP. These results reveal a novel antidepressant mechanism mediated by acute ketamine treatment that may contribute to ketamine’s powerful antidepressant effect. They also suggest that the translocation of Gαs from lipid rafts is a reliable hallmark of antidepressant action that might be exploited for diagnosis or drug development.
Introduction
Major depressive disorder (MDD) is a common mood disorder that effects one in six in the United States and is a leading cause of disability worldwide [1]. Despite decades of rigorous research, no clear etiology or molecular pathophysiology of MDD exists. A focus on monoamines has guided research and drug development, and the vast majority of marketed antidepressants act on monoaminergic reuptake, catabolism or receptors. However, the rationale for this focus is weakened by the disconnect between the quick-acting effect of antidepressants on monoaminergic targets (typically within a few hours of administration) and the therapeutically relevant response, which requires as long as 8 weeks [2]. Ketamine is defined classically, as a noncompetitive N-methyl-d-aspartate receptor (NMDAR) antagonist. While ketamine has regulatory approval as an anesthetic, it also has rapid antidepressant effects that last from 3 days to two weeks following a single intravenous infusion [3, 4]. Ketamine has catalyzed research into new antidepressant mechanisms of action and proffered hope for a new class of rapid-acting antidepressants. Many of ketamine's proposed antidepressant mechanisms of action have been reviewed [5–7], and its use remains controversial [8]. Briefly, leading theories posit NMDAR antagonism increases synthesis of brain-derived neurotrophic factor (BDNF), a process mediated by decreased phosphorylation of eukaryotic elongation factor 2 (eEF2) and subsequent activation of the mTOR pathway by BDNF activation of TrkB receptor [9, 10]. Moreover, ketamine increases synaptogenesis, dendritic spine density, and increases α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) throughput and expression [9, 11]. The precise antidepressant mechanism for ketamine remains unclear and may involve targets other than NMDAR antagonism, given that other NMDAR antagonists do not provide quick-acting, robust, and persistent antidepressant effects in animals and humans [12, 13]. Moreover, (R)-ketamine has fourfold less affinity for the NMDAR than (S)-ketamine yet (R)-ketamine’s antidepressant effects are more potent over a longer period in animal models of depression [14].
One commonality between chronic treatment with commonly used antidepressants and ketamine is increased synaptogenesis and increased expression of BDNF in animals and cell cultures. While these are seen after extended treatment in rats or cells, they appear subsequent to a single infusion of ketamine [5, 9, 15]. On a cellular and molecular level, increased cAMP has been associated with antidepressant action, which upregulates neurotrophic factors and increases synaptogenesis [16–18]
A recent PET study observed a global reduction in cAMP in the brain of depressed subjects that rebounded to control levels after successful antidepressant treatment [19]. Consistent with these observations, our studies suggest that treatment with all classes of antidepressants increases cAMP by augmenting functional coupling of Gαs and adenylyl cyclase after translocating Gαs from lipid raft domains into non-raft regions [20–23]. Lipid rafts are microdomains in the plasma membrane rich in sphingolipids, cholesterol, and cytoskeletal proteins [24]. The rigid, cholesterol-dependent order within lipid rafts serve as an organizer, to regulate cellular signaling. Gαs is singly palmitoylated on its N-terminus, which targets the protein to lipid rafts [25] where its ability to associate with adenylyl cyclase is diminished [26]. Indeed, production of cAMP is diminished when Gαs is localized to lipid raft microdomains [26]. Following from the above, human post-mortem brain tissue of depressed suicides show Gαs preferentially localized to lipid rafts [21]. Taken together, these studies suggest that some antidepressant effects are mediated by elevating cAMP levels subsequent to a more facile interaction between Gαs and adenylyl cyclase in non-raft regions of the plasma membrane.
Studies suggesting a mechanism underlying ketamine action and the subsequent ketamine-induced synaptogenesis and production of BDNF are conflicting [9, 10, 13, 27]. Thus, we questioned if Gαs translocation and subsequent increase in cAMP may play a role in ketamine’s antidepressant action. We sought to determine if ketamine translocates Gαs from lipid raft microdomains to non-raft domains in a manner similar to, but much more rapidly than, established antidepressants. In this study, we show that a single, 15-min, exposure of C6 glioma cells or primary astrocytes to clinically relevant concentrations of ketamine induces selective redistribution of Gαs. Furthermore, parallel to, but more rapidly than monoamine targeted antidepressants, ketamine exposure elicits sustained increase in cellular cAMP attributable to the result of Gαs translocation from lipids rafts. A ketamine metabolite known to not bind NMDARs has effects similar to ketamine and ketamine exerts these effects in cells where NMDARs have been knocked down. These observations lend credence to the hypothesis that ketamine may have both immediate and delayed effects, employing different mechanisms.
Methods
Cell culture and drug treatments
C6 cells were cultured in DMEM, 4.5 g of glucose/L and 10% newborn calf serum (Hyclone Laboratories, Logan, UT) at 37 °C in humidified 5% CO2 atmosphere to a confluence of ~60% before drug treatments. Treatment with ketamine was for either 15 min or 24 h. Treatment with DAMGO, nor-BNI, MK-801, memantine, and AP-V was for 15–30 min. After treatment, cells were rinsed twice with pre-warmed phosphate-buffered saline (PBS) to remove debris and wash away unbound drugs. An aliquot of 1 μM of the cAMP competitive inhibitor cAMPS-Rp was applied just subsequent to ketamine treatment.
TX100 lipid raft isolation
Cells were washed and harvested in ice-cold 1× PBS then C6 cells were extracted in 1 mL of ice-cold lysis buffer (10 mM HEPES pH 7.4; 150 mM NaCl; 1 mM DTT; 1% Triton X-100; Protease inhibitor cocktail). Following 30 min incubation on ice, lipid rafts were collected as previously described [24]. Lipid raft pellets were reconstituted in 50 μL of TME buffer (10 mM Tris HCl, 1 mM MgCl2, 1 mM EDTA pH 7.5, 1 mM DTT, protease inhibitors). Protein content was determined by absorbance at 280 nm on a nanodrop.
Detergent-free sucrose density gradient lipid raft isolation
C6 cells were placed on ice and scraped in detergent-free Tricine buffer (250 mM sucrose, 1 mM EDTA, 20 mM Tricine, pH 7.4). The material was homogenized and centrifuged at low speed (1500 × g for 5 min at 4 °C) to precipitate nuclear fraction. The supernatant was collected, mixed with 30% percoll in tricine buffer and subjected to ultracentrifugation for 45 min (Beckmann SW55-Ti rotor, 109,400 × g, at 4 °C) to collect plasma membrane fraction (PM). PMs were collected and sonicated (3×30 s bursts). The sonicated material was mixed with 60% sucrose (to a final concentration of 40%), overlaid with a 35–5% step sucrose gradient and subjected to overnight ultracentrifugation (Beckman SW55-Ti rotor, 109,400 × g at 4 °C). Fractions were collected every 400 μL from the top sucrose layer and proteins were precipitated using 0.25 volume TCA-deoxycholic acid in double distilled water, 0.1% deoxycholic acid was used to solubilize protein precipitates. Each fraction was loaded into gels by volume.
Fluorescence recovery after photobleaching
C6 cells were transfected or infected with GFP-Gαs and cells expressing GFP-Gαs were analyzed as described previously [23]. For infection protocols, GFP-Gαs was inserted into a baculoviral vector by Montana Molecular - Bozeman MT and used according to the manufacturers directions. One hundred fifty data points, ~300 ms apart (including ten pre-bleach values) were measured for each cell. Zeiss Zen software was used to calculate fluorescence recovery after photobleaching (FRAP) recovery half-time utilizing a one-phase association fit, correcting for total photobleaching of the analyzed regions.
SDS-PAGE and western blotting
Western blotting was carried out as described [26]. Antibodies used were: anti-Gαs monoclonal antibody (NeuroMab clone N192/12, Davis, CA, USA, catalog #75–211), caveolin-1 (BD Biosci #610059), β-actin (SIGMA Clone AC-74), p-CREB (Cell Signaling #9198), CREB (Cell Signaling #9197), BDNF (Cell Signaling #3987), NMDAR1 (Cell signaling #5704). Blots were imaged using Chemidoc computerized densitometer (Bio-Rad, Hercules, CA, USA) and quantified by ImageLab 3.0 software (BioRad, Hercules, CA, USA). In all experiments, the original gels are either visualized using BioRad stainfree technology to normalize protein loading or normalized to β-actin.
Depletion of NR1 NMDA subunit
Expression of NR1 was inhibited with a SMARTpool: ONTARGET plus Grin1 (24408) siRNA (Catalog # L-080174–02-0005) and scrambled control siRNA (Dharmacon, Inc, Pittsburg, PA). Briefly, C6 cells at 40% confluence were transfected with 50 nM siRNA using DharmaFECT-1. The expression level of NR1 was determined by Western blot analysis.
Primary astrocyte culture
P3 wistar rats were sacrificed and brains removed and placed into a HBSS. Olfactory bulb and cerebellum were removed, and the remaining cortex was cut into small pieces with 2.5% trypsin mixed with the tissue and incubated at 37 °C for 30 min. Tissue was then centrifuged at 300 × g and supernatant aspirated. The pellet was resuspended in astrocyte plating medium (DMEM, high glucose + 10% heat-inactivated fetal bovine serum + 1% Penicillin/Streptomycin) and seeded at 10 × 106 cells per T-75 flask. Medium was changed every 2 days until cells reached confluency. To obtain an enriched astrocyte culture, microglia were removed by shaking flasks for 180 rpm for 30 min and discarding medium. Oligodendrocyte precursor cells were removed by shaking flasks at 240 rpm for 6 h. Remaining astrocytes were seeded into flasks at 5 × 105 density. Medium was changed every 2–3 days and astrocytes allowed to mature for 14 days before experimentation.
Viral Infection and cAMP quantification
C6 cell32s were grown on glass bottom microscope dishes and infected with (1.09 × 109 VG/mL) cADDIS BacMam virus encoding the green upward cAMP sensor (Montana Molecular, Bozeman, MT, USA), supplemented with sodium butyrate at a final concentration of 2 mM, and grown for 24–26 h before live imaging under a ×40 objective on a Zeiss 880 microscope. Cells were serum starved with 1% serum for 2–3 h before drug treatments. Images were taken every 30 s. Average responses from 4 to 10 cells were selected randomly from the visual field and fluorescence was normalized to baseline fluorescence for each experiment.
cAMP dose–response assay
C6 cells were plated in 96-well black-sided clear-bottom plates (Costar 3603, Corning Inc.) 24 h before measurement at a density of 48,000 cells per well. At time of plating, each well was infected with (2.18 × 108 VG/mL) cADDIS BacMam virus Green cADDis upward cAMP sensor and supplemented with sodium butyrate at a final concentration of 2 mM. Final volume of each well was brought up to a volume of 140 μL with culture media (DMEM supplemented with 10% newborn calf serum). Twenty-four hours after plating, media was replaced with fresh culture media, with or without Ketamine at a final concentration of 10 μM. After 15 min, the culture media was replaced with 200 μL DPBS. GFP signal intensity was determined on a Biotek Synergy H4 plate reader (Biotek Instruments Inc.) using inbuilt monochromator with excitation set to 488 nM and emission set to 525 nM. Wells were read from the bottom with detector set to 90% sensitivity. Each condition was measured in duplicate wells for each experiment. Blank subtracted baseline measurements were obtained for each well before isoproterenol challenge. Wells containing 200 μL DPBS were used for blank subtraction. Following baseline measurement, either isoproterenol or vehicle (ddH2O) was added to each well and GFP signal intensity was measured.
Statistical analysis
Western blot bands were quantified using Biorad image lab software. Control values were set to one and compared to treatment values. The graphs are represented with either fold change or percent change with p values. Data are represented from at least three biological replicate experiments. Statistical significant differences (p < 0.05) were determined by unpaired t-test or two-way ANOVA, or if variances were unequal a two-way Kruskal—Wallis test was performed followed by Dunn’s post hoc test for multiple comparisons. Unpaired t-test for control vs treatment conditions were performed followed by Welch’s correction. In cAMP dose—response experiments F0 was defined as the mean signal intensity of the vehicle + vehicle condition. Isoproterenol EC50 and maximal efficacy were calculated by fitting the data to a standard agonist concentration versus response curve (Hill slope = 1). For time course experiments, signal intensity was measured with a 20 s read interval for the duration of the experiment. Results are represented as mean ± S.E.M. All statistical analysis was performed with the Prism version 5.0 software package for statistical analysis (GraphPad Software Inc., San Diego, CA).
Results
Brief ketamine treatment redistributes plasma membrane Gαs into non-raft regions of the plasma membrane of C6 glioma cells
Gαs localization in lipid rafts is decreased after 3-day treatment with 10 μM antidepressants in C6 cells and 3-week treatment in several regions of rat brain [20, 23]. Likewise, ketamine concentration rises to ~10 μM in the brains of rats after they have been treated with an antidepressant dose [13]. To test if ketamine-induced a similar redistribution, C6 cells were treated for 15 min or 24 h with 10 μM ketamine and lipid raft fractions were isolated using a detergent-free sucrose density gradient method. The results show ketamine liberated Gαs from lipid rafts after as little as 15 min treatment (Fig. 1a). Additional experiments were restricted to 15-min treatment as this is sufficient time to observe a phenotypic antidepressant response in cells after ketamine administration. This time course represents an appropriate juxtaposition of the rapid onset of antidepressant effects seen in humans compared with the delayed therapeutic onset of traditional antidepressant compounds [3, 4].
Fig. 1.
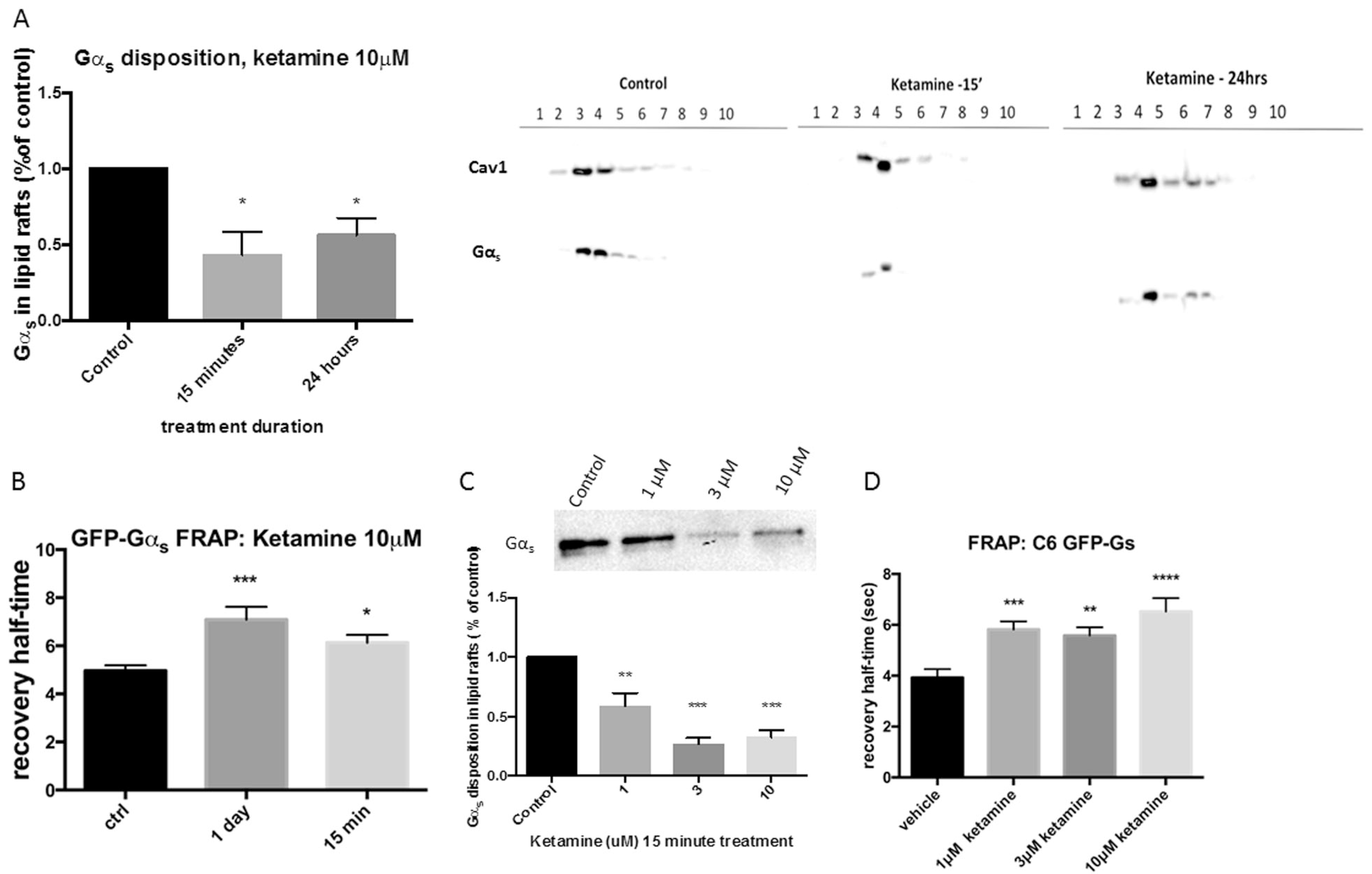
Ketamine decreases Gαs lipid raft localization. a C6 cells were treated with 10 μM ketamine for 15 min or 24 h and lipid raft fractions isolated and probed for Gαs. Both groups show a statistical decrease of Gαs in lipid raft fractions indicating ketamine-mediated Gαs translocation from lipid rafts into non-raft regions of the plasma membrane, blots were re-probed for caveolin-1 to confirm lipid raft fractions. The histogram on the left represents the average lipid raft localization of 4 experiments b Gαs -GFP monoclonal C6 cells were treated for 15 min or 24 h and Gαs lateral mobility analyzed by fluorescence recovery after photobleaching (FRAP), which revealed a statistical increase of recovery half-time consistent with augmented association of Gαs and adenylyl cyclase. c Dose—response of ketamine-mediated Gαs translocation was analyzed by TX100 lipid raft isolation and d FRAP, both methods reveal a dose dependency of ketamine for Gαs translocation, which occurred at clinically relevant levels of drug (n ≤ 4) *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Translocation of Gαs from lipid rafts was also determined using fluorescence recovery after photobleaching (FRAP). FRAP of GFP-Gαs from antidepressant treated C6 cells results in decreased lateral membrane diffusion due to its increased association between GFP-Gαs and adenylyl cyclase. Treatment (3 days) of C6 cells with all tested antidepressants translocate GFP-Gαs from lipid rafts as indicated by FRAP [16]. Data presented here record an “antidepressant signature” for ketamine after only 15 min treatment (Fig. 1b), comparable to 3-day treatment with “classic” antidepressants. These data corroborate the data derived from lipid-raft isolation (Fig. 1a).
Ketamine translocates Gαs from lipid rafts at clinically relevant concentrations
Patients are commonly treated with one 0.5 mg/kg infusion of ketamine over 40 min, where ketamine serum levels after 10 min and 30 min increase to 1.3 μM and 1.0 μM, respectively [28]. To determine if ketamine translocation of Gαs from lipid rafts occurs at clinically relevant concentrations, C6 cells were exposed to 1, 3, and 10 μM ketamine for 15 min and lipid rafts were isolated using TX100 and sucrose density protocol to allow equal protein and all groups to be loaded on one gel. There was a dose-dependent Gαs exodus from lipid rafts at concentrations achieved in human subjects (Fig. 1c). These biochemical data for Gαs translocation are corroborated by FRAP (Fig. 1d).
Ketamine-induced translocation of Gαs returns to baseline after 24 h
The duration of ketamine’s antidepressant effects are highly variable, lasting from 24 h to 2 weeks, with an average remission of depressive symptoms for one week [3, 4]. We sought to determine the duration of Gαs translocation to non-raft microdomains after one 15-minute treatment with ketamine. C6 cells were treated with 10 μM ketamine and collected 15 min, 1, 6, 12, and 24 h afterwards, lipid rafts were isolated and Gαs quantified by immunoblotting. Translocation of Gαs from lipid rafts was sustained significantly for 12 h after transient 15-min treatment, returning to baseline after 24 h (Fig. 2), suggesting rapid, transient effects, similar to those seen in human subjects, in the cellular model system.
Fig. 2.
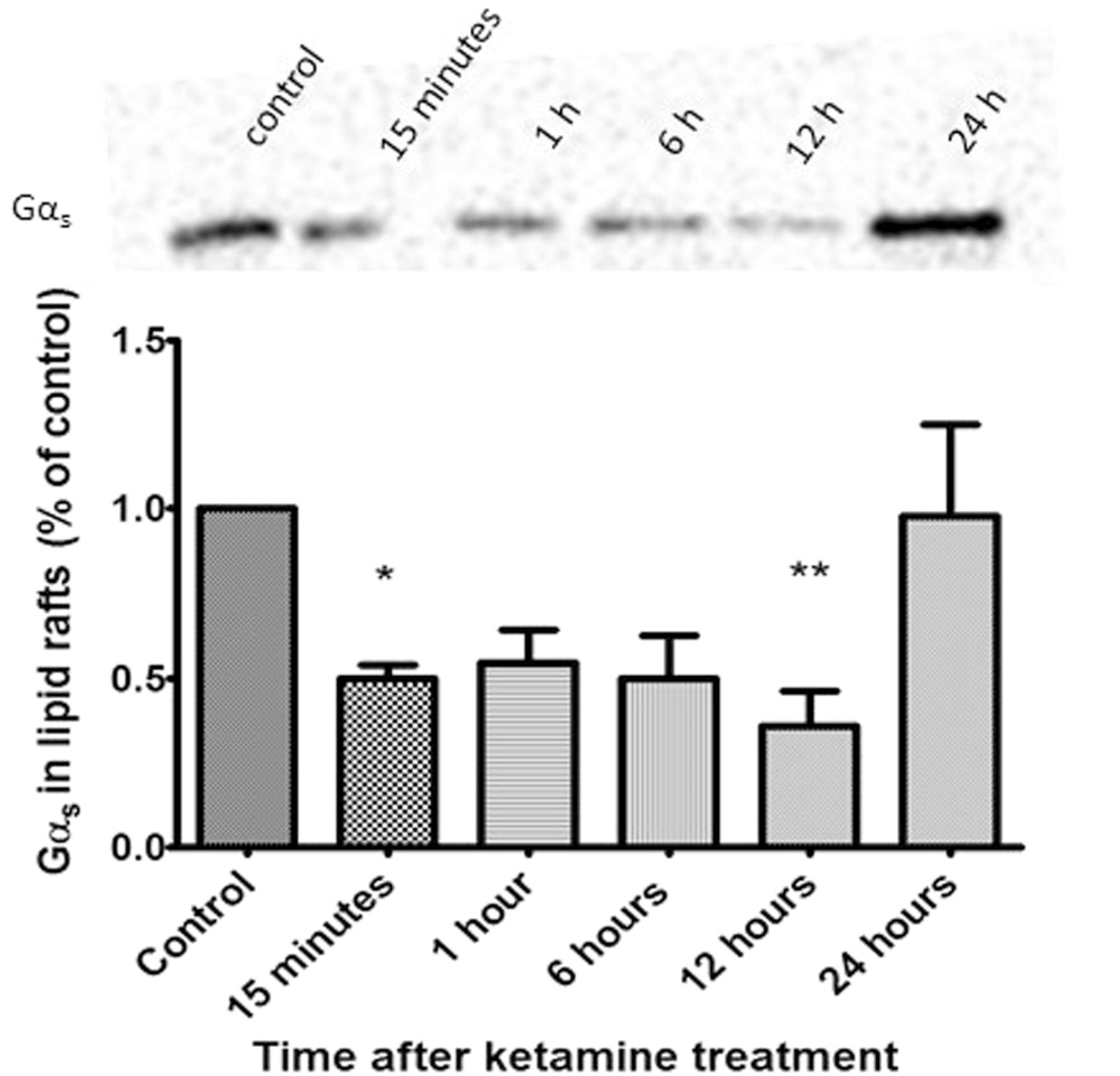
Gαs lipid raft localization returns 24 h after ketamine withdrawal. C6 cells were treated with ketamine for 15 min and drug was washed off. Cells were collected at 15 min, 1, 6, 12 and 24 h afterward, lipid rafts were isolated and probed for Gαs. Data reveal the localization of Gαs in non-raft regions is maintained for 12 h after 15-minute treatment (n = 4). *p < 0.05; **p < 0.01. Ketamine effects were no longer evident by 24 h after washout
Other NMDA antagonists, MOR agonists, or KOR antagonist do not mediate Gαs translocation from lipid rafts
C6 cells lack monoamine transporters yet show delayed effects of antidepressant treatment [29]. Given that C6 cells express NMDARs, the canonical target of ketamine’s anesthetic action, we questioned whether Gαs lipid raft exodus was a result of NMDAR antagonism. To test this, C6 cells were treated with 10 μM of the NMDA antagonists MK-801, memantine or AP-V for 15 min and Gαs lipid raft localization was evaluated via TX100 and sucrose density gradient lipid raft isolation. No other NMDAR antagonist had a significant effect on Gαs lipid raft localization suggesting that ketamine may be acting on a target other than the NMDA receptors (Fig. 3a). Again, these data were corroborated with FRAP after a 15-min treatment and no other NMDAR antagonist altered Gαs rate of lateral membrane diffusion (Fig. 3b) These data agree with reports suggesting targets other than NMDA receptors are responsible for ketamine’s antidepressant action [12].
Fig. 3.
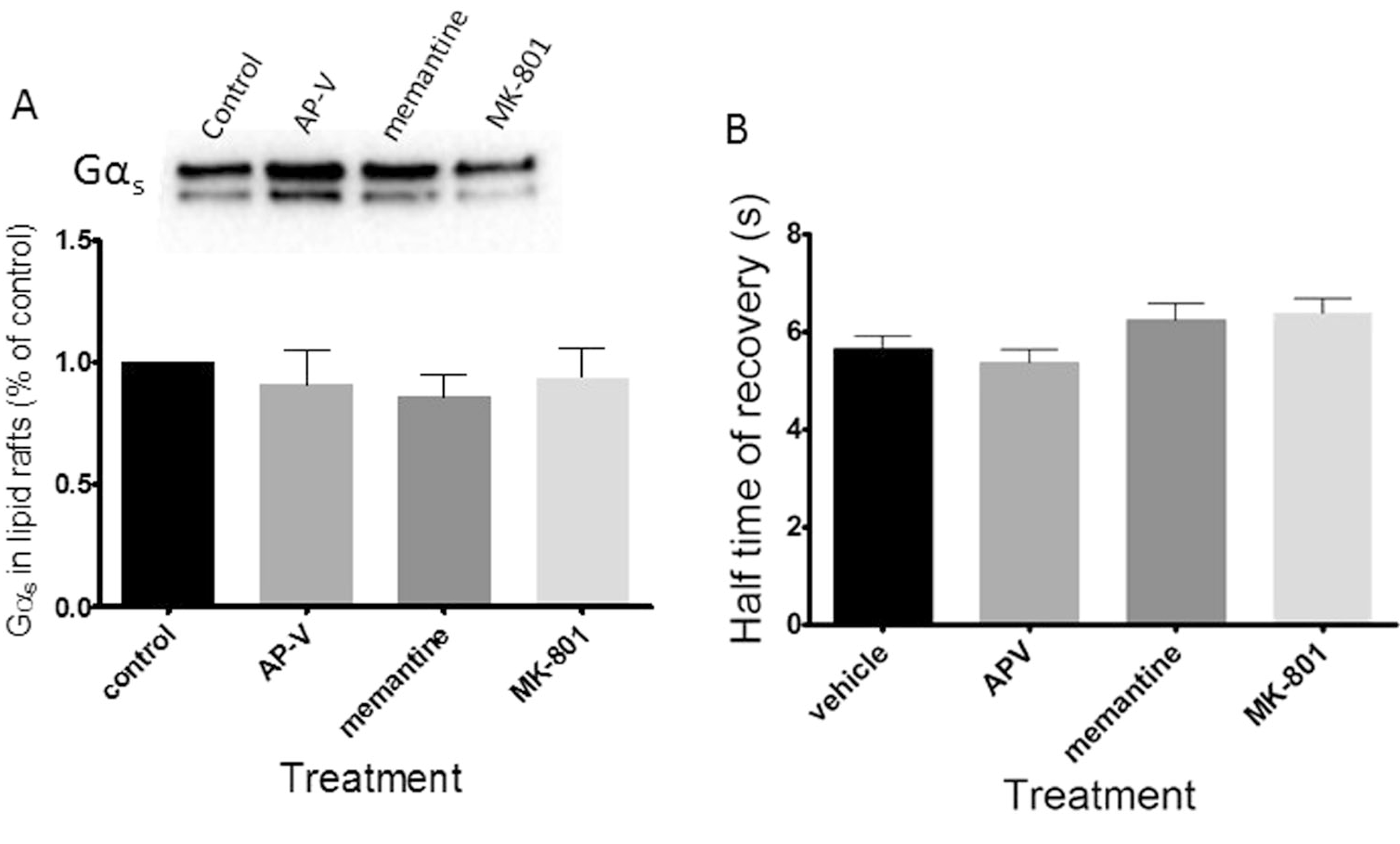
Other NMDA antagonist do not mediate Gαs translocation from lipid rafts. C6 cells were treated with NMDAR antagonists AP-V, memantine, and MK-801 for 15 min and Gαs translocation was analyzed by (a) lipid raft isolation (n = 3) and (b) FRAP (n ≥ 48). Both methods reveal no effect upon Gαs lipid raft localization, indicating ketamine-mediated Gαs translocation may result from an NMDAR-independent target of ketamine
It has been suggested that ketamine’s antidepressant properties may involve μ-opioid receptor (MOR) agonism or κ-opioid receptor antagonism (KOR) [8]. To determine if either action influences Gαs plasma membrane localization C6 cells expressing Gαs-GFP were treated with 10 μM [D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin (DAMGO-a MOR antagonist) or 100 nM Norbinaltorphimine (nor-BNI-a KOR antagonist) and FRAP was evaluated (Fig. S1). Neither drug influenced fluorescencerecovery of Gαs-GFP, suggesting ketamine MOR agonism or KOR antagonism is not involved in rapid, ketamine-mediated plasma membrane redistribution of Gαs.
Ketamine enhances isoproterenol elicited cAMP accumulation in NR1 knockdown C6 cells
As antidepressant treatment translocates Gαs from lipid rafts, it increases association of Gαs with adenylyl cyclase, increasing cellular cAMP production [22]. To test if ketamine had a similar effect, we measured cAMP in live C6 cells using exchange factor directly activated by cAMP (EPAC)-based fluorescent biosensors [30]. No difference in basal fluorescence between control and ketamine-treated cells was observed (Fig. 4a). However, when cells were treated with ketamine for 15 min and then stimulated by the Gαs-coupled agonist isoproterenol (C6 cells have endogenous β adrenergic receptors) after drug washout, ketamine-treated cells show both a quicker and more robust increase in fluorescence, consistent with an enhanced coupling of Gαs and adenylyl cyclase (Fig. 4b). These data agree with previous reports, which show the requirement of an activator of Gαs to detect changes in cAMP production after antidepressant treatment [22]. To investigate ketamine-mediated cAMP production independent of its canonical target, the NMDAR was ablated. The NR1 subunit was knocked down with siRNA (Fig. S2), preventing proper assembly of the NMDA receptor complex [31]. The ketamine-treated NR1 knockdown group shows an increase in cAMP similar to ketamine-treated groups in which NMDAR is present (Fig. 4b). These data further support an NMDAR-independent antidepressant target of ketamine. To determine if ketamine influences Gαs-coupled GPCR potency and efficacy, the isoproterenol mediated cAMP accumulation C6 cells was examined. C6 cells were seeded in a 96-well plate, treated with ketamine, and challenged with increasing doses of isoproterenol. Dose—response reveals ketamine treatment increases, significantly, percent stimulation (efficacy) over baseline (180 ± 10) compared to control (130 ± 11) while EC50 (potency) of ketamine (−9.0 ± 0.23) compared to (−8.2 ± 0.28) was unaffected (Fig. 4c). These results suggest that Gαs is coupling more effectively to adenylyl cyclase.
Fig. 4.
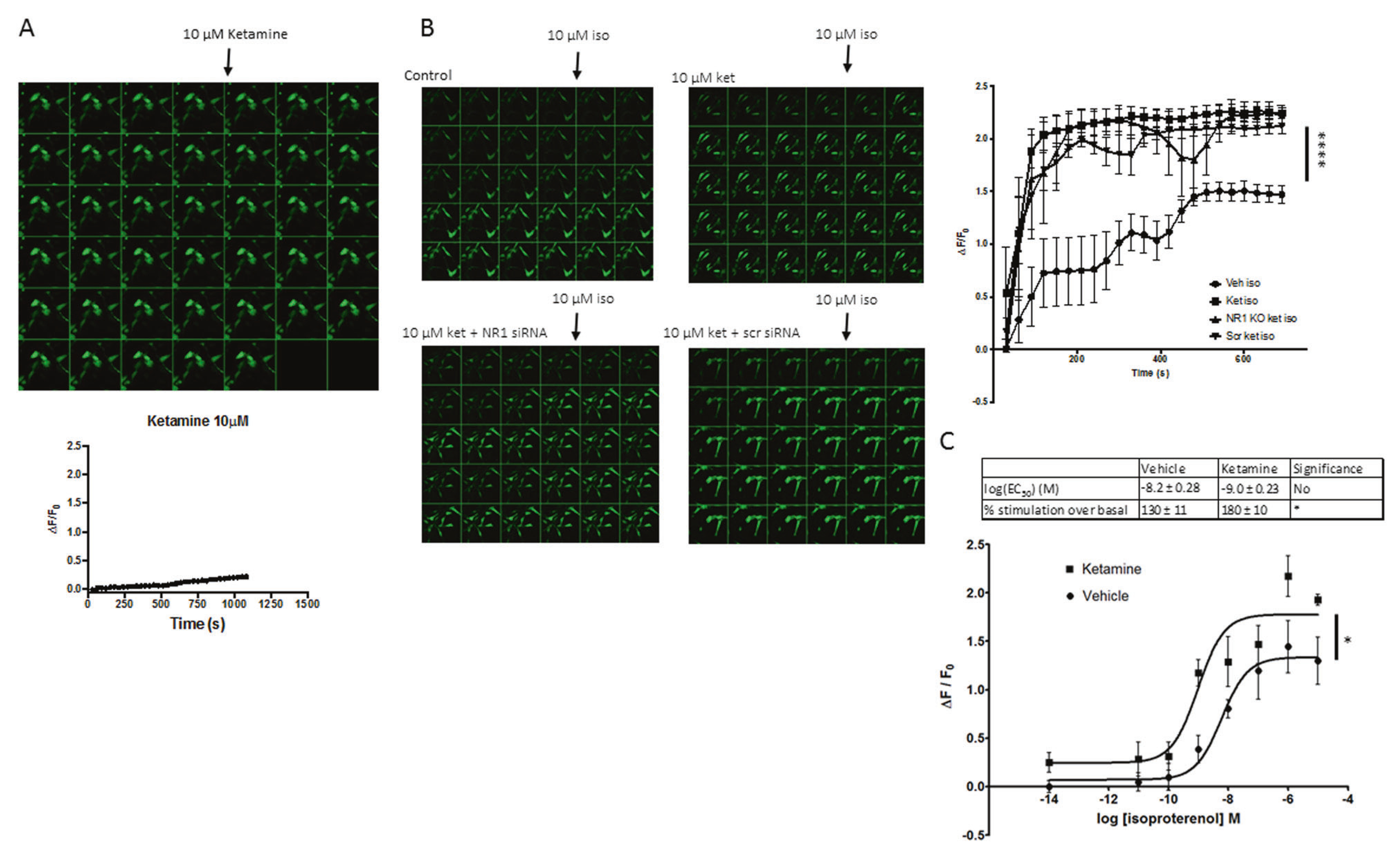
Ketamine treatment results in increased accumulation of cAMP, independent of NMDARs. a C6 cells were infected with (1.09 × 109 VG/mL) cADDIS virus and imaged every 30 s and 10 μM ketamine was added after the fourth frame (n = 2). b Clockwise from top left; vehicle pretreatment with addition of isoproterenol; ketamine pretreatment with isoproterenol added after frame 4; NR1 siRNA pretreatment, ketamine pretreatment with addition of isoproterenol; scrambled siRNA pretreatment, ketamine pretreatment with the addition of isoproterenol. All ketamine pretreated groups showed a quicker and more robust increase in fluorescence indicating relieved inhibition of lipid rafts upon Gαs and a more facile interaction of Gαs and adenylyl cyclase and NMDAR-independent effects of ketamine (n = 4). c C6 cells in a 96-well plate were infected with (2.18 × 108 VG/mL) cADDIS virus and treated with 10 μM ketamine or vehicle for 15 min. Vehicle or isoproterenol was added to each well and GFP signal intensity was measured. These data reveal a statistically significant increase in efficacy of adenylyl cyclase activation but not agonist potency. (n = 3) *p < 0.05; ****p < 0.0001
Transient ketamine treatment promotes sustained phosphorylation of CREB
Activation of protein kinase A (PKA) by cAMP results in the phosphorylation of cAMP response element-binding protein (CREB) at serine-133 (Ser-133). Phosphorylated CREB (pCREB) then translocates to the nucleus where it acts as a transcription factor for genes involved in growth and survival, neuroprotection, and synaptic plasticity [18, 32]. CREB is both upregulated and phosphorylated at Ser-133 after chronic antidepressant treatment in animals and cultured cells [18]. We sought to evaluate the effect of ketamine-mediated cAMP elevation on CREB phosphorylation. To test this, C6 cells were treated with 10 μM ketamine for 15 min, drug was washed off and cells harvested 15 min, 1, 2, 6, and 24 h after treatment. The data are consistent with a sustained increase of pCREB after a transient exposure to ketamine with significant increases shown at 15 min and again at 2 h (Fig. 5a). Total cellular CREB content remains constant. Increased pCREB is aligned, temporally, with the observed increase of cAMP production. Interestingly, increase in cAMP-dependent CREB phosphorylation was achieved without the addition of a Gαs-coupled agonist. This may be the result from activation of endogenous GPCRs by agonists present in serum or an increase of cAMP below the range of the sensor [30].
Fig. 5.
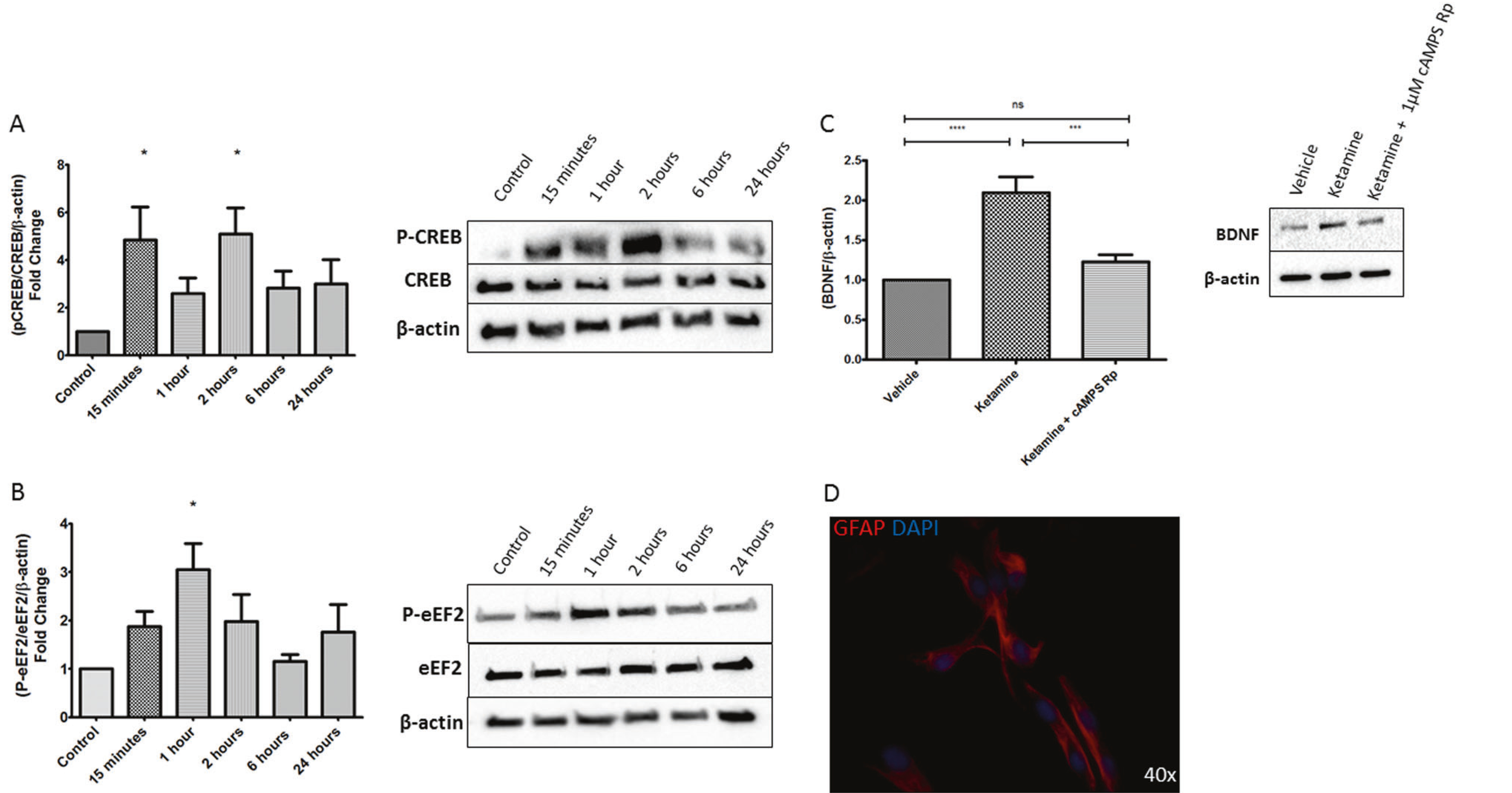
Ketamine increases phosphorylation of PKA-associated proteins and BDNF in primary astrocytes. Western blot analysis of cAMP/PKA-associated proteins, C6 cells were treated for 15 min with 10 μM ketamine and collected at the indicated time point and probed for a phosphorylation of CREB at Ser-133 which showed elevated levels for 24 h and statistically significant increases at 15 min and 2 h. The histogram represents the average of CREB phosphorylation from 5 determinations. The blot is a representative determination. B Phosphorylation of eEF2 at Thr-56 showed increased levels for 24 h reaching after reaching statistical significance at 1 h (n = 4) c Primary astrocytes were treated with ketamine or ketamine plus 1 μM cAMPS-Rp and cells collected 24 h later, the ketamine-treated group showed an increase in BDNF, which was abolished by the cAMP antagonist d staining for GFAP confirmed primary astrocytes. (n = 5), *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Transient ketamine treatment increases phosphorylation of eukaryotic elongation factor 2 (eEF2) in C6 cells
Monteggia et al. have shown that NMDAR blockade by ketamine and MK801 results in decreased threonine-56 (Thr-56) phosphorylation of eEF2 by attenuating the activity of calcium and calmodulin dependent eEF2 kinase (eEF2k) via reduced calcium influx [10]. However, PKA phosphorylates eEF2K to increase eEF2 Thr-56 phosphorylation [33]. Given the observed ketamine-mediated increase in cAMP, we sought to determine the effect of ketamine on p-eEF2. To evaluate p-eEF2, C6 cells were treated with ketamine for 15 min, drug was washed off and cells harvested after 15 min, 1, 2, 6, and 24 h. The data indicate a sustained increase in p-eEF2 on Thr-56 after transient treatment with ketamine, paralleling increased levels of cAMP. A statistically significant increase of p-eEF2 occurs 1 h after drug washout (Fig. 5b). Total eEF2 protein remains constant (Fig. 5b). These data indicate that eEF2k may be phosphorylated by PKA after ketamine treatment in C6 cells.
Ketamine increases BDNF in a cAMP-dependent manner in primary astrocytes
Numerous reports indicate that chronic treatment with antidepressants increase BDNF mRNA and protein levels in the rodent brain and in cell cultures [15, 18, 34]. Emerging evidence also indicates that acute ketamine treatment results in increased levels of BDNF [9, 10, 13] and, ketamine treatment does not elicit an antidepressant behavioral response in mice lacking forebrain BDNF [10]. Given PKA phosphorylation of CREB has been shown to increase the transcription of BDNF, we sought to determine the effects of these cellular events. C6 cells were treated with 10 μM ketamine for 15 min and were collected after either 1 h or 24 h. Only at 24 h after ketamine treatment was there a significant increase in BDNF indicating the possibility of a cAMP/PKA/pCREB-mediated transcriptional-dependent increase (Fig. S3).
Next, we sought to establish the relationship of ketamine-induced cAMP increase with that of the ketamine-induced increase in BDNF. Mature primary rat astrocytes were treated (15 min) with ketamine alone or ketamine plus the cell permeable cAMP competitive antagonist cAMPs-Rp and collected after 24 h. Staining for GFAP confirmed presence of astrocytes (Fig. 5d). The data show a significant increase in BDNF 24 h after ketamine treatment, which was attenuated by 1 μM cAMPs-Rp (Fig. 5c). These data suggest a ketamine-induced, cAMP-dependent, production of BDNF.
The ketamine metabolite, (2R,6R)-hydroxynorketamine increases cAMP accumulation
Recent reports indicate that the ketamine metabolite (2R,6R)-hydroxynorketamine (2R,6R)-HNK produces rapid and robust antidepressant effects through increased expression of BDNF, GluA1, GluA2, and dephosphorylation of eEF2k in synaptosomes, through an unknown mechanism. Extensive binding displacement studies have shown that these effects are independent of NMDAR antagonism as (2R,6R)-HNK lacks affinity for the NMDAR [13]. Suzuki et al. have suggested a possible role of NMDAR antagonism for (2R,6R)-HNK, yet this remains controversial [35]. Hence, we tested whether (2R,6R)-HNK decreases translocation of Gαs-GFP similarly to ketamine. A 15-minute treatment with (2R,6R)-HNK had effects similar to ketamine (Fig. 6a). To determine the functional consequences of augmented Gαs and adenylyl cyclase coupling, C6 cells were infected with the EPAC-based fluorescent biosensor, pretreated with 10 μM (2R,6R)-HNK for 15 min, drug washed off, stimulated with isoproterenol and fluorescence was measured. Indeed, (2R,6R)-HNK robustly increased cAMP (Fig. 6b) indicating a possible role of cAMP in the antidepressant effects of (2R,6R)-HNK.
Fig. 6.
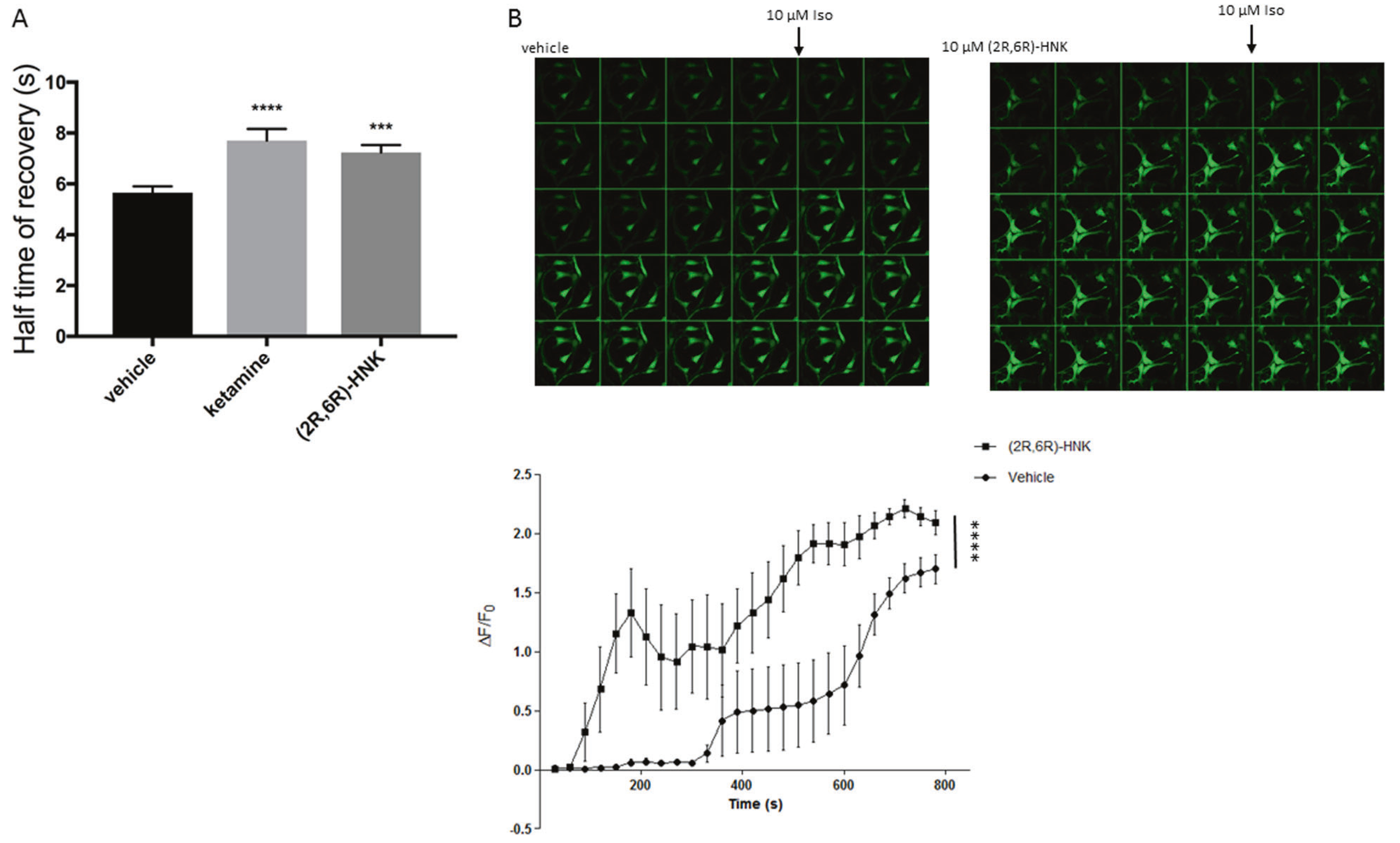
(2 R,6 R)-Hydroxynorketamine mediates attenuated Gαs lipid raft localization and cAMP accumulation similarly to ketamine. a Gαs-GFP monoclonal C6 cells were treated for 15 min with 10 μM (2 R,6 R)-HNK and Gαs lateral mobility analyzed by FRAP (n ≥ 75), which revealed an increase of recovery half-time, suggesting augmented association of Gαs and adenylyl cyclase. b C6 cells were infected with (1.09 × 109 VG/mL) cADDIS virus, pretreated with 10 μM (2R,6R)-HNK, imaged every 30 s and 10 μM isoproterenol was added after the fourth frame. (n = 4), ***p < 0.001; ****p < 0.0001
Discussion
Data presented here suggest astrocytic cAMP-dependent BDNF may be involved in antidepressant actions of ketamine. Some of the effects of ketamine (e.g., Gαs translocation and cAMP production) were immediate and others (e.g., CREB phosphorylation and BDNF production) were delayed. Ketamine has a metabolic half-life of ~6 h, while remission of depressive symptoms on average persist for 1–2 weeks, suggesting deployment of both rapid and sustained cellular pathways for antidepressant effects [3, 4]. To the best of the authors’ knowledge, this is the first report to show cAMP-dependent production of BDNF 24 h after ketamine treatment in astrocytes. Reports are conflicting on the timeframe of increased BDNF expression; some showing increased BDNF production only after 24 h, while others observe this within 30 min [9, 10, 13, 27]. The method of tissue collection and data presented here may explain this controversy in the literature, given isolation of synaptosomes would limit glia derived BDNF. Moreover, the cAMP-dependent effect may be specific to glia, as reports on primary neurons and animals show antidepressant doses of ketamine decrease phosphorylation of eEF2 and show conflicting reports on the effect of phosphorylation of CREB [10, 13, 36, 37].
The inability of NMDAR antagonists other than ketamine to elicit the Gαs biosignature, combined with these cellular antidepressant hallmarks persisting after reduced expression of NMDARs suggests that ketamine actions are NMDAR-independent, at least in astrocytes and C6 cells. This is further reinforced by the positive effect seen with (2R,6R)-HNK since it does not bind the NMDAR at 10 μM, yet still elicits antidepressant biochemical and behavioral responses [13]. This report also contrasts studies, where NMDAR antagonists other than ketamine elicit a similar biochemical antidepressant response to ketamine [9, 10]. However, ketamine may act at both neurons and glia, and it is possible that NMDAR are relevant for the former.
Note that accumulating evidence suggests glia contribute to the pathophysiology of MDD. Several post-mortem histological investigations of depressed subjects report reduced astrocyte numbers in brain regions implicated in depression, including the prefrontal cortex, dorsolateral prefrontal cortex, orbitofrontal cortex, and hippocampus. This is consistent with impaired astrocyte function in the pathophysiology of MDD [38]. Furthermore, PET studies showing global reduction of cAMP in the brains of depressed subjects cannot differentiate between cell types [19]. The role of cAMP in astrocytes or other glia in MDD and the antidepressant response has been largely unexplored.
Given the delayed increase in BDNF expression in astrocytes compared to studies performed in neurons, the NMDAR-independent effects of ketamine in translocating Gαs from lipid rafts may contribute to the maintenance of the antidepressant action of ketamine. BDNF is an essential component of antidepressant action, and glia synthesize and secrete a variety of neurotrophic factors, including BDNF. Astrocyte-derived BDNF is involved in synaptogenesis, dendritic arborization, and increased dendritic spine density [38]. One may speculate that a major loss of neurotrophic producing cells, or a loss of astrocytic neurotrophic production would result in depression. Parallel to this concept, increased astrocytic neurotrophic production may have antidepressant effects. Indeed, preclinical data suggests overexpressing BDNF in mouse hippocampal astrocytes produces antidepressant effects [39]. Data presented herein suggest cAMP-dependent BDNF production may play a role in the antidepressant actions of ketamine. Recently, we reported that the translocation of Gαs from lipid rafts was mediated by disruption of lipid raft tubulin/ Gαs complexes after sustained treatment with a variety of antidepressant compounds [40]. Unlike previously tested compounds, ketamine’s action is dramatically more rapid, suggesting a different portal to the translocation of Gαs. Like many lipophilic anesthetic compounds ketamine partitions into the plasma membrane [41] and may interact directly with lipids or proteins in the plasma membrane to influence the localization of Gαs. Indeed, other anesthetics have been shown to alter plasma membrane localization of transmembrane proteins within minutes of treatment [42].
We have suggested that the translocation of Gαs from lipid rafts is a cellular hallmark of antidepressant action and it may well provide a biosignature with which to identify compounds with antidepressant potential. The notion that ketamine displays this signature along a time course relevant for its antidepressant effects is intriguing. Thus, it is possible that these results not only reveal a mechanism for ketamine action, but also suggest a cAMP-dependent mechanism of action of many classes of antidepressants.
Supplementary Material
Acknowledgements
This research is supported by VA Merit Award BX00149 (MMR) and NIH R01AT009169 (MMR). NHW and JS were supported by T32 MH067631 and HS by an AHA postdoctoral fellowship. (2R,6R)-HNK was gifted by the NIH.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41380-018-0083-8) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest MMR has received research support from Eli Lilly and Lundbeck, Inc. and is consultant to Otsuka Pharmaceuticals. He also has ownership in Pax Neuroscience. The remaining authors declare that they have no conflict of interest.
References
- 1.Kessler RC, Bromet EJ. The epidemiology of depression across cultures. Annu Rev Public Health. 2013;34:119–38. 10.1146/annurev-publhealth-031912-114409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am J Psychiatry. 2006;163:1905–17. 10.1176/appi.ajp.163.11.1905 [DOI] [PubMed] [Google Scholar]
- 3.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4. 10.1016/S0006-3223(99)00230-9 [DOI] [PubMed] [Google Scholar]
- 4.Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64. 10.1001/archpsyc.63.8.856 [DOI] [PubMed] [Google Scholar]
- 5.Abdallah CG, Sanacora G, Duman RS, Krystal JH. Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med. 2015;66:509–23. 10.1146/annurev-med-053013-062946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lener MS, Niciu MJ, Ballard ED, Park M, Park LT, Nugent AC, et al. Glutamate and gamma-aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol Psychiatry. 2016;81:1–12. 10.1016/j.biopsych.2016.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62: 63–77. 10.1016/j.neuropharm.2011.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanacora G, Schatzberg AF. Ketamine: promising path or false prophecy in the development of novel therapeutics for mood disorders? Neuropsychopharmacology. 2015;40:259–67. 10.1038/npp.2014.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N mTOR-dependent synapse formation. Science. 2010;329: 959–65. 10.1126/science.1190287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. 10.1038/nature10130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maeng S, Zarate CA, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of α-amino-3-hydroxy-5-methylisox-azole-4-propionic acid receptors. Biol Psychiatry. 2008;63: 349–52. 10.1016/j.biopsych.2007.05.028 [DOI] [PubMed] [Google Scholar]
- 12.Newport DJ,Div M,Carpenter LL,Mcdonald WM,Potash JB, Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172: 950–66. 10.1176/appi.ajp.2015.15040465. [DOI] [PubMed] [Google Scholar]
- 13.Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533:481–6. 10.1038/nature17998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang C, Shirayama Y, Zhang J, Ren Q, Yao W, Ma M, et al. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. 2015;5:e632 10.1038/tp.2015.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malberg JE, Blendy JA. Antidepressant action: to the nucleus and beyond. Trends Pharmacol Sci. 2005;26:631–8. 10.1016/j.tips.2005.10.005 [DOI] [PubMed] [Google Scholar]
- 17.Gass P, Riva MA. CREB neurogenesis and depression. Bioessays. 2007;29:957–61. 10.1002/bies.20658 [DOI] [PubMed] [Google Scholar]
- 18.Dwivedi Y, Pandey GN. Adenylyl cyclase-cyclic AMP signaling in mood disorders: role of the crucial phosphorylating enzyme protein kinase A. Neuropsychiatr Dis Treat. 2008;4(1 A):161–76. 10.2147/NDT.S2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujita M, Richards EM, Niciu MJ, Ionescu DF, Zoghbi SS, Hong J, et al. cAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor HHS public access. Mol Psychiatry. 2017;22:754–9. 10.1038/mp.2016.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toki S, Donati RJ, Rasenick MM. Treatment of C6 glioma cells and rats with antidepressant drugs increases the detergent extraction of Gαs from plasma membrane. J Neurochem. 1999;73:1114–20. 10.1046/j.1471-4159.1999.0731114.x [DOI] [PubMed] [Google Scholar]
- 21.Donati RJ, Dwivedi Y, Roberts RC, Conley RR, Pandey GN, Rasenick MM. Postmortem brain tissue of depressed suicides reveals increased Gαs localization in lipid raft domains where it is less likely to activate adenylyl cyclase. J Neurosci. 2008;28:3042–50. 10.1523/JNEUROSCI.5713-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, & Rasenick MM (2010). Chronic treatment with escitalopram but not R-citalopram translocates Gαs from lipid raft domains and potentiates adenylyl cyclase: a 5-hydroxytryptamine transporter-independent action of this antidepressant compound. J Pharmacol Exp Ther. 2010;332: 977–84. 10.1124/jpet.109.162644.sponse [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Czysz AH, Schappi JM, Rasenick MM. Lateral diffusion of Gαs in the plasma membrane is decreased after chronic but not acute antidepressant treatment: role of lipid raft and non-raft membrane microdomains. Neuropsychopharmacology. 2014;40:1–8. 10.1038/npp.2014.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–40. 10.1038/nrn2059 [DOI] [PubMed] [Google Scholar]
- 25.Wedegaertner PB, Chu DH, Wilson PT, Levis MJ, Bourne HR. Palmitoylation is required for signaling functions and membrane attachment of Gαq and Gαs. J Biol Chem. 1993;268:25001–8. [PubMed] [Google Scholar]
- 26.Allen JA, Yu JZ, Dave RH, Bhatnagar A, Roth BL, Rasenick MM. Caveolin-1 and lipid microdomains regulate Gαs trafficking and attenuate Gαs /adenylyl cyclase signaling. Mol Pharmacol. 2009;76:1082–93. 10.1124/mol.109.060160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popp S, Behl B, Joshi JJ, Lanz TA, Spedding M, Schenker E, et al. In search of the mechanisms of ketamine’s antidepressant effects: how robust is the evidence behind the mTor activation hypothesis [version 1; referees: 1 approved, 1 approved with reservations]. F1000Research. 2016;5:634 10.12688/f1000research.8236.1 [DOI] [Google Scholar]
- 28.Sos P, Klirova M, Novak T, Kohutova B, Horacek J, Palenicek T. Relationship of ketamine’s antidepressant and psychotomimetic effects in unipolar depression. Neuro Endocrinol Lett. 2013;34:287–93. [PubMed] [Google Scholar]
- 29.Erb SJ, Schappi JM, & Rasenick MM (2016). Antidepressants accumulate in lipid rafts independent of monoamine transporters to modulate redistribution of the G protein, Gαs. J Biol Chem. 2016;291:19725–33. 10.1074/jbc.M116.727263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tewson PH, Martinka S, Shaner NC, Hughes TE, Quinn AM. New DAG and cAMP sensors optimized for live-cell assays in automated laboratories. J Biomol Screen. 2016;21:298–305. 10.1177/1087057115618608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulbrich MH, Isacoff EY. Rules of engagement for NMDA receptor subunits. Proc Natl Acad Sci USA. 2008;105:14163–8. 10.1073/pnas.0802075105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duman RS, Malberg J, Thome J. Neural plasticity to stress and antidepressant treatment. Biol Psychiatry. 1999;46:1181–91. 10.1016/S0006-3223(99)00177-8 [DOI] [PubMed] [Google Scholar]
- 33.Browne GJ, Proud CG. Regulation of peptide-chain elongation in mammalian cells. Eur J Biochem. 2002;5368:5360–8. 10.1046/j.1432-1033.2002.03290.x. October [DOI] [PubMed] [Google Scholar]
- 34.Coppell AL, Pei Q, Zetterström TSC. Bi-phasic change in BDNF gene expression following antidepressant drug treatment. Neuropharmacology. 2003;44:903–10. 10.1016/S0028-3908(03)00077-7 [DOI] [PubMed] [Google Scholar]
- 35.Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, Monteggia LM. Effects of a ketamine metabolite on synaptic NMDAR function. Nature. 2017;546:E1–E3. 10.1038/nature22084 [DOI] [PubMed] [Google Scholar]
- 36.Xue W, Wang W, Gong T, Zhang H, Tao W, Xue L, et al. PKA-CREB-BDNF signaling regulated long lasting antidepressant activities of Yueju but not ketamine. Sci Rep. 2016;6:26331 10.1038/srep26331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Réus GZ, Stringari RB, Ribeiro KF, Ferraro AK, Vitto MF, Cesconetto P, et al. Ketamine plus imipramine treatment induces antidepressant-like behavior and increases CREB and BDNF protein levels and PKA and PKC phosphorylation in rat brain. Behav Brain Res. 2011;221:166–71. 10.1016/j.bbr.2011.02.024 [DOI] [PubMed] [Google Scholar]
- 38.Wang Q, Jie W, Liu JH, Yang JM, Gao TM. An astroglial basis of major depressive disorder? An overview. Glia. 2017;65:1227–50. 10.1002/glia.23143 [DOI] [PubMed] [Google Scholar]
- 39.Quesseveur G, David DJ, Gaillard MC, Pla P, Wu MV, Nguyen HT, et al. BDNF overexpression in mouse hippocampal astrocytes promotes local neurogenesis and elicits anxiolytic-like activities. Transl Psychiatry. 2013;3:e253 10.1038/tp.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh H, Wray N, Schappi JM, & Rasenick MM (2018). Disruption of lipid-raft localized Gαs /tubulin complexes by antidepressants: a unique feature of HDAC6 inhibitors, SSRI and tricyclic compounds. Neuropsychopharmacology. 2018; 1–11. 10.1038/s41386-018-0016-x [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Jerabek H, Pabst G, Rappolt M, Stockner T. Membrane-mediated effect on ion channels induced by the anesthetic drug ketamine. J Am Chem Soc. 2010;132:7990–7. 10.1021/ja910843d [DOI] [PubMed] [Google Scholar]
- 42.Bademosi AT, Steeves J, Karunanithi S, Zalucki OH, Gormal RS, Liu S, et al. Trapping of Syntaxin1a in presynaptic nanoclusters by a clinically relevant general anesthetic. Cell Rep. 2018;22: 427–40. 10.1016/j.celrep.2017.12.054 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.