

Abstract
Introduction
Plasma-based circulating tumor DNA (ctDNA) is an established biomarker for molecular profiling with emerging applications in disease monitoring in multiple tumor types, including non-small cell lung cancer (NSCLC). However, determinants of ctDNA shedding and correlation with tumor burden are incompletely understood, particularly in advanced-stage disease.
Methods
We retrospectively analyzed ctDNA-based and tissue-based genomic data and imaging from 144 patients with NSCLC. Tumor burden was quantified with CT and brain MRI for the overall cohort, and 18F-FDG PET/CT in a subset of patients.
Results
There was a moderate but statistically significant correlation between ctDNA variant allele frequency (VAF) and multiple imaging measures of tumor burden such as CT volume (rho = 0.34, p ≤ 0.0001) and metabolic tumor volume (rho = 0.36, p = 0.003). This correlation was strongest in KRAS mutant tumors (rho = 0.56, p ≤ 0.001), followed by TP53 mutants (rho = 0.43, p ≤ 0.0001), and weakest in EGFR mutated (EGFR+) tumors (rho = 0.24, p = 0.077). EGFR+ tumors with EGFR copy number gain (CNG) had significantly higher VAF than EGFR+ without CNG (p ≤ 0.00001). In multivariable analysis, TP53 and EGFR mutations, visceral metastasis, and tumor burden, were independent predictors of increased ctDNA shedding.
Conclusions
Levels of detectable ctDNA were impacted not only by tumor burden, but also by tumor genotype. The genotype-specific differences observed may be due to variations in DNA shedding and/or cellular turnover. These findings have implications for the emerging use of ctDNA in NSCLC disease monitoring and early detection.
Keywords: lung cancer, ctDNA, liquid biopsy, genotype, PET/CT, tumor volume, cancer detection
INTRODUCTION
Plasma-based circulating tumor DNA (ctDNA) is a highly specific non-invasive biomarker that is increasingly used in multiple solid tumor types for molecular profiling1, 2 It is also emerging as a promising marker for disease monitoring 3–8 and predicting response to systemic therapy 9–11. Specifically in non-small cell lung cancer (NSCLC), targeted next-generation sequencing of ctDNA is clinically used as a “liquid biopsy” to identify EGFR, ALK, ROS1, BRAF, MET, RET, ERBB2, and other oncogenic targets for therapy in advanced-stage disease 12, 13. Early changes in ctDNA levels of advanced NSCLC patients treated with immunotherapy and targeted therapy have also been shown to be prognostic 9–11, 14 Additionally, ctDNA-derived tumor mutation burden (TMB) has shown promise as a predictive biomarker for immunotherapy response 15–18. In early-stage NSCLC, ultra-sensitive ctDNA assays have enabled on-going development of ctDNA as a biomarker by our group and others for detection of molecular residual disease (MRD) after resection 3, 4, 19 and for prediction of pathologic response to neoadjuvant immune checkpoint blockade 10.
Despite these advances, our understanding of critical features relating to ctDNA dynamics, such as determinants of shedding and correlation with tumor burden, remains incomplete. Tumor characteristics that may impact ctDNA levels include size / tumor burden, tumor type, histology, anatomic location, and metabolic activity 1, 20. The relative contribution of each of these factors and associated characteristics such as genomic subtypes are not well defined. In early-stage lung cancer, plasma ctDNA concentration (as measured by the variant allele frequency, VAF) has been shown to correlate with radiographic tumor volume 3, 4, high Ki67, and presence of lymphovascular invasion 3, using highly customized ctDNA assays. However, there are conflicting data regarding histology as a predictor of ctDNA levels 3, 21. Furthermore, a quantitative correlation between ctDNA VAF with tumor burden in advanced NSCLC has not been described.
In this study, we sought to comprehensively characterize clinicopathologic, anatomical and functional imaging, and genomic correlates of ctDNA shedding in advanced NSCLC.
MATERIALS AND METHODS
Patients
We reviewed genomic profiling results of advanced NSCLC patients that underwent clinical ctDNA testing at M.D. Anderson Cancer Center between November 2015 and July 2017. 144 consecutive cases with evaluable imaging were selected, prioritizing EGFR and KRAS mutant cases to ensure sufficient representation for statistical comparisons of these major genomic subtypes. ctDNA testing was obtained generally prior to initiating treatment for newly diagnosed patients or upon disease progression for patients undergoing treatment. Each patient in the study cohort only had a single ctDNA testing time point included in this analysis. This study was conducted in accordance with the institutional review board at The University of Texas M.D. Anderson Cancer Center (PA13-0589 and PA16-0061).
Cell-free circulating tumor DNA assay
ctDNA sequencing was performed with Guardant360 (Guardant Health; Redwood City, CA), a proprietary ctDNA NGS assay that detects single nucleotide variants (SNV) in 70-73 genes as well as select copy number amplifications, indels, and fusions (Supplemental Table S1) 22, 23. The majority of samples (about 80%) were tested by a 70-gene panel; samples after November 2016 were generally analyzed with the expanded (73-gene) panel. At least 5 ng of cell free DNA was required for hybrid capture-based next generation paired-end sequencing of 160 – 170 base pair DNA strands with average coverage of 8,000x-15,000x depending on the test version. Germline variants were quantitatively excluded, as previously described 22, 23.
Imaging measurements of tumor burden
Tumor burden for each case was quantified using radiographic tumor measurements from CT, 18F-FDG PET-CT, and/or brain MRI scans obtained as part of standard clinical care. Only patients with a CT and/or PET-CT scan within 30 days of ctDNA sample collection were included. RECIST sum of longest diameters (SLD) and tumor volume calculated by semi-automated contouring on CT (syngo.via, Siemens; Malvern, PA) were measured for each case. For cases with available PET imaging, metabolic tumor volume (MTV), maximum SUV (SUVmax), and Total Lesion Glycolysis (TLG) were also obtained. MTV and TLG have shown prognostic utility in various malignancies, including NSCLC 24 MTV and TLG were measured by (MIM v.6, MIM Software; Cleveland, OH). TLG was defined as the product of mean SUV and MTV 25. SLD was defined as the sum of the longest diameters of up to five target lesions (maximum two per organ, including short axis diameters of lymph nodes with short axis ≥ 1.5 cm per RECIST v1.1) 26.
Statistical analysis
For all statistical analyses, ctDNA VAF for each sample was defined as the maximum VAF of the ctDNA alterations detected. All somatic alterations were considered, including variants of unknown significance (VUS) and synonymous mutations. The Wilcoxon rank-sum test and Kruskal-Wallis test were used to compare ctDNA VAF. Spearman’s correlation coefficients were determined to assess the relationship between ctDNA VAF and tumor measurements. All tests were two-sided and a p value ≤ 0.05 was considered to be statistically significant. Statistical analyses were conducted using R (version 3.3.2, Vienna, Austria). This study was conducted in accordance with the institutional review board at The University of Texas M.D. Anderson Cancer Center.
RESULTS
Baseline characteristics and summary statistics
The overall study cohort is mostly comprised of previously treated patients (68.1%) that generally had progressive disease at the time of ctDNA testing (Table 1). The prevalence of histologic and major genomic subtypes in the cohort is generally consistent with known prevalence in NSCLC, though, given our study design, EGFR-mutated cases are relatively over-represented (40% vs 17% in adenocarcinoma cases of the Pan-Lung Cancer NGS study 27). For each case, maximum VAF detected in the sample was generally low (median 2.0%, range 0 – 72.5%) and median tumor burden as approximated by RECIST SLD was 6.3 cm (0 – 19.2 cm) and by CT volume was 27.4 cm3 (0 – 330.5 cm3).
Table 1:
Baseline Characteristics of Overall Study Cohort
| Characteristic (n = 144) | Number (%) |
|---|---|
| Median age (range) | 64 (28 – 96) |
| Sex | |
| Female | 78 (54.2) |
| Male | 66 (45.8) |
| Histology | |
| Adenocarcinoma | 112 (77.8) |
| Squamous cell | 31 (21.5) |
| Other NSCLC | 1 (0.7) |
| Alteration a | |
| TP53 mutation | 77 (53.5) |
| KRAS mutation | 33 (22.9) |
| EGFR mutation | 57 (39.6) |
| ALK fusion | 4 (2.8) |
| Treatment status | |
| Treatment naïve | 46 (31.9) |
| Active treatment b | 77 (53.5) |
| Prior treatment c | 21 (14.6) |
| Immunotherapy exposure | |
| No | 118 (81.9) |
| Yes | 26 (18.1) |
Sum is greater than number of patients due to co-mutations
Systemic treatment within 1 month prior to ctDNA testing
Systemic treatment more than 1 month prior to ctDNA testing but within last 2 years
Of the patients with NCCN guideline recommended biomarkers28 detected by ctDNA, nine patients (6%) had MET amplification, six (4%) had EGFR exon 20 insertion, four (3%) had MET exon 14 skipping, two (1%) had HER2 amplification, one (1%) had HER2 exon 20 insertion, and one (1%) had NTRK1 fusion (Supplemental Table S4). In the overall study cohort, seven patients (4.9%) had no detectable ctDNA somatic alteration. Eighty patients (55.6%) had prior tissue molecular profiling results available that helped inform their overall mutation status (e.g. TP53, KRAS, or EGFR mutated). In those cases, the overall mutation status was determined by the union of the ctDNA and tissue somatic mutations.
Radiographic quantifications of tumor burden are associated with ctDNA shedding
We quantified tumor burden using five different approaches: RECIST SLD, CT volume, metabolic tumor volume (MTV), SUVmax, and Total Lesion Glycolysis (TLG). Within the subgroup of patients that had both CT and PET imaging evaluable (n = 69), each of these radiographic measurements positively correlated with ctDNA VAF, with Spearman’s rho ranging from 0.273 (SUVmax) to 0.414 (RECIST SLD); Supplemental Fig. S1 and Fig. 1. When assessing the overall advanced-stage cohort, CT volume had the strongest correlation (Spearman’s rho = 0.336, p ≤ 0.00001; Fig. 1) and was an independent predictor of ctDNA shedding in a stepwise multivariable regression accounting for visceral metastasis and genomic subtype (Table 2). While the positive correlation between VAF and CT volume was seen across all of the genomic subtypes analyzed, it was strongest in KRAS mutant cases (rho = 0.56, p ≤ 0.001) and weakest in AGFR-mutated tumors (rho = 0.24, p = 0.077); Supplemental Fig. S2.
Figure 1.
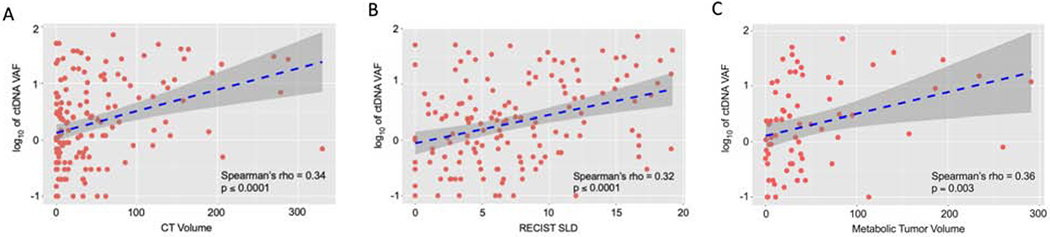
Correlation between ctDNA VAF and tumor burden as approximated by (A) CT volume (cm3) (B) RECIST SLD (cm) in the overall cohort and (C) MTV (cm3) in the PET subgroup; Spearman’s rho correlation.
Table 2:
Univariate and multivariate analysis of ctDNA VAF
| Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|
| Characteristic | HR (95% CI) | P-value | HR 95% CI | P-value |
| Histology | 0.94 (0.70 – 1.27) | 0.694 | ||
| Immunotherapy exposure | 1.03 (0.75 – 1.43) | 0.845 | ||
| Visceral metastasis | 1.66 (1.27 – 2.19) | < 0.001 | 1.48 (1.16 – 1.91) | 0.002 |
| CNS metastasis | 1.33 (1.00 – 1.79) | 0.053 | ||
| RECIST SLD | 1.05 (1.03 – 1.08) | < 0.0001 | ||
| CT volume | 1.00 (1.00 – 1.00) | < 0.0001 | 1.00 (1.00 – 1.00) | < 0.001 |
| TP53+ | 1.58 (1.25 – 2.00) | < 0.001 | 1.45 (1.16 – 1.80) | 0.001 |
| EGFR+ | 1.19 (0.92 – 1.53) | 0.179 | 1.36 (1.08 – 1.71) | 0.009 |
| KRAS+ | 1.22 (0.91 – 1.64) | 0.178 | ||
Visceral metastasis is associated with increased ctDNA shedding
Patients with visceral metastasis (defined in this study as hepatic, adrenal, renal, or splenic metastasis) had larger tumor burden (as approximated by CT volume, Wilcoxon p ≤ 0.001; figure not shown) and increased ctDNA VAF (Wilcoxon rank sum test p ≤ 0.001; Fig. 2). Stepwise multivariate Cox regression modeling, accounting for mutation status, and tumor burden, affirmed visceral metastasis as an independent predictor of increased ctDNA shedding (Table 2). Notably, there were numerically higher ctDNA levels observed in patients with the CNS as at least one metastatic site (Wilcoxon p = 0.100; Fig. 2c), though CNS metastasis was not significant in the final regression model. Furthermore, we assessed the impact on ctDNA VAF due to tumor distribution not reflected in RECIST SLD: pleural effusions (moderate or large) and bilateral sub-centimeter pulmonary nodules (< 10 nodules). There was no statistically significant increase in ctDNA shedding in the presence of those tumor burden characteristics. Additionally, we detected no significant impact on ctDNA shedding due to histology (adenocarcinoma vs squamous cell carcinoma).
Figure 2.
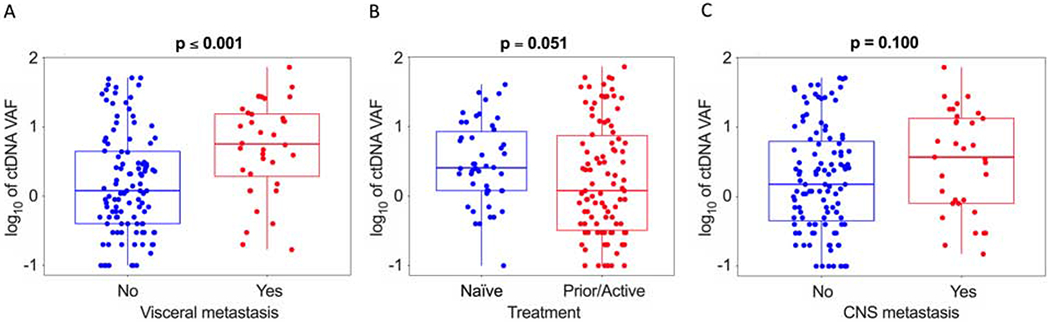
Impact of (A) visceral metastasis, (B) treatment status, and (C) CNS metastasis on ctDNA VAF. Wilcoxon rank-sum test.
Oncogenic mutations in EGFR and TP53 are associated with increased ctDNA shedding
We next compared ctDNA shedding in subgroups defined by the three most common genomic alterations in our cohort: KRAS, TP53, and EGFR. The KRAS and TP53 subgroups consisted of all patients with non-synonymous somatic mutations in those genes. Only patients with canonical EGFR driver mutations (e.g. L858R, exon 19 deletion, G719X, S768I, L861Q, and T790M resistance mutation; no exon 20 insertions) were included in the EGFR subgroup. Significantly higher ctDNA VAF was observed in TP53 mutant vs wild-type tumors (Wilcoxon p ≤ 0.001), while numerically higher VAF were observed for the EGFR, and KRAS subgroups (p = 0.17 and p = 0.16 respectively; Fig. 3a). To verify that these observations were not due to differences in tumor burden, we divided CT tumor volume into tertiles (low, mid, and high volume) and assessed VAF. Median CT volume for the low, mid, and high tertiles were 2.6 cm3, 28.3 cm3, and 95.6 cm3, respectively. For the TP53-mutated and EGFR-mutated subgroups, numerically higher VAF was observed across the tumor burden spectrum as approximated by CT volume (Fig. 3b). In the multivariate analysis, TP53-mutated and EGFR-mutated status were affirmed as strong, independent predictors of increased ctDNA shedding (Table 2).
Figure 3.
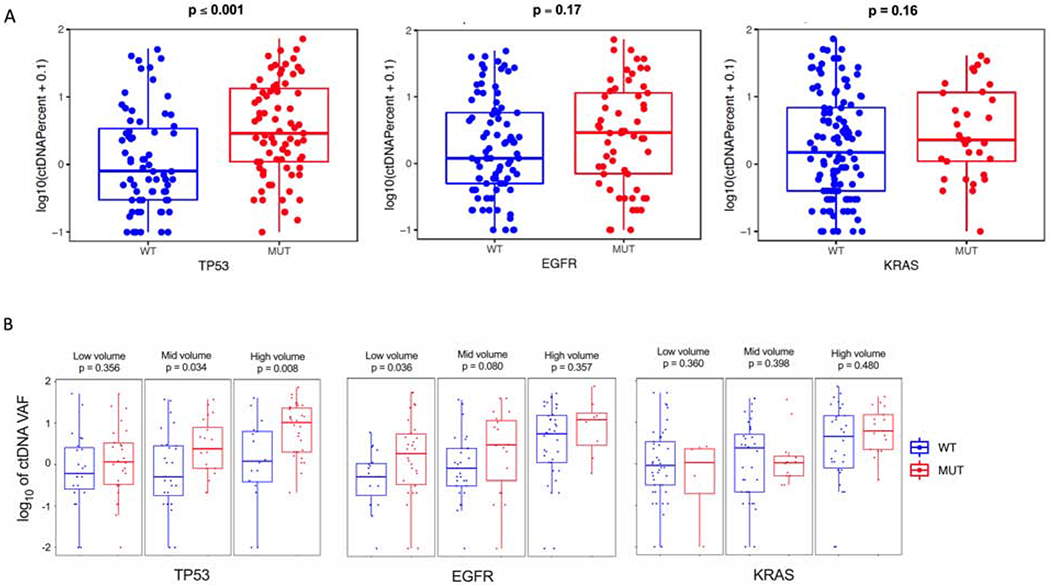
ctDNA VAF comparison across genomic subtypes. (A) Wilcoxon rank-sum test for TP53, EGFR, and KRAS mutated tumors and (B) Wilcoxon rank-sum test in TP53, EGFR, and KRAS mutated cases by CT volume (low [0-10 cm3], mid [10-50 cm3], high [> 50 cm3]).
Tumors with EGFR mutations are known to commonly have copy number gains (CNGs); observed in 28.3% of the EGFR+ cases in this cohort. We examined the impact of CNGs on ctDNA levels in EGFR+ tumors. EGFR+/CNG+ tumors had a significantly higher VAF than EGFR+/CNG-tumors (Tukey’s HSD p ≤ 0.00001) (Fig. 4a) . Furthermore, EGFR+/CNG-tumors had a VAF comparable to EGFR wt/CNG-tumors, suggesting that EGFR CNGs were an important contributor to the increased VAF in EGFR+ tumors. Furthermore, to determine whether concurrent TP53 mutations may be driving increased shedding in the EGFR+ tumors, we compared EGFR+/TP53 mutant vs EGFR+/TP53 wt tumors. TP53 co-mutation had no significant additive effect on ctDNA VAF (Fig. 4b).
Figure 4.

Impact of co-alterations on EGFR-mutated shedding. (A) Impact of EGFR copy number gain and (B) TP53 co-mutation on EGFR-mutated shedding; ANOVA comparison.
Patients undergoing active treatment may have decreased ctDNA shedding and thus potentially impact these genotype-specific findings. To further evaluate this potential interaction, we performed additional subgroup analyses with (1) patients that were treatment-naïve and (2) patients that had not received treatment within a month prior to ctDNA testing (treatment-naïve plus “prior treatment” as defined in Table 1). These analyses affirmed differential ctDNA shedding in TP53 and EGFR mutants; though with diminished statistical significance, as expected due to smaller sample size of the subgroups (Supplemental Fig. S3 – S6, Table S7).
DISCUSSION
As clinical uses of ctDNA continue to rapidly mature and incorporate more challenging applications such as MRD detection, disease monitoring, and cancer screening and localization, it is critical to further our understanding of tumor factors that may contribute to ctDNA shedding heterogeneity. In lung cancer, insight about specific histologic, anatomic, tumor burden, and genomic determinants of ctDNA shedding remains incomplete. In this retrospective analysis, we leverage comprehensive patient-level data of an advanced-stage NSCLC cohort with ctDNA testing results to further our understanding of the relationship between ctDNA detection and key clinicopathological features such as tumor volume and genotype.
A strong correlation between ctDNA VAF and tumor volume has previously been demonstrated in early-stage NSCLC 3, 4. However, how this correlation holds in advanced-stage disease is not well understood, particularly since volumetrics and other comprehensive measures of metastatic tumor burden are not routinely reported in the clinical setting. We observed that ctDNA VAF positively correlated with metastatic tumor burden as quantified by various radiographic measures including functional assessments of metabolic tumor volume (MTV), SUV, and TLG. To our knowledge, this is the largest analysis of ctDNA correlation with comprehensive radiographic tumor metrics in advanced NSCLC to date. It can be reasonably speculated that non-functional imaging such as CT scans may not be able to accurately measure systemic tumor burden, since observed radiographic lesions may be comprised of necrotic cells or non-neoplastic cells (e.g. stromal, inflammatory) 29 It is therefore notable that tumor burden approximated by anatomical CT volumetrics was comparable to PET-based volumetrics in ctDNA fraction correlation. Furthermore, the ubiquitous RECIST SLD measurement also appeared to comparably approximate overall tumor burden, as correlated by ctDNA fraction.
While positive, the modest correlation observed between VAF and tumor burden in our cohort suggests that other factors also significantly impact ctDNA shedding. Our findings that visceral metastasis predicts for increased ctDNA shedding is consistent with prior studies correlating liver metastasis with increased ctDNA levels in NSCLC 30, 31. While vascular accessibility may facilitate tumor ctDNA release, poor tumor vascularization may also contribute to ctDNA shedding in the setting of hypoxia and cell death.
Additionally, liver metastasis, in particular, may possibly be associated with high ctDNA VAF because of attenuation in the hepatic clearance of ctDNA, since the liver is the primary mechanism of ctDNA removal 32 In contrast, histology, brain metastasis, and significant pleural effusions or bilateral sub-centimeter pulmonary nodules did not appear to independently impact ctDNA levels in our study.
We observed genotype-specific differences in ctDNA levels in our cohort, potentially highlighting underlying differences in tumor biology mediated by metabolic activity and DNA copy number alterations. The finding that TP53 mutations independently predict for increased ctDNA shedding, even after accounting for tumor burden and visceral metastasis, may reflect the increased cellular turnover or metabolic activity of these tumors. TP53 mutation (or overexpression) has been considered a marker of poor prognosis but this assessment has been hampered by an evolving understanding of how best to accurately evaluate TP53 function and status 33, 34 Moreover, our observations about increased ctDNA shedding in EGFR mutated tumors and possible interactions due to concomitant EGFR copy number gains have implications for the use of ctDNA as a biomarker in this important genomic subgroup. Focal amplifications of specific chromosomal regions, such as EGFR CNGs, result in an increased fraction of detectable ctDNA in plasma, thus potentially over-estimating tumor volume and biasing ctDNA-derived measures of clonality 35 and tumor mutation burden, a biomarker with predictive utility across multiple tumor types 15–18, 36
Our findings of differential shedding also have ramifications for one of the major emerging clinical applications of ctDNA: cancer detection. Our group and others have shown that ultra-sensitive ctDNA assays can effectively detect MRD after curative intent therapy in localized NSCLC, thus identifying patients with highest risk for subsequent relapse 3, 4, 19 Other ctDNA-based platforms further optimize DNA fragment size and methylation analyses to enable detection of minimal levels of ctDNA and thus potential population-scale cancer screening 37, 38. Clinical validation of these emerging ctDNA applications will need to evaluate and potentially stratify for heterogeneity in the various determinants of shedding in order to accurately demonstrate clinical utility. For example, early detection of recurrence may be missed in patients presenting primarily with pleural effusions or sub-centimeter pulmonary nodules. Conversely, overrepresentation in the number of patients presenting with visceral metastasis or tumors with CNGs may exaggerate the ability of a particular assay to detect small tumor volumes. Additionally, since clinical trials required to prospectively validate cancer screening tests are enormously time and resource intensive 39, 40, in-silico modeling and simulation are useful for projecting ctDNA test performance characteristics such as minimal tumor detection volumes 41. Observations about determinants of ctDNA shedding are critical to inform these models.
Our study has several possible limitations. Since our cohort is mostly comprised of previously treated patients (68%), generally with progressive disease at the time of ctDNA testing, there may be a higher prevalence of resistance alterations (e.g. MET amplifications) that may impact ctDNA levels but not accounted for in the current analysis. Additionally, our KRAS-mutated cohort reflects the known heterogeneity of KRAS in lung adenocarcinoma. We have previously shown that KRAS subgroups (characterized by inactivation of TP53, STK11, KEAP1, and CDKN2A/B) indeed have distinct cancer biology 42. A larger KRAS cohort may better delineate potential ctDNA shedding differences between these KRAS subgroups. Finally, while the clinical ctDNA assay used in our study bioinformatically excludes variants associated with clonal hematopoiesis (CH), the potential inclusion of these non-tumor-related somatic variants in our analyses cannot be definitively quantified since matching white blood cells were not available for further DNA sequencing.
Despite these limitations, our findings strongly suggest that ctDNA shedding is not only impacted by tumor burden but also by anatomic location and genomic subtype. These findings further the understanding of ctDNA biology in NSCLC and have implications for the rapidly emerging use of ctDNA in NSCLC MRD, disease monitoring, and early detection.
Supplementary Material
Figure S1. ctDNA VAF correlation with tumor burden in patients with PET scan. Correlation between ctDNA VAF and (A) RECIST SLD (cm) (B) CT volume (cm3) (C) SUVmax (D) TLG (cm3).
Figure S2. ctDNA VAF correlation with tumor burden across genomic subtypes, as approximated by CT volume (cm3); Spearman’s rho correlation in TP53, EGFR, and KRAS mutated cases.
Figure S3. Impact of (A) visceral metastasis and (B) treatment status on ctDNA VAF in treatment-naïve subgroup. Wilcoxon rank-sum test.
Figure S4. ctDNA VAF comparison across genomic subtypes in treatment-naïve subgroup. (A) Wilcoxon rank-sum test for TP53, EGFR, and KRAS mutated tumors and (B) Wilcoxon rank-sum test in TP53, EGFR, and KRAS mutated cases by CT volume (low [0-10 cm3], mid [10-50 cm3], high [> 50 cm3]).
Figure S5. Impact of (A) visceral metastasis and (B) treatment status on ctDNA VAF in the combined treatment-naïve + prior treatment subgroup. Wilcoxon rank-sum test.
Figure S6. ctDNA VAF comparison across genomic subtypes in the combined treatment naïve + prior treatment subgroup. (A) Wilcoxon rank-sum test for TP53, EGFR, and KRAS mutated tumors and (B) Wilcoxon rank-sum test in TP53, EGFR, and KRAS mutated cases by CT volume (low [0-10 cm3], mid [10-50 cm3], high [> 50 cm3]).
Acknowledgments
Conflicts of Interest: VKL reports advisory role fees from Takeda, Seattle Genetics, Bristol-Myers Squibb and research funding from Bristol-Myers Squibb. VMR is an employee and stockholder of Guardant Health, Inc. RBL reports advisory role fees from Guardant Health, Inc. and is a stockholder. JR reports ownership interest in and advisory role fees from Genprex. DLG reports advisory role fees from AstraZeneca, GlaxoSmithKline, Sanofi and Janssen and research funding from Janssen, Takeda, Ribon Therapeutics, Astellas and AstraZeneca. VAP reports honoraria from F. Hoffman-La Roche, research funding from AstraZeneca, BMS, Eli Lilly, Novartis, Merck, F. Hoffman-La Roche, Nektar Therapeutics, Janssen, Bristol-Myers-Squibb, Checkmate, Incyte, advisory role fees from AstraZeneca, BMS, Eli Lilly, Novartis, Merck, F. Hoffman-La Roche, Nektar Therapeutics, Janssen, Bristol-Myers-Squibb, AbbVie, Araxes, Arrys Therapeutics, Bolt Therapeutics, Clovis Oncology, Exelixis, G2 Innovation, Gritstone, Ideaya, Leeds Biolabs, Loxo Oncology, Takeda, Tesaro, TRM Oncology. JZ reports research funding from Merck, Johnson and Johnson, and consulting fees from Bristol-Myers Squibb, Johnson and Johnson, AstraZeneca, Geneplus, OrigMed, Innovent. JVH reports advisory role fees from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Catalyst, EMD Serono, Foundation Medicine, Hengrui Therapeutics, Genentech, GSK, Guardant Health, Eli Lilly, Merck, Novartis, Pfizer, Roche, Sanofi, Seattle Genetics, Spectrum, Takeda, research funding from AstraZeneca, GlaxoSmithKline, Spectrum, and royalties and licensing fees from Spectrum. The remaining authors have no conflicts of interest to report.
Funding/Support: We thank the GEMINI team for their research support. This study was funded by Lung SPORE P50CA07907 (The University of Texas Southwestern Medical Center and The University of Texas MD Anderson Cancer Center), NIH CCSG CA016672, and the Gil and Dody Weaver Foundation. We would also like to acknowledge the generous philanthropic contributions to the University of Texas MD Anderson Cancer Center Moon Shots Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017;17:223–238. [DOI] [PubMed] [Google Scholar]
- 2.Zill OA, Mortimer S, Banks KC, et al. Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. Journal of Clinical Oncology 2016;34:LBA11501–LBA11501. [Google Scholar]
- 3.Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reinert T, Henriksen TV, Christensen E, et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Li L, Cohen JD, et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Murillas I, Schiavon G, Weigelt B, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 2015;7:302ra133. [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Gong Y, Lam VK, et al. Deep sequencing of circulating tumor DNA detects molecular residual disease and predicts recurrence in gastric cancer. Cell Death Dis 2020;11:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raja R, Kuziora M, Brohawn PZ, et al. Early Reduction in ctDNA Predicts Survival in Patients with Lung and Bladder Cancer Treated with Durvalumab. Clin Cancer Res 2018. [DOI] [PubMed] [Google Scholar]
- 10.Anagnostou V, Forde PM, White JR, et al. Dynamics of Tumor and Immune Responses during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Res 2019;79:1214–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phallen J, Leal A, Woodward BD, et al. Early Noninvasive Detection of Response to Targeted Therapy in Non-Small Cell Lung Cancer. Cancer Res 2019;79:1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.US Food and Drug Administration. cobas EGFR Mutation Test v2. Available at https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm504540.htm. Accessed March 31, 2018
- 13.National Comprehensive Cancer Network. Non–Small Cell Lung Cancer, Version 6.2020. Available at https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed July 22 2020.
- 14.Zhou C, Imamura F, Cheng Y, et al. Early clearance of plasma EGFR mutations as a predictor of response to osimertinib and comparator EGFR-TKIs in the FLAURA trial. Journal of Clinical Oncology 2019;37:9020–9020. [Google Scholar]
- 15.Gandara DR, Paul SM, Kowanetz M, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med 2018;24:1441–1448. [DOI] [PubMed] [Google Scholar]
- 16.Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 2018;24:1449–1458. [DOI] [PubMed] [Google Scholar]
- 17.Aggarwal C, Thompson JC, Chien AL, et al. Baseline Plasma Tumor Mutation Burden Predicts Response to Pembrolizumab-based Therapy in Patients with Metastatic Non-Small Cell Lung Cancer. Clin Cancer Res 2020;26:2354–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizvi NA, Cho BC, Reinmuth N, et al. Durvalumab With or Without Tremelimumab vs Standard Chemotherapy in First-line Treatment of Metastatic Non-Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam V, Tran H, Vasquez M, et al. MA23.02 Circulating Tumor DNA Analysis with a Novel Variant Classifier for Recurrence Detection in Resected, Early-Stage Lung Cancer. Journal of Thoracic Oncology 2018;13:S438. [Google Scholar]
- 20.Diaz LA Jr., Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 2014;32:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen KZ, Lou F, Yang F, et al. Circulating Tumor DNA Detection in Early-Stage Non-Small Cell Lung Cancer Patients by Targeted Sequencing. Sci Rep 2016;6:31985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lanman RB, Mortimer SA, Zill OA, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One 2015;10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Odegaard JI, Vincent JJ, Mortimer S, et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin Cancer Res 2018;24:3539–3549. [DOI] [PubMed] [Google Scholar]
- 24.Im HJ, Pak K, Cheon GJ, et al. Prognostic value of volumetric parameters of (18)F-FDG PET in non-small-cell lung cancer: a meta-analysis. Eur J Nucl Med Mol Imaging 2015;42:241–251. [DOI] [PubMed] [Google Scholar]
- 25.Larson SM, Erdi Y, Akhurst T, et al. Tumor Treatment Response Based on Visual and Quantitative Changes in Global Tumor Glycolysis Using PET-FDG Imaging. The Visual Response Score and the Change in Total Lesion Glycolysis. Clin Positron Imaging 1999;2:159–171. [DOI] [PubMed] [Google Scholar]
- 26.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 27.Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 2016;48:607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ettinger DS, Wood DE. Non-Small Cell Lung Cancer (Version 1.2021). Available at https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed Nov 29, 2020.
- 29.Morbelli S, Alama A, Ferrarazzo G, et al. Circulating Tumor DNA Reflects Tumor Metabolism Rather Than Tumor Burden in Chemotherapy-Naive Patients with Advanced Non-Small Cell Lung Cancer: (18)F-FDG PET/CT Study. J Nucl Med 2017;58:1764–1769. [DOI] [PubMed] [Google Scholar]
- 30.Sacher AG, Paweletz C, Dahlberg SE, et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol 2016;2:1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aggarwal C, Thompson JC, Black TA, et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elshimali YI, Khaddour H, Sarkissyan M, et al. The clinical utilization of circulating cell free DNA (CCFDNA) in blood of cancer patients. Int J Mol Sci 2013;14:18925–18958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitsudomi T, Hamajima N, Ogawa M, et al. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: a meta-analysis. Clin Cancer Res 2000;6:4055–4063. [PubMed] [Google Scholar]
- 34.Campling BG, el-Deiry WS. Clinical implications of p53 mutations in lung cancer. Methods Mol Med 2003;75:53–77. [DOI] [PubMed] [Google Scholar]
- 35.Blakely CM, Watkins TBK, Wu W, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet 2017;49:1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 2019;30:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lennon AM, Buchanan AH, Kinde I, et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020;369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu MC, Oxnard GR, Klein EA, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Annals of Oncology 2020;31:745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Koning HJ, van der Aalst CM, de Jong PA, et al. Reduced Lung-Cancer Mortality with Volume CT Screening in a Randomized Trial. N Engl J Med 2020;382:503–513. [DOI] [PubMed] [Google Scholar]
- 40.Janes S, Dickson J, Devaraj A, et al. P1.11-19 Trial in Progress: Cancer Screening Study With or Without Low Dose Lung CT to Validate a Multi-Cancer Early Detection Blood Test. Journal of Thoracic Oncology 2019;14:S523. [Google Scholar]
- 41.Avanzini S, Kurtz DM, Chabon JJ, et al. A mathematical model of ctDNA shedding predicts tumor detection size. bioRxiv 2020:2020.2002.2012.946228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5:860–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. ctDNA VAF correlation with tumor burden in patients with PET scan. Correlation between ctDNA VAF and (A) RECIST SLD (cm) (B) CT volume (cm3) (C) SUVmax (D) TLG (cm3).
Figure S2. ctDNA VAF correlation with tumor burden across genomic subtypes, as approximated by CT volume (cm3); Spearman’s rho correlation in TP53, EGFR, and KRAS mutated cases.
Figure S3. Impact of (A) visceral metastasis and (B) treatment status on ctDNA VAF in treatment-naïve subgroup. Wilcoxon rank-sum test.
Figure S4. ctDNA VAF comparison across genomic subtypes in treatment-naïve subgroup. (A) Wilcoxon rank-sum test for TP53, EGFR, and KRAS mutated tumors and (B) Wilcoxon rank-sum test in TP53, EGFR, and KRAS mutated cases by CT volume (low [0-10 cm3], mid [10-50 cm3], high [> 50 cm3]).
Figure S5. Impact of (A) visceral metastasis and (B) treatment status on ctDNA VAF in the combined treatment-naïve + prior treatment subgroup. Wilcoxon rank-sum test.
Figure S6. ctDNA VAF comparison across genomic subtypes in the combined treatment naïve + prior treatment subgroup. (A) Wilcoxon rank-sum test for TP53, EGFR, and KRAS mutated tumors and (B) Wilcoxon rank-sum test in TP53, EGFR, and KRAS mutated cases by CT volume (low [0-10 cm3], mid [10-50 cm3], high [> 50 cm3]).