

Abstract
Alkenes, ethers, and alcohols account for a significant percentage of bulk reagents available to the chemistry community. The petrochemical, pharmaceutical, and agrochemical industries each consume gigagrams of these materials as fuels and solvents each year. However, the utilization of such materials as building blocks for the construction of complex small molecules is limited by the necessity of prefunctionalization to achieve chemoselective reactivity. Herein, we report the implementation of efficient, sustainable, diaryl ketone hydrogen-atom transfer (HAT) catalysis to activate native C–H bonds for multicomponent dicarbofunctionalization of alkenes. The ability to forge new carbon–carbon bonds between reagents typically viewed as commodity solvents provides a new, more atom-economic outlook for organic synthesis. Through detailed experimental and computational investigation, the critical effect of hydrogen bonding on the reactivity of this transformation was uncovered.
Graphical Abstract
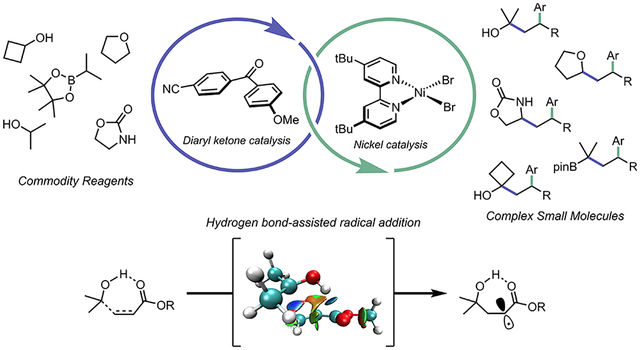
INTRODUCTION
Among the most prevalent materials available to the chemistry community are hydrocarbons (alkanes and alkenes) and their oxygenated derivatives (alcohols and ethers). Despite their ubiquity in nature and chemical industries, the preparation of complex small molecules from these commodity materials remains a pertinent synthetic challenge. Prefunctionalization, typically requiring multistep sequences, is often necessary for promoting selective reactivity of simple building blocks. In an effort to overcome the inefficiency of non-native functional group installation, several research programs have been initiated that focus on the selective functionalization of C–H bonds.1
Despite significant advancements in this arena, multicomponent C–H functionalizations are still exceedingly rare.2 This stands in stark contrast to the advancements in the field of multicomponent, transition metal-mediated cross-couplings that utilize prefunctionalized substrates.3,4 Dicarbo-functionalizations (DCF) are a subset of multicomponent reactions that employ an alkene or alkyne as a “linchpin”, facilitating the regioselective construction of two C–C bonds. Early work in nickel-catalyzed DCF employed aryl- and alkylmetallic coupling partners (e.g., organozinc and organo-boron reagents).5–9 Significant advancement in functional group compatibility has been achieved in nickel-catalyzed DCF reactions by leveraging radical-based mechanisms.10 More recently, photoredox/Ni dual catalysis has dramatically expanded the suite of installable fragments by exploiting a well-developed library of divergent radical precursors.11–18 Dicarbofunctionalization of styrenes has also been achieved via radical-polar crossover mechanisms.19,20
Although synthetically enabling, even the most modern DCF reactions do not address the longstanding challenge of utilizing commodity reagents in multicomponent reactions, instead relying on prefunctionalized radical precursors that typically exhibit poor atom economy. To address these limitations, we sought to develop a radical-based DCF strategy enabled by the selective homolysis of C–H bonds. At the outset, various intermolecular hydrogen-atom transfer (HAT) catalysts (e.g., tertiary amines or tungsten-based salts) were considered to achieve the desired transformation, but these either require activation via organometallic photocatalysts or lack sufficient chemoselectivity.21–26 Notably, diaryl ketones have been identified as effective HAT catalysts that are photoactive, sustainable, and chemoselective, thus overcoming the inherent challenges of the aforementioned HAT agents. In this vein, diaryl ketone HAT catalysis has been used in the hydroalkylation of activated alkenes as well as the arylation of C–H bonds via Ni-catalyzed cross-coupling.27–31 We envisioned that the merger of these two fundamental transformations, achieved by synergistic diaryl ketone/Ni dual catalysis, could overcome the inherent limitations of prefunctionalized radical precursors in multicomponent cross-coupling reactions. In addition to developing this transformation, we performed quantum mechanical calculations to provide insight into the mechanism and the origin of selectivity of this protocol.
RESULTS AND DISCUSSION
We envisioned that the desired hydrogen atom transfer-mediated DCF would operate via the mechanism outlined in Figure 1. Photoexcitation of a diaryl ketone (I) would access the triplet-state diradical II. This persistent radical would perform selective homolysis of an activated C–H bond (BDE = 90.4 kcal/mol; see SI Figure S6 for full details), generating the desired carbon-centered radical (IV) with concomitant formation of ketyl radical III. In turn, this alkyl radical would undergo regioselective Giese addition to an activated alkene V, leading to the corresponding radical adduct VI. The newly formed radical adduct (VI) could be captured by a Ni0 species (VII), and the resulting NiI complex would undergo subsequent oxidative addition with an aryl halide, affording IX (blue pathway). Alternatively, the aryl halide could first undergo oxidative addition with the Ni0 species prior to radical capture (VII to X; green pathway). After reversible square planar-tetrahedral isomerism, VI could then add to the productive tetrahedral NiII intermediate X,32 forming complex IX. Reductive elimination from IX would produce the desired DCF product XII and NiI complex XI.33 Finally, single-electron transfer (SET) from ketyl radical III to XI would regenerate I and VII, respectively, completing both catalytic cycles.
Figure 1.
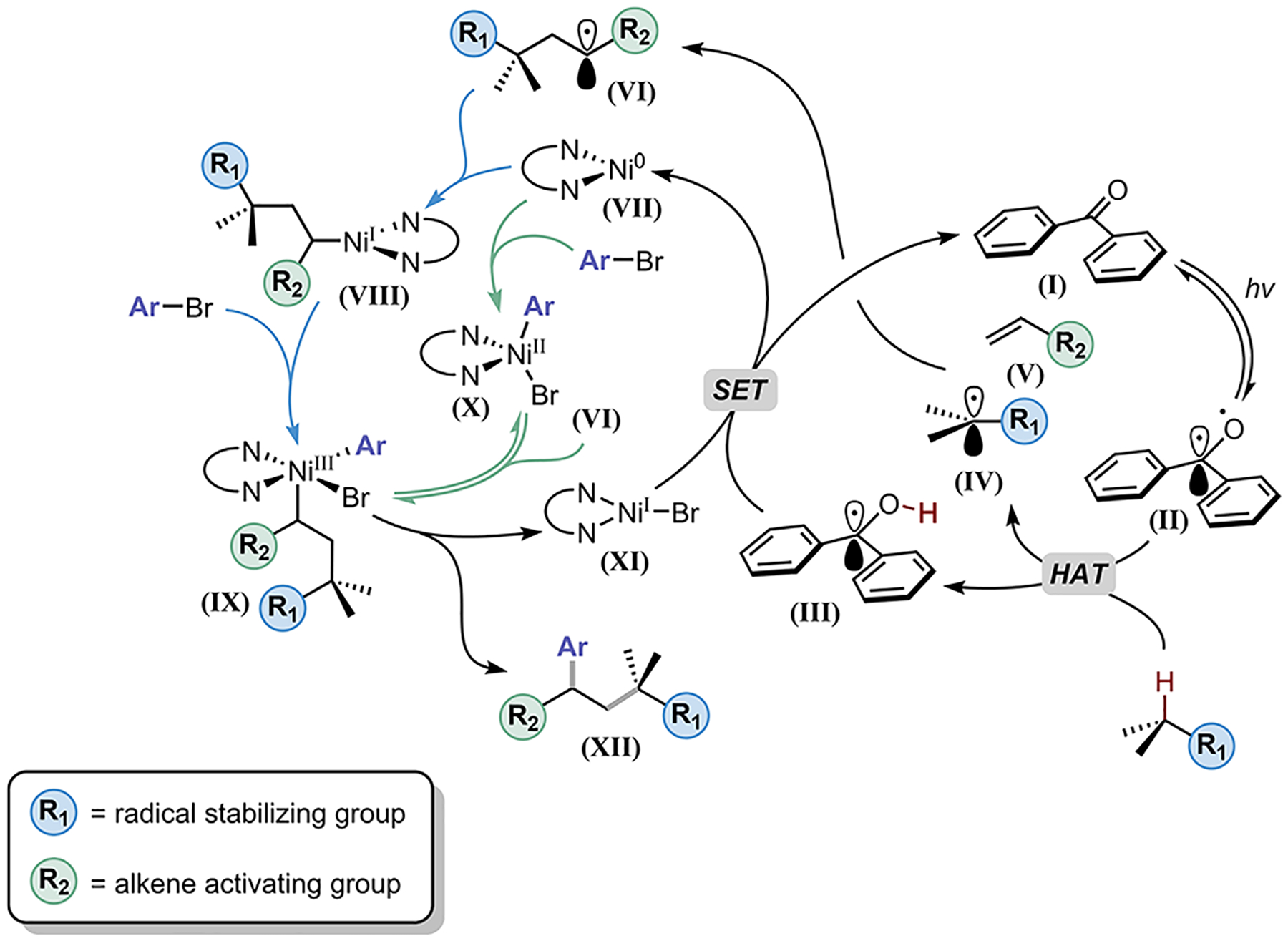
Proposed mechanism for three-component HAT-mediated photoredox/nickel dual-catalyzed DCF of alkenes. BDE were calculated at the (U)B3LYP-D3/def2-TZVPP//(U)B3LYP-D3/def2-SVP level of theory.
We began optimization of the described DCF reaction with a survey of several different HAT catalysts, which revealed that diaryl ketones bearing one electron-rich and one electron-poor arene were the most effective.27 We presume that such catalysts have a longer-lived triplet state because of captodative radical stabilization.34 Notably, quantum mechanical calculations revealed a general trend between the DCF product yield (determined by crude 1H NMR) and the calculated triplet state energy of a range of benzophenone derivatives (SI Table S6). Nonpolar, aprotic solvents, such as benzene and α,α,α-trifluorotoluene (TFT), were critical for the successful formation of the desired product; in all other cases, high ratios of alkene hydroalkylation were observed.
With a suitable set of conditions established (see SI for full optimization details), the scope of each reaction component was evaluated (Table 1). Use of cyclopentyl methyl ether (CPME) as the C–H precursor provided good yields across a range of aryl halides; however, i-PrOH was found to be superior in several instances. Arene substructures that contained other activated C–H bonds were accommodated under the standard conditions, but fared better when the loading of the C–H precursor was increased to that of a cosolvent with TFT (1:1 v/v). In general, aryl bromides bearing electron withdrawing groups resulted in the best yields of DCF products, while electron-neutral and electron-donating arenes returned some starting material and protodebromination byproducts. However, incorporation of electron-neutral and electron-donating aryl iodides at extended reaction times provided significantly improved yields (SI Table S5). A range of bifunctional arenes were evaluated with excellent chemoselectivity, generating products that could serve as linchpins for subsequent diversification of the arene region (17–19). An arene derived from the GABA-receptor antagonist, Flumazenil, was incorporated into the DCF reaction with moderate yield (24). In addition to the diverse array of aryl halides, selected heteroaryl halides were competent in the DCF reaction (25–31).
Table 1.
Scope of Three-Component HAT/DCFf

|
Values indicate yield of the isolated product. pin = pinacolato. Unless otherwise noted: aryl halide (1 equiv, 0.5 mmol), alkene (2 equiv, 1.0 mmol), C–H precursor (15 equiv, 7.5 mmol), Ni 2 (10 mol %, 0.05 mmol), benzophenone 1 (20 mol %, 0.10 mmol), K2HPO4 (2 equiv, 1.0 mmol), TFT (0.1 M), 24 h, irradiating with 390 nm Kessil lamp.
Using the corresponding aryl iodide.
Using a 1:1 v/v ratio of C–H precursor/TFT.
Reaction time extended to 48 h.
Using 4 equiv of alkene.
Isolated as the corresponding carboxylic acid after treatment with HCl in 1,4-dioxane.
We then turned our attention to the scope of C–H precursors. Several ether and thioether structures were utilized with good success (32–39). In the case of C–H precursors that generated secondary radicals, some of the aryl bromide was consumed via two-component cross-coupling with the C–H precursor, consistent with previous studies using alkyl radicals generated from organotrifluoroborates.13 However, increased loading of the alkene provided synthetically useful yields of the DCF product. Free alcohols performed extraordinarily well in the targeted reaction (40–44). These DCF products bear both a nucleophilic hydroxyl, as well as an electrophilic ester, offering several complementary avenues for downstream functionalization. To that end, lactonization of selected DCF products was accomplished with excellent yields (S5–S7). Several α-amido radicals were also quite effective in this protocol (45–47). Gratifyingly, utilization of 2-oxazolidinone as a C–H precursor generated only an α-amido substituted product (47), demonstrating the inherent selectivity of the benzophenone catalyst. Attempts to use alkyl amines as C–H precursors furnished the DCF product in only low yields, likely because of undesired interactions between the excess amine and the Ni catalyst. Finally, substrate 48 was generated from C–H abstraction alpha to a pinacol boronate, which, to our knowledge, is the first example of a photochemically mediated HAT of an α-boronate C–H bond. This example is particularly useful as a synthetic building block given the well-established array of transformations of alkyl boronates.35
In addition to the model acrylate, a range of sterically and electronically dissimilar vinyl functional groups were incorporated into the DCF protocol (49–52). Furthermore, a variety of internal alkenes were also competent, providing the DCF products with superb diastereoselectivity (53–56). Utilizing dimethyl fumarate as a radical acceptor produced two stereoisomers; the syn diastereomer was obtained as the anticipated hydroxy succinate 61, while the anti diastereomer underwent spontaneous lactonization to provide 62, as previously observed with 29. Finally, we prepared a collection of more complex substrates (57–59) based on natural product scaffolds. The rapid assembly of substrates containing such highly functionalized motifs validates the application of this DCF strategy in the synthesis of complex molecular architectures.
In an effort to lend further support for the mechanism outlined in Figure 1, a series of control reactions were conducted. In particular, we focused on understanding the role of the excited-state benzophenone in the C–H abstraction step. We considered alternative mechanistic scenarios such as the liberation of bromide radical from 2 via direct photochemical excitation or triplet-state energy transfer from 1. However, in the absence of 1, the desired DCF product was not observed when irradiating with either 390 or 300 nm light sources (SI Figure S2). To rule out the action of bromide radicals in the formation of the DCF product, we utilized Ni(dtbbpy)(NO3)2 in conjunction with 4-iodobenzonitrile, which, as anticipated, resulted in a good yield of the desired product (SI Figure S3). Because hydrogen atom abstraction via iodide radical is considerably endothermic (SI Figure S3), we conclude that the mechanism of DCF product formation does not depend on the participation of halide radicals. An intermolecular competition reaction using i-PrOH and i-PrOH-d7 resulted in a 3.6:1 ratio of 40 to 40-d6, indicative of an exothermic radical C–H abstraction (SI Figure S4).36 As an unsymmetrical ether, CPME allowed us to study the innate HAT selectivity in the DCF protocol. In addition to the desired DCF product, we also observed a cross-coupling product derived from the generation of a primary α-alkoxy radical as a minor byproduct. SI Figure S7 outlines a detailed computational investigation of the HAT selectivity of 1 and subsequent mechanistic pathways.
Because of the potential for alkyl radicals to undergo either direct two-component cross-coupling (CC) or three-component DCF, DLPNO–CCSD(T) and dispersion-corrected DFT calculations were used to provide further insight into the mechanism and selectivity (Figure 2; see Supporting Information for full computational details). Such calculations have recently been used by our group to study the mechanism of a nickel-catalyzed photoredox single-electron Tsuji-Trost reaction37 and provide accuracy at the level of coupled cluster theory at the cost of DFT calculations.38 Consistent with previous results,32 calculations showed that both the Ni0/NiII/NiIII and Ni0/NiI/NiIII mechanisms are energetically feasible for two- and three-component pathways, (SI Figure S8 and S9), as observed in related systems,32 and converge on the singlet, square planar complex 1C (Figure 2, inset). Two plausible mechanisms for two-component cross-coupling (CC) of radical fragment A are outlined in Figure 2 (left; blue/green pathways). After formation of the singlet, square planar complex 1C, this species is expected to equilibrate rapidly under photocatalytic conditions to the triplet, tetrahedral complex 3C.32,39,40 On the basis of prior work,34 alkyl radical A is expected to add to either the “axial” position of 1C (blue pathway, via A-TS-B, barrier of 12.8 kcal/mol) or the “equatorial” position of 3C (green pathway, via A-TS-B′, barrier of 11.5 kcal/mol), the latter being more energetically favorable. On the basis of these calculations, at low concentration of alkene, alkyl radical A is expected to produce the direct (two component) cross-coupling product (Pcc) via the green pathway. However, at high concentration of alkene, alkyl radical A is expected to undergo irreversible Giese addition to methyl acrylate via A-TS-D, which is isoenergetic to the barrier for metalation of A (A-TS-B′). We also considered radical addition of A to Ni0-bound alkene but found this pathway less likely because of the inherent higher concentration of alkene in the system with respect to nickel, as well as the low barrier for the oxidative addition of aryl bromide to the Ni0 center (see SI Figure S10 for more detailed discussion). Finally, subsequent metalation of D to 3C affords the key NiIII intermediate E, which will quickly undergo reductive elimination (via E-TS-F) to form the desired, three-component product (PDCF) and bromo-NiI species F. Notably, both metalation (via A-TS-B′) and Giese addition (via A-TS-D) of radical A were found to be irreversible, and their transition states higher in energy than the subsequent reductive eliminations (B′-TS-F and E-TS-F, respectively). Thus, these calculations demonstrate that the kinetic product selectivity for the competing pathways (DCF vs CC) is determined by the relative energy difference between the metalation (A-TS-B′) versus Giese addition (A-TS-D) transition states and, given the small energetic difference between these, is highly dependent on the relative concentration of nickel to alkene. Analysis of a secondary α-ether system (THF, SI Figure S11) is also in agreement with these findings.
Figure 2.
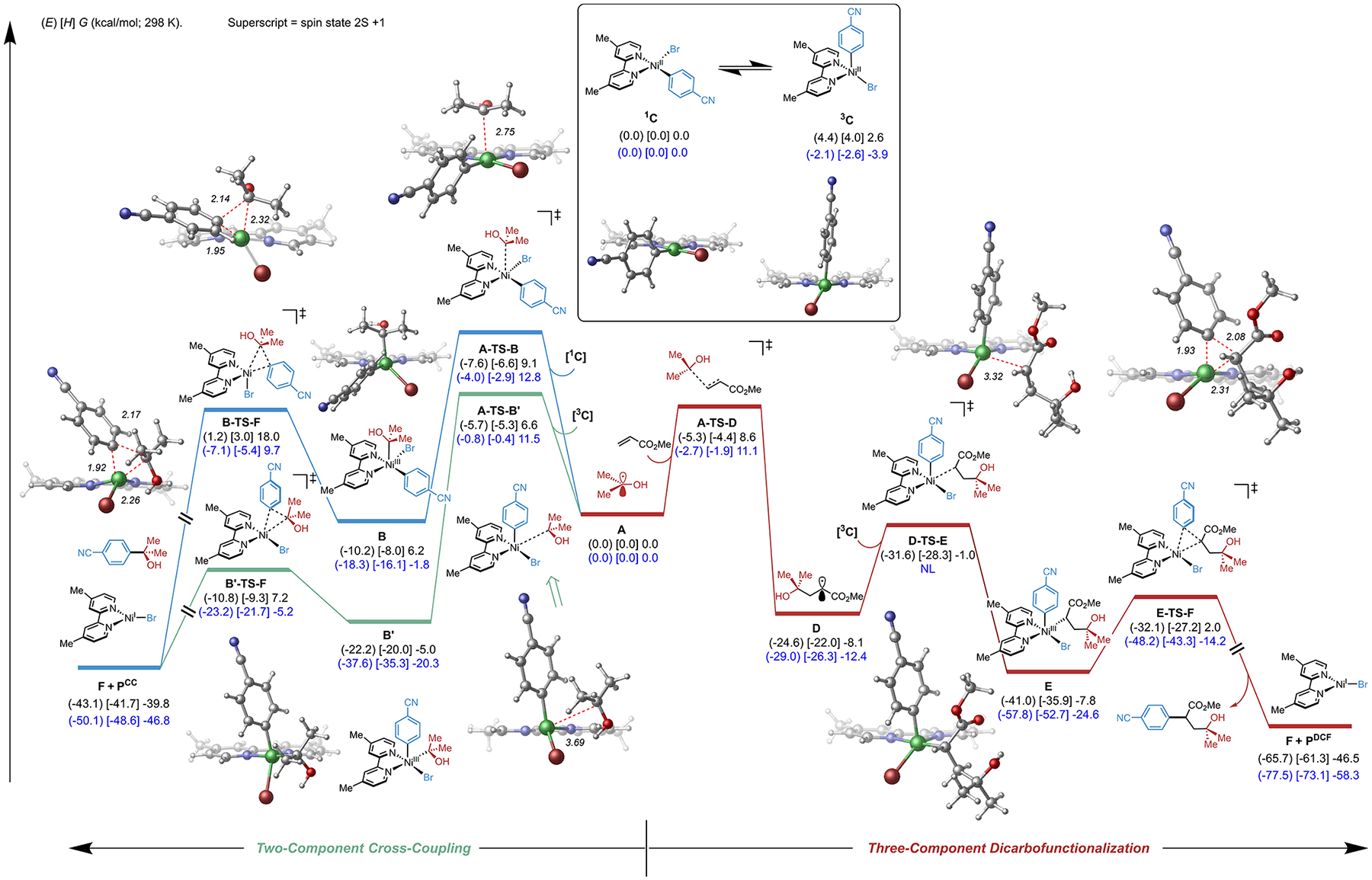
Computational analysis of the divergent reactivity of alkyl radical in the isopropyl alcohol system. Electronic (in parentheses), enthalpy (in bracket), and free energies (kcal/mol; 298 K) given were calculated at the (U)B3LYP-D3/def2-TZVPP-CPCM(benzene)//(U)B3LYP-D3/def2-SVP-CPCM(benzene) (black) and DLPNO–CCSD(T)/def2-TZVPP//(U)B3LYP-D3/def2-SVP-CPCM(benzene) (blue) levels of method.
We then examined the DCF:CC selectivity for a range of sterically and electronically unique alkyl radicals. As was established previously by our group and others, tertiary radicals are reluctant to undergo direct metalation by nickel32,41 and in the presence of an activated alkene, undergo exclusive Giese addition (Figure 2, red pathway). Under similar conditions in our previous report,13 we observed that direct two-component CC was favored over three-component DCF (~3:1 NMR ratio) for unstabilized, secondary alkyl radicals generated from organotrifluoroborates. In contrast, secondary α-alkoxy radicals gave approximately equimolar ratios of DCF to CC products (Figure 3A, entries 4–9). These results suggest that the presence of an α-oxygen increases the nucleophilic character of the radical, accelerating the Giese addition. Notably, the secondary radical generated from EtOH gave a significantly higher DCF:CC ratio than cyclic α-alkoxy radicals. The DCF selectivity was also severely eroded in the case of the α-thio-substituted radical derived from tetrahydrothiophene (entry 10). Satisfyingly, α-amido radicals exhibited the highest DCF:CC selectivity of all the examined secondary radicals (entry 11). Further, we found that the trends of experimentally observed DCF:CC selectivities is also consistent with the computed barriers (SI Table S7). That is, in general, the DCF:CC selectivity arises from competitive alkyl radical Giese addition versus metalation to the tetrahedral NiII species, which could be operative in related transformations involving secondary α-heterosubstituted alkyl radicals.
Figure 3.
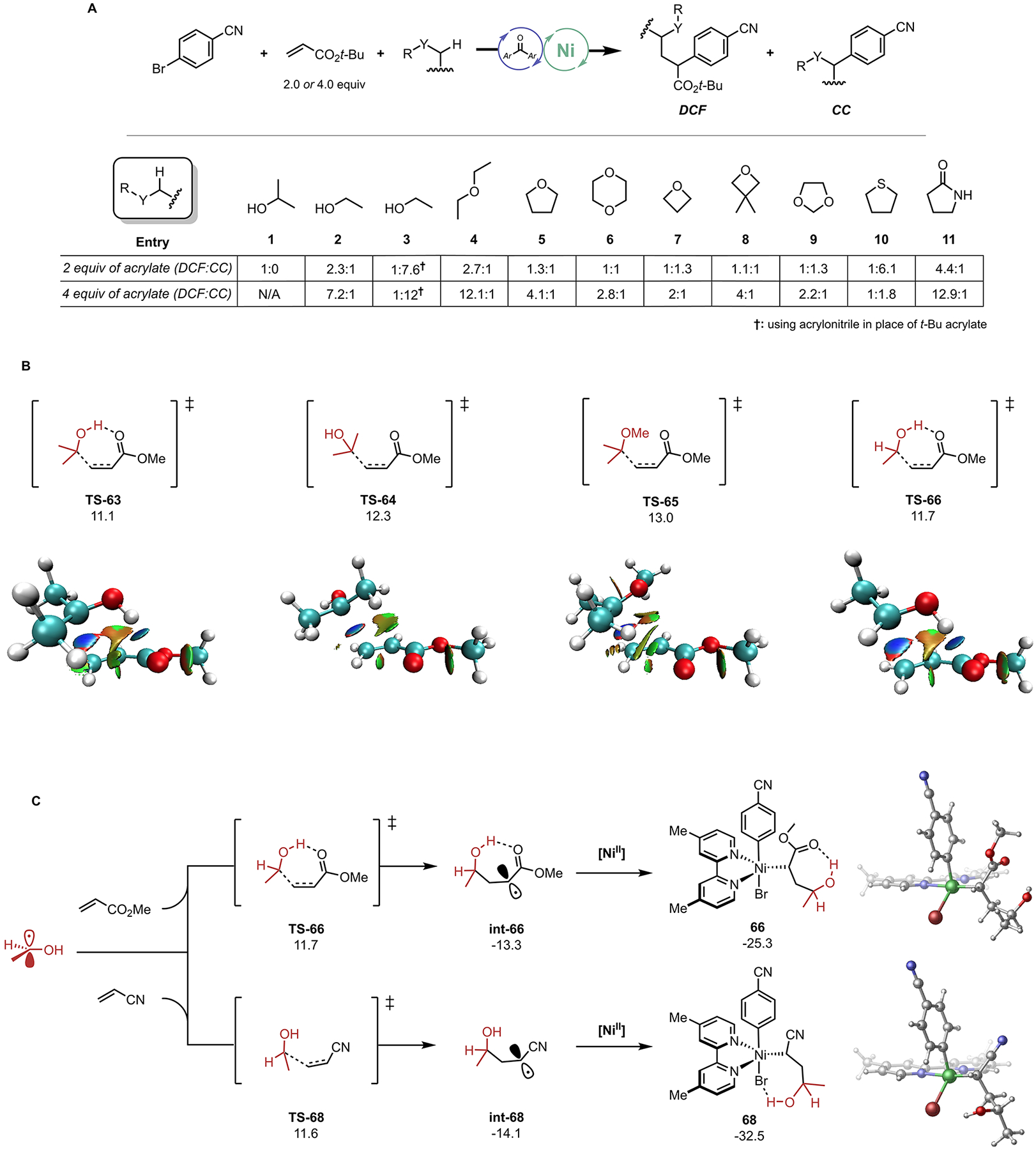
Selectivities of various secondary radicals for competing DCF and CC pathways. (A) Values indicate ratio of DCF to CC product determined by crude 1H NMR. Reaction conditions: 4-bromobenzonitrile (1 equiv, 0.1 mmol), tert-butyl acrylate or acrylonitrile (2 equiv or 4 equiv, 0.2 or 0.4 mmol), C–H precursor (15 equiv, 1.5 mmol), Ni 2 (10 mol %, 0.01 mmol), benzophenone 1 (20 mol %, 0.02 mmol), K2HPO4 (2 equiv, 0.2 mmol), TFT (0.1 M), 24 h, irradiating with 390 nm Kessil lamp. (B) NCI plots and the computed energetics (free energy, kcal/mol) of the Giese addition transition states of various alkyl radicals to acrylate. (C) Proposed hydrogen bonding increases the rate of Giese-type addition vs direct metalation in the reaction of secondary ethoxy radical. Free energies (kcal/mol; 298 K) given were calculated at the DLPNO–CCSD(T)/def2-TZVPP//(U)B3LYP-D3/def2-SVP-CPCM(benzene) level of method.
Given the unique reactivity exhibited by secondary α-hydroxy radicals (e.g., derived from EtOH), we speculated that hydrogen bonding could influence the selectivity for the two competing pathways. Prior studies revealed that the presence of hydrogen bonding significantly accelerates radical addition of alcohols into alkenes,22,42,43 presumably by weakening the α-hydroxy C–H bond in the presence of exogenous hydrogen bond acceptors. However, there are very few computational studies that examine the role of hydrogen bonding between the radical component and Giese acceptor.44 We hypothesized that this type of hydrogen bonding interaction could play a paramount role in controlling selectivity for this DCF protocol. On the basis of our calculations, we attribute the increased DCF selectivity in entry 2 compared with entries 5–9 to an accelerated Giese addition via hydrogen bonding as demonstrated by the NCI plots (Figure 3B).45 In the case of the α-alkoxy systems, the higher barrier for Giese addition (owing to the lack of hydrogen bonding) and the lower barrier for direct radical addition to the Ni center results in increased formation of the CC product (see SI Table S7 for the computed barriers of each system). Further support for the influence of hydrogen bonding was found when replacing tert-butyl acrylate with acrylonitrile as the Giese acceptor (entry 2 vs 3). The dramatic erosion of DCF to CC selectivity could be attributed to the inability of acrylonitrile to engage in hydrogen bonding with the hydroxyl group. However, considering the similar barrier of Giese addition for acrylonitrile and an acrylate (Figure 3C, TS-66 vs TS-68), we propose that the carbonyl of the acrylate “locks” the hydroxyl via an intramolecular hydrogen bond, facilitating efficient arylation of α-carboxy radical int-66. Upon closer inspection of the NiIII intermediates that result from metalation of radical adducts int-66 and int-68, we found that the hydroxyl group remains oriented toward the carbonyl in 66, while pointing to Br atom in 68. We presume that the alternate orientation of the hydroxyl in complex 68 negatively impacts the arylation step, leading to hydroalkylation of acrylonitrile. The exact mechanistic details concerning the formation of this hydroalkylation product have not yet been fully elucidated. Interestingly, the α-thio-substituted radical (Figure 3A, entry 10) shows a low barrier for metalation, but a much higher barrier for Giese addition, which makes the generation of cross-coupling product more favorable (SI Table S7). Overall, these results convincingly demonstrate an unappreciated yet critical effect of hydrogen bonding in controlling product formation in metallaphotoredox cross-couplings.
In an effort to understand the hydrogen bonding interactions between various radical and alkene components in the Giese addition step more fully, we performed hydroalkylation competition reactions (Figure 4). Isopropanol preferentially engaged acrylates and acrylonitrile in hydroalkylation in comparison to the less electrophilic acrylamide (Figure 4A; entries 1 and 2). However, i-PrOH underwent hydroalkylation with only a slight preference for acrylonitrile over adamantyl acrylate. Complete selectivity for addition to acrylonitrile over adamantyl acrylate was observed using diisopropyl ether as a C–H precursor (Figure 4A, entry 4). The calculations in Figure 4C rationalize the dissimilar selectivities of alcohols and ethers observed in the hydroalkylation competition reactions. In particular, we attribute the significant decrease in ΔΔG‡CN/CO2R for i-PrOH compared to diisopropyl ether to the presence of hydrogen bonding between the hydroxyl and the acrylate. Addition of a chaotropic reagent (MgCl2) in entry 5 resulted in high selectivity for hydroalkylation of acrylonitrile with i-PrOH. We attribute the dissimilar selectivities in entries 3 and 5 to the disruption of hydrogen bonding between i-PrOH and adamantyl acrylate when MgCl2 is present. A control reaction demonstrated that hydroalkylation of adamantyl acrylate with i-PrOH is not inhibited by the presence of excess MgCl2. Furthermore, a similar trend in chemoselectivity for i-PrOH and diisopropyl ether was observed in competition reactions between acrylonitrile and phenyl vinyl sulfone, which is again attributed to the presence/absence of hydrogen bonding in the Giese addition transition states (entries 6 and 7).
Figure 4.
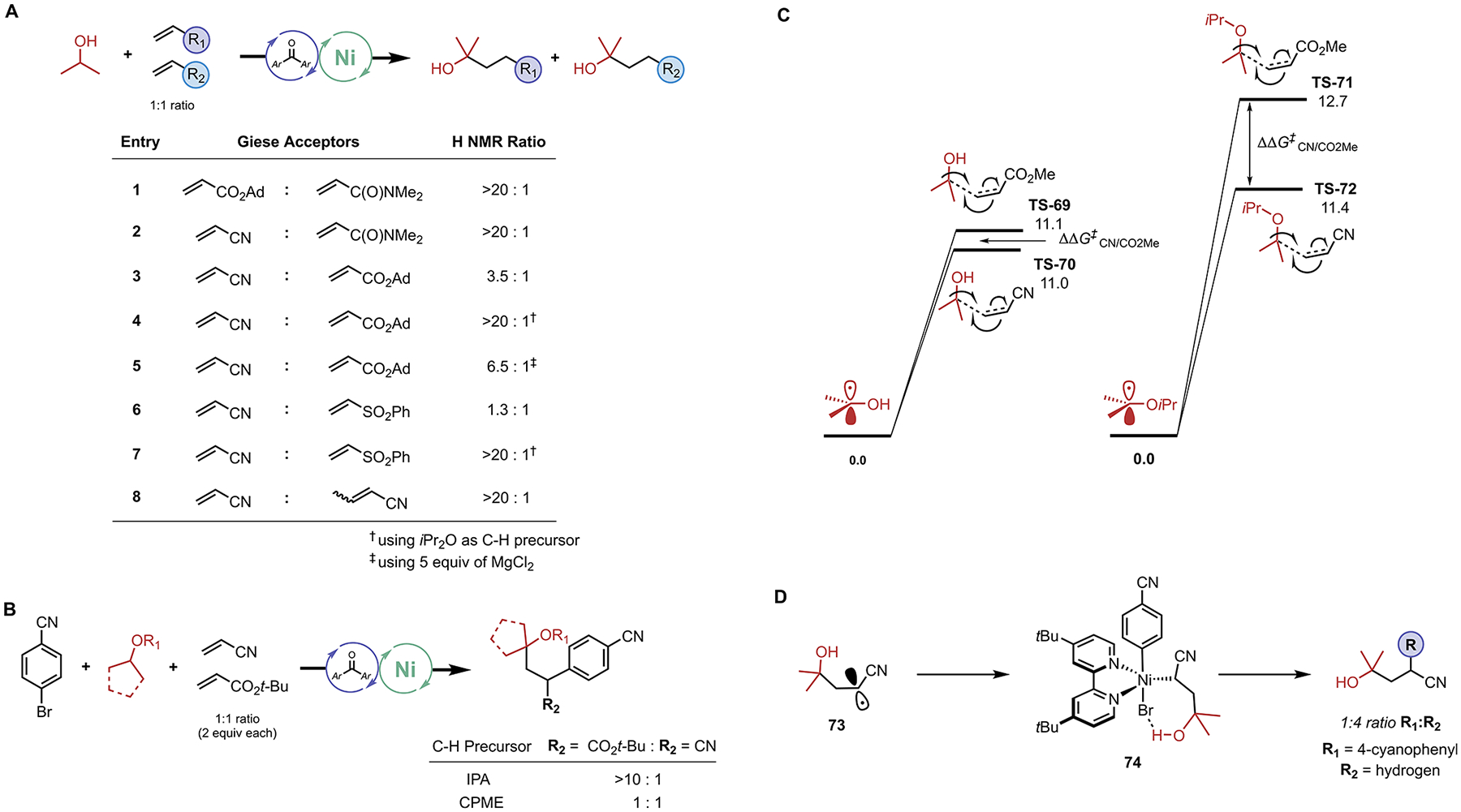
Competition reactions and related computational details. (A) Hydroalkylation competition reactions, alkenes (1 equiv each, 0.1 mmol), C–H precursor (15 equiv, 1.5 mmol), Ni 2 (10 mol %, 0.01 mmol), benzophenone 1 (20 mol %, 0.02 mmol), TFT (0.1 M), irradiating with 390 nm Kessil lamp for 30–90 min, reactions carried out to ~25% conversion. (B) DCF competition reactions, standard reaction conditions as in Table 1. (C) Computed energetics for the Giese addition processes. Free energies (kcal/mol; 298 K) given were calculated at the DLPNO–CCSD(T)/def2-TZVPP//(U)B3LYP-D3/def2-SVP-CPCM (benzene) level of method. (D) Result of reaction of i-PrOH/acrylonitrile/p-BrC6H4CN under standard reaction conditions.
As evident in entry 8, the presence of β-substitution on the alkene significantly retards the rate of Giese addition. Next, equimolar ratios of acrylonitrile and tert-butyl acrylate were subjected to the standard DCF conditions using either i-PrOH or CPME as the C–H precursor (Figure 4B). When i-PrOH was used as the C–H precursor, the ratio of DCF products greatly favored the acrylate. However, CPME exhibited an equivalent ratio of DCF products. Because of these unexpected results, we conducted a DCF reaction of i-PrOH with acrylonitrile under standard conditions, which produced an atypically high ratio of hydroalkylation/DCF products (Figure 4D). On the basis of these experiments and calculated transition state energies in Figure 4C, it is apparent that the ratio of DCF products in these complex reaction systems is not solely governed by the rates of each Giese addition. As such, we conclude that the arylation step is significantly impacted by the steric and electronic nature of Giese adducts (69–72) as well as the presence or absence of hydrogen bonding in NiIII complexes (66668, 74). We are currently pursuing further detailed calculations (including quasi-classical molecular dynamic simulations) to probe this hypothesis and these results will be reported in due course.
Finally, we employed 1-cyclopropyl ethanol as a C–H precursor, anticipating that the resultant alkyl radical could trigger ring-opening of the α-cyclopropane ring prior to undergoing Giese addition and/or metalation (Figure 5A). To our astonishment, the major product from the reaction with tert-butyl acrylate under standard conditions was the three-component cyclopropane-containing adduct 75. An additional product, a [3 + 2] cycloaddition adduct (76), was isolated as well. This product was presumably generated from cyclopropane ring opening followed by Giese addition and cis-selective 5-exo-trig cyclization to achieve a cyclopentane-containing α-hydroxy radical (vide infra), which could undergo subsequent DCF reactivity (Figure 5B).46 Similar reactivity was observed in our previous DCF report when employing an organotrifluoroborate derived from (S)-verbenone, which produced a [4 + 2] cycloaddition adduct.13 Under identical conditions, reaction with acrylonitrile produced some cyclopropane-containing DCF product (78). However, the major product obtained in this system was a two-component cross-coupled product (77), which stemmed from α-cyclopropyl radical ring opening. Computed energetics for these systems (Figure 5C) show that ring opening of the α-cyclopropyl radical (TS-81) is competitive with Giese addition to either acrylate or acrylonitrile and thus account for the mixture of both cyclopropyl DCF products (75 and 78) and various ring opened products (76 and 77). We propose that 76 and 77 stem from primary radical 81 (generated from reversible cyclopropane ring opening), which could undergo either Giese addition to the corresponding alkene or metalation to the Ni center (not calculated). The formation of 77 as the major product in the reaction with acrylonitrile suggests that TS-82 is higher than the barrier for metalation of 81. Conversely, the absence of 77 and the formation of 76 in the reaction with tert-butyl acrylate is rationalized by the significantly lower barrier of Giese addition of 81 to the acrylate. As demonstrated by the transition state structure (TS-84), hydrogen bonding between 81 and acrylate is responsible for the accelerated Giese addition leading to 84. We therefore conclude that hydrogen bonding is again a prevailing factor in determining product selectivity in this DCF protocol.
Figure 5.
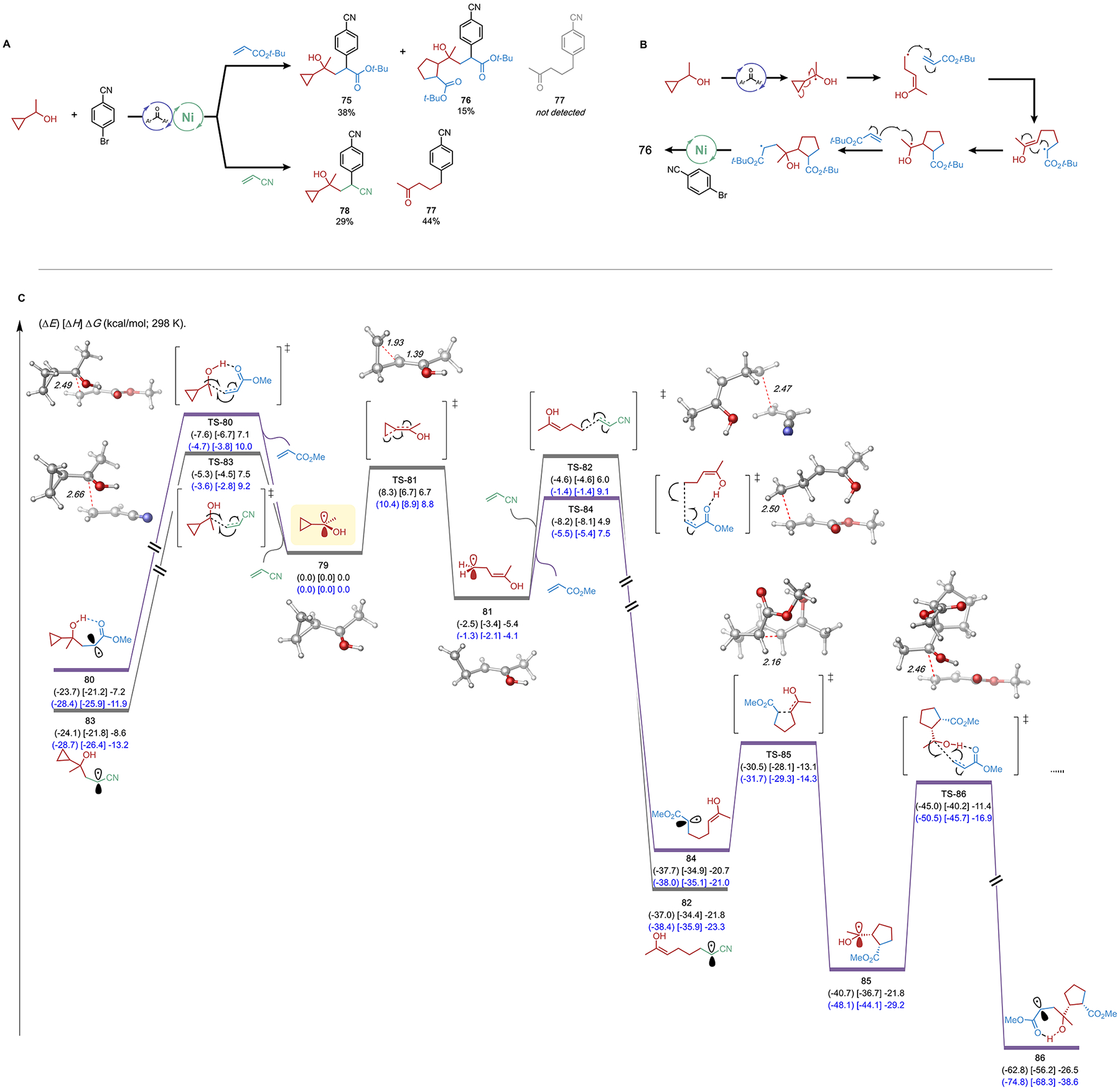
Experiments with 1-cyclopropylethanol as C–H precursor. (A) DCF reactions with 1-cyclopropylethanol; standard reaction conditions as in Table 1. (B) Proposed pathway of the formation of cyclopentane-containing DCF product. (C) Computational analysis of the competing ring-opening and Giese addition of tertiary radical in acrylate and acrylonitrile systems. Electronic (in parentheses), enthalpy (in bracket), and free energies (kcal/mol; 298 K) given were calculated at the (U)B3LYP-D3/def2-TZVPP-CPCM(benzene)//(U)B3LYP-D3/def2-SVP-CPCM-(benzene) (black) and DLPNO–CCSD(T)/def2-TZVPP//(U)B3LYP-D3/def2-SVP-CPCM(benzene) (blue) levels of method.
CONCLUSIONS
A novel multicomponent DCF strategy has been developed that relies on a sustainable and chemoselective mode of HAT catalysis. New insights into the nature and effect of hydrogen bonding in radical/alkene reactivity as well as metallaphotoredox cross-coupling have been discovered. We anticipate that not only will these findings provide a new and efficient strategy for small molecule synthesis but also that the mechanistic insights will prompt further discovery in the area of radical reactivity mediated by hydrogen bonding.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Charles W. Ross, III (University of Pennsylvania) for obtaining HRMS data. We gratefully acknowledge Dr. Mike Gao and Dr. Pat Carroll (University of Pennsylvania) for acquiring X-ray crystal structures. We acknowledge Frontier Scientific and Johnson-Matthey for fine chemicals donations and Kessil Lighting for lights used in this study.
Funding
The authors are grateful for the financial support provided by NIGMS (R35 GM 131680 to G.A.M.). O.G. acknowledges the NIGMS NIH (R35GM137797) for funding and UMD Deepthought2, MARCC/BlueCrab HPC clusters, and XSEDE (CHE160082 and CHE160053) for computational resources. M.W.C. is grateful for the financial support provided by NSF Graduate Research Fellowships. The NIH supplements awards 3R01GM118510–03S1 and 3R01GM087605–06S1, and Vagelos Institute for Energy Science and Technology supported the purchase of NMR spectrometers used in this study
Footnotes
Supporting Information The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c13077.
Experimental details and spectral data (PDF)
Accession Codes
CCDC 2046230 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c13077
The authors declare no competing financial interest.
Contributor Information
Mark W. Campbell, Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, United States
Mingbin Yuan, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Viktor C. Polites, Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, United States.
Osvaldo Gutierrez, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States;.
Gary A. Molander, Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, United States;.
REFERENCES
- (1).Davies HML; Morton D Recent Advances in C-H Functionalization. J. Org. Chem 2016, 81 (2), 343–350. [DOI] [PubMed] [Google Scholar]
- (2).Wan JP; Gan L; Liu YY Transition metal-catalyzed C-H bond functionalization in multicomponent reactions: a tool toward molecular diversity. Org. Biomol. Chem 2017, 15 (43), 9031–9043. [DOI] [PubMed] [Google Scholar]
- (3).Jensen KH; Sigman MS Mechanistic approaches to palladium-catalyzed alkene difunctionalization reactions. Org. Biomol. Chem 2008, 6 (22), 4083–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Derosa J; Apolinar O; Kang T; Tran VT; Engle KM Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chemical Science 2020, 11 (17), 4287–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Derosa J; Tran VT; Boulous MN; Chen JS; Engle KM Nickel-Catalyzed beta,gamma-Dicarbofunctionalization of Alkenyl Carbonyl Compounds via Conjunctive Cross-Coupling. J. Am. Chem. Soc 2017, 139 (31), 10657–10660. [DOI] [PubMed] [Google Scholar]
- (6).Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science 2016, 352 (6287), 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gu JW; Min QQ; Yu LC; Zhang XG Tandem Difluoroalkylation-Arylation of Enamides Catalyzed by Nickel. Angew. Chem., Int. Ed 2016, 55 (40), 12270–12274. [DOI] [PubMed] [Google Scholar]
- (8).KC S; Dhungana RK; Shrestha B; Thapa S; Khanal N; Basnet P; Lebrun RW; Giri R Ni-Catalyzed Regioselective Alkylarylation of Vinylarenes via C(sp(3))-C(sp(3))/C(sp(3))-C-(sp(2)) Bond Formation and Mechanistic Studies. J. Am. Chem. Soc 2018, 140 (31), 9801–9805. [DOI] [PubMed] [Google Scholar]
- (9).Chierchia M; Xu PL; Lovinger GJ; Morken JP Enantioselective Radical Addition/Cross-Coupling of Organozinc Reagents, Alkyl Iodides, and Alkenyl Boron Reagents. Angew. Chem., Int. Ed 2019, 58 (40), 14245–14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Garcia-Dominguez A; Li ZD; Nevado C Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2017, 139 (20), 6835–6838. [DOI] [PubMed] [Google Scholar]
- (11).Zhu C; Yue HF; Chu LL; Rueping M Recent advances in photoredox and nickel dual-catalyzed cascade reactions: pushing the boundaries of complexity. Chemical Science 2020, 11 (16), 4051–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Garcia-Dominguez A; Mondal R; Nevado C Dual Photoredox/Nickel-Catalyzed Three-Component Carbofunctionalization of Alkenes. Angew. Chem., Int. Ed 2019, 58 (35), 12286–12290. [DOI] [PubMed] [Google Scholar]
- (13).Campbell MW; Compton JS; Kelly CB; Molander GA Three-Component Olefin Dicarbofunctionalization Enabled by Nickel/Photoredox Dual Catalysis. J. Am. Chem. Soc 2019, 141 (51), 20069–20078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Guo L; Tu HY; Zhu SQ; Chu LL Selective, Intermolecular Alkylarylation of Alkenes via Photoredox/Nickel Dual Catalysis. Org. Lett 2019, 21 (12), 4771–4776. [DOI] [PubMed] [Google Scholar]
- (15).Guo L; Song F; Zhu SQ; Li H; Chu LL syn-Selective alkylarylation of terminal alkynes via the combination of photoredox and nickel catalysis. Nat. Commun 2018, 9, 69049 DOI: 10.1038/s41467-018-06904-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sun SZ; Duan YY; Mega RS; Somerville RJ; Martin R Site-Selective 1,2-Dicarbofunctionalization of Vinyl Boronates through Dual Catalysis. Angew. Chem., Int. Ed 2020, 59 (11), 4370–4374. [DOI] [PubMed] [Google Scholar]
- (17).Mega RS; Duong VK; Noble A; Aggarwal VK Decarboxylative Conjunctive Cross-coupling of Vinyl Boronic Esters using Metallaphotoredox Catalysis. Angew. Chem., Int. Ed 2020, 59 (11), 4375–4379. [DOI] [PubMed] [Google Scholar]
- (18).Zheng SL; Chen ZM; Hu YY; Xi XX; Liao ZX; Li WR; Yuan WM Selective 1,2-Aryl-Aminoalkylation of Alkenes Enabled by Metallaphotoredox Catalysis. Angew. Chem., Int. Ed 2020, 59, 17910–17916. [DOI] [PubMed] [Google Scholar]
- (19).Wang X; Han YF; Ouyang XH; Song RJ; Li JH The photoredox alkylarylation of styrenes with alkyl N-hydroxyphthalimide esters and arenes involving C-H functionalization. Chem. Commun 2019, 55 (97), 14637–14640. [DOI] [PubMed] [Google Scholar]
- (20).Ouyang XH; Li Y; Song RJ; Hu M; Luo SL; Li JH Intermolecular dialkylation of alkenes with two distinct C(sp(3))-H bonds enabled by synergistic photoredox catalysis and iron catalysis. Science Advances 2019, 5 (3), eaav9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Native functionality in triple catalytic cross-coupling: sp(3) C-H bonds as latent nucleophiles. Science 2016, 352 (6291), 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jeffrey JL; Terrett JA; MacMillan DWC O-H hydrogen bonding promotes H-atom transfer from alpha C-H bonds for C-alkylation of alcohols. Science 2015, 349 (6255), 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Capaldo L; Ravelli D Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem 2017, 2017 (15), 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Le C; Liang YF; Evans RW; Li XM; MacMillan DWC Selective sp(3) C-H alkylation via polarity-match-based cross-coupling. Nature 2017, 547 (7661), 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC Direct arylation of strong aliphatic C-H bonds. Nature 2018, 560 (7716), 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sarver PJ; Bacauanu V; Schultz DM; DiRocco DA; Lam YH; Sherer EC; MacMillan DWC The merger of decatungstate and copper catalysis to enable aliphatic C(sp(3))-H trifluoromethylation. Nat. Chem 2020, 12 (5), 459–467. [DOI] [PubMed] [Google Scholar]
- (27).Shen YY; Gu YT; Martin R sp(3) C-H Arylation and Alkylation Enabled by the Synergy of Triplet Excited Ketones and Nickel Catalysts. J. Am. Chem. Soc 2018, 140 (38), 12200–12209. [DOI] [PubMed] [Google Scholar]
- (28).Dewanji A; Krach PE; Rueping M The Dual Role of Benzophenone in Visible-Light/Nickel Photoredox-Catalyzed C-H Arylations: Hydrogen-Atom Transfer and Energy Transfer. Angew. Chem., Int. Ed 2019, 58 (11), 3566–3570. [DOI] [PubMed] [Google Scholar]
- (29).Abadie B; Jardel D; Pozzi G; Toullec P; Vincent JM Dual Benzophenone/Copper-Photocatalyzed Giese-Type Alkylation of C(sp(3))-H Bonds. Chem. - Eur. J 2019, 25 (70), 16120–16127. [DOI] [PubMed] [Google Scholar]
- (30).Zhu DL; Young DJ; Li HX Carbonyl-Photoredox/Metal Dual Catalysis: Applications in Organic Synthesis. Synthesis 2020, 52 (23), 3493–3510. [Google Scholar]
- (31).Xu S; Chen H; Zhou Z; Kong W Three-Component Alkene Difunctionalization via Direct and Selective Activation of Aliphatic C-H Bonds. Angew. Chem., Int. Ed 2021, DOI: 10.1002/anie.202014632. [DOI] [PubMed] [Google Scholar]
- (32).Yuan MB; Song ZH; Badir SO; Molander GA; Gutierrez O On the Nature of C(sp(3))-C(sp(2)) Bond Formation in Nickel-Catalyzed Tertiary Radical Cross-Couplings: A Case Study of Ni/Photoredox Catalytic Cross-Coupling of Alkyl Radicals and Aryl Halides. J. Am. Chem. Soc 2020, 142 (15), 7225–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereo-convergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137 (15), 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Viehe HG; Janousek Z; Merenyi R; Stella L The captodative effect. Acc. Chem. Res 1985, 18 (5), 148–154. [Google Scholar]
- (35).Sandford C; Aggarwal VK Stereospecific functionalizations and transformations of secondary and tertiary boronic esters. Chem. Commun 2017, 53 (40), 5481–5494. [DOI] [PubMed] [Google Scholar]
- (36).Simmons EM; Hartwig JF On the Interpretation of Deuterium Kinetic Isotope Effects in C-H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem., Int. Ed 2012, 51 (13), 3066–3072. [DOI] [PubMed] [Google Scholar]
- (37).Matsui JK; Gutierrez-Bonet A; Rotella M; Alam R; Gutierrez O; Molander GA Photoredox/Nickel-Catalyzed Single-Electron Tsuji-Trost Reaction: Development and Mechanistic Insights. Angew. Chem., Int. Ed 2018, 57 (48), 15847–15851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Liakos DG; Guo Y; Neese F Comprehensive Benchmark Results for the Domain Based Local Pair Natural Orbital Coupled Cluster Method (DLPNO-CCSD(T)) for Closed- and Open-Shell Systems. J. Phys. Chem. A 2020, 124 (1), 90–100. [DOI] [PubMed] [Google Scholar]
- (39).Shields BJ; Kudisch B; Scholes GD; Doyle AG Long-Lived Charge-Transfer States of Nickel(II) Aryl Halide Complexes Facilitate Bimolecular Photoinduced Electron Transfer. J. Am. Chem. Soc 2018, 140 (8), 3035–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ting SI; Garakyaraghi S; Taliaferro CM; Shields BJ; Scholes GD; Castellano FN; Doyle AG (3)d-d Excited States of Ni(II) Complexes Relevant to Photoredox Catalysis: Spectroscopic Identification and Mechanistic Implications. J. Am. Chem. Soc 2020, 142 (12), 5800–5810. [DOI] [PubMed] [Google Scholar]
- (41).Primer DN; Molander GA Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer. J. Am. Chem. Soc 2017, 139 (29), 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gawlita E; Lantz M; Paneth P; Bell AF; Tonge PJ; Anderson VE H-bonding in alcohols is reflected in the C alpha-H bond strength: Variation of C-D vibrational frequency and fractionation factor. J. Am. Chem. Soc 2000, 122 (47), 11660–11669. [Google Scholar]
- (43).Tian YF; Liu ZQ H-Bonding-promoted radical addition of simple alcohols to unactivated alkenes. Green Chem. 2017, 19 (21), 5230–5235. [Google Scholar]
- (44).Domingo LR; Perez P Global and local reactivity indices for electrophilic/nucleophilic free radicals. Org. Biomol. Chem 2013, 11 (26), 4350–4358. [DOI] [PubMed] [Google Scholar]
- (45).Lu T; Chen FW Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem 2012, 33 (5), 580–592. [DOI] [PubMed] [Google Scholar]
- (46).Amador AG; Sherbrook EM; Yoon TP Enantioselective Photocatalytic 3 + 2 Cycloadditions of Aryl Cyclopropyl Ketones. J. Am. Chem. Soc 2016, 138 (14), 4722–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.