

Abstract
Vascular EGF receptors (EGFR) influence function and structure of arterial vessels. In genetic mouse models we described the role of vascular smooth muscle (VSMC) EGFR for proper physiological function and structure as well as for pathophysiological alterations by obesity or angiotensin II. As the importance of endothelial (EC) EGFR in vivo is unknown, we analyzed the impact of EC-EGFR knockout in a conditional mouse model on vascular and renal function under control condition as well as in obesity and in comparison to VSMC-KO. Heart and lung weight, blood pressure and aortic transcriptome (determined by RNA-seq) were not affected by EC-EGFR-KO. Aortic reactivity to α1-adrenergic stimulation was not affected by EC-EGFR-KO contrary to VSMC-EGFR-KO. Endothelial-induced relaxation was reduced in abdominal aorta of EC-EGFR-KO animals, whereas it was enhanced in VSMC-EGFR-KO animals. Mesenteric arteries of EC-EGFR-KO animals showed enhanced sensitivity to α1-adrenergic stimulation, whereas endothelial-induced relaxation and vessel morphology were not affected. Renal weight, histomorphology, function (albumin excretion, serum creatinine, fractional water excretion) or transcriptome were not affected by EC-EGFR-KO, likewise in VSMC-EGFR-KO. High fat diet (HFD) over 18 weeks induced arterial wall thickening, renal weight increase, creatininemia, renal and aortic transcriptome alterations with a similar pattern in EC-EGFR-WT and EC-EGFR-KO animals by contrast to the previously reported impact of VSMC-EGFR-KO. HFD induced endothelial dysfunction in abdominal aortae of EC-EGFR-WT, which was not additive to the EC-EGFR-KO-induced endothelial dysfunction. As shown before, VSMC-EGFR-KO prevented HFD-induced endothelial dysfunction. HFD-induced albuminuria was less pronounced in EC-EGFR-KO animals and abrogated in VSMC-EGFR-KO animals. Our results indicate that EC-EGFR, in comparison to VSMC-EGFR, is of minor and opposite importance for basal renovascular function as well as for high fat diet-induced vascular remodeling and renal end organ damage.
Subject terms: Cardiovascular biology, Medical research, Preclinical research, Obesity
Introduction
The epidermal growth factor receptor (EGFR/ErbB1), a member of the ErbB-receptor tyrosine kinase family, is activated by binding e.g. epidermal growth factor (EGF) or heparin bound EGF (HB-EGF) and modulates signaling pathways that affect cell differentiation, migration and matrix homeostasis1. EGFR can also be transactivated by receptors for vasoactive substances, thereby contributing to vascular tone, dysfunction and remodeling2,3 as a heterologous transducer for non-EGFR ligands.
The relevance of vascular smooth muscle cells (VSMC)-EGFR for basal vascular function, for angiotensin II (AII)- or obesity-induced structural and functional vascular remodeling, as well as for complete renal end organ damage succeeding vascular remodeling, was shown4,5. These and other studies document the importance of VSMC-EGFR in cardiovascular health and disease3,6–10. In addition, systemic inhibitors of EGFR-kinase activity improved vascular function and vascular wall homeostasis in diabetic animals7–13. Thus, vascular EGFR could not only be of pathogenetic but also of therapeutic cardiovascular importance when conventional therapies fail, having pharmacological tools already at hands. To evaluate this concept further, the mechanistic role of vascular EGFR should be understood, meaning that the respective relevance of VSMC- and endothelial cell (EC)- EGFR needs to be determined.
Hitherto, the importance of endothelial EGFR is not as well understood as the one of VSMC. However, there is evidence from pharmacological studies for a modulatory role of EGFR in normal tissue, besides its enhanced expression in tumor endothelium14 or during the development of endothelial dysfunction3. But, because these studies could not differentiate between VSMC- and EC-EGFR, investigations in a conditional KO mouse model are required.
Recently, we established a knockout (KO) mouse model with deletion of the EGFR in VSMC15 that confirmed the involvement of VSMC-EGFR in basal blood pressure homeostasis, ageing-related vascular changes, acute AII vascular responsiveness15 and pathological AII action in vivo4. Furthermore, this model unveiled the importance of VSMC-EGFR for obesity-induced vascular and renal damage5. The present study investigates in vivo and ex vivo the role of EC-EGFR under basal conditions as well as during high fat diet-induced obesity/diabetes mellitus type 2 (DMT2) using the (B6.Cg-Tg(Tek-cre)1Ywa/J = Tie2CRE)16 mouse model to generate EGFR deletion, with respect to vascular remodeling, vascular gene expression and renal damage. Furthermore, we compare the possible importance of EC-EGFR with the previously investigated role of VSMC-EGFR, assuming that EC-EGFR is of less importance. The data presented confirm this hypothesis under basal as well as under pathological condition.
Materials and methods
All mouse experiments were approved by the local government (Landesverwaltungsamt Sachsen-Anhalt, Germany, Az.: 505.6.3-42502-2-1389 MLU_G; Veterinäramt Stadt Halle, Germany; Bescheid T16/2019) and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the ARRIVE guidelines and under consideration of the 3R-principle.
Animals
As already described before4,5,15 mice were kept at constant temperature of 22 ± 2 °C, relative humidity of 30–60%, under a 12/12 h light–dark cycle with ad libitum access to water and standard chow. Knock-out in endothelial cells was achieved by mating EGFRflox/flox C57BL/6 mice (originally provided by Maria Sibilia, Vienna, Austria) with B6.Cg-Tg(Tek-cre)1Ywa animals16 from Jackson Laboratory (Stock No: 008863, C57BL/6J genetic background). Inducible knock out for EGFR in VSMC was created by mating EGFRflox/flox C57BL/6 mice (originally provided by Maria Sibilia, Vienna, Austria) with B6.FVB-Tg(Myh11-cre/ERT2)1Soff mice (= iSMMHC-Cre+/− C57BL/6N mice, Jackson laboratory, stock no: 019079, C57BL/6N genetic background, originally provided by Stefan Offermanns, Bad Nauheim, Germany)4,5,15. As controls (wild type, WT) mice without LoxP-sites but carrying the Cre-recombinase under control of the iSMMHC-promotor and treated with tamoxifen (= VSMC-model) or carrying the Cre-recombinase under control of the Tie2-promotor (= EC-model) were employed. HFD (60% fat share of energy versus 10% in SFD) was started at 6 weeks of age. Genotyping was performed on tail biopsies by PCR as previously described2. Knockout was confirmed by determination of the excision of EGFR exon 1 by genomic PCR. Successful excision of the floxed DNA-fragment in the EGFR gene, comprising exon 1, was determined with DNA extracted from lung or aortic tissue applying the following protocol: denaturation at 94 °C for 3 min followed by 35 cycles with denaturation at 94 °C for 30 s, annealing at 66,1 °C for 45 s and extension at 72 °C for 4 min. Final extension was performed at 72 °C for 4.5 min. The reaction mix contained additional 2 mM Mg2Cl. Forward primer: GGGTGACGTGTTCCCATTCA, reverse primer: AAAGTTTGCTACCGGCCTCA. Product length without excision = 2579 bp, product length with excision = 549 bp. DNA was extracted by proteinase K digestion and salt precipitation. PCR products were analysed by agarose gel electrophoresis followed by iodidium bromide staining (Supplementary methods SM01). To confirm reduced EGFR mRNA expression we performed quantitative RT-PCR (qRT-PCR) in lung tissue. RNA was isolated using InviTrap spin tissue RNA mini kit (Invitek Molecular GmbH) following manufacturer’s instructions. DNA contamination was removed (DNAse I, New England Biolabs) and reverse transcription (RT) was performed using random primers and Super-Script II reverse transcriptase (Invitrogen, Life Technologies) according to manufacturer’s instructions. 1 µL of the obtained cDNA was used in RT-qPCR (AriaMx Real-Time PCR System, Agilent Technologies). EGFR forward primer: GACCTTCACATCCTGCCAGT. EGFR reverse primer: GCATGGAGGTCAGTCCAGTT. qPCR efficiency was > 90%. The relative mRNA expression of the genes of interest was calculated according to the 2-ΔΔCt method, using the 18S RNA signal for normalization. 18S forward primer: GTAACCCGTTGAACCCCATT. 18S reverse primer: CCATCCAATCGGTAGTAGCG. Each sample was analyzed as triplicate. Results are shown in Supplementary methods SM02, expressed as mean difference between wild-type and knock out ± standard error of mean. Breeding of the animals and assignment to experimental groups was performed randomly by place holder numbers before knowledge regarding the animals was obtained. During further experimentation the genotype of the animals was blinded by pseudonymisation (assignment of numbers). Due to the differences in weight gain, the type of diet could not be blinded.
For body and organ weight analysis as well as for blood pressure measurements by Millar catheter of the VSMC-model new animal cohorts (WT-SFD, WT-HFD, KO-SFD, KO-HFD) were generated. For the aortic ring force development split into thoracic and abdominal aorta a separated reanalysis of thoracic and abdominal aortic rings investigated previously was performed5.
EGFR ELISA
In order to obtain an initial indication regarding the relative expression of EGFR in endothelial cells compared to vascular smooth muscle cells, we quantified EGFR protein in primary cells of human origin (Supplementary methods SM03). For additional referencing we included primary human mesangial cells (smooth muscle like cell type), renal proximal tubule cells (epithelial cell type) and HEK293 cell. EGFR expression was determined by an EGFR sandwich ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer’s protocol. Cells were lysed at 4 °C in extraction buffer [10 mM Tris (pH 7.4), 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 1%Triton X-100, 10%glycerol, 0.1%SDS, 0.5%deoxycholate, protease inhibitor cocktail (1:1000)]. Protein content was determined using the BCA reagent from Pierce (Rockford, IL). SM03 shows that EGFR expression was lowest, but clearly distinct from zero, in endothelial cells.
Invasive measurement of blood pressure
Intravasal blood pressure measurements (diastolic, systolic, mean) were performed in anaesthetized (80 mg/kg body weight (BW) ketamine and 120 mg/kg BW xylazine, Sigma-Aldrich, St. Louis, USA) mice as described before15. The right jugular vein was cannulated for infusion of 2% bovine serum albumin (Sigma-Aldrich, Steinheim, Germany) in Ringer lactate solution (for 1 L: NaCl 5.9 g, KCl 0.3 g, CaCl2 0.22 g, Na-Lactate 2.8 g) at 4 µl/g BW/min. A Millar catheter (size 1F, Millar Instruments, Houston, USA) was inserted into the right carotid artery. After a 20-min stabilization period systolic, diastolic and mean blood pressure were measured (PowerLab data acquisition systems, Spechbach, Germany) and pulse pressure was calculated with the LabChart7 software (ADI instruments, Spechbach, Germany). To analyze the impact of volume load on blood pressure a bolus of 50 µl Ringer Lactate with a subsequent infusion of 100 µl Ringer lactate solution (within one min, infusion rate 6 ml/h) was infused via the jugular vein catheter and the blood pressure was analyzed for 10 min. Subsequently, to analyze the reactivity of blood pressure upon phenylephrine (PE), a bolus of 100 µg PE / kg BW was given followed by an infusion of 100 µl Ringer lactate solution as described above within a minute (infusion rate: 6 ml/h). The change in blood pressure was analyzed for a subsequent 20 min time period.
Measurement of aortic ring force
Wire myography was performed using a system of DMT (Aarhus, Denmark), according to the protocols published before4,5,15. Aortic rings were equilibrated in modified aerated Krebs–Ringer solution (20% O2, 5% CO2) at 37 °C for 30 min. At the beginning and the end of the equilibration the physiological salt solution was changed once, followed by the application of a strain resulting in a force of 12mN4,15. This strain resulted in a similar change in vessel circumference (dL) and similar effective pressure values in both genotypes and was applied for 10 min prior to the first substance application. Wall stress and effective pressure were calculated as described by Mulvany and Halpern17. After each measurement, the chambers were flushed five times with Krebs–Ringer-solution, achieving an approximately 100,000 fold dilution of the substance, before a new reagent was tested. This did not apply for the relaxants carbamoylcholine chloride (carbachol) and S-Nitroso-N-acetyl-DL-penicillamine (SNAP). These substances were given at the point of stable force development of the previously administered vasoconstrictor.
Pressurized mesenteric arteries
Pressure myography was performed using a system of DMT (Aarhus, Denmark)4. After mice were killed, the mesenteric bed was removed and transferred to cold (4 °C) physiological salt solution. Mesenteric (third or fourth order) arteries were mounted on glass cannulas to allow perfusion at physiological pressures (inlet pressure 60 mmHg, outlet pressure 45 mmHg). Vessels were superfused continuously with Krebs–Henseleit solution (20% O2, 5% CO2; pH, 7.4; 37 °C) composed of (mmol/l): 119 NaCl, 4.7 KCl, 25 NaHCO3, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, 0.03 EDTA and 11.1 glucose. The wake-up procedure was carried out according to the manufacturer’s instructions by stepwise pressurizing to 20, 40, 60, 80, or 100 mm Hg using servo control system. Diameter of the vessels was measured with a video microscope (Zeiss Axiovert, Oberkochen, Germany) and a data acquisition and analysis system (Danish Myo Technology A/S, DK-8382 Hinnerup, Denmark). A 60-mmol/l KCl challenge was performed after 45 to 60 min equilibration and before any other interventions.
Harvesting of organs
Mice were sacrificed by cervical dislocation. Livers, kidneys, lungs, hearts and aortas were excised, carefully freed from adjacent tissues, and weighed. Tibia length was measured for normalization of organ weights. Parts of the tissues were immediately snap frozen in liquid nitrogen while parts were fixed in 5% paraformaldehyde solution. Tissues were dehydrated in increasing concentrations of methanol or isopropanol. After embedding in paraffin, 4 µm sections were cut4.
Histomorphometric analysis
Morphometric analysis was performed as described before2,4 in a blinded way (pseudonymisation by number assignment). Sections were double-blinded investigated. Glomerular damage was assessed as Bowman and glomerular area, glomerular cellularity (nuclei per glomerulus) and glomerulosclerosis in kidney sections stained with periodic acid-Schiff’s reagent. At least 30 glomeruli per kidney were evaluated, and the given values are the mean per animal. The degree of glomerulosclerosis was determined using a semiquantitative scoring method18. Glomeruli were selected randomly and scored as follows: grade 0, normal; grade 1, sclerotic area 25% of total glomerular area; grade 2, sclerotic area 25%-50%; grade 3, sclerotic area 50%-75%; grade 4, sclerotic area 75%-100%. For media-to-lumen ratio media thickness was measured at ten different locations within the vessel wall and divided by the internal circumference of the aorta according to Liang19. Fibrosis was analyzed by staining with Sirius red followed by quantitative microscopic determination of the relative fibrotic area.
Next generation sequencing and gene enrichment analysis
Total RNA was isolated as described20. Paired-end sequencing (2 × 150 bp) was performed with a HiSeq Illumina System by Novogene (UK) Co., Ltd (Cambridge, UK) in the same way as in our previous study5. Libraries were prepared with indexed adapters. Quality control was performed on the data provided by the service company (adaptors already clipped, fastQC, v0.11.3, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Read mapping was done with Tophat2 (2.0.14)21 (mouse genome GRCm38/mm10) and counting was done with featureCounts (1.4.6)22. Genes were annotated with BiomaRt v93 (R package v2.36.1)23. Normalization and differential expression analysis were performed using R package EdgeR (3.20.8)24 from Bioconductor (https://www.bioconductor.org/). The counts were normalized using the “trimmed mean of M values” (TMM) method. A false discovery rate (FDR) of 0.05 was used to determine if genes were significantly regulated. Additional filters such as FPM > 5 and Fold Change > 1.5 were applied. Raw data are publicly available on the GEO (Gene Expression Omnibus) database (https://www.ncbi.nlm.nih.gov/geo). GEO IDs: GSE144838 for the VSMC model and GSE158197 for the EC model.
Gene ontology enrichment analysis was performed with g:Profiler (http://biit.cs.ut.ee/gprofiler/;25) and Ingenuity Pathway Analysis (IPA) software (Qiagen, Hilden, Germany) was used for functional analysis (including Canonical Pathways, Upstream Regulator and Downstream Effects Analyses; that are features not included in g:Profiler) on the lists of regulated genes (results of the differential expression analyses). Their Ensembl identifiers were mapped to networks available in the software database. For the canonical pathway analysis, enriched pathways were ranked according to how relevant they were for the genes provided as input. Multiple testing was performed using the Benjamini–Hochberg (B–H) procedure. Analyses were corrected for multiple testing as described for the corresponding tools.
Determination of albumin, creatinine and glucose
Glucose consumption was assessed by commercially available kits (Glucose (HK) assay kit, Sigma, Darmstadt, Germany) according to the manufacturer’s instructions26. Albumin was determined by ELISA (Bethyl Laboratories, Montgomery, Texas, USA). Creatinine was determined enzymatically by the creatinase method (Diazyme, Dresden, Germany).
Statistics
Data are presented as mean ± standard error of mean. As described before5 2-way ANOVA (SFD vs HFD and WT vs KO) was performed, followed by post hoc testing. Student´s T-Test or Mann–Whitney Rank Sum Test were used as applicable according to pre-test data analysis by SigmaPlot 12.5. A p-value < 0.05 was considered significant. N = number of animals or vessels. Biometrical planning was performed under consideration of the 3R-principle with α = 0.05 and β = 0.8. Data from all experiments that proceeded technically according to plan were included into the analyses.
Ethcs approval and consent to participate
All mouse experiments were approved by the local government (Landesverwaltungsamt Sachsen-Anhalt, Germany, Az.: 505.6.3-42502-2-1389 MLU_G; Veterinäramt Stadt Halle, Germany; Bes-cheid T16/2019) and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the ARRIVE guidelines and under consideration of the 3R-principle.
Results
Systemic parameters
EC-EGFR-KO animals were born at a mendelian ratio and showed no gross abnormalities. There was no difference in body weight, tibia length or blood glucose compared to their wildtype littermates as well as to the VSMC-EGFR-WT animals (Fig. 1A–C, Supplementary Fig. SF01), except the known slightly higher blood glucose concentration27 under standard fat diet (SFD), most probably attributable to the genetic background. High fat diet (HFD) led to a similar increase in body weight and blood glucose in EC-EGFR-WT, EC-EGFR-KO, VSMC-EGFR-WT and VSMC-EGFR-KO (Fig. 1a–C, Supplementary Fig. SF01). Analysis of organ weights (Fig. 1A, Supplementary Fig. SF01) revealed no differences between the genotypes under control conditions for heart and kidney. There was a slight trend towards higher heart and kidney weights in EC-EGFR-WT compared to VSMC-EGFR-WT. HFD induced no change in heart weight but an increase in renal weight (normalized to tibia length (TL) or heart weight) that was aggravated in VSMC-EGFR-KO (EC: Fig. 1D,F, VSMC: Supplementary Fig. SF01). Scattered analysis shows that a susceptible subgroup of animals with substantially enhanced renal weight (> 25 mg/mm TL) was responsible for this effect (Fig. 2). This effect was much less pronounced in EC-EGFR-KO animals (Fig. 2, Supplementary Fig. SF02). Lung weight was not affected neither by genotype nor HFD in the EC-EGFR model (Fig. 2, Supplementary Figs. SF02 and SF03). By contrast, VSMC-EGFR-KO induced an increase in lung weight in a susceptible subgroup under SFD. HFD did not aggravate this effect (Fig. 2, Supplementary Fig. SF03).
Figure 1.
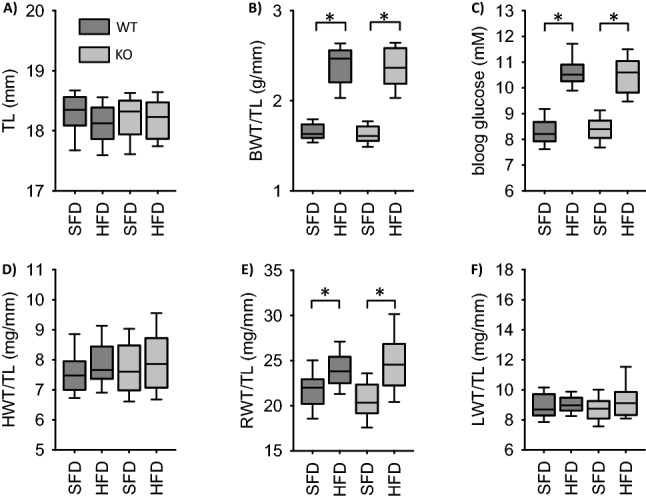
Influence of high fat diet on body weight (A,B), blood glucose (C) and heart (D), kidney (E) and lung (F) weight in EC-EGFR-WT and EC-EGFR-KO animals. Number of animals of the EC-model: N(WT, SFD) = 29, N(WT, HFD) = 30, N(KO, SFD) = 38, N(KO, HFD) = 39. Number of animals of the VSMC-model: N(WT, SFD) = 44, N(WT, HFD) = 38, N(KO, SFD) = 58, N(KO, HFD) = 54.*p < 0.05. SFD standard diet, HFD high fat diet, BWT body weight, TL tibia length, HWT heart weight, RWT renal weight, LWT lung weight, VSMC vascular smooth muscle cell.
Figure 2.

Scatter plots showing the correlation between body weight (BWT) and heart (HWT; A,B,G,H), kidney (RWT; C,D,I,J) or lung (LWT; E,F,K,L) weights in the four genotypes under standard fat diet (SFD) and high fat diet (HFD). TL = tibia length. Number of animals for the EC-model: N(WT, SFD) = 29, N(WT, HFD) = 30, N(KO, SFD) = 38, N(KO, HFD) = 39. Number of animals for the VSMC-model: N(WT, SFD) = 44, N(WT, HFD) = 38, N(KO, SFD) = 58, N(KO, HFD) = 54.
By contrast to VSMC-EGFR-KO15, blood pressure was not significantly different between EC-EGFR-KO and EC-EGFR-WT under SFD (Supplementary Fig. SF04). HFD induced a slight increase in systolic and mean blood pressure in EC-EGFR-WT and EC-EGFR-KO animals.
Functional vascular parameters
Isometric force measurements in aortic rings (conductance vessel) revealed no difference of action for the α1-adrenergic agonist, phenylephrine, in rings from EC-EGFR-KO as compared to EC-EGFR-WT animals under SFD (Supplementary Fig. SF05), in contrast to VSMC-EGFR-KO, as shown previously5. Reduction in phenylephrine induced-force generation during HFD was not prevented by EC-EGFR-KO.
HFD and EGFR-KO reduced KCl-induced contraction in the thoracic aorta from the VSMC-EGFR model but not in abdominal or thoracic aortae from EC-EGFR animals (Supplementary Fig. SF06). In the abdominal aorta, the U46619-induced (thromboxane analogue) contraction of the abdominal aorta was affected neither by genotype nor by HFD. In thoracic aortae, the knockout of EGFR in EC enhanced and knockout in VSMC reduced the effect of U46619 (Supplementary Fig. SF07).
Endothelium-dependent relaxation in response to carbachol was slightly reduced in abdominal but not thoracic aortae from EC-EGFR-KO (Fig. 3). VSMC-EGFR-KO also led to a slight yet opposite effect in abdominal aorta. The impact of the NO-donor SNAP was not affected by genotype (Fig. 3). Surprisingly, aortae from EC-EGFR-WT animals responded to HFD with a mild endothelial dysfunction only in the abdominal aorta, an effect that was absent in abdominal aortae from EC-EGFR-KO animals, probably due to the preexisting mild endothelial dysfunction (Fig. 4A–D). The data from thoracic aortae indicate a trend for enhanced HFD-susceptibility in EC-EGFR-KO animals (Fig. 4). HFD reduced the action of carbachol in abdominal and thoracic aortae from WT but not from KO animals of the VSMC-EGFR model (Fig. 4E–H), as also reported previously5. SNAP-induced relaxation was not affected (Supplementary Figs. SF08 and SF09).
Figure 3.

(A,B) Isometric relaxation of thoracic and abdominal aortic rings of EC-EGFR-KO and EC-EGFR-KO under SFD in response to carbachol. (C–F) Effect of HFD on isometric relaxation of thoracic (C,E) and abdominal (D,F) aortic rings in response to carbachol in EC-EGFR-WT (C,D) and EC-EGFR-KO (E,F) animals. Number of animals N(WT, SFD) = 10, N(WT, HFD) = 8, N(KO, SFD) = 8, N(KO, HFD) = 11. (G,H) Isometric relaxation of thoracic and abdominal aortic rings of VSMC-EGFR-KO and VSMC-EGFR-KO under SFD in response to carbachol. (I–L) Effect of HFD on isometric relaxation of thoracic (I,K) and abdominal (J,L) aortic rings in response to carbachol in VSMC-EGFR-WT (I,J) and VSMC-EGFR-KO (K,L) animals. Number of animals N(WT, SFD) = 10, N(WT, HFD) = 8, N(KO, SFD) = 10, N(KO, HFD) = 10. *p < 0.05 versus respective control. SFD standard diet, HFD high fat diet, WT wildtype, KO knockout, EC endothelial cell, VSMC vascular smooth muscle cell.
Figure 4.
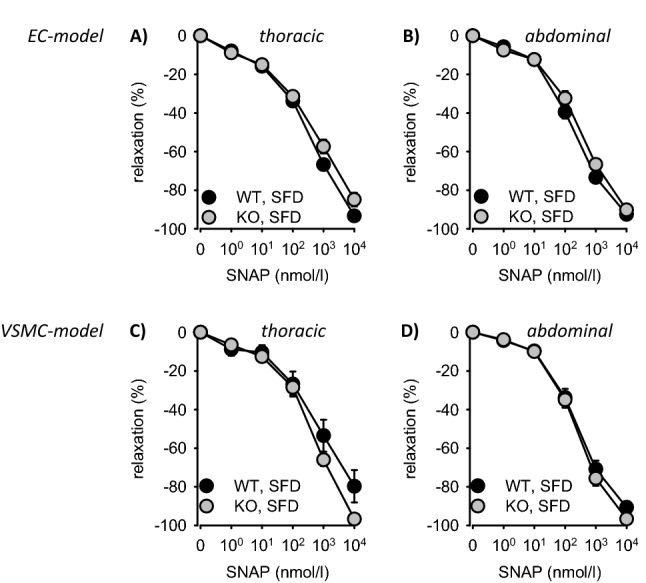
(A,B) Isometric relaxation of thoracic and abdominal aortic rings of EC-EGFR-KO and EC-EGFR-KO under SFD in response to SNAP. (C,D) Isometric relaxation of thoracic and abdominal aortic rings of VSMC-EGFR-KO and VSMC-EGFR-KO under SFD in response to SNAP. Number of animals in the EC-model: N(WT, SFD) = 10, N(KO, SFD) = 8. Number of animals in the VSMC-model: N(WT, SFD) = 10, N(KO, SFD) = 10. *p < 0.05 versus respective control. SFD standard diet, HFD high fat diet, WT wiltype, KO knockout, EC endothelial cell, VSMC vascular smooth muscle cell.
Pressure myography in mesenteric arteries (vessels with myogenic tone) showed reduced vessel diameter and increased wall-to-lumen ratio (inward remodeling) induced by HFD independent of the EC-genotype (Fig. 5). Carbachol induced relaxation was less pronounced in the EC-EGFR model (Fig. 5) compared to the VSMC-EGFR model under SFD (see Fig. 2 in5), indicating reduced endothelial function. EC-EGFR-KO or HFD induced no further reduction of endothelial function. The effect of SNAP was not significantly different between the various groups (Fig. 5; Fig. 2 in5).
Figure 5.

Pressure myography in mesenteric arteries of the EC-EGFR model. (A) Contraction in reponse to 20 mmol/l KCl. (B) Contraction in response to 100 nmol/l U46619. (C) Diameter of mesenteric arteries. (D) Wall-to-lumen ratio of mesenteric arteries. (E) Carbachol-induced dilatation. (F) SNAP-induced dilatation. (G) Phenylephrine-induced contraction. Number of animals N(WT, SFD) = 8, N(WT, HFD) = 10, N(KO, SFD) = 10, N(KO, HFD) = 14. *p < 0.05. SFD standard diet, HFD high fat diet, WT wildtype, KO knockout. *p < 0.05. #p < 0.05 versus WT, SFD.
As reported before, neither EGFR knockout nor HFD altered the effect of phenylephrine-induced contraction of mesenteric arteries in the VSMC-EGFR model (see Fig. 2 in5). In contrast, EGFR knockout enhanced phenylephrine-sensitivity in the EC-EGFR model (Fig. 5). HFD reduced the sensitivity to phenylephrine in mesenteric arteries from EC-EGFR-KO but not from EC-EGFR-WT animals.
Aortic gene expression analysis
The effect of VSMC-EGFR-KO on aortic transcriptome has been reported before5. EC-EGFR-KO induced no differential expression of mRNAs under SFD (Supplementary Table ST01, Supplementary Fig. SF10) in aortas. In EC-EGFR-WT animals HFD induced only mild transcriptome alterations (3 genes were affected according to the results of differential expression analysis, with FDR 0.05, Fold Change > 1.5, FPM > 5) (Supplementary Fig. SF10). Of those three genes only Cyp2e1 was differentially expressed in EC-EGFR-KO animals treated with HFD. These data, together with the functional results above, strongly suggest that EC-EGFR is not of major importance for basal vascular function or structure—in contrast to VMSC-EGFR2,4,5,15.
Renal parameters
Knockout of EGFR in EC did not affect the parameters investigated under SFD (Fig. 6). In EC-EGFR-WT animals HFD led to increased serum creatinine levels and albuminuria, similar to the effects reported previously for VSMC-EGFR-WT5. Knockout of EC-EGFR had no impact on the HFD induced elevated serum creatinine but attenuated albuminuria (Fig. 6). Fractional water excretion was not affected by HFD or EC-EGFR KO. The HFD-induced increase in renal weight in KO animals was accompanied by moderate histological changes, including glomerular sclerosis in both genotypes (Supplementary Fig. SF11). Proximal tubule hypertrophy or an increase in glomerular size were not observed (Supplementary Fig. SF11).
Figure 6.
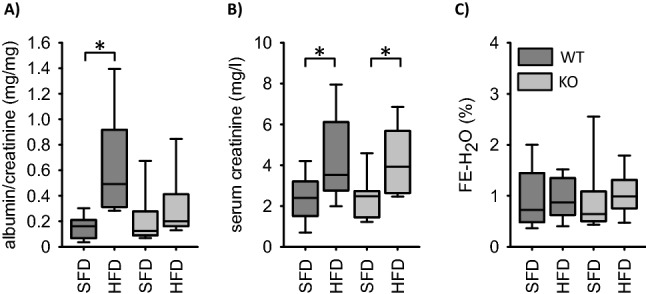
Impact of HFD-diet on functional renal parameters in the EC-EGFR-model. (A) Urinary albumin excretion. (B) Serum creatinine concentrations. (C) Fractional water excretion (FE). Number of animals: N = 10 for each experimental group. *p < 0.05. SFD standard diet, HFD high fat diet, WT wildtype, KO knockout.
Renal gene expression analysis
In EC-EGFR-KO, no mRNA was expressed differently under SFD. HFD induced differential expression of 310 genes in WT and 284 genes in KO animals with an overlap of 222 mRNAs and more up- than downregulated mRNAs (Supplementary Table ST02 and Supplementary Fig. SF12A). The results of Gene Ontology Term (GO) enrichment analysis, i.e. altered lipid metabolism, was not affected by EC-EGFR knockout (Supplementary Table ST03) in contrast to the previously reported effect of VSMC-EGFR-KO5. Furthermore, upstream regulator analysis by IPA® also provided no evidence for a different impact of HFD on EC-EGFR-KO as compared to EC-EGFR-WT animals (Supplementary Fig. SF12B).
Discussion
From our results, we conclude that EC-EGFR is of minor importance for basal physiological renovascular function or HFD-induced alterations compared to VSMC-EGFR. Gross organ morphology, aortic and renal transcriptome, mesenteric artery or renal function were not affected substantially by EC-EGFR-KO under SFD. We detected a slight endothelial dysfunction in the abdominal aorta from EC-EGFR-KO animals under SFD that was not observed in thoracic aorta or mesenteric arteries, indicating limited systemic impact of the KO. This conclusion is supported by the fact that blood pressure did not differ between EC-EGFR-KO and WT animals. Of course, we do not exclude the possibility of a local regulatory importance of EC-EGFR, for example during metabolic blood flow control. Regarding the interpretation of results obtained with EC-KO models the problem of non-EC expression in pan-EC-KO models, as reviewed by Payne et al.28 has to be considered. Non-EC-expression in haematopoietic cells has been reported for the pan-EC models Tie2, VE-cadherin and Flk1. In cases when substantial KO effects are observed a possible contribution of haematopoietic cells must be discussed. Because EGFR-KO in our model exerted no major effect, the conclusions that EGFR-KO is of minor importance is still valid in our opinion. Furthermore, it has to be considered that we used the non-inducible Tie2-CRE model in this initial study, in order to investigate whether major effects of EC-EGFR-KO can be observed, whereas for VSMC-EGFR we previously had used a non-inducible and an inducible CRE-system2,4,5,15 with similar results for the basal effects. In case of major effects in the Tie2-CRE model confirmation in an inducible system (e.g. the tamoxifen-inducible VE-cadherin-CRE model28) should be performed. However, so far the effects observed are only mild in the Tie2-CRE-model. Nevertheless, we started with the implementation of the inducible VE-cadherin-KO model for EC-EGFR-KO. Unfortunately, the knock-down efficiency was very variable in our first cohorts. We will try to improve the protocol for EC-EGFR-KO so that this model can eventually be used to study EC-EGFR further in future studies.
Possibly, VSMC-KO and EC-KO play opposite roles in abdominal aorta under basal conditions, because VSMC-EGFR-KO led to enhanced endothelial dependent and EC-EGFR-KO to reduced relaxation (Fig. 3). As this effect of EC-EGFR-KO was not observed in thoracic aorta or mesenteric arteries from EC-EGFR-KO mice, presumably it is of minor systemic relevance (see also Supplementary Fig. SF04). Furthermore, we observed a slight increase in phenylephrine sensitivity of mesenteric arteries in EC-EGFR-KO animals, which was not observed in the aorta of EC-EGFR-KO animals or in mesenteric arteries from VSMC-EGFR-KO animals. Yet, we could not show an acute systemic impact of phenylephrine, as the acute increase in blood pressure was not affected by EC-EGFR-KO (Supplementary Fig. SF13). HFD-induced cardiovascular effects are virtually identical in EC-EGFR-WT and EC-EGFR-KO animals, with the exception of the mild endothelial dysfunction of abdominal aorta that was not observed in EC-EGFR-KO, most probably due to the already preexisting endothelial dysfunction (Fig. 3). Moreover, HFD-induced albuminuria was less pronounced in EC-EGFR-KO animals whereas the HFD-induced decrease in estimated glomerular filtration rate (from serum creatinine) was not prevented. These finding keeps the option of a partial protection of the glomerular filtration barrier without prevention of overall HFD-induced vascular remodeling, which is supported by mesenteric artery wall thickening. Altogether, these findings are in contrast to VSMC-EGFR that plays an important role under physiological and pathophysiological conditions, as reported previously2,4,5,15.
The limited impact of EC-EGFR on vessel wall homeostasis is also reflected by the lack of changes of the aortic mRNA-transcriptome in EC-EGFR-KO animals under SFD. Of course our data do not exclude alterations in mRNA expression restricted to endothelial cells, due to the fact that endothelial cells represent only a minority of vascular wall cell population. This question has to be addressed using isolated endothelial cells in future studies.
HFD induced virtually no changes of the aortic transcriptome in EC-EGFR-WT animals, unlike in the VSMC-EGFR model5. When we compared the transcriptome of aortas from WT, SFD animals of the VSMC-EGFR model with the EC-EGFR model, we observed distinct differences with respect to the extracellular matrix, indicating a more “fibrotic” vascular type already under control conditions (Supplementary Figs. SF14 and S15). This fits to suggested functional consequences of the slightly different genetic background we had to use for the two models27,29–31, because B6.Cg-Tg(Tek-cre)1Ywa animals are of C57BL/6J background and B6.FVB-Tg(Myh11-cre/ERT2)1Soff of C57BL/6N background (https://www.jax.org/strain/019079, section Details, Development). It has been reported that this difference can lead to subtle yet relevant phenotypic differences, comprising higher blood glucose concentrations, glucose intolerance, lower insulin secretion and lower oxygen consumption in C57BL/6 J, possibly due to the Nnt mutation27. This is of course a limitation of our study with respect to HFD, because a comprised glucose tolerance in WT animals might disguise obesity-induced alterations, although our data show that the animals developed obesity and hyperglycemia.
In contrast to the vascular phenotype, HFD-induced renal alterations were comparable in EC-EGFR-WT and VSMC-EGFR-WT animals, reflecting the metabolic syndrome (reduced estimated glomerular filtration, albuminuria, glomerulosclerosis and a shift in the transcriptome in response to the enhanced lipid load). However, EC-EGFR-KO was almost without effect on HFD-induced alterations, in contrast to VSMC-EGFR-KO, as reported before5. The less pronounced albuminuria in EC-EGFR-KO animals suggests a certain protective effect but cannot be explained or classified at the moment. Because the impairment of estimated glomerular filtration as well as glomerular sclerosis was not prevented, we assume that there is not major protective effect.
The scattered analysis of organ weights in a larger population indicates that, even under standardized laboratory conditions, there are subgroups of more susceptible animals, especially in the VSMC-EGFR model. This was evident for the effect of HFD on renal weight in VSMC-EGFR-KO for the effect of EGFR-KO in VSMC on lung weight already under SFD. Assuming that the enhanced lung weight results from congestion, these data suggest a yet unrecognized importance of VSMC-EGFR for the cardiopulmonary system in a subgroup of animals. The underlying mechanisms for the difference in susceptibility as well as for the pathological lung phenotype are not known and should be investigated in more depth in future studies in order to identify potential genetic risk variants.
Conclusions
In summary, our results show that EC-EGFR, in comparison to VSMC-EGFR, is of minor importance for basal vascular and renal function, as well as for HFD-induced functional vascular remodeling, endothelial dysfunction and renal end-organ damage. Thus, therapeutic targeting strategies aiming at vascular EGFR should put their conceptual focus on VSMC because major EC-derived effects are unlikely.
Supplementary Information
Abbreviations
- AII
Angiotensin II
- DMT2
Diabetes mellitus type 2
- EC
Endothelial cell
- EGFR
EGF receptor
- ErbB1
EGF receptor
- FDR
False discovery rate
- FPM
Fragments per million
- GO
Gene ontology
- HFD
High-fat diet
- IPA
Ingenuity Pathway Analysis
- KO
Knock out
- PAS
Periodic acid–Schiff’s reagent
- SFD
Standard- fat diet
- SNAP
S-nitroso-N-acetyl-DL-penicillamine
- TL
Tibia length
- VSM
Vascular smooth muscle
- VSMC
Vascular smooth muscle cell
- WT
Wild-type
Author contributions
C.S.: isometric force measurements, histological and phenotypical evaluation, qPCR measurements, data analysis, manuscript proofreading. B.S.: design of mouse breeding strategy, phenotypical evaluation, contribution to the experimental design, data analysis, blood pressure measurement, manuscript proofreading. A.N. and V.D.: handling and analysis of RNA-seq data. S.R.: pressure myography, clinical chemistry, phenotypical evaluation. S.M.: isometric force measurements, clinical chemistry. C.W.: histological evaluation. M.G.: development of the conceptual and experimental design of the study, data analysis, wrote the manuscript, responsible for funding.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was funded by the Deutsche Forschungsgemeinschaft (DFG GE905/19-1 and 2, GE905/24-1).
Data availability
Datasets generated during and/or analysed during the current study are available in the gene expression omnibus database with the study identity GSE144838 and GSE158197. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144838) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158197). All further data generated or analysed during this study are included in this published article and its supplementary information files.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Barbara Schreier and Christian Stern.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-86587-3.
References
- 1.Bublil EM, Yarden Y. The EGF receptor family: Spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 2007;19:124–134. doi: 10.1016/j.ceb.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Schreier B, Rabe S, Schneider B, Bretschneider M, Rupp S, Ruhs S, et al. Loss of epidermal growth factor receptor in vascular smooth muscle cells and cardiomyocytes causes arterial hypotension and cardiac hypertrophy. Hypertension. 2013;61:333–340. doi: 10.1161/HYPERTENSIONAHA.112.196543. [DOI] [PubMed] [Google Scholar]
- 3.Forrester SJ, Kawai T, O'Brien S, Thomas W, Harris RC, Eguchi S. Epidermal growth factor receptor transactivation: Mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu. Rev. Pharmacol. Toxicol. 2016;56:627–653. doi: 10.1146/annurev-pharmtox-070115-095427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schreier B, Hünerberg M, Mildenberger S, Rabe S, Bethmann D, Wickenhauser C, et al. Deletion of the EGF receptor in vascular smooth muscle cells prevents chronic angiotensin-II-induced arterial wall stiffening and media thickening. Acta Physiol. 2017;222:e12996. doi: 10.1111/apha.12996. [DOI] [PubMed] [Google Scholar]
- 5.Stern C, Schreier B, Nolze A, Rabe S, Mildenberger S, Gekle M. Knockout of vascular smooth muscle EGF receptor in a mouse model prevents obesity-induced vascular dysfunction and renal damage in vivo. Diabetologia. 2020;63:2218–2234. doi: 10.1007/s00125-020-05187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makki N, Thiel KW, Miller FJ., Jr The epidermal growth factor receptor and its ligands in cardiovascular disease. Int. J. Mol. Sci. 2013;14:20597–20613. doi: 10.3390/ijms141020597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schreier B, Gekle M, Grossmann C. Role of epidermal growth factor receptor in vascular structure and function. Curr. Opin. Nephrol. Hypertens. 2014;23:113–121. doi: 10.1097/01.mnh.0000441152.62943.29. [DOI] [PubMed] [Google Scholar]
- 8.Belmadani S, Palen DI, Gonzalez-Villalobos RA, Boulares HA, Matrougui K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes. 2008;57:1629–1637. doi: 10.2337/db07-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benter IF, Yousif MHM, Griffiths SM, Benboubetra M, Akhtar S. Epidermal growth factor receptor tyrosine kinase-mediated signalling contributes to diabetes-induced vascular dysfunction in the mesenteric bed. Br. J. Pharmacol. 2005;145:829–836. doi: 10.1038/sj.bjp.0706238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benter IF, Benboubetra M, Hollins AJ, Yousif MHM, Canatan H, Akhtar S. Early inhibition of EGFR signaling prevents diabetes-induced up-regulation of multiple gene pathways in the mesenteric vasculature. Vascul. Pharmacol. 2009;51:236–245. doi: 10.1016/j.vph.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Benter IF, Sarkhou F, Al-Khaldi AT, Chandrasekhar B, Attur S, Dhaunsi GS, et al. The dual targeting of EGFR and ErbB2 with the inhibitor Lapatinib corrects high glucose-induced apoptosis and vascular dysfunction by opposing multiple diabetes-induced signaling changes. J. Drug Target. 2015;23:506–518. doi: 10.3109/1061186X.2015.1057150. [DOI] [PubMed] [Google Scholar]
- 12.Li R, Uttarwar L, Gao B, Charbonneau M, Shi Y, Chan JSD, et al. High glucose up-regulates ADAM17 through HIF-1a in mesangial cells. J. Biol. Chem. 2015;290:21603–21614. doi: 10.1074/jbc.M115.651604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi T, Eguchi S. The epidermal growth factor receptor: a missing link between endoplasmic reticulum stress and diabetic complications? Hypertension. 2012;60:20–21. doi: 10.1161/HYPERTENSIONAHA.112.197038. [DOI] [PubMed] [Google Scholar]
- 14.Amin DN, Hida K, Bielenberg DR, Klagsbrun M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006;66:2173. doi: 10.1158/0008-5472.CAN-05-3387. [DOI] [PubMed] [Google Scholar]
- 15.Schreier B, Hünerberg M, Rabe S, Mildenberger S, Bethmann D, Heise C, et al. Consequences of postnatal vascular smooth muscle EGF-receptor deletion on acute angiotensin II action. Clin. Sci. 2016;130:19–33. doi: 10.1042/CS20150503. [DOI] [PubMed] [Google Scholar]
- 16.Kisanuki YY, Hammer RE, Ji M, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 17.Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.RES.41.1.19. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Babínkoví D, Huang J, Swain GM, Wang DH. Deletion of transient receptor potential vanilloid type 1 receptors exaggerates renal damage in deoxycorticosterone acetate-salt hypertension. Hypertension. 2008;52:264–270. doi: 10.1161/HYPERTENSIONAHA.108.110197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang M, Ekblad E, Lydrup ML, Nilsson BO. Combined lack of estrogen receptors alpha and beta affects vascular iNOS protein expression. Cell Tissue Res. 2003;313:63–70. doi: 10.1007/s00441-003-0731-3. [DOI] [PubMed] [Google Scholar]
- 20.Schreier B, Dohler M, Rabe S, Schneider B, Schwerdt G, Ruhs S, et al. Consequences of epidermal growth factor receptor (ErbB1) loss for vascular smooth muscle cells from mice with targeted deletion of ErbB1. Arterioscler. Thromb. Vasc. Biol. 2011;31:1643–1652. doi: 10.1161/ATVBAHA.111.223537. [DOI] [PubMed] [Google Scholar]
- 21.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2013;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 23.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, et al. g:Profiler-a web server for functional interpretation of gene lists (2016 update) Nucleic Acids Res. 2016;44:W83–W89. doi: 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polovic M, Dittmar S, Hennemeier I, Humpf HU, Seliger B, Fornara P, et al. Identification of a novel lncRNA induced by the nephrotoxin ochratoxin A and expressed in human renal tumor tissue. Cell. Mol. Life Sci. 2018;75:2241–2256. doi: 10.1007/s00018-017-2731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fontaine DA, Davis DB. Attention to background strain is essential for metabolic research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes. 2016;65:25. doi: 10.2337/db15-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Payne S, De VS, Neal A. Endothelial-specific Cre mouse models. Arterioscler. Thromb. Vasc. Biol. 2018;38:2550–2561. doi: 10.1161/ATVBAHA.118.309669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simon MM, Greenaway S, White JK, Fuchs H, Vr G-D, Wells S, et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013;14:R82. doi: 10.1186/gb-2013-14-7-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreth K, Fischer R, Fuchs H, Vr G-D, Wurst W, Katus HA, et al. High-throughput phenotypic assessment of cardiac physiology in four commonly used inbred mouse strains. J. Comp. Physiol. B. 2014;184:763–775. doi: 10.1007/s00360-014-0830-3. [DOI] [PubMed] [Google Scholar]
- 31.Kawashita E, Ishihara K, Nomoto M, Taniguchi M, Akiba S. A comparative analysis of hepatic pathological phenotypes in C57BL/6J and C57BL/6N mouse strains in non-alcoholic steatohepatitis models. Sci. Rep. 2019;9:204. doi: 10.1038/s41598-018-36862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets generated during and/or analysed during the current study are available in the gene expression omnibus database with the study identity GSE144838 and GSE158197. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144838) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158197). All further data generated or analysed during this study are included in this published article and its supplementary information files.