

Abstract
The most frequent fetal birth defect associated with prenatal Zika virus (ZIKV) infection is brain calcification, which in turn may potentially affect neurological development in infants. Understanding the mechanism could inform the development of potential therapies against prenatal ZIKV brain calcification. In perivascular cells, bone morphogenetic protein (BMP) is an osteogenic factor that undergoes maturation to activate osteogenesis and calcification. We show that ZIKV infection of cultivated primary human brain pericytes triggers BMP2 maturation, leading to osteogenic gene expression and calcification. We observed extensive calcification near ZIKV+ pericytes of fetal human brain specimens and in vertically transmitted ZIKV+ human STAT2-knock-in mouse pup brains. ZIKV infection of primary pericytes stimulated BMP2 maturation, inducing osteogenic gene expression and calcification that were completely blocked by anti-BMP2/4 neutralizing antibody. Not only did ZIKV NS3 expression alone induce BMP2 maturation, osteogenic gene expression and calcification, but purified NS3 protease also effectively cleaved pro-BMP2 in vitro to generate biologically active mature BMP2. These findings highlight ZIKV-induced calcification where the NS3 protease subverts the BMP2-mediated osteogenic signaling pathway to trigger brain calcification.
Keywords: Zika virus, flaviviruses, fetal brain calcification, BMP2, osteogenic signaling, NS3 protease
The ZIKV outbreak in the Americas and in Southeast Asia was a major global health concern due to its association with congenital birth defects1–4. ZIKV is a single-stranded RNA virus of the Flaviviridae family. The virus was first discovered in Uganda in 1947 and is thought to have spread from Africa through equatorial Asia5. Along the way the virus diversified, so that now two genetic lineages, African lineage and Asian lineage, are widely recognized.
The ZIKV genome encodes a large polyprotein that is translationally processed into three structural proteins: capsid (Cap), precursor membrane (PrM), envelope (Env) and seven non-structural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) by host and viral NS3 protease enzymes. The ZIKV NS3 that is essential for viral replication is comprised of two distinct domains, the N-terminal protease and the C-terminal helicase. ZIKV NS3 protease domain is a trypsin-like serine protease with a classical catalytic triad (H51, D75, S135) with high specificity for substrate with dibasic P1 and P26. In addition, ZIKV NS3 protease impairs host interferon (IFN) signaling pathway and apoptotic cell death by degrading STING and Jak1, respectively7,8, indicating that NS3 has a crucial role in both viral replication and host antiviral immune evasion.
ZIKV infection in immunocompetent WT mice develops viremia but does not recapitulate adult and fetal human congenital pathologies. To date, the mouse models of ZIKV infection are immunocompromised interferon alpha receptor 1 knockout (IFNAR−/−) mice and anti-IFNAR1 neutralizing antibody-treated mice9. ZIKV infection of these IFN-deficient young mice induces pathogenesis like high mortality and premature embryonic death, which practically prevents the precise investigation of ZIKV-induced birth defects. Since ZIKV NS5 antagonizes IFN signaling by degrading human STAT2, not mouse STAT2, human STAT2 knockin (hSTAT2 KI) mice carrying the replacement of mouse STAT2 with human STAT2 have been generated10. ZIKV infection of pregnant hSTAT2 KI mice led to virus spread to placenta and fetal brain, mimicking human ZIKV infection without mortality10.
ZIKV is known to be transmitted primarily by the bite of infected mosquitoes. However, human-to-human vertical transmission and sexual activity have been well documented, resulting in prenatal infection of fetus and congenital ZIKV syndrome. Clinically, congenital ZIKV syndrome is known to be associated with severe microcephaly, brain calcification, and congenital contractures. Interestingly, ZIKV-induced brain calcification is one of the most frequent observations in infants born with prenatal infection, mainly localizing at the gray matter-white matter junctions (confirmed infections = 88%, presumed infections = 100%), basal ganglia (65%) and thalamus (64%)11–13. Pathological fetal brain calcification is a process of abnormal deposition of calcium-phosphate crystals, resulting in sequential neurological defects. In fact, ~30% of Brazilian cohort of ZIKV-exposed children exhibiting brain calcifications showed delayed childhood neurodevelopment13,14. Despite serious repercussions in fetal development, there have not been any studies to understand the molecular detail of ZIKV-induced brain calcification.
Calcification is a process of abnormal calcium-phosphate deposition in soft tissues. The bone morphogenetic protein (BMP), one branch of the transforming growth factor-β (TGF-β) superfamily, is the major morphogen that promotes vascular and basal ganglia calcifications as well as physiological bone and cartilage formation. In fact, BMP2 has been associated with the osteoblastic transdifferentiation of mesenchymal stem cells in blood vessels15. Moreover, a number of other cell types, including the CD146+/platelet-derived growth factor receptor beta-positive (PDGFR-β+) pericytes, utilize BMP2 signal pathway to induce calcification. Upon cleavage of full-length BMP2 by furin-type proteases16, mature BMP2 is secreted and initiates BMP Receptor I/II-mediated signal transduction, leading to the phosphorylation of Smad1/5/9. The phosphorylated Smad1/5/9 and Smad4 complexes subsequently translocate to the nucleus where they induce expressions of a master osteogenic transcription factor, Runt-related transcription factor 2 (RUNX2), and its downstream osteogenic gene, osterix/Sp7(OSX), as well as important effector genes, such as bone-type tissue non-specific alkaline phosphatases (ALPL; also known as TNAP), dentin matrix protein (DMP1) and podoplanin (PDPN; also known as Gp38)17. Thus, abnormal activation of BMP signaling pathway results in aberrant vascular calcification associating with a number of diseases.
In this study, we addressed the molecular mechanism of ZIKV-induced brain calcification. Using human ZIKV+ fetal brain specimens and immunocompetent hSTAT2KI mouse model, we showed that ZIKV infection of PDGFRβ+ perivascular cells induced osteogenic gene expression and calcification. The ZIKV NS3 protease effectively cleaved pro-BMP2/4 to generate biologically active mature BMP2 that induced osteogenic gene expression and calcification. This study discovers a novel viral regulation of osteogenesis where the ZIKV NS3 protease targets BMP2/4-mediated osteogenic signaling pathway, which ultimately contributes to pathological calcification in the fetal brains.
Results
Perivascular cells are in close proximity to calcium deposits in ZIKV+ fetal brain specimens
Computerized Tomography (CT) neuroimaging revealed scattered parenchymal calcifications in Brazilian ZIKV+ infants with severe ventriculomegaly and microcephaly (Fig. 1a). Von Kossa stain of the ZIKV+ fetal brain specimens from Rio de Janeiro, Brazil also showed extensive calcium depositions that were partially overlapped with hematoxylin eosin (H&E) staining, suggesting the presence of cytopathic effect as a result of ZIKV infection (Fig. 1b). Acellular microvascular calcium deposits were also observed in these ZIKV+ fetal brains (Extended Data Fig. 1a). Although there is a clear clinical association between ZIKV infection and brain calcification, how ZIKV infection induces pathological calcifications remains unclear. To determine which cell types were responsible for the calcification in ZIKV+ human fetal brains, we performed in situ hybridization (ISH) of ZIKV RNAs and immunohistochemistry (IHC) of key cellular biomarkers such as Neuronal nuclear Antigen (NeuN) for neurons, Glial Fibrillary Acidic Protein (GFAP) for astrocytes18, and platelet-derived growth factor-beta receptor (PDGFRβ) for pericytes19,20. Serial sectioning of fetal human brain showed that calcification lesions were surrounded by PDGFRβ-positive pericytes lining the blood vessels (Fig. 1c, Extended Data Fig. 1b and 1c). Strikingly, PDGFRβ+ perivascular cells near von Kossa-positive calcification lesions were strongly positive for ZIKV RNAs, further suggesting that ZIKV-infected perivascular cells are, at least in part, highly associated with brain calcification (Fig. 1d).
Fig. 1. Calcification in brain specimens from ZIKV-infected fetuses.

(a) CT scan of calcifications (red arrows) in the subcortical white matter in a 9-month-old infant infected with ZIKV during second trimester of pregnancy. (b) Brain tissue sections (10μm) from ZIKV-positive human fetuses derived from stillbirths were stained with Von Kossa (top) and H&E (bottom). (c) Serial brain sections from ZIKV-positive human fetal brain tissue were stained with Von Kossa (calcium; black), anti-PDGFRβ (pericytes), anti-GFAP (astrocytes), or anti-NeuN (neurons). Images were taken at 10x magnification (top panel) and 20x magnification (bottom panel) (d) Von Kossa and RNAScope duplex in-situ hybridization with ZIKV RNA and PDGFRβ mRNA (pericytes) in serially sectioned ZIKV-positive human fetal brain tissues. Brightness and contrast were adjusted in magnified insert. Red arrows indicate PDGFRβ+/ ZIKV+ pericytes. The data are representative images from five ZIKV-positive fetal donors.
ZIKV infection of primary pericytes induces canonical osteogenic signaling and calcification in vitro
To evaluate the role of pericytes in ZIKV-induced calcifications, human CD146+/CD31−/CD45− fetal pericytes were infected with ZIKV in vitro (Fig. 2a). This showed that the pericytes were effectively infected with ZIKV (African strain MR766, or Asian strain H/PF/2013 or PRVABC59) (Fig. 2b and 2c). High levels of viral RNAs and infectious viral particles were detected in CD146+/CD31−/CD45− fetal pericytes within 3 days post-infection (dpi) with all three ZIKV strains (Fig. 2b and 2c). Notably, Asian ZIKV strains continuously replicated at high levels at 8 dpi, and infectious viruses were detected even at 14 dpi (Fig. 2c). While ZIKV-MR766 replicated faster than ZIKV-H/PF/2013 and ZIKV-PRVABC59, ZIKV-MR766 RNAs were not detected after 3 dpi and infectious virus titers rapidly declined over time (Fig. 2b and 2c). Consistently, African ZIKV strain (MR766 or IbH30656)-infected pericytes showed high levels of cell death, whereas Asian ZIKV strain (H/PF/2013 or PRVABC59)-infected pericytes showed minimal cell death (Extended Data Fig. 2a). Overall, these data showed that human CD146+/CD31−/CD45− fetal pericytes were highly susceptible to ZIKV replication: the African ZIKV strain rapidly replicated in fetal pericytes causing high cell death, whereas Asian ZIKV strain persistently replicated in fetal pericytes with low cell death.
Fig. 2. Osteogenic gene expression and calcification in ZIKV+ fetal pericytes.

(a) Fetal pericytes (CD146+/CD31−) were infected with African strain ZIKV-MR766, Asian strains ZIKV-H/PF/2013 and PRVABC59 at MOI of 0.5 or treated with medium without virus (mock). Blue cell population is unstained control, red cell population is stained with CD146 and CD31 antibody. See Supplementary Data Fig. 1 for gating strategy. (b) At 1–4 and 8 dpi, cells were harvested for viral RNA analysis (n=4). (c) Infectious virus titer was quantified in supernatants (n=6 per group) collected from 1–4, 8 and 14 dpi. (d) At 3, 4 and 8 dpi, mature BMP2 protein from supernatants of mock- and ZIKV-infected pericytes was measured (n=8). (e) Osteogenic gene expression in fetal pericytes was normalized to GAPDH and presented as fold changes relative to mock controls (n=6 per group). (f-g) Alizarin red staining and Alizarin red concentration (mM) of ZIKV-infected fetal pericytes after 14 dpi (n=4). Data are presented as mean ± SEM, using two-way ANOVA followed by Sidak’s multiple comparisons test (e) or one-way (g) ANOVA with Tukey’s posttest. *P< 0.05, **P< 0.01, ***P< 0.001 and ****P< 0.0001. Data in d, e and g are presented as mean ± SEM in box plots showing the upper (75%) and lower (25%) quartiles, with the horizontal line as the median and the whiskers as the maximum and minimum values observed. Exact P values in g compared between Mock and ZIKV H/PF/2013 (P= 0.0046) and between Mock and PRVABC59 (P= 0.0030). Data are representative of three independent experiments.
BMP-mediated osteogenic signal pathway has been shown to contribute to calcifications17,21–24. Infection of fetal pericytes with ZIKV-H/PF/2013 led to an increase of secreted mature BMP2 in the supernatants, but not in that of mock or ZIKV-MR766-infected pericytes (Fig. 2d). In addition, infection of fetal pericytes with ZIKV-H/PF/2013 induced significantly high expression of BMP2 compared to BMP4, BMP6, BMP7 and BMP9 as well as the BMP antagonist, Noggin (NOG), though their activities were not measured (Fig. 2e and Extended Data Fig. 2b). Consistently, expressions of the central osteogenic transcription factor genes RUNX2 and OSX, as well as the downstream mineralization genes ALPL, DMP1 and PDPN were markedly induced upon ZIKV-H/PF/2013 infection (Fig. 2e). In contrast, African ZIKV-MR766 or ZIKV-IbH30656 infection, which showed massive cell death by 3 dpi, exhibited little or no change of their expressions (Fig. 2e, Extended Data Fig. 2a and 2c). Correspondingly, Alizarin Red S staining found aberrant calcification in fetal pericytes infected with ZIKV-H/PF/2013 or ZIKV-PRVABC59 but not with mock or ZIKV-MR766 (Fig. 2f, 2g and Extended Data Fig. 2d). Interestingly, infection of neuroblastoma cell line or astrocytoma cell line with either ZIKV-MR766 or ZIKV-H/PF/2013 induced extensive cell death without calcification (Extended Data Fig. 2e and 2f). In addition, ZIKV infection induced BMP2, OSX, DMP1 and PDPN expression in primary fetal brain-derived pericytes, but not in primary fetal brain-derived astrocytes (Extended Data Fig. 2g). RNAseq analysis of ZIKV-infected peripheral neurons also showed no increase of BMP2, OSX, DMP1 and PDPN expression (Extended Data Fig. 2h)25. These results suggest that ZIKV infection of fetal pericytes, but not fetal astrocytes or neurons, results in the induction of osteogenic gene expression and calcification.
ZIKV-induced brain calcifications in immunocompetent hSTAT2KI ZIKV mice
Immunocompetent transgenic hSTAT2KI mice that carry the replacement of mouse Stat2 with human STAT2 was developed to closely recapitulate human ZIKV pathogenesis10. To investigate ZIKV-induced brain calcification in vivo, three-week-old hSTAT2KI mice were subcutaneously (s.c.) infected with ZIKV-MR766 or ZIKV-PRVABC59 near the sagittal suture areas and their brain tissues were collected either at 4, 8 and 11 dpi to determine osteogenic gene expressions, or at 21 dpi for Alizarin Red S calcium staining. While higher level of ZIKV RNA was detected in ZIKV-MR766-infected brains than in ZIKV-PRVABC59-infected brains (Extended Data Fig. 3A), ZIKV-PRVABC59 infection, not ZIKV-MR766-infection, led to the significant increase of early osteogenic genes (BMP2, RUNX2, OSX, and ALPL) at 4 and 8 dpi and late osteocytic genes (DMP1 and PDPN) at 11 dpi (Extended Data Fig. 3b). Correspondingly, ZIKV-PRVABC59 infected mice showed aberrant brain calcifications (Extended Data Fig. 3c). In contrast, severe tissue damages were observed in the brains of ZIKV-MR766-infected mice with little or no calcium deposition as shown with Alizarin red staining (Extended Data Fig. 3c). These results demonstrate that Asian ZIKV infection can trigger activated osteogenic signaling and brain calcifications in an immunocompetent mouse model.
Recent clinical follow-up study on severe cases of 68 ZIKV-exposed children who exhibited fetal abnormalities has identified brain calcifications in 99% of these children13. To further demonstrate in vivo ZIKV-induced calcification through maternal-fetal vertical transmission, we infected immunocompetent hSTAT2KI time-mated pregnant dams with ZIKV-PRVABC59 intraperitoneally (i.p.) at mouse embryonic day 13.5 (E13.5) and harvested the pups’ brains at post-delivery day (PD) 28 (Fig. 3a). ZIKV-exposed pups’ brains showed high osteogenic gene expression compared to PBS-exposed control mice (Fig. 3b). Strikingly, ZIKV protein and aberrant brain calcium deposits were readily detected in the mid-brain regions of pups’ brain at PD28 (Fig. 3c and 3d). These suggest that ZIKV is vertically transmitted from pregnant dams to their pups, leading to fetal brain calcifications.
Fig. 3. Vertical transmission of ZIKV induces brain calcification in mice.
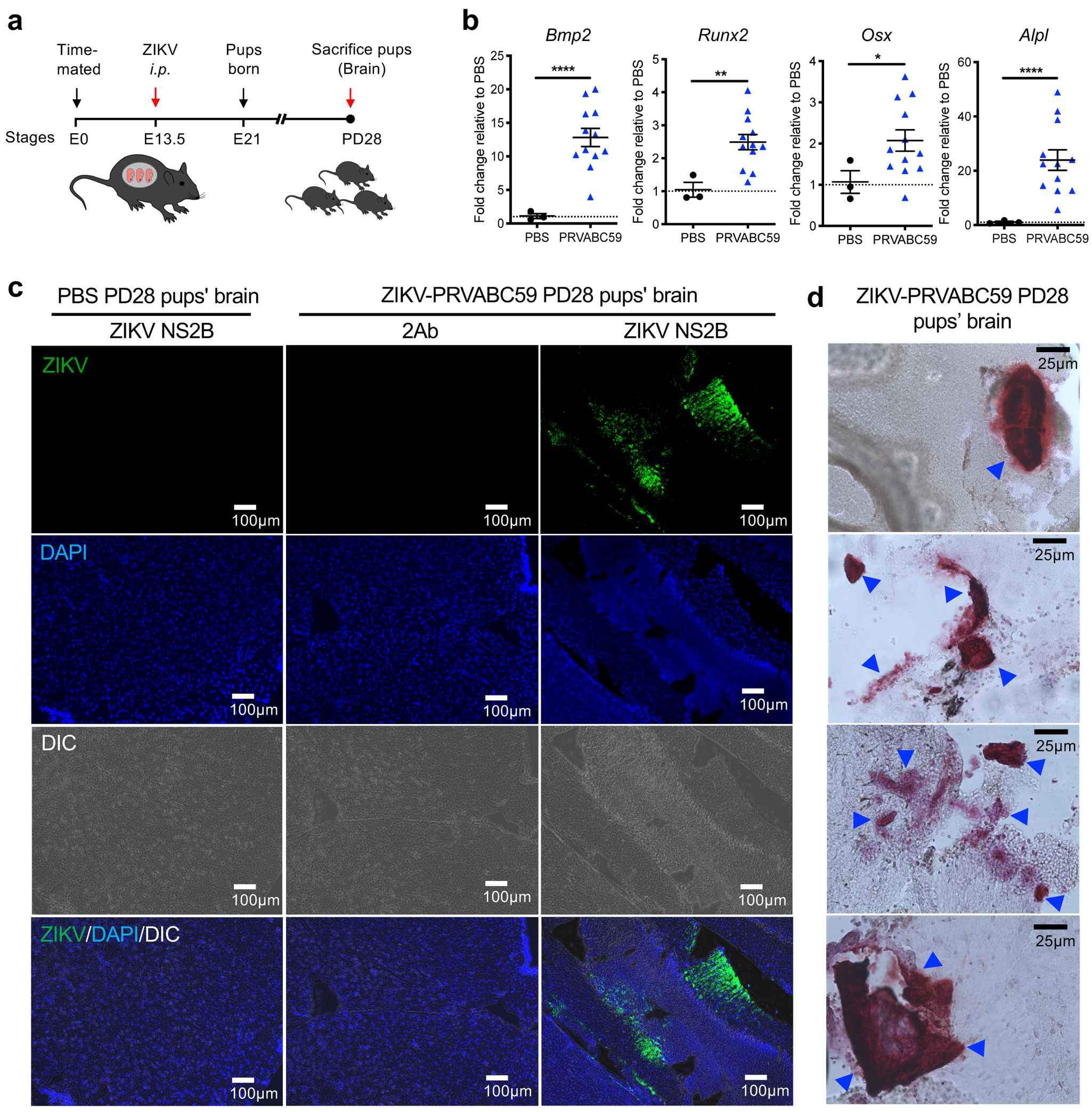
(a) Schematic depicting the timeline for our ZIKV-vertical transmission mouse model. Time-mated immunocompetent hSTAT2 KI pregnant dams (8 to 12 weeks old) were i.p infected with PBS (mock control) or ZIKV PRVABC59 at mouse embryonic day 13.5 (E13.5) and pups were born between 19 to 21 days later. On post-delivery day (PD) 28, ZIKV-infected hSTAT2KI pups were sacrificed and brains were harvested for RNA extraction or cryo-sectioning. (b) Osteogenic gene expression in mock- (n =3) or ZIKV-infected hSTAT2KI pup brains (n=12) were normalized to GAPDH and presented as fold changes relative to mock controls. (c) At PD28, brain sections of pups were stained with DAPI (blue) and anti-NS2B antibody. (d) Brain sections were stained with Alizarin red for calcium deposit. Blue arrows indicate the locations of prominent calcium deposition. Data in b-d are representative of two independent experiments. Data in b are presented as mean ± SEM, using two-tailed unpaired Student t-test. *P< 0.05, **P< 0.01 and ****P< 0.0001. Exact P values in b compared between PBS and ZIKV PRVABC59 (Bmp2 P= < 0.0001; Runx2 P= 0.0027; Osx P= 0.0180; Alpl P= < 0.0001).
ZIKV-induced calcifications through BMP2-mediated osteogenic signaling pathway
Since soft tissue calcification has been shown to be elicited by cellular processes that closely resemble the physiological bone formation17, ZIKV-susceptible human bone-derived U2OS cells were used to dissect the molecular mechanism of ZIKV-induced calcification26,27. Similar to primary fetal pericytes (Fig. 2b and 2c), U2OS cells highly supported ZIKV-MR766 or ZIKV-H/PF/2013 infection and replication (Fig. 4a and 4b). ZIKV-MR766 infection, but not ZIKV-H/PF/2013 infection, led to extensive cell death (Fig. 4c). Interestingly, both ZIKV-MR766 and ZIKV-H/PF/2013 infection of U2OS cells immediately increased mature BMP2 expressions in the supernatants at 2 and 3 dpi (Fig. 4d). These results suggest that while both ZIKV-MR766 and ZIKV-H/PF/2013 infection increase mature BMP2 in supernatants, only ZIKV-H/PF/2013 infection continuously increases mature BMP2 due to its weaker cytopathic activity.
Fig. 4. ZIKV-induced calcification is dependent on BMP2 signaling.

(A-B) U2OS cells were infected with ZIKV at an MOI of 0.5. (a) Supernatants from mock- and ZIKV-infected cultures were harvested for viral plaque assays (n=8 biologically independent samples per group), and (b) cells were tested for viral RNA (n=4 biologically independent samples per group). Data in b are presented as mean ± SEM in box plots showing the upper (75%) and lower (25%) quartiles, with the horizontal line as the median and the whiskers as the maximum and minimum values observed. (c) At 1–4 dpi, cell viability was determined by flow cytometry. See Supplementary Data Fig. 1 for gating strategy. (d) At 1–3 dpi, mock- and ZIKV-infected U2OS (MR766: MR; H/PF/2013: HPF) and their supernatants were collected, and TCA precipitated for immunoblot analysis with anti-BMP2 and Ponceau staining. (e) Phosphorylated SMAD1/5/9, total SMAD1 and ZIKV NS3 from whole cell lysates were detected by immunoblotting with indicated antibodies. (f-h) Inhibition of BMP2 activity. Mock- or ZIKV-infected U2OS cells were treated with 2 μg/mL of mouse IgG isotype control or human BMP2/4 neutralizing antibody. (f) At 4 dpi, cells were harvested for osteogenic gene expressions, normalized to GAPDH and presented as fold changes relative to mock controls (n=3 biologically independent cells per group). (g) At 21 dpi, cells were stained with Alizarin red stain (calcium) and (h) Alizarin red stain concentration was quantified (n=6). All results are representative of biological independent replicates from two independent experiments. Data are presented as mean ± SEM, using two-tailed unpaired Student t-test (f), two-way ANOVA followed by Sidak’s multiple comparisons test (b), or one-way ANOVA with Tukey’s posttest (h). *P< 0.05, **P< 0.01, ***P< 0.001 and ****P< 0.0001. Exact P values in f compared between IgG and Nab group (BMP2 P= 0.0034; RUNX2 P= 0.0267; OSX P= 0.0054; DMP1 P= 0.0113 and PDPN P= 0.0105). Significance differences of the means in h comparing between ZIKV-H/PF/2013 +IgG and ZIKV-H/PF/2013 +Nab group (P= <0.0001)
BMP2 transduces signals by binding to complexes of type I and II serine/threonine kinase receptors which induce the phosphorylation of Smad1/5/9 to activate canonical signaling, ultimately leading to expression of the osteogenic genes RUNX2, OSX, DMP1, PDPN and BMP228. Indeed, ZIKV-H/PF/2013 infection apparently induced a high level of SMAD1/5/9 phosphorylation and increased expressions of RUNX2, OSX, ALPL, DMP1, PDPN and BMP2, whereas ZIKV-MR766 infection did not (Fig. 4e, Extended Data Fig. 4a, 4b and 4c). Strikingly, treatment of anti-BMP2/4 neutralizing antibody not only drastically suppressed ZIKV-H/PF/2013-induced expression of osteogenic genes RUNX2, OSX, DMP1, PDPN and BMP2 (Fig. 4f) but also completely abrogated ZIKV-H/PF/2013-induced calcium deposition (Fig. 4g and 4h) without significant effect on the virus load (Extended Data Fig. 4d). In addition, anti-BMP2/4 neutralizing antibody treatment of human fetal brain pericytes also showed suppression of BMP2, RUNX2, OSX, DMP1 and PDPN osteogenic gene expressions (Extended Data Fig. 4e). These findings strongly indicate that Asian ZIKV strain infection triggers BMP2-driven osteogenic signals, resulting in calcifications.
ZIKV NS3 protease cleaves pro-BMP2 into mature form, promoting osteogenic signal
ZIKV encodes a polyprotein that is co- and post-translationally processed into three structural proteins: capsid (Cap), precursor membrane (PrM), envelope (Env) and seven non-structural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5). To identify which viral proteins contributed to triggering osteogenic signaling upon ZIKV infection, individual ZIKV protein was expressed in fetal primary pericytes by recombinant lentivirus infection29. Interestingly, ZIKV NS3 expression alone was sufficient to markedly increase BMP2, RUNX2, OSX, DMP1 and PDPN mRNA levels (Fig. 5a). ZIKV NS3 is a serine protease/helicase protein essential for viral polyprotein proteolysis during viral replication. In contrast to full-length NS3 wild type (NS3WT), a protease defective NS3 mutant (NS3Mut) carrying the H51A/S135A mutations was unable to elicit osteogenic responses and calcification in fetal pericytes (Fig. 5b and 5c), as well as in U2OS cells (Extended Data Fig. 5).
Fig. 5. ZIKV NS3 serine protease upregulates osteogenic signaling in fetal pericytes.

(a) Primary fetal pericytes transduced with lentivirus encoding individual ZIKV protein. Lenti-transduced cells were harvested for analysis of osteogenic gene expression normalized to GAPDH and expressed as fold changes relative to vector control (n=8). (b) Schematic of ZIKV NS3 protease WT and H51A/S135A mutant (ZIKV NS3Mut). (c) Osteogenic gene expressions in WT and mutant (Mut) NS3-transduced pericytes (n=4). Data in a and c are presented as mean ± SEM in box plots showing the upper (75%) and lower (25%) quartiles, with the horizontal line as the median and the whiskers as the maximum and minimum values observed. Data are presented as mean ± SEM, using two-tailed unpaired Student t-test. **P< 0.01, ***P< 0.001, ****P< 0.0001. Exact P values in c compared between WT and Mut NS3 group (BMP2 P= 0.0012; RUNX2 P= 0.0013; OSX P= 0.0002; DMP1 P= 0.0030; PDPN P= <0.0001). Data are representative of three independent experiments.
Full-length BMP2 has a dibasic sequence motif which is assumed to be cleaved by furin-type convertase in order to generate mature BMP2 for secretion to the extracellular compartment30–32. Since ZIKV-induced osteogenic response and calcification were dependent on mature BMP2 activity (Fig. 4g and 4h), we tested whether ZIKV NS3 serine protease activity was able to promote endogenous BMP2 processing in U2OS and furin-deficient CHO-FD11 cells (Extended Data Fig. 6a). Expression of NS3WT resulted in considerable increase of mature BMP2 in the supernatants of U2OS and CHO-FD11 cells, whereas expression of the protease-defective NS3Mut did not (Fig. 6a). Consequently, expression of NS3WT, not NS3Mut, resulted in extensive calcium deposition (Fig. 6b). Next, we performed an in vitro cleavage assay of purified human pro-BMP2 with ZIKV NS3 protease (bZiPro) (Extended Data Fig. 6b and 6c)6,33. Indeed, bacterially purified ZIKV NS3 protease was able to cleave full-length BMP2 (~60 kDa) into mature forms (~18 kDa and 22 kDa) in vitro as efficiently as commercial furin protease (Fig. 6c and 6d). In addition, ZIKV NS3 protease-cleaved BMP2 was able to induce SMAD1/5/9 phosphorylation as strongly as furin-cleaved BMP2, demonstrating active biological activity of NS3-cleaved BMP2 (Fig. 6e). Apart from BMP2, BMP4 has been associated with osteogenic signaling and vascular calcification22,34. In fact, the mature forms of both BMP2 and BMP4 share high amino acid homology of 86% and furin-like cleavage site at the KR residues (Extended Data Fig. 6c). Indeed, ZIKV NS3 protease was able to cleave full-length BMP4 (~60 kDa) into mature forms (~18 kDa and 22 kDa) in vitro as efficiently as commercial furin protease (Extended Data Fig. 6e). Overall, these results indicate that ZIKV NS3 serine protease enzymatically processes pro-BMP2 to biologically active mature forms, ultimately inducing BMP2-mediated SMAD1/5/9 phosphorylation and osteogenic signaling.
Fig. 6. Purified ZIKV NS3 serine protease processes pro-BMP2 to biologically active mature form to induce osteogenic signaling.
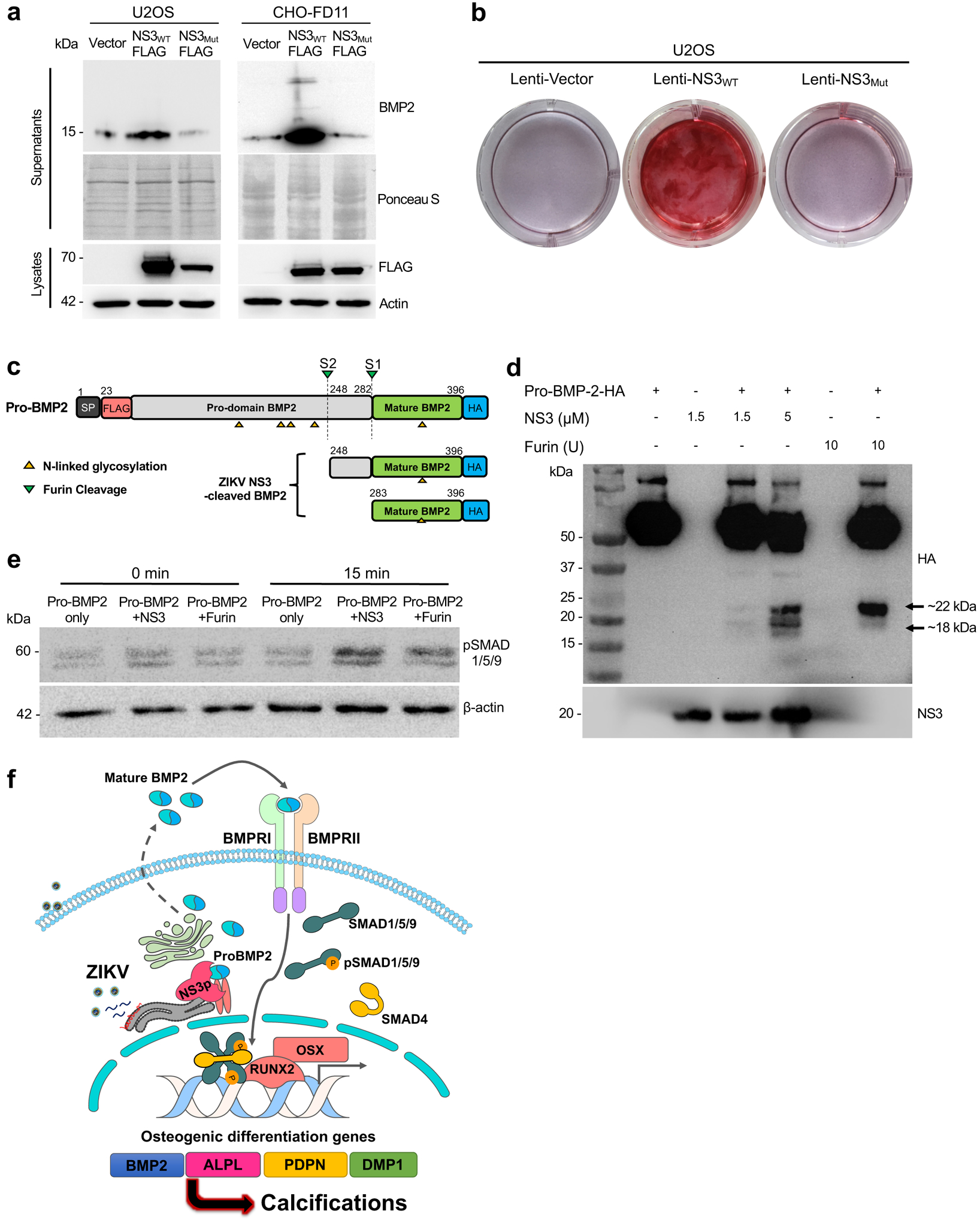
(a) At 3 days after transfection with Flag-tagged WT or mutant NS3, the supernatants and lysates of U2OS cells were subjected to immunoblotting analysis or Ponceau staining. (b) U2OS stably-expressing WT or mutant NS3 were cultured and stained with Alizarin red. (c) Schematic of predicted BMP2 cleavage products. (d) In vitro cleavage assay of carboxyl terminal HA-tagged purified human BMP2 and purified ZIKV NS3 protease were performed at 37 °C for 3 h, followed by immunoblot analysis. (e) Cleaved BMP2 products from in vitro reaction were added to U2OS cells for 0 or 15 min and cells were harvested for immunoblot analysis. Data are representative of two independent experiments. (f) Schematic model of ZIKV NS3 protease-induced BMP2 maturation and osteogenic gene expression for fetal brain calcification. ZIKV NS3 protease efficiently cleaves pro-BMP2 to generate secreted mature BMP2 that induces its receptor-mediated osteogenic gene expression and calcification.
Discussion
Brain calcifications are frequently reported neurologic finding in ZIKV-infected babies born during or in the aftermath of recent outbreaks2,35–37. These brain calcifications are linked to severe developmental defects such as motor disorders, cognitive disability, eye abnormalities, hearing deficits as well as seizures13,38–45. A follow-up brain imaging in children with congenital ZIKV syndrome after one year showed no further increase in number or size of calcifications but these children still presented neurologic repercussions such as epilepsy and feeding disorders, demonstrating potential pathologic consequences of ZIKV-induced brain calcifications during early childhood development44,46. Yet to date, there has been no studies on the molecular mechanisms of ZIKV-induced brain calcifications.
Although human neural-lineage cells such as neural progenitor cells, astrocytes, oligodendrocytes and neurons have been well studied for ZIKV infection47, perivascular cells have not yet been studied for ZIKV infection and calcification48,49. Since pericytes are essential in the development and maintenance of the neurovascular unit, the degeneration of these pericytes has been strongly implicated in neuropsychiatric diseases such as primary familial brain calcification50,51. Interestingly, pericytes have also been shown to exhibit stem cell potential similar to mesenchymal stem cells in vitro and in vivo49,52,53. For instance, microvascular pericytes express early osteogenic marker RUNX2 and form bone-line mineralized matrix in vivo, suggesting that pericytes function as osteogenic precursors. Herein, we found that ZIKV+ brain tissues from postmortem human fetuses in 2015–2016 Brazil outbreak showed specific perivascular calcium-phosphate deposition and that PDGFRβ+ pericytes resided closely to these calcified brain vasculatures. These suggest that perivascular cells are, at least in part, involved with brain calcification in response to ZIKV infection.
In vitro ZIKV infection of primary human fetal-derived pericytes robustly induced osteogenic gene expression and calcification even in the absence of osteogenic media, suggesting that ZIKV infection alone is sufficient to trigger osteogenic differentiation of perivascular cells. Interestingly, ZIKV-induced calcification is lineage-specific to Asian ZIKV, but not to African ZIKV, likely due to the persistent replication and low cytopathic activity of Asian ZIKV. Consistent with previous reports54–56, Asian ZIKV tends to infect poorly, leading to less cytotoxicity and infectious virion production compared to the African strain counterpart which causes extensive cell death. Furthermore, African ZIKV has been demonstrated to induce high inflammatory responses and cell death compared to Asian ZIKV57,58. Thus, these intrinsic differences may explain why Asian ZIKV-infection, but not African ZIKV infection, leads to the calcification of perivascular cells.
Calcification is a biological process that may utilize canonical BMP-SMAD signal pathway, mimicking bone formation. In early embryogenesis, the localized concentration gradients of BMPs are essential for the development of skeletal and nervous systems. Mature BMPs bind to their receptor to induce SMAD1/5/9 phosphorylation, leading to expression of master osteogenic regulators RUNX2 and OSX, and effector proteins ALPL, DMP1 and PDPN to trigger calcification17. In fact, the processed pro-domain of BMPs such as BMP2 also showed bone inducing properties59. To date, there are over 20 types of BMPs, but only BMP2, BMP4, BMP6, BMP7 and BMP9 have strong osteogenic properties. Interestingly, RNA-seq analysis of ZIKV-infected human neuro-progenitor cells showed increased BMP2/4 expression, but not other BMPs60. Consistently, we also found that ZIKV-infected fetal pericytes showed increased expression of BMP2 and BMP4, but not the other BMPs. In addition, we showed that ZIKV NS3 protease effectively processed full-length BMP2 to generate biologically active secreted mature BMP2 that leads to calcification, which was drastically suppressed by anti-BMP2/4 neutralizing antibody. In addition, BMP2/4 have been shown not only to repress neurogenesis and oligodendrogliogenesis during late embryogenesis and postnatal neural development, but also to strongly induce osteogenesis in cells of mesenchymal origins61–64. In human ZIKV-positive brains, most calcification lesions were primarily associated with pericytes, suggesting that the BMP2 released from infected pericytes activates osteogenic programs in an autocrine mode in vivo rather than paracrine mode. Hence, this study suggests that the ZIKV NS3 protease subverts the BMP2-mediated osteogenic developmental signaling pathway to trigger aberrant brain development and calcifications (Fig. 6f).
Full-length BMP2, similar to full-length BMP4, are assumed to be cleaved by a furin-type convertase at its dibasic site K/R to generate two glycosylated 18–22 kDa mature forms which are secreted to the extracellular compartment30–32. As ZIKV NS3 protease domain is a trypsin-like serine protease with high specificity for substrate with dibasic P1 and P26, it efficiently cleaved pro-BMP2/4 to generate two mature forms of BMP2/4, suggesting that BMP2 and BMP4 could play critical roles in ZIKV-induced osteogenic signaling. Previous studies have been shown that ZIKV NS3 protease targets STING and Jak1 to impair host IFN signaling pathway and apoptotic cell death, respectively7,8. Thus, this study identifies pro-BMP2/4 as a previously undescribed NS3 substrate to induce BMP2/4-mediated osteogenic signal transduction for brain calcification.
Historically, as brain calcifications have often been interpreted as dead brain tissues which calcify following injury, the molecular mechanism behind brain calcifications has not been explored until now to our knowledge. Thus, this study demonstrates the molecular mechanism of how Asian ZIKV has evolved to exploit BMP2-mediated osteogenic signaling mechanisms, which ultimately contributes to pathological calcification in the fetal brains. This study may also shed insights into the pathophysiological process generating calcifications in other congenital infections.
Methods
Human fetal brain tissues
Paraffin blocks containing pathology brain tissue specimens from five cases (one stillbirth and four neonatal deaths due to in utero infection with ZIKV following maternal infection in pregnancy) and three healthy brain tissue specimens from neonatal deaths unrelated to ZIKV infection were obtained from the southeastern state of Rio de Janeiro, Brazil. Brain tissues were formalin-fixed and paraffin-embedded prior to sectioning and immunohistochemical staining. The babies were all born at term expect two of them who were born prematurely at 32 weeks. Deceased infants were confirmed positive for ZIKV by either PCR or in situ hybridization in liver and spleen. All pregnant mothers had reported typical symptoms of ZIKV infection between the 4th and 16th gestational weeks. ZIKV RNA was detected in maternal blood, amniotic fluid and/or samples of postmortem neonatal liver and spleen. In all five cases, Dengue and Chikungunya virus infection were excluded by ELISA and PCR. All cases were tested negative for TORCH-like conditions (Toxoplasmosis, Others, Rubella, Cytomegalovirus, Herpes virus, and Syphilis, HIV and parvovirus B19).
Viruses
African ZIKV strains MR766 (ATCC; Passage 5; Vero cells; Uganda, 1947) and IbH30656 (ATCC; Passage 2; Vero cells; Nigeria, 1968), as well as Asian ZIKV strain PRVABC59 (ATCC; Passage 2; Vero cells; Puerto Rico, 2015) virus stocks were purchased from ATCC. Asian ZIKV strain H/PF/2013 (Passage 2; Vero cells; French Polynesia, 2013) was kindly provided by Dr. Michael Diamond (Washington University School of Medicine). All ZIKV virus stocks were propagated in Vero cell line (ATCC CCL-81, ATCC) and supernatant was harvested at 3–5 dpi. The viral titers were determined by plaque assays on Vero cells. Vero cells were authenticated by morphology check, growth curve analysis, and validated to be mycoplasma-free with Hoechst staining.
Animals and infection
Immunocompetent hSTAT2KI mice were bred and maintained within the animal unit of University of Southern California (California, USA). 21-day-old male and female mice, of equal sex distribution, were infected subcutaneously (s.c.) above sagittal suture with 2×105 pfu (plaque forming units) of either ZIKV-MR766 (Uganda strain, 1947) or ZIKV-PRVABC59 (Puerto Rico strain, 2015) diluted in phosphate-buffered saline (PBS) to a volume of 20μL. Mock-infected mice were inoculated with PBS alone. Mice were weighed every 24h. At indicated time points of infection, brains were collected for RNA analysis or immunohistochemistry. For pregnant immunocompetent hSTAT2KI infection, 8 to12-weeks-old timed-mated pregnant dams were infected intraperitoneally (i.p.) with PBS (mock) or 2×105 pfu ZIKV-PRVABC59 diluted in PBS to a volume of 200μL at mouse embryonic day 13.5 (E13.5). Animal studies were carried out in animal facility managed and maintained by the University of California (USC) Animal Resource Center. Facilities were maintained at an acceptable range of 68–79 °F at a humidity of 30–70% on a 12 h dark/12 h light cycle. All procedures used in this study complied with federal and institutional guidelines enforced by the USC Institutional Animal Care and Use Committee (IACUC) and were granted institutional approval after veterinary and committee review.
Bacterial strains, mammalian cell lines and primary cells
E. coli Top10 were grown in LB (Lenox, Sigma) medium for genetic manipulations with ampicillin (100 μg/ml). HEK293T, U2OS, U251, SK-N-SH, Vero E6 cells were purchased from ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Seradigm) and 100 U/ml penicillin and 100 μg/ml streptomycin (1% Pen/Strep, Gibco). CHO FD11 cells were kindly provided by Dr. Stephen Leppla (NIAID, NIH). ZIKV NS3 stably expressing U2OS cells were generated by transduction with lentivirus generated from pCDH-NS3–3xFlag-puro construct, followed by selection with puromycin (1 μg/ml, Gibco). Human fetal bone tissues were obtained from Novogenix Laboratories following informed consent and elective termination, and primary fetal CD146+/CD31−/CD45− pericytes were isolated from bone marrow as previously described65. Primary fetal brain vascular pericytes (#1200) and fetal astrocytes (#1800) were obtained from ScienCell Research Laboratories. All cells were maintained at 37°C with 5% CO2. To inhibit BMP2/4 activity, cells were treated with 2 μg/mL of purified mouse IgG1 isotype control (Clone 11711, R&D Systems, #MAB002) or human BMP2/4 neutralizing antibody (Clone 100230, R&D Systems, #MAB3552100).
Plasmid constructs
Infectious clone of PRVABC59, kindly provided by Dr. Ren Sun (University of California, Los Angeles) was used as DNA template for PCR amplification of individual viral proteins. Amplified PCR products were cloned into lentiviral pCDH-puro vector with C-terminal 3xFlag tag. The pDONR223-BMP2 construct was purchased from Addgene. To generate full-length recombinant BMP2 proteins, modifications were made to BMP2 construct by introduction of Flag tag after signal peptide (aa1–22) and was subsequently re-cloned onto pIRES-puro vector with C-terminal HA tag. bZiPro construct was purchased from Addgene. All plasmids sequences were analyzed using SnapGene v5.2.3.
Immunohistochemical staining
Fetal human brain and mouse brain tissues were fixed in 4% paraformaldehyde (PFA) and embedded in paraffin. Ten-micron sections were dewaxed, rehydrated and stained with hematoxylin and eosin (H&E), Alizarin Red S, Von Kossa stain or RNAscope. RNAscope in situ hybridization was performed with specific ZIKV-H/PF/2013 polyprotein probes (RNAscope Probe-V-ZIKA-pp; ACDbio, 463781) and human PDGFRβ probes (RNAscope Probe-Hs-PDGFRB-C2; ACDbio, 548991-C2) according to manufacturer’s protocol. Followed by immunohistochemical staining with anti-human PDGFRβ (1:100 dilution; clone 28E1, Cell Signaling Technology, #3169S), GFAP (1:50 dilution; clone GA5, Cell Signaling Technology, #3670S), NeuN (1:50 dilution; Millipore, #ABN78) antibodies and counterstained with hematoxylin.
Flow cytometry
Surface staining of fetal pericytes was performed with CD31-AF488 (1:100 dilution; clone WM59, BioLegend, #303110) and CD146-AF647 (1:100 dilution; clone P1H12, BioLegend, #361014) antibodies. Zombie Violet fixable viability kit (BioLegend, #423113) was used for fixable live/dead staining as per manufacturer instructions. FACS acquisitions was performed on BD FACSCanto II (BD Biosciences), using BD FACSDIVA v 6.0. All FACS data were analyzed using FlowJo v10.0 software.
RNA extraction, viral load and gene expression analyses
Total RNA extractions were performed with RNeasy mini kit (Qiagen) according to manufacturer’s instructions. RNA concentration was determined by NanoDrop 1000 spectrophotometer (Thermo Scientific). Extracted total RNA were reverse-transcribed with iScript cDNA synthesis kit (Biorad) according to the manufacturer’s instructions. Specific ZIKV NS1 primers and probe targeting conserved NS1 region across all 4 ZIKV strains (MR766, IbH30656, H/PF/2013 and PRVABC59) were designed for viral load detection. Standard curve (101 to 108 NS1 copies/μl) was generated from serial dilutions of plasmid expressing MR766 NS1 protein. Viral load and gene expression qRT-PCR were performed with 10 ng of cDNA/well with SsoAdvanced Universal Probe Supermix (BIO-RAD) or SsoAdvanced Universal SYBR Green Supermix (BIO-RAD), respectively. All qRT-PCR reactions were performed on BIO-RAD CFX96 Touch Real-Time PCR Detection System on 96-well plates. All qRT-PCR data were analyzed with CFX Manager 3.1 software. Viral load in specimens were interpolated from standard curve with Prism Graphpad software. All viral load qRT-PCR performed in this study included mock controls in which no CT values could be detected. Gene expression fold change was calculated with the ΔΔCt method on Microsoft Excel. Briefly, ΔΔCt = ΔCt(ZIKV-infected)–ΔCt(mock control) with ΔCt = Ct(gene-of-interest)—Ct(housekeeping gene-GAPDH). The fold change for each gene is calculated as 2−ΔΔCt. All primers and probe sequences used in this study is listed in Supplementary Table 1.
Plaque assay
Vero E6 cells were seeded into 24-well plates at a density of 1 × 105 cells per well and cultured to confluence. Serially diluted samples (10−1 to 10−6) were added to cell monolayers and incubated for 2 h at 37 °C in a 5% CO2 incubator. Cells were then overlaid with DMEM (Gibco) containing 5% FBS and 1% agarose (Sigma Aldrich, Sydney, Australia) and incubated at 37 °C for 4 days in a 5% CO2 incubator. The cells were fixed with 10% neutral buffered formalin solution (Sigma-Aldrich) and plaques were visualised by staining with 0.1% crystal violet. Viral titres were expressed as plaque forming units per millilitre.
ELISA
Human BMP2 ELISA assay (R&D systems, #DY355) were performed according to manufacturer’s instructions.
Alizarin Red staining and quantification
Cells were washed twice with PBS, fixed in 4% PFA, washed twice with dH2O followed by incubation with Alizarin Red S stain for 30 min at room temperature with gentle shaking. Stained cells were rinsed in PBS for 10 min with gentle agitation for 3 times. For quantification of alizarin red staining, cells were incubated with 800 μl of 10% (v/v) acetic acid for 30 min with gentle agitation. The monolayer was then scraped from the plate and transferred with 10% (v/v) acetic acid to a 1.5 mL microcentrifuge tube. The slurry was vortexed for 30 s, overlaid with 500 μL mineral oil (Sigma) and heated to 85 °C for 10 min, and transferred to ice for 5 min. The slurry was centrifuged at 20,000g for 15 min and supernatant removed to a new 1.5 mL microcentrifuge tube. Then 200 μL of 10% (v/v) ammonium hydroxide was added to neutralize the acid and absorbance readings were taken at 405 nm with FilterMax F5 multi-mode microplate reader (Molecular Devices).
Immunoblotting analysis
Whole-cell lysates (WCLs) were lysed with RIPA lysis buffer and measured for protein concentration by Bradford protein assay (Thermo Fisher) to equalize protein loading. Proteins (25–50 μg) were resolved on SDS-PAGE gels and transferred to PVDF membrane by semi-dry transfer at 25 V for 45 min (Trans-Blot Turbo, Bio-Rad). All membranes were blocked in 5% milk in Tris buffered saline with Tween20 (TBST, pH 8.0, Sigma) and probed overnight with indicated antibodies in 5% milk or 5% BSA in TBST. Primary antibodies include: rabbit anti-BMP2 (1:1500 dilution; EPR20807, Abcam, #ab214821), rabbit anti-phospho-SMAD1 (Ser463/465)/ SMAD5 (Ser463/465)/ SMAD9 (Ser465/467) (1:1000; clone D5B10, Cell Signaling, #13820S), rabbit anti-SMAD1 (1:1000 dilution; Cell Signaling, #9743S), rabbit anti-ZIKV NS3 (1:3000 dilution; GeneTex, #GTX133309), mouse anti-Flag (1:3000 dilution; Sigma, #F7425), mouse anti-HA (1:1000 dilution; clone 16B12, BioLegend, #901503) and mouse anti-β-actin (1:3000 dilution; Santa Cruz, #sc-47778). Appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-rabbit IgG-HRP; 1:2000 dilution, Cell Signaling Technology, #7074S or anti mouse IgG-HRP; 1:2000 dilution, Cell Signaling Technology, #7076V) were incubated on membranes in 5% milk in TBST at room temperature for 1 h. Protein bands were developed with HyGLO enhanced chemiluminescence (ECL) reagent (Denville Scientific), imaged on ChemiDoc Touch (Bio-Rad) and analyzed with Image Lab v5.2.1. All source gels with uncropped blots are available in the manuscript. Proteins in supernatants were precipitated with trichloroacetic acid (TCA)-acetone extraction. Briefly, 0.15% deoxycholate (DOC, Sigma) was added to supernatants to a final concentration of 0.015% and incubated on ice for 30 min. TCA (100%, Sigma) was added to supernatants to a final concentration of 20% and incubated overnight at 4 °C. Precipitated proteins were centrifuged at maximum speed for 15 min and wash twice in ice-cold acetone. Protein pellets were air-dried and resuspended in 2x SDS loading buffer.
Transient expression of viral proteins
Recombinant lentiviruses expressing individual viral proteins were generated by co-transfection of HEK293T with Lipofectamine 2000 (Thermo Fisher) with pCDH constructs expressing individual viral protein, along with packaging plasmids – pGag/Pol, pRev and pVSV-G for 2 days. Primary fetal pericytes were transduced with lentiviruses for 3 days prior to harvesting. U2OS and CHO FD11 cells were transfected with Lipofectamine 3000 reagent (Thermo Fisher) for 3 days. All transfections were performed according to manufacturer’s protocols.
Recombinant protein purification
To generate recombinant full-length BMP2 and BMP4 protein, CHO FD11 cells were transfected with either pIRES-full-length BMP2-HA or pIRES-full-length BMP4-HA plasmid with standard polyethylenimine (PEI) transfection procedures. Cells were collected at 48 h post-transfection followed by PBS washing. Cell pellets were re-suspended and lysed in TritonX-100 lysis buffer containing 150 mM NaCl, 1% TritonX-100, 50 mM Tris-HCl (pH 7.5), supplemented with complete protease inhibitor EDTA-free cocktail (Roche) for 1 h at 4°C with gentle agitation. Whole-cell extracts (WCEs) were centrifuged at maximum speed and filtered through 0.45 μm polyethersulfone (PES) filter (Thermo Fisher), followed by pre-clear with Sepharose beads rotating at 4°C for 1 h. Pre-cleared WCEs were incubated overnight with EZview Red anti-Flag M2 affinity gel (Sigma) at 4°C. Recombinant BMP2 or BMP4 protein was eluted with 3xFlag peptide (Sigma). To generate recombinant NS3 protease, bZiPro construct was transformed into E. coli BL21 (DE3) competent cells and grown in LB medium. Protein expression was induced overnight at 18 °C by addition of IPTG to 1 mM final concentration when the OD600 reached 0.8. Bacterial cells were harvested by centrifugation and re-suspended in lysis buffer containing 20 mM sodium phosphate (pH 7.8), 500 mM NaCl, 2 mM β-mercaptoethanol, 10 mM imidazole, 5% glycerol. Cells were lysed by sonication followed by centrifugation and filtered through 0.45 μm PES filter. WCEs were incubated with Nickel-NTA agarose (Thermo Fisher) for 1 h at 4 °C with rotation and eluted from 10 ml purification column. Eluted proteins were desalted using Sephadex G-25 PD-10 desalting column (GE Healthcare). Thrombin CleanCleave kit (Sigma) was used to remove 6xHis tag from purified recombinant NS3 protease according to manufacturer’s protocol.
In vitro NS3 protease cleavage assay
In vitro cleavage assays were performed by incubating purified recombinant BMP2 or BMP4 protein with purified ZIKV NS3 protease or furin (NEB) in protease assay buffer containing 20 mM Tris-HCL (pH 8,5), 10% glycerol, 0.01% TritonX-100) at 37 °C for 3 h. Cleavage assays were terminated by the addition of 6x Laemmli SDS sample buffer (Boston Bioproducts) with 50 mM DTT.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 7.0 and GraphPad Prism 8.4 software with *P<0.05, ** P<0.01, *** P<0.001 and **** P<0.0001. For analyses between 2 groups, two-tailed unpaired Student’s t-test or Mann-Whitney U-test were used. For comparisons among more than 2 groups, either one-way ANOVA with Tukey’s posttest or Kruskal-Wallis test with Dunn’s posttest, and two-way ANOVA with Tukey’s or Sidak’s posttest were used. All data were assessed for Gaussian distribution using the D’Agostino–Pearson normality test before analysis with these parametric tests. Data (mean ± SEM) were presented in box-and-whiskers plot showing upper (75%) and lower (25%) quartiles, with horizontal line as median and whiskers as maximum and minimum values observed.
Extended Data
Extended Data Fig. 1. Perinatal ZIKV infection in brain tissues of deceased fetuses exhibits pronounced calcifications.

(a) Paraffin-embedded brain tissue sections (10μm) from ZIKV-positive deceased human fetuses were stained with Von Kossa (red arrow). (b) Immunohistochemical staining for NeuN (neurons), PDGFRβ (pericytes) or GFAP (astrocytes) were performed using sequential brain sections of ZIKV-positive human fetal brain tissue. Arrows indicate NeuN-, PDGFRβ- or GFAP-positive cells (c) RNAScope duplex in-situ hybridization with ZIKV RNA and PDGFRβ mRNA of healthy human fetal brain tissues. Black arrows indicate PDGFRβ-positive cells lining the blood vessels. Neither non-specific staining ZIKV RNA nor calcium deposit was detected in healthy tissues. Data in a-c are examined from biologically independent human specimens (healthy n=3; ZIKV n=5) and are representative of two independent repeats.
Extended Data Fig. 2. Asian ZIKV infection, but not African ZIKV elicits in vitro calcification in pericytes.

Human fetal pericytes were infected with different ZIKV strains at MOI of 0.5 or mock (PBS) as control (n=4 biologically independent cells per group). (a) At 3 dpi, cell viability was quantified. See Supplementary Data Fig. 1 for gating strategy. (b) BMP4, BMP6, BMP7, BMP9 and NOG expression between ZIKV MR766- and ZIKV H/PF/2013-infected pericytes at 1, 3 and 4 dpi (n=6 biologically independent cells per group). Data in a and b are presented as mean ± SEM in box plots showing the upper (75%) and lower (25%) quartiles, with the horizontal line as the median and the whiskers as the maximum and minimum values observed. (c) Osteogenic gene expression between ZIKV MR766- and ZIKV IbH30656-infected pericytes was normalized to GAPDH and expressed as fold change relative to mock controls (n=3 biologically independent cells per group). Data are presented as mean ± SEM. (d) At 8 and 14 dpi, mock- or ZIKV-infected human fetal pericytes were subjected to Alizarin red staining for calcium deposition. Data are representative of two independent experiments. (e-f) Alizarin red staining were performed on mock- or ZIKV-infected SK-N-SH and U251 at 14 and 21 dpi. Data are representative of four independent experiments. (g) Human primary fetal pericytes (n=5) and astrocytes (n=6) were infected with PBS (mock) or ZIKV PRVABC59 at MOI of 0.5. At 3 dpi, osteogenic gene expressions were normalized to GAPDH and expressed as fold changes relative to mock controls. Data are presented as mean ± SEM and are representative of two independent experiments. (h) RNAseq data generated from Nature Neuroscience; 2017; 20(9); p1209–1212, doi: 10.1038/nn.4612 (GEO ID: GSE87750) was reanalyzed for osteogenic gene expression. Human primary peripheral neurons were infected with ZIKV PRVABC59 of MOI 0.4 and harvested at 3 dpi. Statistical analyses were performed using one-way ANOVA with Tukey’s posttest (a and c). ***P< 0.001 and ****P< 0.0001. Exact P values in a compared between ZIKV MR766 and ZIKV H/PF/2013 group (P= 0.0008), ZIKV MR766 and ZIKV PRVABC59 group (P= 0.0005), ZIKV IBH30656 and ZIKV H/PF/2013 group (P= <0.0001) and ZIKV IBH30656 and ZIKV PRVABC59 group (P= <0.0001).
Extended Data Fig. 3. ZIKV infection of 3-weeks-old hSTAT2KI mice resulted in brain calcification.

Three-week-old hSTAT2KI were s.c. infected with ZIKV.(a) At 8 dpi, ZIKV RNA was quantified in the brain (MR766 n=7; PRVABC59 n=5). (b) Osteogenic gene expression in mock- or ZIKV-infected hSTAT2KI brains (PBS n=5; MR766 n=6; PRVABC59 n=6) were normalized to GAPDH and presented as fold changes relative to mock controls. (c) Brain sections were stained with H&E or Alizarin red for calcium deposit. Black dotted line in the magnified insert indicates vasculature and blue dotted line in the magnified insert indicates prominent calcification sites. Data are presented as mean ± SEM, using Mann-Whitney U-test (a) or two-way ANOVA followed by Tukey’s multiple comparisons test (b). *P< 0.05, **P< 0.01 and ***P< 0.001. Exact P values in a compared between ZIKV MR766 and ZIKV PRVABC59 group (P= 0.0051). Data in a-c are representative of three independent experiments.
Extended Data Fig. 4. ZIKV infection of osteoblastic-like cells increase osteogenic gene expression.

(a-b) Mock- and ZIKV-infected U2OS cells were harvested at 2 and 4 dpi respectively for osteogenic gene expression. (c) Band intensity of pSMAD1/5/9 from U2OS whole cell lysate. (d) At day 1–4, IgG or nAb-treated and mock or ZIKV-infected U2OS cells (n=6) were harvested for RNA extraction and viral load against ZIKV NS1 RNA was quantified using qRT-PCR (N.D.; Not detected) (e) Human primary fetal brain pericytes are infected with ZIKV PRVABC59 or PBS (mock control) that were treated with 2 μg/mL of mouse IgG isotype control or human BMP2/4 neutralizing antibody. At 4 dpi, cells were harvested for osteogenic gene expressions, normalized to GAPDH and presented as fold changes relative to mock controls (n=4/group). Data in a, b, d and e are presented as mean ± SEM in box plots showing the upper (75%) and lower (25%) quartiles, with the horizontal line as the median and the whiskers as the maximum and minimum values observed. Data are analyzed using Kruskal-Wallis test followed by Dunn’s multiple comparisons (a), Mann-Whitney U-test (b), or two-tailed unpaired Student t-test (e). *P< 0.05, **P< 0.01, ***P< 0.001. Exact P values in b compared between Mock and ZIKV H/PF/2013 (BMP2 P= 0.0002; RUNX2 P= 0.0011; OSX P= 0.0002; ALPL P= 0.0002; DMP1 P= 0.0002 and PDPN P= 0.0002). Exact P values in e compared between IgG-treated and Nab-treated group (BMP2 P= 0.0054; RUNX2 P= 0.0005; OSX P= 0.0085; DMP1 P= 0.0099 and PDPN P= 0.0005). Data are representative of two independent experiments.
Extended Data Fig. 5. Overexpression of ZIKV NS3 protease induced osteogenic gene expression in osteoblastic-like cells.

U2OS cells (n=6) were transiently transfected with plasmids encoding individual ZIKV genes for 3 days. Transfected cells were harvested for qRT-PCR analysis of osteogenic gene expressions. Gene expressions were normalized to GAPDH and expressed as fold changes relative to vector control. Data are presented as mean ± SEM in box plots showing the upper (75%) and lower (25%) quartiles, with the horizontal line as the median and the whiskers as the maximum and minimum values observed. Data are representative of three independent experiments.
Extended Data Fig. 6. ZIKV NS3 protease is highly conserved across different lineages.

(a) Sequence alignment comparison between BMP2 gene in human (homo sapiens; amino acid residues 1–396) and hamster (Cricetulus; amino acid residues 1–399). (b) Purity of BL21 strain-derived recombinant ZIKV NS3 protease, CHO-FD11 cells-derived full-length BMP2 and BMP4 were determined by coomassie blue stain. MW: molecular weight. (c) Sequence alignment comparison between human BMP2 (amino acid residues 1–396) and BMP4 (amino acid residues 1–408). (d) Sequence alignment of NS3 protease domain (amino acid residues 1–177) was compared across two African ZIKV (MR766, IbH30656) and two Asian ZIKV (PRVABC59 and H/PF/2103). The red-colored letters indicate protease catalytic triad. (e) In vitro cleavage assay of carboxyl terminal HA-tagged purified human BMP4 and purified ZIKV NS3 protease were performed at 37 °C for 3 h, followed by immunoblot analysis. Data in b and e are representative of three independent repeats.
Supplementary Material
Acknowledgements
This work was partly supported by K99DE028573 (W.C.), CA200422, CA251275, AI140705, AI140705S, AI140718, AI152190, AI116585, AI116585S, DE023926, DE027888, DE028521 and KGM9942011 (J.U.J.), AI069120, AI056154, AI078389 and AI28697 (G.C.), AI129534-01, AI298847-01 (K.N.S.), AI140718 (J.U.J., G.C. and K.N.S.), Departamento de Ciência e Tecnologia (DECIT/25000.072811/2016-17) do Ministério da Saúde do Brasil and Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior CAPES/ 88887.116627/2016-01 (P.B.), and NIH/NIAID grants U19AI118610 and R21AI129486 (A.G.-S.).
Footnotes
Data availability
The gene expression data presented in Extended Data Fig. 2h of this manuscript are reanalyzed from RNA-seq data reported by Oh et al (Nature Neuroscience; 2017; 20(9); p1209–1212, doi: 10.1038/nn.4612) and deposited in GEO, GSE87750. Data supporting the findings of this study are available from the corresponding author upon reasonable request. All qRT-PCR primers and probe sequences used in this study are listed in Supplementary Table 1. Source data are provided with this paper.
Competing interests
Dr. Jae U Jung is a scientific adviser of the Vaccine Stabilization, a California corporation
References
- 1.Bonaldo MC et al. Isolation of Infective Zika Virus from Urine and Saliva of Patients in Brazil. PLoS Negl Trop Dis 10, e0004816, doi: 10.1371/journal.pntd.0004816 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brasil P et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N Engl J Med 375, 2321–2334, doi: 10.1056/NEJMoa1602412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao-Lormeau VM et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387, 1531–1539, doi: 10.1016/S0140-6736(16)00562-6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duffy MR et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 360, 2536–2543, doi: 10.1056/NEJMoa0805715 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Wang L et al. From Mosquitos to Humans: Genetic Evolution of Zika Virus. Cell Host Microbe 19, 561–565, doi: 10.1016/j.chom.2016.04.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phoo WW et al. Structure of the NS2B-NS3 protease from Zika virus after self-cleavage. Nat Commun 7, 13410, doi: 10.1038/ncomms13410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Y et al. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins. Cell Discov 3, 17006, doi: 10.1038/celldisc.2017.6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding Q et al. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc Natl Acad Sci U S A 115, E6310–E6318, doi: 10.1073/pnas.1803406115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith DR et al. Neuropathogenesis of Zika Virus in a Highly Susceptible Immunocompetent Mouse Model after Antibody Blockade of Type I Interferon. PLoS Negl Trop Dis 11, e0005296, doi: 10.1371/journal.pntd.0005296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorman MJ et al. An Immunocompetent Mouse Model of Zika Virus Infection. Cell Host Microbe 23, 672–685.e676, doi: 10.1016/j.chom.2018.04.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soares de Oliveira-Szejnfeld P et al. Congenital Brain Abnormalities and Zika Virus: What the Radiologist Can Expect to See Prenatally and Postnatally. Radiology 281, 203–218, doi: 10.1148/radiol.2016161584 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Li H, Saucedo-Cuevas L, Shresta S & Gleeson JG The Neurobiology of Zika Virus. Neuron 92, 949–958, doi: 10.1016/j.neuron.2016.11.031 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Pool KL et al. Association Between Neonatal Neuroimaging and Clinical Outcomes in Zika-Exposed Infants From Rio de Janeiro, Brazil. JAMA Netw Open 2, e198124, doi: 10.1001/jamanetworkopen.2019.8124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mulkey SB et al. Sequential Neuroimaging of the Fetus and Newborn With In Utero Zika Virus Exposure. JAMA Pediatr 173, 52–59, doi: 10.1001/jamapediatrics.2018.4138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Speer MY et al. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res 104, 733–741, doi: 10.1161/CIRCRESAHA.108.183053 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felin JE et al. Nuclear variants of bone morphogenetic proteins. BMC Cell Biol 11, 20, doi: 10.1186/1471-2121-11-20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pillai IC et al. Cardiac Fibroblasts Adopt Osteogenic Fates and Can Be Targeted to Attenuate Pathological Heart Calcification. Cell Stem Cell 20, 218–232.e215, doi: 10.1016/j.stem.2016.10.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu H et al. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat Neurosci 22, 47–56, doi: 10.1038/s41593-018-0298-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubrac A et al. NCK-dependent pericyte migration promotes pathological neovascularization in ischemic retinopathy. Nat Commun 9, 3463, doi: 10.1038/s41467-018-05926-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montagne A et al. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat Med 24, 326–337, doi: 10.1038/nm.4482 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Goswami R et al. Expression of osteogenic molecules in the caudate nucleus and gray matter and their potential relevance for Basal Ganglia calcification in hypoparathyroidism. J Clin Endocrinol Metab 99, 1741–1748, doi: 10.1210/jc.2013-3863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boström K et al. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest 91, 1800–1809, doi: 10.1172/JCI116391 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Derwall M et al. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler Thromb Vasc Biol 32, 613–622, doi: 10.1161/ATVBAHA.111.242594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L et al. Inhibition of Vascular Calcification. Arterioscler Thromb Vasc Biol 38, 2382–2395, doi: 10.1161/ATVBAHA.118.311546 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Oh Y et al. Zika virus directly infects peripheral neurons and induces cell death. Nat Neurosci 20, 1209–1212, doi: 10.1038/nn.4612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corry J, Arora N, Good CA, Sadovsky Y & Coyne CB Organotypic models of type III interferon-mediated protection from Zika virus infections at the maternal-fetal interface. Proc Natl Acad Sci U S A 114, 9433–9438, doi: 10.1073/pnas.1707513114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rausch K et al. Screening Bioactives Reveals Nanchangmycin as a Broad Spectrum Antiviral Active against Zika Virus. Cell Rep 18, 804–815, doi: 10.1016/j.celrep.2016.12.068 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SJ et al. Pyruvate Dehydrogenase Kinase 4 Promotes Vascular Calcification via SMAD1/5/8 Phosphorylation. Sci Rep 5, 16577, doi: 10.1038/srep16577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang Q et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell, doi: 10.1016/j.stem.2016.07.019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui Y, Jean F, Thomas G & Christian JL BMP-4 is proteolytically activated by furin and/or PC6 during vertebrate embryonic development. EMBO J 17, 4735–4743, doi: 10.1093/emboj/17.16.4735 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leighton M & Kadler KE Paired basic/Furin-like proprotein convertase cleavage of Pro-BMP-1 in the trans-Golgi network. J Biol Chem 278, 18478–18484, doi: 10.1074/jbc.M213021200 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Künnapuu J, Björkgren I & Shimmi O The Drosophila DPP signal is produced by cleavage of its proprotein at evolutionary diversified furin-recognition sites. 106, 8501–8506, doi: 10.1073/pnas.0809885106 %J Proceedings of the National Academy of Sciences (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Z et al. Crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science 354, 1597–1600, doi: 10.1126/science.aai9309 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Dhore CR et al. Differential expression of bone matrix regulatory proteins in human atherosclerotic plaques. 21, 1998–2003 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Melo AS et al. Congenital Zika Virus Infection: Beyond Neonatal Microcephaly. JAMA Neurol 73, 1407–1416, doi: 10.1001/jamaneurol.2016.3720 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Moore CA et al. Characterizing the Pattern of Anomalies in Congenital Zika Syndrome for Pediatric Clinicians. JAMA Pediatr 171, 288–295, doi: 10.1001/jamapediatrics.2016.3982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Driggers RW et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N Engl J Med 374, 2142–2151, doi: 10.1056/NEJMoa1601824 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Yamada M et al. High frequency of calcification in basal ganglia on brain computed tomography images in Japanese older adults. Geriatr Gerontol Int 13, 706–710, doi: 10.1111/ggi.12004 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Koyama S et al. Clinical and radiological diversity in genetically confirmed primary familial brain calcification. Sci Rep 7, 12046, doi: 10.1038/s41598-017-11595-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Livingston JH, Stivaros S, Warren D & Crow YJ Intracranial calcification in childhood: a review of aetiologies and recognizable phenotypes. Dev Med Child Neurol 56, 612–626, doi: 10.1111/dmcn.12359 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Nielsen-Saines K et al. Delayed childhood neurodevelopment and neurosensory alterations in the second year of life in a prospective cohort of ZIKV-exposed children. Nat Med 25, 1213–1217, doi: 10.1038/s41591-019-0496-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adachi K et al. Early Clinical Infancy Outcomes for Microcephaly and/or SGA Zika-exposed Infants. Clin Infect Dis, doi: 10.1093/cid/ciz704 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Einspieler C et al. Association of Infants Exposed to Prenatal Zika Virus Infection With Their Clinical, Neurologic, and Developmental Status Evaluated via the General Movement Assessment Tool. JAMA Netw Open 2, e187235, doi: 10.1001/jamanetworkopen.2018.7235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopes Moreira ME et al. Neurodevelopment in Infants Exposed to Zika Virus In Utero. N Engl J Med 379, 2377–2379, doi: 10.1056/NEJMc1800098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zin AA et al. Screening Criteria for Ophthalmic Manifestations of Congenital Zika Virus Infection. JAMA Pediatr 171, 847–854, doi: 10.1001/jamapediatrics.2017.1474 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petribu NCL et al. Follow-up brain imaging of 37 children with congenital Zika syndrome: case series study. BMJ 359, j4188, doi: 10.1136/bmj.j4188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bayless NL, Greenberg RS, Swigut T, Wysocka J & Blish CA Zika Virus Infection Induces Cranial Neural Crest Cells to Produce Cytokines at Levels Detrimental for Neurogenesis. Cell Host Microbe 20, 423–428, doi: 10.1016/j.chom.2016.09.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian X, Brookes O & Battaglia G Pericytes from Mesenchymal Stem Cells as a model for the blood-brain barrier. Sci Rep 7, 39676, doi: 10.1038/srep39676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crisan M et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3, 301–313, doi: 10.1016/j.stem.2008.07.003 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Zarb Y et al. Ossified blood vessels in primary familial brain calcification elicit a neurotoxic astrocyte response. Brain 142, 885–902, doi: 10.1093/brain/awz032 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanlandewijck M et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480, doi: 10.1038/nature25739 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Crisan M et al. Perivascular multipotent progenitor cells in human organs. Ann N Y Acad Sci 1176, 118–123, doi: 10.1111/j.1749-6632.2009.04967.x (2009). [DOI] [PubMed] [Google Scholar]
- 53.Xu J et al. Human perivascular stem cell-derived extracellular vesicles mediate bone repair. Elife 8, doi: 10.7554/eLife.48191 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cataneo AHD et al. The citrus flavonoid naringenin impairs the in vitro infection of human cells by Zika virus. Sci Rep 9, 16348, doi: 10.1038/s41598-019-52626-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goodfellow FT et al. Strain-Dependent Consequences of Zika Virus Infection and Differential Impact on Neural Development. Viruses 10, doi: 10.3390/v10100550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simonin Y, van Riel D, Van de Perre P, Rockx B & Salinas S Differential virulence between Asian and African lineages of Zika virus. PLoS Negl Trop Dis 11, e0005821, doi: 10.1371/journal.pntd.0005821 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foo SS et al. Asian Zika virus strains target CD14(+) blood monocytes and induce M2-skewed immunosuppression during pregnancy. Nat Microbiol, doi: 10.1038/s41564-017-0016-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tripathi S et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog 13, e1006258, doi: 10.1371/journal.ppat.1006258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hillger F, Herr G, Rudolph R & Schwarz E Biophysical comparison of BMP-2, ProBMP-2, and the free pro-peptide reveals stabilization of the pro-peptide by the mature growth factor. J Biol Chem 280, 14974–14980, doi: 10.1074/jbc.M414413200 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Tang H et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 18, 587–590, doi: 10.1016/j.stem.2016.02.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao M et al. BMP2-SMAD signaling represses the proliferation of embryonic neural stem cells through YAP. J Neurosci 34, 12039–12048, doi: 10.1523/JNEUROSCI.0486-14.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakashima K et al. BMP2-mediated alteration in the developmental pathway of fetal mouse brain cells from neurogenesis to astrocytogenesis. Proc Natl Acad Sci U S A 98, 5868–5873, doi: 10.1073/pnas.101109698 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheng H et al. Osteogenic activity of the fourteen types of human bone morphogenetic proteins (BMPs). J Bone Joint Surg Am 85, 1544–1552, doi: 10.2106/00004623-200308000-00017 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Zhang M, Ngo J, Pirozzi F, Sun YP & Wynshaw-Boris A Highly efficient methods to obtain homogeneous dorsal neural progenitor cells from human and mouse embryonic stem cells and induced pluripotent stem cells. Stem Cell Res Ther 9, 67, doi: 10.1186/s13287-018-0812-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Corselli M et al. Perivascular support of human hematopoietic stem/progenitor cells. Blood 121, 2891–2901, doi: 10.1182/blood-2012-08-451864 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.