

Abstract
Dataset is the basis of deep learning model development, and the success of deep learning models heavily relies on the quality and size of the dataset. In this work, we present a new data preparation protocol and build a large fragment-based dataset Frag20, which consists of optimized 3D geometries and calculated molecular properties from Merck Molecular Force Field (MMFF) and DFT at B3LYP/6-31G* level of theory for more than half a million molecules composed of H, B, C, O, N, F, P, S, Cl, Br with no larger than 20 heavy atoms. Based on the new dataset, we develop robust molecular energy prediction models using a simplified PhysNet architecture for both DFT-optimized and MMFF-optimized geometries, which achieve better than or close to chemical accuracy (1 kcal/mol) on multiple test sets, including CSD20 and Plati20 based on experimental crystal structures.
Graphical Abstract
INTRODUCTION
Molecular energy calculation is crucial for conformation analysis and structure-based drug design. However, accurate calculation achieved by high-level quantum mechanical calculations1 can be computationally demanding even for small molecules,2 while the application of more computational efficient molecular mechanical methods2–5 is limited by the accuracy of force fields. Recently, an appealing alternative is to obtain the molecular energy using deep learning models.6–56 Deep learning models can extract the high-level atom or molecule representation from raw data using multiple nonlinear layers and provide reliable predictions with much less computational cost.57 The success of deep learning methods heavily relies on the data quality, and the dataset covering broad chemical space is necessary for developing a robust model with good generalization ability. Therefore, many datasets focusing on different chemical domains have been constructed.13–14, 41–43, 58–66, 91
Most of deep learning models for molecular energy prediction have been developed with the QM9 dataset,59, 64, 66 which was built using organic drug-like molecules from a subset of GDB-17.66 QM9 encompasses equilibrium structures and molecular properties calculated using DFT method at B3LYP/6-31G(2df, p) level of theory for 133,885 molecules composed of H,C,N,O,F with no larger than nine heavy atoms, and it has become a classic benchmark dataset for deep learning models. However, the applicability of deep learning models to predict molecular energies based on DFT optimized geometries would be significantly limited due to the computational cost of DFT geometry optimizations. To address this problem, in our recent study, we built several datasets based on QM9 which provide both DFT calculated properties and MMFF optimized geometries, and developed deep learning models that can achieve 0.34 kcal/mol MAE and 0.79 kcal/mol MAE on QM9 when using DFT optimized geometries and MMFF optimized geometries as inputs, respectively.38 However, our model’s performance dropped significantly on an external conformation test set composed of molecules with 10–12 heavy atoms and functional groups that have not been covered in QM9. Recently, Glavatskikh et al. also pointed out that QM9 lacks chemical diversity after detailed bond distance analysis and functional groups analysis.67 Thus, to make further progress in developing more robust and applicable deep learning models for molecular energy prediction using 3D geometries, larger and more diverse molecular datasets are needed.
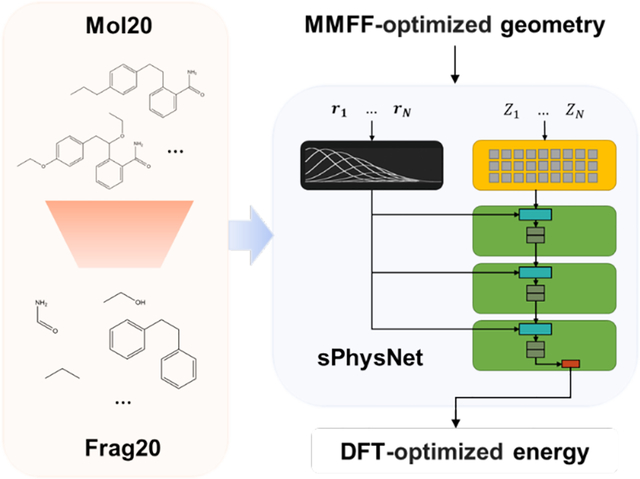
In this work, we presented a new data preparation protocol and built a fragment-based dataset Frag20. Frag20 is built using commercially available and publicly reported molecules from ZINC68–69 and PubChem70 database, and it has mainly made improvements from the following three aspects: 1. Molecule size and element coverage: Frag20 includes more than half a million molecules with no larger than 20 heavy atoms and covers common elements (H, B, C, N, O, F, P, S, Cl, Br) in organic drug-like compounds. 2. Chemical diversity and chemical space coverage: in the construction of Frag20, representative and diverse molecules are selected using Murcko fragmentation71 and extended functional groups (EFGs). 3. Geometries and properties: Frag20 provides geometries and molecular properties calculated using both Merck Molecular Force Field (MMFF) and DFT at B3LYP/6-31G* level of theory. Thus, Frag20 can be used to develop deep learning models that can make predictions based on MMFF-optimized geometries. Besides Frag20, we also constructed Plati20 and CSD20 using protein-bound ligand molecules from Platinum dataset72 and crystal structures from Cambridge Structure Database (CSD)73 to evaluate model’s generalization performance.
Based on datasets with both DFT and MMFF-optimized geometries, we have built robust molecular energy prediction models using simplified PhysNet.47 PhysNet was originally written in TensorFlow86 and we reimplemented it in PyTorch85. All results shown below involving PhysNet are from the PyTorch implementation. PhysNet has achieved the state-of-the-art performance on QM9 through a deep neural network architecture that incorporates long-range terms explicitly, which should be desirable for large molecule energy predictions. We did a grid search over hyperparameters as well as slightly modified its model architecture and found a simplified PhysNet (sPhysNet) with nearly doubled training speed while maintaining the similar performance. Our developed deep learning models can predict DFT level energy using MMFF- as well as DFT-optimized geometries and can achieve better than or close to chemical accuracy (MAE of 1 kcal/mol) on multiple test sets. Corresponding source codes and data sets are available on the web at: https://www.nyu.edu/projects/yzhang/IMA.
DATASET
Dataset is the basis of deep learning model development. Here we constructed Frag20, which includes more than half a million fragments with no larger than 20 heavy atoms. In addition, we built Plati20 and CSD20 datasets as two external test sets, as shown in Table 1.
Table 1.
Datasetsa Used for Machine Learning Model Development and Evaluation. Frag20, Plati20 and CSD20 datasets are newly developed in this work.
| Name | Source | #Heavy Atoms | Atom Type | #Mols/Confs | Geometry | Property |
|---|---|---|---|---|---|---|
| QM9a | GDB-9 | [1, 9] | H, C, O, N, F | 133,885 | B3LYP/6-31G(2df,p) | B3LYP/6-31G(2df,p) |
| QM9Mb | MMFF | |||||
| Frag20 | ZINC & PubChem | [1, 20] | H, B, C, O, N, F, P, S, Cl, Br | 565,438/566,296 | MMFF & B3LYP/6-31G* | B3LYP/6-31G* |
| eMol9b | eMolecules & GDB-9 | [1, 9] | H, C, O, N, F | 9959/88,234 | ||
| Plati20 | Platinum | [10, 20] | H, C, O, N, F | 401/20,972 | ||
| CSD20 | CSD | [2, 20] | H, C, O, N, F, P, S, Cl, Br | 33,572/39,816 |
QM9 is constructed by Ramakrishnan et al.59
A. Frag20 Dataset
Frag20 includes representative and diverse fragments with no larger than 20 heavy atoms. Figure 1 illustrates the data preparation protocol for Frag20, and it mainly includes four steps: data preprocessing, molecule fragmentation, molecule selection, and 1D (SMILES) to 3D (geometry) labeling.
Figure 1.
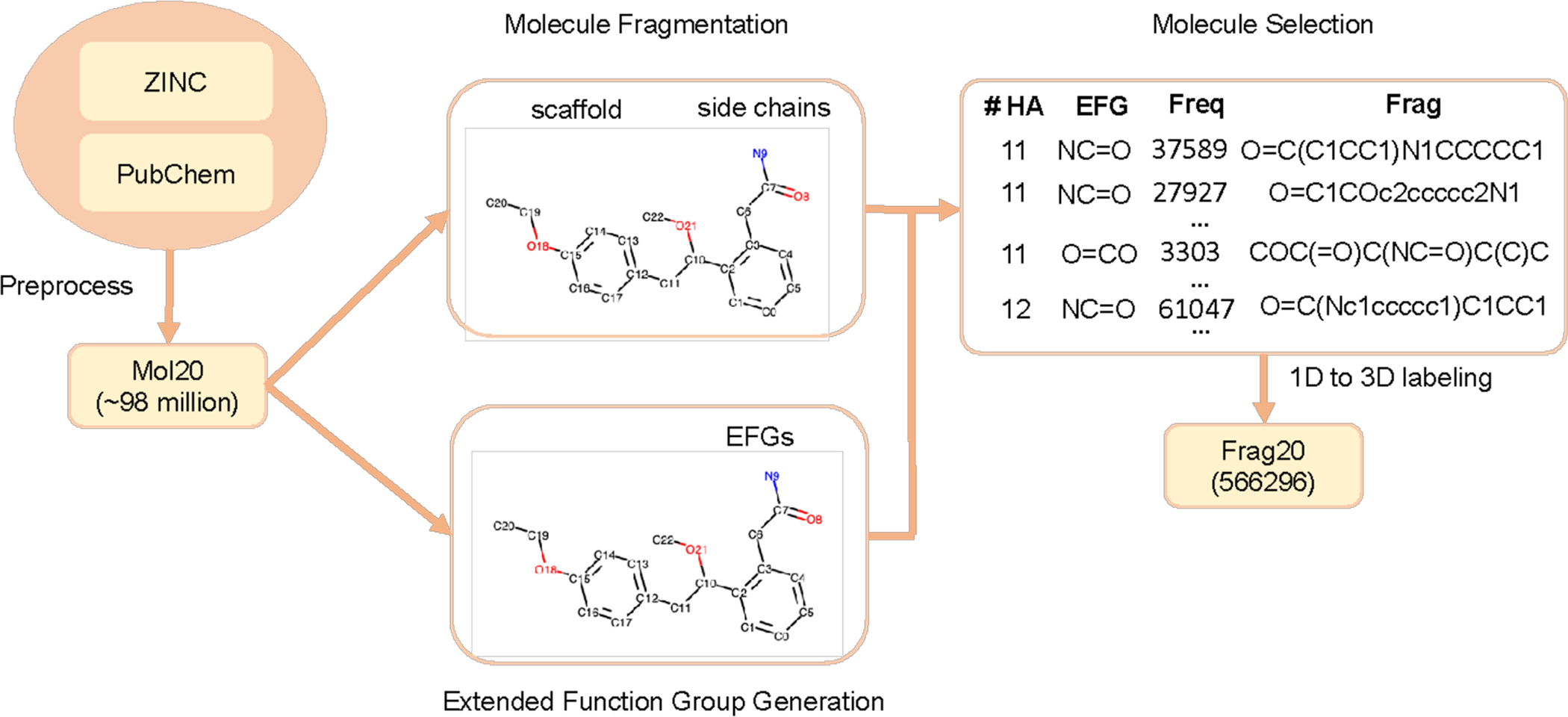
Data Preparation Protocol for Frag20. Frag20 is built based on ZINC and PubChem, and after data preprocessing, we first created Mol20. In molecule fragmentation, each molecule was cut into scaffold and side chains which are colored differently. To select molecules, extended functional group (EFG) library has been generated based on Mol20. EFG can be used to fully describe a molecule through chemical groups. Here, different color means different EFGs. Molecule selection is based on the number of heavy atoms, EFG, and fragment frequency. After 1D (SMILES) to 3D (geometry) labeling, we finally built Frag20.
Data Preprocessing
Frag20 is built based on commercially available and publicly reported molecules from ZINC and PubChem databases. The ZINC database is for virtual screening, and we downloaded more than 1 billion SMILES strings for molecules with molecular weight no larger than 400 Daltons and LogP no larger than 5 from the ZINC 15.75 Similarly, we downloaded around 96 million SMILES from PubChem70. We first merged two datasets and removed duplicates and then filtered molecules to only keep molecules with no larger than 20 heavy atoms and composed of H, B, C, N, O, F, P, S, Cl and Br. Following the SMILES cleaning procedure described in the recent work,76 we also removed stereochemistry and only kept the largest fragment after stripping salts. Our initial Mol20 dataset includes SMILES for 98,449,207 molecules.
Molecule Fragmentation
Due to the huge number of molecules in Mol20 (~98 million), it would be intractable for us to conduct QM calculations for all molecules. Therefore, we decomposed the molecules into fragments and built our fragment-based dataset to cover molecular pieces. Here, we used Murcko fragmentation,71 and each molecule was cut into the scaffold, which is a ring system with linker atoms, and the side chains (Figure 1). Hydrogen atoms have been added to the cut positions to convert the fragments into the completed molecules. Molecules which cannot pass Murcko fragmentation were removed. After molecule fragmentation, the dataset size was reduced to around 9 million (8,659,028), which is 1/10 of the original Mol20 size. The distribution of fragments for different number of heavy atoms is shown in Figure S1 of the Supporting Information (SI). There are still huge number of molecules when the number of heavy atoms increases. For example, we have around 1.3 million molecules with 20 heavy atoms.
Molecule Selection
To further reduce the number of molecules with larger than 10 heavy atoms, we selected molecules based on an extended functional group (EFG) library. EFG extends the traditional chemical functional group definition88 by including chemical groups formed only by carbon atoms, and hence the whole molecule can be described using EFGs (Figure 1). The generation of extended functional groups has been implemented into a python package (EFGs, https://github.com/HelloJocelynLu/EFGs). We generated an EFG library for our initial Mol20 with frequency percentage cutoff of 0.1 and only kept the Top 10% most frequent EFGs from Mol20. Our EFG library includes 4,520 different EFGs and covers 99.9% of molecules in Mol20. To select diverse molecules, we first divided datasets into several subsets with different number of heavy atoms. Then for each EFG in the EFG library, we ranked molecules containing corresponding EFG using their fragment frequencies and selected the ones with high fragment frequencies until the number of unique molecules meets the selection rate times the original subset size. Here, the selection rates have been predefined to make sure that we would not include too many large molecules that are computational demanding in subsequent QM calculations, and they are gradually decreasing from 10% to 1% for molecules with 11–20 heavy atoms.
1D to 3D labelling: Geometry Generation
As shown in Figure 2, the first step in 1D to 3D labeling pipeline is to generate 3D geometry for each molecule since the original data only provides 1D SMILES. Here, we randomly generated 1 conformation for each molecule using ETKDG method from RDKit.77–78 Molecules that failed in the conformation generation process were excluded.
Figure 2.
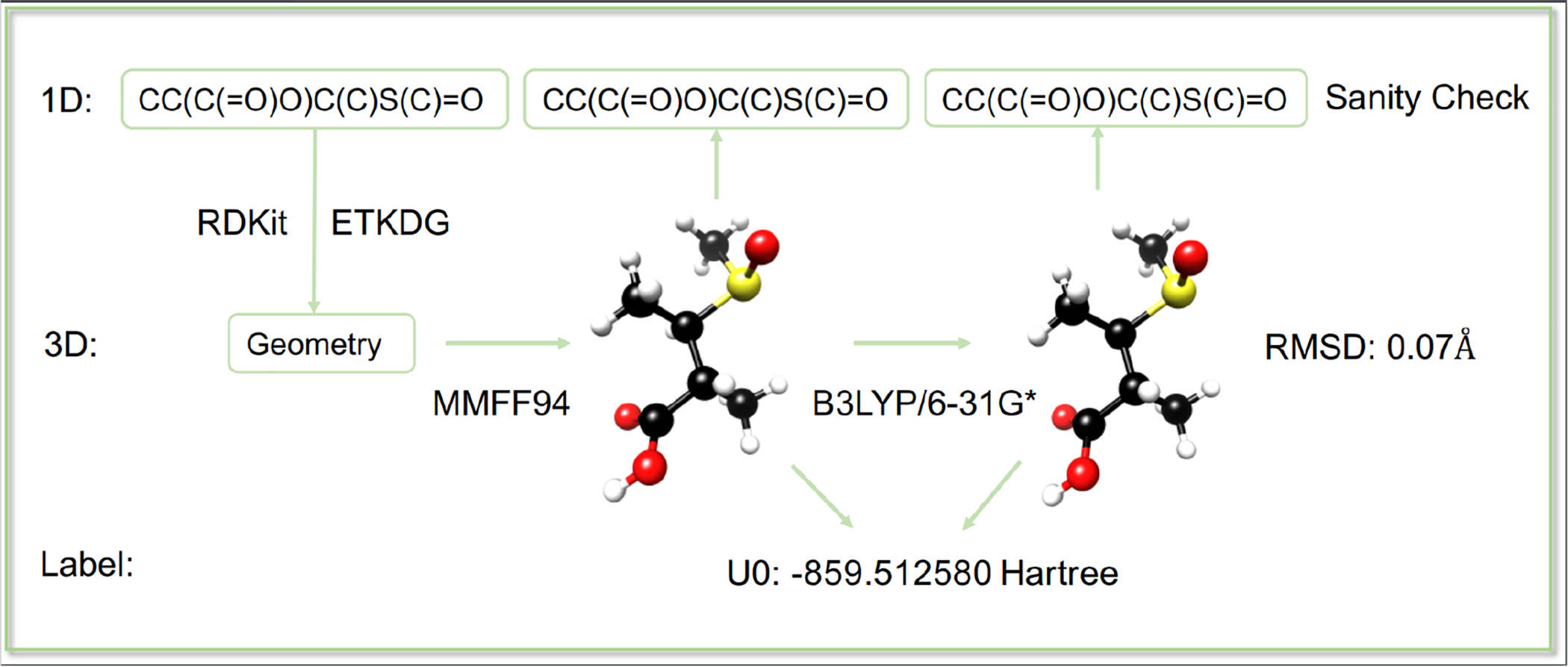
1D (SMILES) to 3D (Geometry) Labeling Pipeline.
1D to 3D labelling: MMFF and QM Calculation
For each molecule, we optimized its geometry using MMFF94 (MMFF)3 implemented in RDKit. Based on MMFF optimized geometry, QM geometry optimization and frequency calculation have been performed using Gaussian09 with DFT method at B3LYP/6-31G* level of theory.79 All molecules failed in QM calculation have been removed. Hence, for each molecule, our dataset provides two type of geometries optimized in both MMFF and DFT and the corresponding DFT level electronic and thermodynamic properties. There are 5,786 molecules failed in MMFF optimization, and we used Universal force field (UFF)5 to optimize these molecules. Since different force field methods have been applied, we only used molecules with MMFF optimized geometries in our Frag20-hold out test set and MM-based model development.
1D to 3D labelling: Sanity Check
In the last step, we checked the canonical SMILES for initial molecule, MMFF optimized geometry and QM optimized geometry and only kept the molecules with consistent SMILESs. We also removed molecules with partial charge or radicals to make sure that our dataset only includes neutral molecules.
As shown in Table 1, Frag20 dataset includes MMFF and DFT optimized geometries and calculated molecular properties for 566,296 molecules. Since some fragments become the same after SMILES conversion, the number of unique molecules in our Frag20 is 565,438. The detailed information for molecules with different number of heavy atoms can be found in Table S1. In addition, RMSD of heavy atoms, as a useful measurement to evaluate the difference between 3D structures, was calculated (Table 2 and Figure S2). The whole data preparation process has been implemented into a python package (Frag20Prep, https://github.com/jenniening/Frag20_prepare) which can be adapted for further dataset construction.
Table 2.
RMSD Information for Datasets.
| Dataset | S1a | S2 | ≤0.2b | (0.2, 0.5] | (0.5, 1.0] | (1.0, 1.5] | (1.5, 2.0] | >2.0 |
|---|---|---|---|---|---|---|---|---|
| Frag20 | DFT | MMFF | 325954 | 155923 | 70873 | 11717 | 1598 | 231 |
| Plati20c | Cry | MMFF | 95 | 140 | 129 | 36 | 1 | 0 |
| Cry | DFT | 75 | 131 | 157 | 37 | 1 | 0 | |
| CSD20 | Cry | MMFF | 21888 | 12527 | 4590 | 710 | 90 | 11 |
| Cry | DFT | 26678 | 8855 | 3245 | 834 | 180 | 24 | |
| DFT | MMFF | 27487 | 9292 | 2731 | 261 | 39 | 6 |
S1 is the abbreviation of structure 1 and S2 is the abbreviation of structure 2. Here, RMSD is between S1 and S2.
RMSD Unit is Å.
The RMSD for the crystal structure of protein-bound ligand conformation and optimized structure of Plati20 is the smallest RMSD achieved by all generated conformations of corresponding molecule.
B. Plati20 Dataset
To evaluate our model’s performance on molecular conformation analysis, we prepared the Plati20 dataset based on Platinum, which is a data set composed of high-quality X-ray structures for protein-bound ligand conformations.72 We selected neutral molecules with 10–20 heavy atoms and composed of H, C, O, N, F from Platinum. For each selected compound, up to 300 conformations have been generated using ETKDG78 method and optimized with MMFF3. We removed similar conformations using Butina80 clustering with 0.2Å RMSD cutoff, and mirror-image conformations identified by ArbAlign81 RMSD calculation with consideration of symmetry. After that, we optimized each MMFF optimized conformation using B3LYP/6-31G* to get corresponding DFT-level energy. The final dataset includes 401 unique molecules with 20,972 conformations. For each molecule, we computed the smallest RMSD that has been achieved by all generated and optimized conformations in comparison with the protein-bound ligand structures. As shown in Table 2 and Figure S3, less than 1.0 Å RMSD has been obtained for most molecules (> 90%), which indicates that our employed conformation generation protocol is very reasonable.
CSD20 Dataset
In order to further evaluate our model’s performance, we prepared CSD20 dataset based on Cambridge Structure Database (CSD), which is a curated and comprehensive repository for crystal structures of small organic molecules.73 Here, starting from the crystal structure for each molecule which appears both in Mol20 and CSD, we directly conducted MMFF optimization, DFT optimization and molecular energy calculation. Our constructed CSD20 dataset includes 39,816 molecules with no larger than 20 heavy atoms (C, N, O, F, P, S, Cl, Br). Since some molecules have multiple crystal structures in CSD, the unique number of molecules in CSD20 is 33,572. RMSD between crystal structure and optimized geometry for molecules in CSD20 have been computed and shown in Table 2 and Figure S4.
METHOD
A. Deep Learning Models
In our previous work, we developed DTNN_7ib model based on deep tensor neural network36 which achieved 0.34 kcal/mol MAE on QM9. To overcome the application limitation caused by using DFT optimized geometry as model inputs, we applied transfer learning and built models to predict molecular energy at DFT level using MMFF optimized geometries and atomic vectors learnt from DTNN_7ib.38
Recently, PhysNet has been introduced and it has achieved state-of-the-art performance on QM9 dataset for molecular energy prediction.47 The architecture of PhysNet (Figure 3A) was inspired by both ScheNet82 and HIP-NN28. Similar to many other deep learning models for molecular energy prediction based on 3D geometries, the input of PhysNet includes a nuclear charge vector Z and a pairwise distance matrix. To obtain the initial atom vector , atom nuclear charge vector Zi is mapped to embedding vectors ez composed of learnable parameters. The initial atom vector is passed to Nmodule modules that have the same composition but independent parameters. Each module contains an interaction block, atomic residual blocks and one output block. In the interaction block, the atom vector x is updated by accounting for its local environment as following:
where ul is a learnable parameter vector, f is a neural network which compose of multiple residual layers and one linear layer, and is the message accounting for the local environments. can be obtained by a message pass layer:
where gself is an activation-first linear layer, Ni is a set containing all of atom xi’s neighbors and gneighbor is a neural network calculating the interaction from xi to xj depending on the Radius Basis Function(RBFji) which is a expansion function depending purely on the distance between xi and xj. Detailed process is described in Ref 47. Residual block is used to refine the atom vector in each module, and it adds shortcut connections to enable the neural networks to increase or at least have the similar performance when the depth is increased. Finally, output block is used to compute the atom-wise properties through linear transformation of activated atom vector passed from residual blocks. Each module in PhysNet produces one atom-wise prediction and they are aggregated throughout all modules, finally, molecule-level properties are obtained by summing up every atom in each molecule. PhysNet can predict energy, force, charge and dipole moment at the same time, and hence its loss function is the weighted sum of loss of each term. To make sure the prediction of each module decay hierarchically when the depth of the module increases, a regularization term of nonhierarchical penalty is also added. PhysNet also incorporates long range interaction by adding electrostatic interaction and dispersion correction terms explicitly. Thus, it should be a more suitable model for large molecules compared to DTNN_7ib. PhysNet47 was originally implemented in TensorFlow86. In this work, we have reimplemented it with PyTorch85, which has the same number of trainable parameters as the TensorFlow one (1,293,948) and achieved similar performance and computational efficiency. Furthermore, by exploring model hyperparameters, we found a simplified version of PhysNet (sPhysNet) (Figure 3B, Table 3), which significantly reduced the number of trainable parameters to about 0.74 million while achieved the similar performance on the QM9 dataset. We reduced the number of main modules from 5 to 3, removed one residual layer in the main module and 2 residual layers in interaction layers, while slightly increased the atomic embedding dimension (num_feature) from 128 to 160. Unlike original PhysNet, the output of sPhysNet modules was the output of the last module rather than summing over all outputs.
Figure 3.
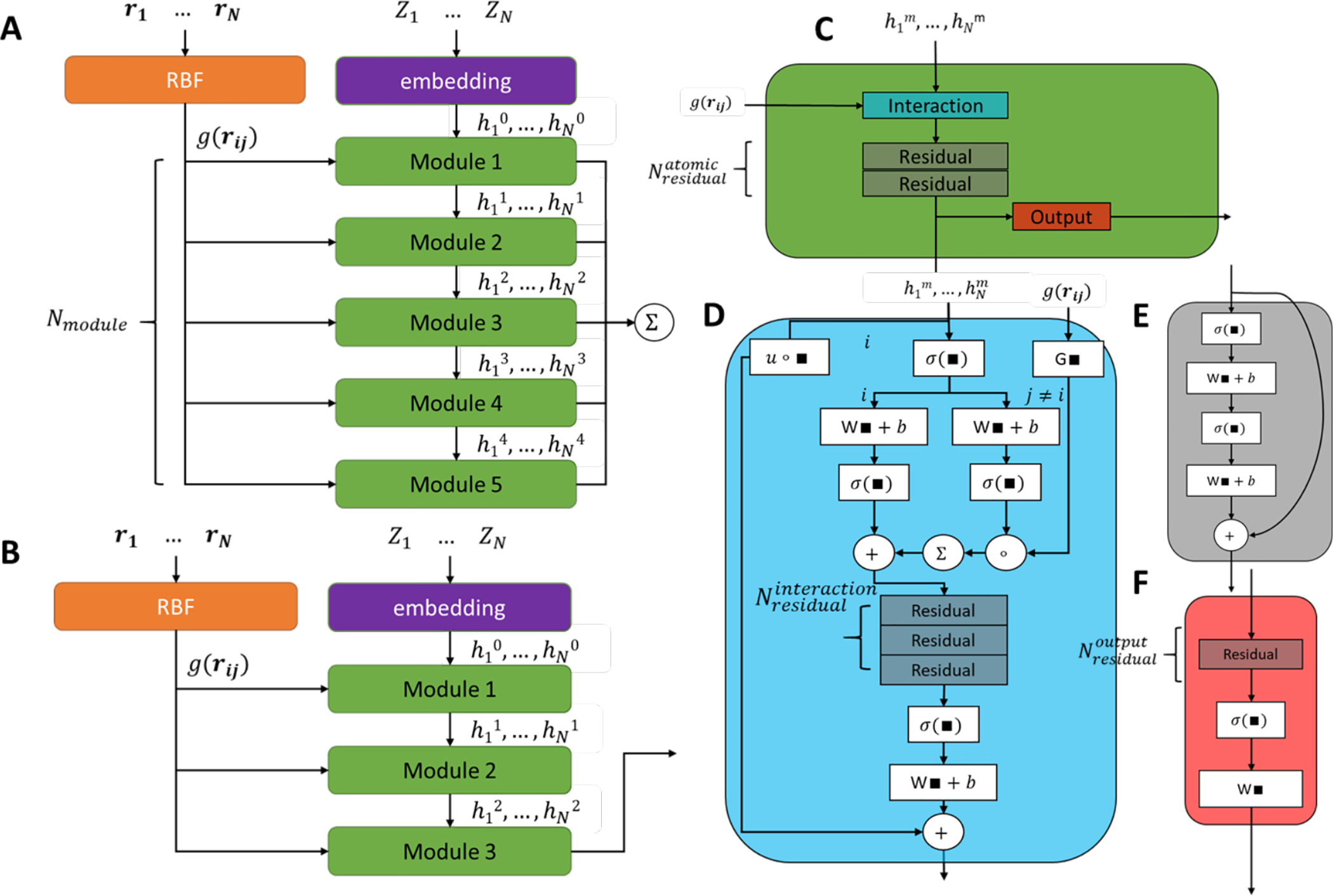
Overview of PhysNet47 and sPhysNet Architecture. (A) Original PhysNet architecture. (B) sPhyNet architecture, noted that the number of modules decreased to 3 and only the last module (Module 3) contributes to the output. (C) A single PhysNet module, consists of an interaction module, residual module(s), and an output module. (D), (E) and (F) are interaction module, residual module and output module, respectively.
Table 3.
Difference in Hyperparameters of PhysNet and sPhysNet.
| Name | PhysNet | sPhysNet |
|---|---|---|
| num_featurea | 128 | 160 |
| Nmodule | 5 | 3 |
| 2 | 1 | |
| 3 | 1 | |
| 1 | 1 | |
| Total trainable params | 1.29M | 0.74M |
num_feature is the length of the atom vector.
For explicit energy terms, we removed DFT-D3 energy term because it is not considered in B3LYP/6-31G* calculations used in 1D to 3D labeling. The final predicted energy therefore changes from:
To:
Where N is number of atoms in the molecule and χ(rij) is a function which approximate 1/rij at long range while avoiding singularity at rij = 0.47 And is the corrected charge of atom i by:
Where Q is the total charge of the system. This correction is necessary to guarantee charge conservation.47
B. Training Protocol
Since Frag20 consists of molecules containing 1 to 20 heavy atoms, we prepared a hold-out test set from Frag20 by randomly selecting 10% of molecules for each heavy atom number. Our Frag20 test set includes 56,636 molecules, validation set includes 1,000 molecules, and all remain 508,660 molecules were used as training set in our final model development.
As mentioned above, we successfully reimplemented PhysNet architecture on PyTorch with similar performance and efficiency, and the rest of work were run on PyTorch version.
Model was trained on a single GPU (P1080, P100, K80 and V100, depending on resource allocation) with batch size 100. We used AMSGrad87 optimizer at learning rate=0.001, betas= (0.9, 0.99), eps=1e-8 and weight decay=0 to optimize the model. The actual model used for validation and testing was a shadow model with the same initialization and exponential moving average over training model parameters.47,89 No early stopping was used, and the model was trained until it reaches 1000 epochs or time limit 36 hours, whichever came first. At the end of each epoch, we test our model on a separate validation set and calculate the loss. If the validation loss was better than the previous lowest validation loss, we saved the model as the best model into disk. In this way, the saved model was the one with lowest validation loss throughout the training.
To build models with MMFF optimized geometries, we restored all weights trained using DFT optimized geometries, and then either retrained the weights in the output block of each module using MMFF optimized geometries (transfer learning), or directly fine-tuned the whole model without any layer-freezing (fine tuning).
When performing model assessment using external CSD20 and Plati20 datasets, we excluded molecules that also exist in Frag20 and eMol9, and final Plati20 and CSD20 used as test sets contained 380 molecules with 19,504 conformations and 36,552 molecules, respectively.
To evaluate model performance, both mean absolute error (MAE) and root mean square error (RMSE) have been used. It should be noted that RMSE is more sensitive for outliers, which are data points with large prediction errors. In addition, percentages of molecules with prediction error larger than 1 kcal/mol and 10 kcal/mol have also been calculated and presented. To assess the conformational energy prediction, we used both absolute error (ErrorA) and relative error (ErrorR). ErrorA measures the MAE and RMSE of predictions for all conformation. In terms of ErrorR, for each molecule, we first computed the MAE and RMSE for the energy difference between each conformational energy and the lowest energy of the molecule, and then averaged the MAEs or RMSEs among all molecules. Besides ErrorR, the success rate for finding the right lowest conformation of all molecules in the test set has also been calculated.
RESULTS
A. Dataset Analysis
Chemical diversity of the dataset can be analyzed using extended functional group. Extended functional group is a generalized version of traditional functional group and it also contains chemical groups formed by only carbon atoms. EFG library was generated based on Mol20 and it includes 4,520 EFGs which can fully cover 99.9% of molecules in Mol20. By checking the existence of each EFG in molecules, we found Frag20 has 3,889 EFGs and its subset Frag9 has 2,486 EFGs (up to 9 heavy atoms), which are much more than 482 EFGs that QM9 has. In addition, some of EFGs with top 100 frequencies in Mol20 such as O=CNO, N-N, and C=NN are not found in QM9. This indicates that our fragmentation process has led to a much more diverse dataset which would facilitate the development of more robust and applicable deep learning models.
B. Molecular Energy Prediction with both DFT and MMFF Optimized Geometries
QM9 dataset has been used as a classic benchmark for deep learning models with DFT optimized geometries. Considering the computational cost of DFT optimizations, the applicability of deep learning models with DFT-optimized geometries as input would be significantly limited. Previously, in order to explore whether MM-optimized geometries can be used for molecular energy prediction, we introduced QM9M and eMol9 datasets, and developed DTNN_7ib based on deep tensor neural network36. Our model can achieve 0.34 kcal/mol MAE on QM9 and 0.79 kcal/mol MAE on QM9M with transfer learning. In this work, we trained both PhysNet and our optimized sPhysNet on QM9 and QM9M datasets with the same training/validation/test splits as for DTNN_7ib, and the results are shown in Table 4. For training on the QM9M dataset, first we restored all weights learned from pre-trained models using DFT optimized geometries, then either only retrained the output block weights in each module using MMFF optimized geometries from QM9M dataset (transfer-learning) or retrain the whole model without any weight freezing (fine-tuning). From Table 4, we can see that sPhysNet has the similar performance as PhysNet, and both models can perform significantly better than DTNN_7ib. The sPhysNet model can achieve 0.19 kcal/mol MAE on QM9 and 0.35 kcal/mol MAE on QM9M with fine-tuning. Since the sPhysNet model is less complicated and more efficient to train than PhysNet and fine-tuning always yields better results than transfer learning alone for molecular energy prediction with MMFF optimized geometries, we mainly focus on the sPhysNet model and fine-tuning in our further model development with Frag20, a significantly larger and diverse dataset.
Table 4.
Performance on QM9 and QM9M Datasets (Training Set Size is 100k)a.
| Architecture | DTNN_7ib | PhysNet | sPhysNet | |||||
|---|---|---|---|---|---|---|---|---|
| Geometry | DFT | MMFF | DFT | MMFF | MMFF | DFT | MMFF | MMFF |
| Training | ETEb | TLc | ETE | TL | FTd | ETE | TL | FTc |
| MAE (kcal/mol) | 0.34 | 0.79 | 0.21 | 0.50 | 0.34 | 0.19 | 0.57 | 0.35 |
| RMSE (kcal/mol) | 0.86 | 1.44 | 0.52 | 1.01 | 0.79 | 0.49 | 1.03 | 0.81 |
| Error > 1kcal/mol | - | - | 1.72% | 11.08% | 6.12% | 4.41% | 14.16% | 6.42% |
| Error > 10kcal/mol | - | - | 0.03% | 0.09% | 0.06% | 0.05% | 0.09% | 0.07% |
Performance of other state-of-the-art models (kcal/mol) as reference: SchNet82: 0.26; HIP-NN28: 0.26; MPNN90: 0.42; DimeNet56: 0.18
ETE refers to end to end training;
TL represents transfer learning;
FT refers to fine-tuning.
Based on the Frag20 dataset, we further explored to develop molecular energy prediction models with sPhysNet. In order to considering conformations, we also added the previously developed eMol9 dataset (see Table 1), which is a conformation dataset and is built using overlapping molecules from QM9 and eMolecules, into our training set. To extensively examine the model’s performance, we not only used Frag20 hold-out test set, but also employed two additional test sets CSD20 and Plati20, which have been newly constructed in this work based on crystal structures (See Table 1). As shown in Table 5, using DFT optimized geometries as input, our trained sPhysNet model can achieve 0.34 kcal/mol MAE for Frag20, 0.82 kcal/mol MAE for CSD20, and 0.72 kcal/mol MAE for Plati 20, and all are better than chemical accuracy of 1.0 kcal/mol. Meanwhile, MAEs of our further fine-tuned sPhysNet model with MMFF-optimized geometries as input are 0.63 kcal/mol, 1.36 kcal/mol and 1.40 kcal/mol respectively for Frag20, CSD20 and Plati20 test sets. Although deep learning models using DFT-optimized geometries as inputs outperform those with MMFF-optimized geometries, the computational cost to obtain DFT optimized geometries is more than thousands of that to obtain MMFF-optimized geometries. To obtain a DFT optimized geometry, which needs to do multiple DFT energy and gradient calculations, is much more expensive than to calculate the DFT energy itself. From this perspective, deep learning models requiring DFT-optimized geometries as inputs have limited value in real applications. Therefore, our results here indicate that to develop deep learning models for predicting molecular energies with force-field optimized geometries as input is a very promising direction while there is still room to be improved, and our trained sPhysNet model based on Frag20 and eMol9 can be utilized as a baseline model for future development to explore chemical space with 3D geometries.
Table 5. (A).
sPhysNet Performance on Molecular Datasets (Training Set Size is ~590k).
| Test set | Metric | DFT-optimized | MMFF-optimized |
|---|---|---|---|
| Frag20 | MAE (kcal/mol) | 0.34 | 0.63 |
| RMSE (kcal/mol) | 0.72 | 1.23 | |
| Error > 1kcal/mol | 5.54% | 16.96% | |
| Error > 10kcal/mol | 0.04% | 0.14% | |
| CSD20 | MAE (kcal/mol) | 0.82 | 1.36 |
| RMSE (kcal/mol) | 1.57 | 2.33 | |
| Error > 1kcal/mol | 24.91% | 42.04% | |
| Error > 10kcal/mol | 0.37% | 0.70% |
DISCUSSION AND CONCLUSION
Deep learning models have achieved considerable progress in molecular energy prediction and their successes are dependent on the size and quality of the training set. In this work, we presented a data preparation protocol based on molecular fragmentation and selection and built a Frag20 dataset which includes more than half million molecules up to 20 heavy atoms. Frag20 shows broad coverage of chemical space and wide diversity of chemical groups which would enhance the performance of deep learning models. With more than 500k molecules in the dataset, Frag20 can also be used to do active learning for uncertainty models including ensemble models42, 53 and Bayesian neural networks83–84. Frag20 provides both DFT and MMFF geometries so that it can be used to develop deep learning models for predicting molecular energies without the dependence on DFT optimized geometries. Furthermore, Frag20 can be used as the basis to develop new molecular datasets to predict other molecular properties, such as solvation effects and molecular spectroscopies. Besides Frag20, we also constructed Plati20 and CSD20 datasets, which are based on protein-bound ligand molecules from Platinum dataset72 and crystal structures from Cambridge Structure Database (CSD)73 respectively, to evaluate model’s generalization performance in potential real applications.
In this work, we have also reimplemented PhysNet, a state-of-the-art deep learning model to predict molecular properties with 3D geometries, with PyTorch. By modifying its model architecture and hyperparameters, we found a simplified PhysNet (sPhysNet), which reduced trainable parameters by about 40%, nearly doubled the training speed while yielded the similar performance in comparison with the original PhysNet model. The sPhysNet model can achieve 0.19 kcal/mol MAE on QM9 and 0.35 kcal/mol MAE on QM9M with fine-tuning, which has significantly improved over our previously developed DTNN_7ib model (0.34 kcal/mol MAE on QM9 and 0.79 kcal/mol MAE on QM9M with transfer learning). Finally, based on both Frag20 and eMol9 datasets, we developed the sPhysNet model to predict molecular energies for MMFF-optimized geometries, which achieved 0.63 kcal/mol, 1.36 kcal/mol and 1.40 kcal/mol respectively for Frag20, CSD20 and Plati20 test sets. Our work further demonstrated that it is a promising direction to develop deep learning models to predict molecular energies with force field based geometries, which would facilitate the efficient exploration of chemical space with 3D geometries.
Supplementary Material
Table 5. (B).
sPhysNet Performance on Plati20 (Training Set Size is ~590k).
| DFT-optimized | MMFF-optimized | ||
|---|---|---|---|
| ErrorA | MAE (kcal/mol) | 0.72 | 1.40 |
| RMSE (kcal/mol) | 1.01 | 2.09 | |
| Error > 1kcal/mol | 26.67% | 47.34% | |
| Error > 10kcal/mol | 0% | 0.25% | |
| ErrorR | MAE (kcal/mol) | 0.41 | 0.80 |
| RMSE (kcal/mol) | 0.50 | 1.00 | |
| Success Rate | 67.49% | 53.10% |
ACKNOWLEDGMENTS
We would like to acknowledge the support by NIH (R35-GM127040) and computing resources provided by NYU-ITS.
Footnotes
Supporting Information
The Supporting Information is available free of charge at:
Figure S1-S5, Tables S1-S2 (PDF).
REFERENCES
- 1.Rossi M; Chutia S; Scheffler M; Blum V Validation Challenge of Density-Functional Theory for Peptides—Example of Ac-Phe-Ala 5 -LysH +. J Phys Chem 2014, 118, 7349–7359. 10.1021/jp412055r. [DOI] [PubMed] [Google Scholar]
- 2.Hawkins PCD Conformation Generation: The State of the Art. J Chem Inf Model 2017, 57, 1747–1756. 10.1021/acs.jcim.7b00221. [DOI] [PubMed] [Google Scholar]
- 3.Halgren TA Merck Molecular Force Field. II. MMFF94 van Der Waals and Electrostatic Parameters for Intermolecular Interactions. J Comput Chem 1996, 17, 520–552. . [DOI] [Google Scholar]
- 4.Halgren TA Merck Molecular Force Field. II. MMFF94 van Der Waals and Electrostatic Parameters for Intermolecular Interactions. J Comput Chem 1996, 17, 520–552. . [DOI] [Google Scholar]
- 5.Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-atom Additive Biological Force Fields. J Comput Chem 2010, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramakrishnan R; Dral PO; Rupp M; Lilienfeld O. A. von. Big Data Meets Quantum Chemistry Approximations: The Δ-Machine Learning Approach. J Chem Theory Comput 11, 2087–2096. 10.1021/acs.jctc.5b00099. [DOI] [PubMed] [Google Scholar]
- 7.Bartók AP; Kondor R; Csányi G Publisher’s Note: On Representing Chemical Environments [Phys. Rev. B 87, 184115 (2013)]. Phys Rev B 2013, 87, 219902. 10.1103/physrevb.87.219902. [DOI] [Google Scholar]
- 8.Bartók AP; Payne MC; Kondor R; Csányi G Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons. Phys Rev Lett 2010, 104, 136403. 10.1103/physrevlett.104.136403. [DOI] [PubMed] [Google Scholar]
- 9.Behler J Atom-Centered Symmetry Functions for Constructing High-Dimensional Neural Network Potentials. J Chem Phys 2011, 134, 074106. 10.1063/1.3553717. [DOI] [PubMed] [Google Scholar]
- 10.Behler J; Parrinello M Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys Rev Lett 2007, 98, 146401. 10.1103/physrevlett.98.146401. [DOI] [PubMed] [Google Scholar]
- 11.Brockherde F; Vogt L; Li L; Tuckerman ME; Burke K; Müller K-R Bypassing the Kohn-Sham Equations with Machine Learning. Nat Commun 2017, 8, 872. 10.1038/s41467-017-00839-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butler KT; Davies DW; Cartwright H; Isayev O; Walsh A Machine Learning for Molecular and Materials Science. Nature 2018, 559, 547–555. 10.1038/s41586-018-0337-2. [DOI] [PubMed] [Google Scholar]
- 13.Chmiela S; Sauceda HE; Müller K-R; Tkatchenko A Towards Exact Molecular Dynamics Simulations with Machine-Learned Force Fields. Nat Commun 2018, 9, 3887. 10.1038/s41467-018-06169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chmiela S; Tkatchenko A; Sauceda HE; Poltavsky I; Schütt KT; Müller K-R Machine Learning of Accurate Energy-Conserving Molecular Force Fields. Sci Adv 2017, 3, e1603015. 10.1126/sciadv.1603015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eickenberg M; Exarchakis G; Hirn M; Mallat S Solid Harmonic Wavelet Scattering: Predicting Quantum Molecular Energy from Invariant Descriptors of 3D Electronic Densities. 31st Conference on Neural Information Processing System 2017, 6543–6552. [Google Scholar]
- 16.Eickenberg M; Exarchakis G; Hirn M; Mallat S; Thiry L Solid Harmonic Wavelet Scattering for Predictions of Molecule Properties. J Chem Phys 2018, 148, 241732. 10.1063/1.5023798. [DOI] [PubMed] [Google Scholar]
- 17.Faber FA; Christensen AS; Huang B; Lilienfeld OA von. Alchemical and Structural Distribution Based Representation for Universal Quantum Machine Learning. J Chem Phys 2018, 148, 241717. 10.1063/1.5020710. [DOI] [PubMed] [Google Scholar]
- 18.Faber FA; Hutchison L; Huang B; Gilmer J; Schoenholz SS; Dahl GE; Vinyals O; Kearnes S; Riley PF; Lilienfeld O. A. von. Prediction Errors of Molecular Machine Learning Models Lower than Hybrid DFT Error. J Chem Theory Comput 2017, 13, 5255–5264. 10.1021/acs.jctc.7b00577. [DOI] [PubMed] [Google Scholar]
- 19.Faber FA; Lindmaa A; Lilienfeld OA von; Armiento, R. Machine Learning Energies of 2 Million Elpasolite (ABC2D6) Crystals. Phys Rev Lett 2016, 117, 135502. 10.1103/physrevlett.117.135502. [DOI] [PubMed] [Google Scholar]
- 20.Ferré G; Haut T; Barros K Learning Molecular Energies Using Localized Graph Kernels. J Chem Phys 2017, 146, 114107. 10.1063/1.4978623. [DOI] [PubMed] [Google Scholar]
- 21.Faber FA; Christensen AS; Huang B; Lilienfeld OA von. Alchemical and Structural Distribution Based Representation for Universal Quantum Machine Learning. J Chem Phys 2018, 148, 241717. 10.1063/1.5020710. [DOI] [PubMed] [Google Scholar]
- 22.Han J; Zhang L; Car R; E W Deep Potential: A General Representation of a Many-Body Potential Energy Surface 2017, arXiv:1707.09571. arXiv.org e-Print archive. https://arxiv.org/abs/1707.01478 [Google Scholar]
- 23.Hansen K; Biegler F; Ramakrishnan R; Pronobis W; von Lilienfeld OA; Müller K-R; Tkatchenko A Machine Learning Predictions of Molecular Properties: Accurate Many-Body Potentials and Nonlocality in Chemical Space. J Phys Chem Lett 2015, 6, 2326–2331. 10.1021/acs.jpclett.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen K; Montavon G; Biegler F; Fazli S; Rupp M; Scheffler M; Lilienfeld O. A. von; Tkatchenko A; Müller K-R Assessment and Validation of Machine Learning Methods for Predicting Molecular Atomization Energies. J Chem Theory Comput 2013, 9, 3404–3419. 10.1021/ct400195d. [DOI] [PubMed] [Google Scholar]
- 25.Hansen K; Biegler F; Ramakrishnan R; Pronobis W; von Lilienfeld OA; Müller K-R; Tkatchenko A Machine Learning Predictions of Molecular Properties: Accurate Many-Body Potentials and Nonlocality in Chemical Space. J Phys Chem Lett 2015, 6, 2326–2331. 10.1021/acs.jpclett.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huo H; Rupp M Unified Representation of Molecules and Crystals for Machine Learning 2017. arXiv:1704.06439. arXiv.org e-Print archive. https://arxiv.org/abs/1704.06439 [Google Scholar]
- 27.Jørgensen PB; Jacobsen KW; Schmidt MN Neural Message Passing with Edge Updates for Predicting Properties of Molecules and Materials 2018. arXiv:1806.03146. arXiv.org e-Print archive. https://arxiv.org/abs/1806.03146 [Google Scholar]
- 28.Lubbers N; Smith JS; Barros K Hierarchical Modeling of Molecular Energies Using a Deep Neural Network. J Chem Phys 2018, 148, 241715. 10.1063/1.5011181. [DOI] [PubMed] [Google Scholar]
- 29.Mills K; Ryczko K; Luchak I; Domurad A; Beeler C; Tamblyn I Extensive Deep Neural Networks for Transferring Small Scale Learning to Large Scale Systems. Chem Sci 2019, 10, 4129–4140. 10.1039/c8sc04578j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montavon G; Rupp M; Gobre V; Vazquez-Mayagoitia A; Hansen K; Tkatchenko A; Müller K-R; Lilienfeld O. A. von. Machine Learning of Molecular Electronic Properties in Chemical Compound Space. New J Phys 2013, 15, 095003. 10.1088/1367-2630/15/9/095003. [DOI] [Google Scholar]
- 31.Podryabinkin EV; Shapeev AV Active Learning of Linearly Parametrized Interatomic Potentials. Comp Mater Sci 2017, 140, 171–180. 10.1016/j.commatsci.2017.08.031. [DOI] [Google Scholar]
- 32.Pronobis W; Tkatchenko A; Müller K-R Many-Body Descriptors for Predicting Molecular Properties with Machine Learning: Analysis of Pairwise and Three-Body Interactions in Molecules. J Chem Theory Comput 2018, 14, 2991–3003. 10.1021/acs.jctc.8b00110. [DOI] [PubMed] [Google Scholar]
- 33.Rowe P; Csányi G; Alfè D; Michaelides A Development of a Machine Learning Potential for Graphene. Phys Rev B 2018, 97, 054303. 10.1103/physrevb.97.054303. [DOI] [Google Scholar]
- 34.Rupp M; Tkatchenko A; Müller K-R; Lilienfeld O. A. von. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning. Phys Rev Lett 2012, 108, 058301. 10.1103/physrevlett.108.058301. [DOI] [PubMed] [Google Scholar]
- 35.Ryczko K; Mills K; Luchak I; Homenick C; Tamblyn I Convolutional Neural Networks for Atomistic Systems. Comp Mater Sci 2018, 149, 134–142. 10.1016/j.commatsci.2018.03.005. [DOI] [Google Scholar]
- 36.Schütt KT; Arbabzadah F; Chmiela S; Müller KR; Tkatchenko A Quantum-Chemical Insights from Deep Tensor Neural Networks. Nat Commun 2017, 8, 13890. 10.1038/ncomms13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schütt KT; Glawe H; Brockherde F; Sanna A; Müller KR; Gross EKU How to Represent Crystal Structures for Machine Learning: Towards Fast Prediction of Electronic Properties. Phys Rev B 2014, 89, 205118. 10.1103/physrevb.89.205118. [DOI] [Google Scholar]
- 38.Lu J; Wang C; Zhang Y Predicting Molecular Energy Using Force-Field Optimized Geometries and Atomic Vector Representations Learned from an Improved Deep Tensor Neural Network. J Chem Theory Comput 2019, 15, 4113–4121. 10.1021/acs.jctc.9b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shapeev AV Moment Tensor Potentials: A Class of Systematically Improvable Interatomic Potentials. Multiscale Model Sim 2016, 14, 1153–1173. 10.1137/15m1054183. [DOI] [Google Scholar]
- 40.Sinitskiy AV; Pande VS Deep Neural Network Computes Electron Densities and Energies of a Large Set of Organic Molecules Faster than Density Functional Theory (DFT) 2018. arXiv:1809.02723. arXiv.org e-Print archive. https://arxiv.org/abs/1809.02723 [Google Scholar]
- 41.Smith JS; Isayev O; Roitberg AE ANI-1: An Extensible Neural Network Potential with DFT Accuracy at Force Field Computational Cost. Chem Sci 2017, 8, 3192–3203. 10.1039/c6sc05720a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith JS; Nebgen B; Lubbers N; Isayev O; Roitberg AE Less Is More: Sampling Chemical Space with Active Learning. J Chem Phys 2018, 148, 241733. 10.1063/1.5023802. [DOI] [PubMed] [Google Scholar]
- 43.Smith JS; Nebgen BT; Zubatyuk R; Lubbers N; Devereux C; Barros K; Tretiak S; Isayev O; Roitberg AE Approaching Coupled Cluster Accuracy with a General-Purpose Neural Network Potential through Transfer Learning. Nat Commun 2019, 10, 2903. 10.1038/s41467-019-10827-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith JS; Roitberg AE; Isayev O Transforming Computational Drug Discovery with Machine Learning and AI. Acs Med Chem Lett 2018, 9, 1065–1069. 10.1021/acsmedchemlett.8b00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsubaki M; Mizoguchi T Fast and Accurate Molecular Property Prediction: Learning Atomic Interactions and Potentials with Neural Networks. J Phys Chem Lett 2018, 9, 5733–5741. 10.1021/acs.jpclett.8b01837. [DOI] [PubMed] [Google Scholar]
- 46.Unke OT; Meuwly M A Reactive, Scalable, and Transferable Model for Molecular Energies from a Neural Network Approach Based on Local Information. J Chem Phys 2018, 148, 241708. 10.1063/1.5017898. [DOI] [PubMed] [Google Scholar]
- 47.Unke OT; Meuwly M PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges. J Chem Theory Comput 2019, 15, 3678–3693. 10.1021/acs.jctc.9b00181. [DOI] [PubMed] [Google Scholar]
- 48.Unke OT; Meuwly M PhysNet: A Neural Network for Predicting Energies, Forces, Dipole Moments, and Partial Charges. J Chem Theory Comput 2019, 15, 3678–3693. 10.1021/acs.jctc.9b00181. [DOI] [PubMed] [Google Scholar]
- 49.Wang R Significantly Improving the Prediction of Molecular Atomization Energies by an Ensemble of Machine Learning Algorithms and Rescanning Input Space: A Stacked Generalization Approach. J Phys Chem C 2018, 122, 8868–8873. 10.1021/acs.jpcc.8b03405. [DOI] [Google Scholar]
- 50.Yao K; Herr JE; Toth DW; Mckintyre R; Parkhill J The TensorMol-0.1 Model Chemistry: A Neural Network Augmented with Long-Range Physics. Chem Sci 2018, 9, 2261–2269. 10.1039/c7sc04934j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L; Han J; Wang H; Car R; E W Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics. Phys Rev Lett 2018, 120, 143001. 10.1103/physrevlett.120.143001. [DOI] [PubMed] [Google Scholar]
- 52.Zhang L; Han J; Wang H; Saidi WA; Car R; E W End-to-End Symmetry Preserving Inter-Atomic Potential Energy Model for Finite and Extended Systems 2018. arXiv:1805.09003. arXiv.org e-Print archive. https://arxiv.org/abs/1805.09003 [Google Scholar]
- 53.Zhang L; Lin D-Y; Wang H; Car R; E W Active Learning of Uniformly Accurate Interatomic Potentials for Materials Simulation. Phys Rev Mater 2019, 3, 023804. 10.1103/physrevmaterials.3.023804. [DOI] [Google Scholar]
- 54.Zubatyuk R; Smith JS; Leszczynski J; Isayev O Accurate and Transferable Multitask Prediction of Chemical Properties with an Atoms-in-Molecules Neural Network. Sci Adv 2019, 5, eaav6490. 10.1126/sciadv.aav6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pronobis W; Schütt KT; Tkatchenko A; Müller K-R Capturing Intensive and Extensive DFT/TDDFT Molecular Properties with Machine Learning. European Phys J B 2018, 91, 178. 10.1140/epjb/e2018-90148-y. [DOI] [Google Scholar]
- 56.Klicpera J; Groß J; Günnemann S Directional Message Passing for Molecular Graphs 2020. arXiv:2003.03123. arXiv.org e-Print archive. https://arxiv.org/abs/2003.03123 [Google Scholar]
- 57.LeCun Y; Bengio Y; Hinton G Deep Learning. Nature 2015, 521, 436–444. 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 58.Blum LC; Reymond J-L 970 Million Druglike Small Molecules for Virtual Screening in the Chemical Universe Database GDB-13. J Am Chem Soc 2009, 131, 8732–8733. 10.1021/ja902302h. [DOI] [PubMed] [Google Scholar]
- 59.Ramakrishnan R; Dral PO; Rupp M; Lilienfeld O. A. von. Quantum Chemistry Structures and Properties of 134 Kilo Molecules. Sci Data 2014, 1, 140022. 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reymond J-L The Chemical Space Project. Accounts Chem Res 2015, 48, 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- 61.Smith JS; Zubatyuk R; Nebgen B; Lubbers N; Barros K; Roitberg AE; Isayev O; Tretiak S The ANI-1ccx and ANI-1x Data Sets, Coupled-Cluster and Density Functional Theory Properties for Molecules. Sci Data 2020, 7, 134. 10.1038/s41597-0200473-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montavon G; Rupp M; Gobre V; Vazquez-Mayagoitia A; Hansen K; Tkatchenko A; Müller K-R; Lilienfeld O. A. von. Machine Learning of Molecular Electronic Properties in Chemical Compound Space. New J Phys 2013, 15, 095003. 10.1088/1367-2630/15/9/095003. [DOI] [Google Scholar]
- 63.Rupp M; Tkatchenko A; Müller K-R; Lilienfeld OA von. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning. Phys Rev Lett 2012, 108, 058301. 10.1103/physrevlett.108.058301. [DOI] [PubMed] [Google Scholar]
- 64.Ramakrishnan R; Hartmann M; Tapavicza E; Lilienfeld O. A. von. Electronic Spectra from TDDFT and Machine Learning in Chemical Space. J Chem Phys 2015, 143, 084111. 10.1063/1.4928757. [DOI] [PubMed] [Google Scholar]
- 65.Smith JS; Isayev O; Roitberg AE ANI-1, A Data Set of 20 Million Calculated off-Equilibrium Conformations for Organic Molecules. Sci Data 2017, 4, 170193. 10.1038/sdata.2017.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruddigkeit L; Deursen R. van; Blum LC; Reymond J-L Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J Chem Inf Model 2012, 52, 2864–2875. 10.1021/ci300415d. [DOI] [PubMed] [Google Scholar]
- 67.Glavatskikh M; Leguy J; Hunault G; Cauchy T; Mota BD Dataset’s Chemical Diversity Limits the Generalizability of Machine Learning Predictions. J Cheminformatics 2019, 11, 69. 10.1186/s13321-019-0391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Irwin JJ; Shoichet BK ZINC — A Free Database of Commercially Available Compounds for Virtual Screening. Cheminform 2005, 36. 10.1002/chin.200516215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Irwin JJ; Sterling T; Mysinger MM; Bolstad ES; Coleman RG ZINC: A Free Tool to Discover Chemistry for Biology. J Chem Inf Model 2012, 52, 1757–1768. 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim S; Chen J; Cheng T; Gindulyte A; He J; He S; Li Q; Shoemaker BA; Thiessen PA; Yu B; Zaslavsky L; Zhang J; Bolton EE PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res 2018, 47, gky1033. 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bemis GW; Murcko MA The Properties of Known Drugs. 1. Molecular Frameworks. J Med Chem 1996, 39, 2887–2893. 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 72.Friedrich N-O; Meyder A; Kops C. de B.; Sommer K; Flachsenberg F; Rarey M; Kirchmair J High-Quality Dataset of Protein-Bound Ligand Conformations and Its Application to Benchmarking Conformer Ensemble Generators. J Chem Inf Model 2017, 57, 529–539. 10.1021/acs.jcim.6b00613. [DOI] [PubMed] [Google Scholar]
- 73.Groom CR; Bruno IJ; Lightfoot MP; Ward SC The Cambridge Structural Database. Acta Crystallogr Sect B Struct Sci Cryst Eng Mater 2016, 72, 171–179. 10.1107/s2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.eMolecules. https://www.emolecules.com/. (accessed Oct 2017).
- 75.Sterling T; Irwin JJ ZINC 15 – Ligand Discovery for Everyone. J Chem Inf Model 2015, 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Winter R; Montanari F; Noé F; Clevert D-A Learning Continuous and Data-Driven Molecular Descriptors by Translating Equivalent Chemical Representations. Chem Sci 2018, 10, 1692–1701. 10.1039/c8sc04175j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.The RDKit: Open-Source Cheminformatics Software. http://www.rdkit.org (accessed Mar 2019).
- 78.Riniker S; Landrum GA Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J Chem Inf Model 2015, 55, 2562–2574. 10.1021/acs.jcim.5b00654. [DOI] [PubMed] [Google Scholar]
- 79.Frisch MJT,GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Gaussian Inc.: Wallingford, CT, 2009. [Google Scholar]
- 80.Butina D Unsupervised Data Base Clustering Based on Daylight’s Fingerprint and Tanimoto Similarity: A Fast and Automated Way To Cluster Small and Large Data Sets. J Chem Inf Comp Sci 1999, 39, 747–750. 10.1021/ci9803381. [DOI] [Google Scholar]
- 81.Temelso B; Mabey JM; Kubota T; Appiah-Padi N; Shields GC ArbAlign: A Tool for Optimal Alignment of Arbitrarily Ordered Isomers Using the Kuhn–Munkres Algorithm. J Chem Inf Model 2017, 57, 1045–1054. 10.1021/acs.jcim.6b00546. [DOI] [PubMed] [Google Scholar]
- 82.Schütt KT; Sauceda HE; Kindermans P-J; Tkatchenko A; Müller K-R SchNet – A Deep Learning Architecture for Molecules and Materials. J Chem Phys 2018, 148, 241722. 10.1063/1.5019779. [DOI] [PubMed] [Google Scholar]
- 83.Ryu S; Kwon Y; Kim WY A Bayesian Graph Convolutional Network for Reliable Prediction of Molecular Properties with Uncertainty Quantification. Chem Sci 2019, 10, 8438–8446. 10.1039/c9sc01992h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y; Lee AA Bayesian Semi-Supervised Learning for Uncertainty-Calibrated Prediction of Molecular Properties and Active Learning. Chem Sci 2019, 10, 8154–8163. 10.1039/c9sc00616h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Paszke A; Gross S; Massa F; Lerer A; Bradbury J; Chanan G; Killeen T; Lin Z; Gimelshein N; Antiga L; Desmaison A; Köpf A; Yang E; DeVito Z; Raison M; Tejani A; Chilamkurthy S; Steiner B; Fang L; Bai J; Chintala S PyTorch: An Imperative Style, High-Performance Deep Learning Library 2019. arXiv:1912.01703. arXiv.org e-Print archive. https://arxiv.org/abs/1912.01703 [Google Scholar]
- 86.Abadi M; Agarwal A; Barham P; Brevdo E; Chen Z; Citro C; Corrado GS; Davis A; Dean J; Devin M; Ghemawat S; Goodfellow I; Harp A; Irving G; Isard M; Jia Y; Jozefowicz R; Kaiser L; Kudlur M; Levenberg J; Mane D; Monga R; Moore S; Murray D; Olah C; Schuster M; Shlens J; Steiner B; Sutskever I; Talwar K; Tucker P; Vanhoucke V; Vasudevan V; Viegas F; Vinyals O; Warden P; Wattenberg M; Wicke M; Yu Y; Zheng X TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems 2016. arXiv:1603.04467. arXiv.org e-Print archive. https://arxiv.org/abs/1603.04467 [Google Scholar]
- 87.Reddi SJ; Kale S; Kumar S On the Convergence of Adam and Beyond 2019. arXiv:1904.09237. arXiv.org e-Print archive. https://arxiv.org/abs/1904.09237 [Google Scholar]
- 88.Ertl P An Algorithm to Identify Functional Groups in Organic Molecules. J Cheminformatics 2017, 9, 36. 10.1186/s13321-017-0225-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Exponential Moving Average from TensorFlow (We reimplemented it in PyTorch). https://www.tensorflow.org/api_docs/python/tf/train/ExponentialMovingAverage (accessed Jan 2021) [Google Scholar]
- 90.Gilmer J; Schoenholz SS; Riley PF; Vinyals O; Dahl GE Neural Message Passing for Quantum Chemistry 2017. arXiv:1704.01212. arXiv.org e-Print archive. https://arxiv.org/abs/1704.01212 [Google Scholar]
- 91.John P. C. St.; Guan Y; Kim Y; Etz BD; Kim S; Paton RS Quantum Chemical Calculations for over 200,000 Organic Radical Species and 40,000 Associated Closed-Shell Molecules. Sci Data 2020, 7, 244. 10.1038/s41597-020-00588-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.