

Abstract
Historically, primordial germ cells (PGCs) have been a good model to study pluripotency. Despite their low numbers and limited accessibility in the mouse embryo, they can be easily and rapidly reprogrammed at high efficiency with external physicochemical factors and do not require transcription factor transfection. Employing this model to deepen our understanding of cell reprogramming, we specifically aimed to determine the relevance of Ca2+ signal transduction pathway components in the reprogramming process. Our results showed that PGC reprogramming requires a normal extracellular [Ca2+] range, in contrast to neoplastic or transformed cells, which can continue to proliferate in Ca2+-deficient media, differentiating normal reprogramming from neoplastic transformation. Our results also showed that a spike in extracellular [Ca2+] of 1-3 mM can directly reprogram PGC. Intracellular manipulation of Ca2+ signal transduction pathway components revealed that inhibition of classical Ca2+ and diacylglycerol (DAG)-dependent PKCs, or intriguingly, of only the novel DAG-dependent PKC, PKCε, were able to induce reprogramming. PKCε inhibition changed the metabolism of PGCs toward glycolysis, increasing the proportion of inactive mitochondria. This metabolic switch from oxidative phosphorylation to glycolysis is mediated by hypoxia-inducible factors (HIFs), given we found upregulation of both HIF1α and HIF2α in the first 48 hours of culturing. PKCε inhibition did not change the classical pluripotency gene expression of PGCs, Oct4, or Nanog. PKCε inhibition changed the histone acetylation of PGCs, with histones H2B, H3, and H4 becoming acetylated in PKCε-inhibited cultures (markers were H2BacK20, H3acK9, and H4acK5K8, K12, K16), suggesting that reprogramming by PKCε inhibition is mediated by histone acetylation.
Keywords: Cellular reprograming, primordial germ cells, pluripotency, calcium signaling, PKC, HIF, histone acetylation
Introduction
Primordial germ cells (PGCs) are gamete precursors during embryonic life. As such, they retain some pluripotency clues, such as alkaline phosphatase activity, stage-specific embryonic antigens, and Lin28, Oct4, Sox2, and Nanog expression (for a review see [1]).
Historically, PGCs have been a good model to study pluripotency because, despite their low numbers (approximately 25 cells per 8.5 days post coitum [dpc] mouse embryo), they can easily be reprogrammed at high efficiency (typically 10% [2], and up to 20% with a combination of factors [3]), and with external physicochemical factors such as FGF2 [4-6], the mitogens retinoic acid and forskolin [7,8], the epigenetic modulator trichostatin A [9], mitogen-activated protein kinase/extracellular-signal-regulated kinase (MAP/ERK) and GSK3 inhibitors [10], and more recently by hypoxia (hypoxia-induced EG-like cells, hi-EGL [2]). Each of these factors alone reprograms unipotent PGCs to pluripotent embryonic germ cells (EGCs), which are similar to embryonic stem cells (ESCs), and induced pluripotent stem cells (iPSCs), with minor differences such as epigenetic state [1,11,12]. Another advantage is that PGC reprogramming is faster than that of iPSC derivation, taking only about a week to complete [6]. Accordingly, using this model to deepen our understanding of cell reprogramming has highlighted several important processes, such as the disconnection of self-renewal and pluripotency acquisition itself, a metabolic shift toward glycolysis having a direct role in reprogramming, the implications of histone acetylation, and the need for a reactive oxygen species (ROS) spike and autophagy for reprogramming [2,11,13].
In a previous study comparing the expression of pluripotent EGCs with PGCs by suppression subtractive hybridization and microarray analysis [2] and National Center for Biotechnology Information Gene Expression Omnibus database depository, accessible through GEO Series accession number GSE61472, we found differences in several genes related to calcium signaling; thus, in our study, we aimed to deepen our understanding of the relevance of Ca2+ signal transduction pathway components in the reprogramming process.
Materials and methods
PGC isolation and culture conditions
We used 8.5-days post coitum (dpc) embryos from Oct4-green fluorescent protein (GFP) transgenic mice of the C57BL/6 strain for this study. The isolation and culture of the PGCs were performed as described previously [2]. Briefly, PGCs were cultured onto a confluent monolayer of nutritious mitomycin C-treated STO cells (Sandoz Thioguanine- and Ouabain-resistant cells, immortalized mouse embryonic fibroblasts), in Dulbecco’s Modified Eagle Medium (DMEM) with 15% embryonic stem cell-qualified fetal bovine serum (FBS) and supplemented with leukemia inhibitory factor (LIF)(Millipore) and stem cell factor (SCF) (R&D Systems), which are essential for PGC survival and proliferation but do not induce reprogramming. As a positive control for reprogramming, either fibroblast growth factor-2 (FGF2) at 1 ng/ml or 3% O2 were used, Cultures were exposed daily to calcium signaling-related soluble factors. Soluble factors were added in decreasing logarithmic concentrations according to the published literature to establish nontoxic ranges. Factors and final concentrations used were as follows: ionomycin at 0.5 µM, flunarizine at 4 µM, thapsigargin at 0.1 nM, trifluoperazine at 20 µM, KN-62 at 0.8 µM, cyclosporin A at 100 nM, staurosporine at 15 nM, BAPTA-AM at 0.1 µM, FR236924 at 10 µM, Ro-31-8220 at 30 nM, and PKCε inhibitory peptide (PIP) at 10 nM. All factors were from SigmaAldrich and were added daily and continuously to the cultures except for trifluoperazine, which was added for 1 h/day and KN-62 for 30 min/day.
Cultures were also exposed to various extracellular Ca2+ concentrations, by using DMEM devoid of Ca2+ and dialyzed (Pierce, ThermoFisher) FBS, and then adding CaCl2 at the desired final concentrations.
Pluripotency and differentiation assays
To identify both isolated PGCs and pluripotent EGC colonies, tissue-nonspecific alkaline phosphatase (TNAP) staining was performed. Colonies of 8 or more cells were regarded as reprogrammed PGCs. A t-test was then performed between experimental conditions, with 3 technical replicates and at least 3 biological replicates. To demonstrate actual pluripotency, embryoid bodies (EBs) and 3 germ layer differentiations were obtained and demonstrated from reprogrammed PGCs as in [13], using the antibodies anti-albumin (Dako) for endoderm, anti-vimentin (Dako) for mesoderm, and anti-cytokeratins AE1/AE3 (Dako) for ectoderm demonstration by immunofluorescence.
Immunofluorescence
Immunofluorescence was performed after fixation using 4% paraformaldehyde at pH 7.4. PGCs were identified by labeling for stage-specific embryonic antigen-1 (SSEA1) (R&D Systems). Double staining together with molecules of interest was performed: hypoxia-inducible factor (HIF) 1α (Abcam), HIF2α (Abcam), Nanog (Abcam), Klf4 (R&D Systems), c-Myc (Santa Cruz Biotechnology), H2BacK20 (Abcam), H3acK9 (Abcam), and H4acK5K8, K12, K16 (Abcam). As controls, the mouse embryonic stem cell line E14Tg2a, monkey Cos7, and mouse NIH3T3 cell lines were used. Images were obtained by confocal microscopy.
Mitochondrial activity
To show mitochondrial activity, flow cytometry was performed after the addition of a mitochondrial JC-1 fluorescent probe (Life Technologies) and subsequent fixation as previously described [13] or with live cells to simultaneously detect Oct4-GFP levels. Cell sorting was also performed based on this design to isolate PGCs by SSEA-1 and measure mitochondrial activity by JC-1 fluorescence.
Gene expression analysis
Cell sorting was also performed to extract RNA for specific gene expression analysis, using the RNeasy Minikit (Qiagen). RNA was converted to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), which was then preamplified using TaqMan PreAmp Master Mix (Applied Biosystems) to reach enough of a sample before performing quantitative polymerase chain reaction (qPCR), using β-actin as the reference gene. Primers were CaMK2γ Forward 5’-3’: acatccaaacattgtgcgtct, Reverse 3’-5’: aaacaactcccctccggtaa; Ppp3cc Forward 5’-3’: accctgcagtttgtgaattttt, Reverse 3’-5’: gaaagccagttgcttggttc; Prkcε Forward 5’-3’: aagaaacaggaaacccctgac, Reverse 3’-5’: acttgtggggcatgttgac; and β-actin Forward 5’-3’: ctgtattcccctccatcgtg, Reverse 3’-5’: aggagtccttctgacccattc. The conditions for qPCR were 10 min at 95°C and then 10 cycles of 15 s at 95°C and 4 min at 60°C in a Biometra TPersonal thermal cycler.
Statistical analysis
Statistical significance in cultures was assessed by Student’s t-test with Fisher modification in at least 3 separate experiments, each performed in triplicate wells. EBs and spontaneous differentiation were performed at least 3 times. Flow cytometry results were assessed by a parametric F-Snedecor test analysis. Significance was considered at P ≤.05.
Results
Extracellular Ca2+ is necessary for PGC survival and reprogramming
Most cells require a strict Ca2+ level for various cellular functions, such as cell migration, invasion, survival, proliferation, differentiation, and apoptosis. As expected, PGC survival was Ca2+-concentration dependent, and PGCs did not survive on media containing less than 1 mM Ca2+ (Figure 1A). Similarly, PGCs were not able to reprogram by the classical method of FGF2 addition until the extracellular Ca2+ concentration reached 2 mM (Figure 1B).
Figure 1.
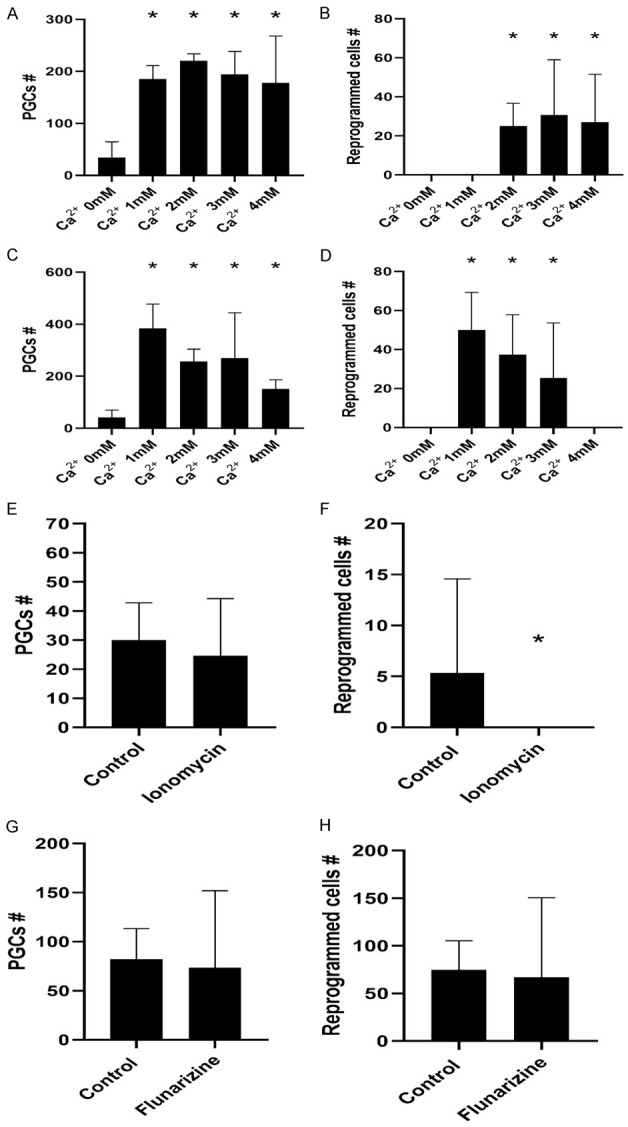
Number of PGCs (A, C, E, G) and reprogrammed EGCs (B, D, F, H) under various culture conditions. (A) Extracellular Ca2+ dose response in reprogramming condition cultures. At least 1 mM concentration is needed for PGC survival; (B) A minimum of 2 mM [Ca2+] is needed to allow for PGC reprogramming; (C) Extracellular Ca2+ dose response in non-reprogramming conditions. Again, at least 1 mM concentration is needed for PGC survival; (D) 1 mM to 3 mM [Ca2+] is enough to reprogram PGCs in non-reprogramming conditions (absence of FGF2); (E) No differences were found in PGC numbers in the presence of ionomycin, demonstrating it is neither toxic nor induces augmented proliferation in cultures at this concentration; (F) Ionomycin prevents PGC reprogramming by a statistically significant reduction in reprogrammed EGC colonies; (G) No differences were found in PGC numbers by the addition of flunarizine, demonstrating that it is neither toxic nor induces augmented proliferation in cultures at this concentration; (H) Flunarizine does not have an effect on PGC reprogramming. An asterisk represents a statistically significant p value < .05.
Extracellular Ca2+ concentrations 1-3 mM reprogram PGCs in the absence of FGF2
Surprisingly, the addition of Ca2+ to the extracellular media in concentrations 1-3 mM reprogrammed PGCs even in the absence of FGF2 (Figure 1C), without affecting PGC survival (Figure 1D). Higher concentrations negatively affected both PGC survival and reprogramming (Figure 1C, 1D).
Ca2+ flux from extracellular to intracellular space prevents reprogramming, whereas blocking Ca2+ entry does not affect reprogramming efficiency
To ascertain whether these narrow Ca2+ concentrations in the medium for reprogramming correlated with the intracellular Ca2+ requirements, we subjected the cultures in reprogramming conditions to ionomycin, a cell membrane permeabilizer that allows Ca2+ flux into the cell; and to the opposite, flunarizine, an antagonist to type T Ca2+/Na+ channels, thus preventing Ca2+ entry.
Ionomycin did not affect PGC survival at 0, 5 µM (Figure 1E), but it did prevent PGC reprogramming (Figure 1F), again suggesting that high intracellular free Ca2+ prevents reprogramming. At higher concentrations, ionomycin was toxic for PGCs (not shown). However, the addition of flunarizine did not affect reprogramming efficiency (Figure 1G, 1H), suggesting that enough Ca2+ had been stored in intracellular compartments in regular media and thus could be released for reprogramming.
Ca2+ release from the endoplasmic reticulum might play a role in reprogramming
To confirm this hypothesis, cultures were exposed to thapsigargin, a compound that liberates Ca2+ from the endoplasmic reticulum, and BAPTA, an intracellular Ca2+ chelator that restricts intracellular free Ca2+ availability. Thapsigargin was toxic for PGCs at 10 nM and at 1 nM (not shown), suggesting that high intracellular Ca2+ levels are toxic for PGCs. At a concentration of 0.1 nM, results were inconsistent with half of the experiments showing reprogramming (Figure S1A, S1B). In reprogramming conditions, thapsigargin did not have any effect (Figure S1A, S1B). In normal range Ca2+ concentration, BAPTA did not affect PGC survival or reprogramming at concentrations between 0.1 and 10 µM (Figure S1C, S1D). These data agree with the increased expression of Atp2a2 in reprogrammed hiEGLs [2] and suggest that endoplasmic reticulum-released Ca2+ levels must be exquisitely fine-tuned for reprogramming.
Ca2+/calmodulin signal transduction pathway inhibition is not implicated in PGC reprogramming
To ascertain the implication of the Ca2+/calmodulin signal transduction family in PGC reprogramming, we inhibited the pathway at various levels: at the calmodulin protein with trifluoperazine, at the calmodulin kinase II with KN-62, and at the calcineurin with ciclosporin A. None of these compounds affected reprogramming efficiency (Figure S2A-F), suggesting that inhibition of this pathway is not implicated. In accordance with these results, quantitative reverse transcription-polymerase chain reaction (qRT-PCR) comparison between PGCs and pluripotent hiEGL levels of Camk2γ and Ppp3cc were only slightly upregulated in hiEGLs (Figure S2G).
Inhibition of the PKC pathways is implicated in PGC reprogramming
To implicate protein kinase C (PKC) in reprogramming, we subjected the cultures to staurosporine, an inhibitor of a wide spectrum of PKCs (all 3 types, conventional isoforms [α, βI, βII and γ], novel isoforms [PKC δ, ε, η, µ and θ], and atypical isoforms [ζ, λ and ι]). Inhibition of PKCs induced PGC reprogramming in the absence of FGF2 (Figure 2A, 2B).
Figure 2.
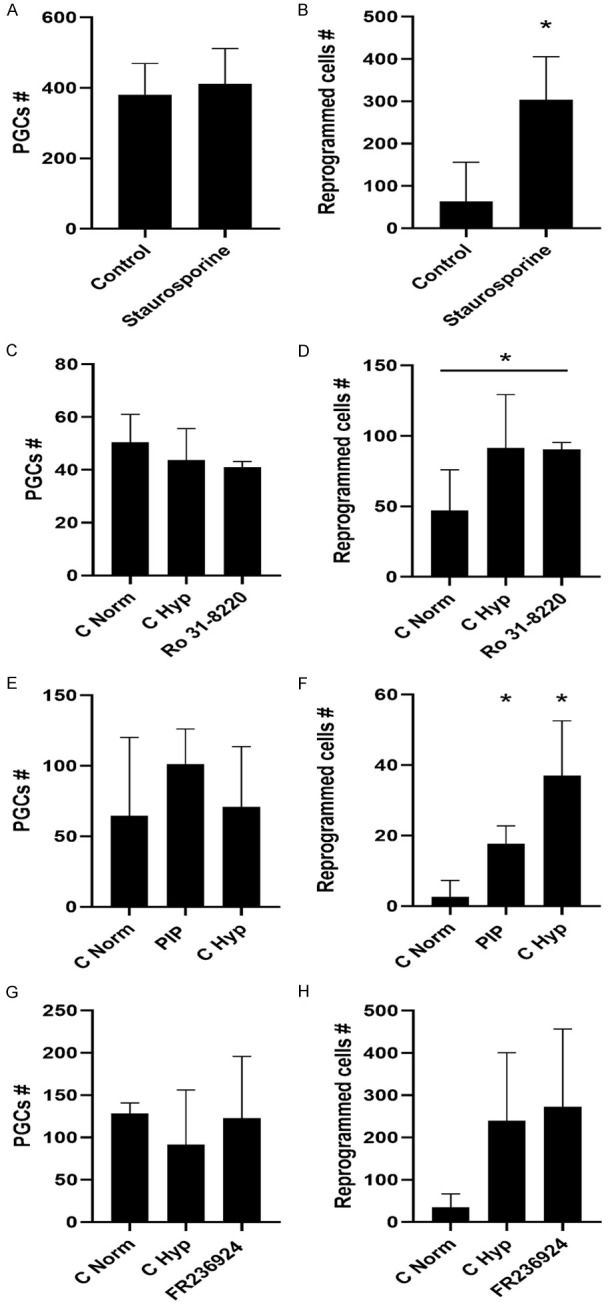
Number of PGCs (A, C, E, G) and reprogrammed EGCs (B, D, F, H) under non-reprogramming conditions (except for G&H) and different pharmacological supplementation. (A, C, E, G) No differences were found in PGC numbers in the presence of staurosporine, Ro-31-8220, PIP, and FR236924, demonstrating that these compounds are neither toxic nor induce augmented proliferation in cultures at the concentrations employed. (B) Staurosporine alone induces PGC reprogramming, by a statistically significant increase in reprogrammed EGC colonies. (D) Ro-31-8220 alone induces PGC reprogramming, by a statistically significant increase in reprogrammed EGC colonies. (F) PIP alone induces PGC reprogramming, by a statistically significant increase in reprogrammed EGC colonies. (H) FR236924 does not have an effect on PGC reprogramming under hypoxic conditions. An asterisk represents a statistically significant p value < .05.
To discern between the various types of PKCs, cultures were exposed to Ro-31-8220, an inhibitor of conventional PKCs (α, βI, βII, γ; Ca2+ and diacylglycerol [DAG]-dependent). Inhibition of such PKCs reprogrammed PGCs (Figure 2C, 2D). In parallel, to ascertain the role of DAG-dependent novel PKCs (δ, ε, η, µ, θ), cultures were exposed to PIP, an inhibitor of the translocation of PKCε from the Golgi to the cytoplasm, thus preventing its binding to DAG and hence its activation. This inhibitor was capable of PGC reprogramming in the absence of FGF2 (Figure 2E, 2F). When PKCε was activated by FR236924 in reprogramming conditions, no effect was observed (Figure 2G, 2H). Accordingly, expression of Prkce was lower in reprogrammed hiEGLs with respect to PGCs by qRT-PCR (Figure S2G). These data indicate that both conventional and novel PKCs might play a role in cell reprogramming.
The fact that a Ca2+-independent PKC isoform inhibition was able to reprogram PGCs was very intriguing Given PKCε was already identified as downregulated in reprogrammed PGCs by microarray analysis and validated by qRT-PCR (Figure S2G), we pursued its role in PGC reprogramming. Colonies generated by PKCε inhibition were actually pluripotent, as demonstrated by EB formation and differentiation into the 3 germ layers: endoderm, mesoderm, and ectoderm (Figure 3).
Figure 3.
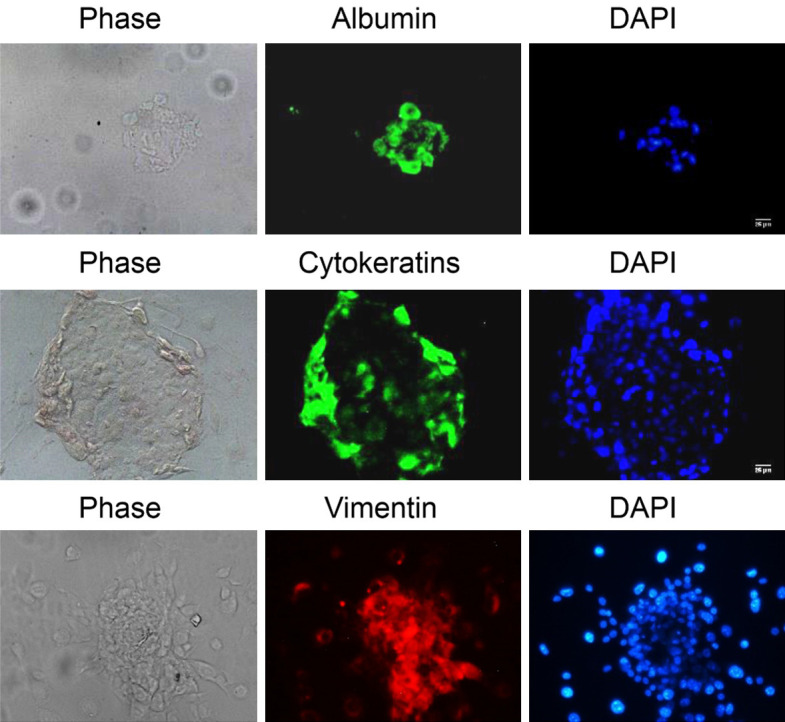
EB formation and spontaneous differentiation of reprogrammed PGCs by PIP exposure. The markers for endoderm, ectoderm, and mesoderm are, respectively, albumin, AE1/AE3 cytokeratins, and vimentin. DAPI (blue) shows every cell nucleus. Scale bars correspond to 25 μm.
PKCε inhibition changed the metabolism of PGCs mediated by HIF upregulation
To further explain how the inhibition of PKCε resulted in PGC reprogramming, we examined the cell metabolism. In particular, it has been previously shown that in reprogramming conditions there is a change from OXPHOS to glycolysis metabolism. In fact, we found that PIP exposure increased the proportion of inactive mitochondria in a similar manner as occurs with hypoxic exposure (Figure 4).
Figure 4.
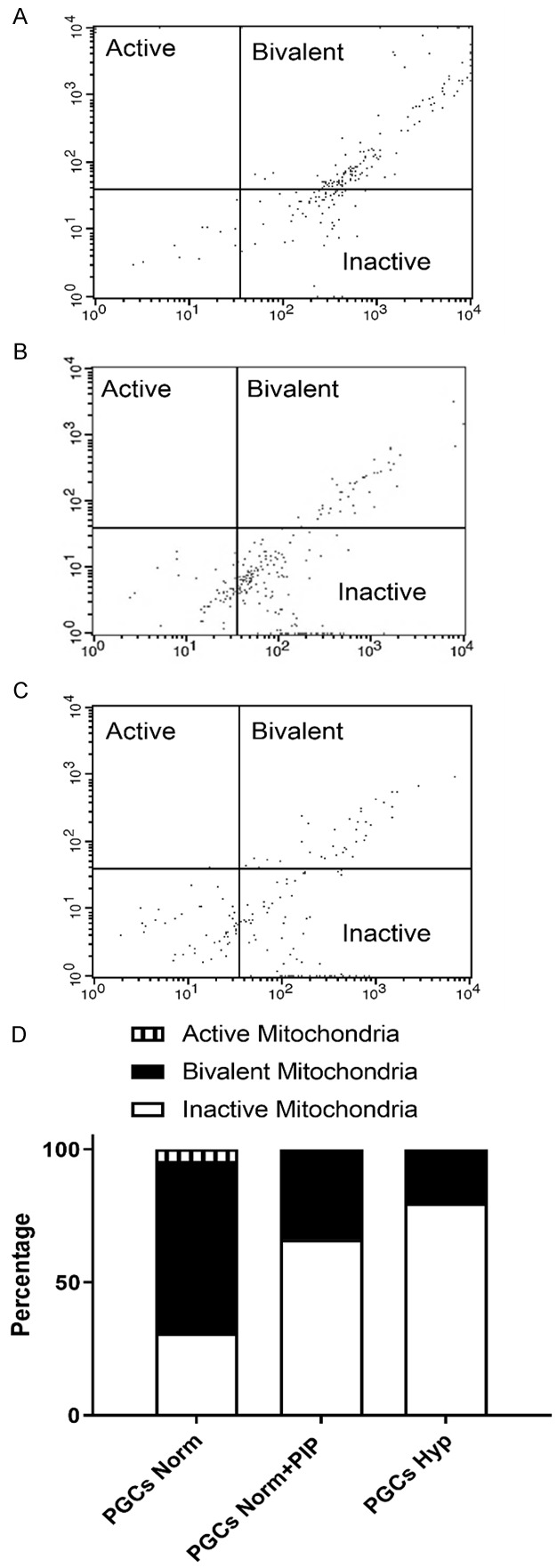
Flow cytometry data of PGC cultures showing the percentage of SSEA1+ cells displaying a green signal from the JC-1 probe (inactive mitochondria), a red signal (active mitochondria), or bivalent (both types) under non-reprogramming normoxia conditions (A), reprogramming hypoxia conditions (B), or PIP-supplemented cultures (C). An increase in inactive mitochondria to the detriment of bivalent mitochondria is observed in normoxia after 5 days of PIP supplementation (shown as percentage in D).
This switch from OXPHOS to glycolysis is probably mediated by HIFs. Given hypoxia-induced embryonic germ-like (hiEGLs) express HIF1α and both hiEGLs and PGCs express HIF2α, we checked whether PGCs reprogrammed with PIP in normoxia shared such a phenotype. This study revealed an increase in HIF1α protein expression (or stabilization) from 48 h onward (Figure 5), decreasing at 6 days of culture (Figure S3A). In the same manner, there was a peak in HIF2α at 48 h (Figure 6) that was lost by the sixth day of culture (Figure S3B). This result suggests that both hypoxia and PKCε inhibition share an upregulation of HIFs that might be responsible for downstream events leading to reprogramming.
Figure 5.

Confocal microscopy images for immunofluorescence against SSEA1 and HIF1α in PGC cultures. Samples include Cos7 cells cultured in normoxic conditions as a negative control and in hypoxia as a positive control. Images also show PGCs cultured in normoxia (Norm), hypoxia (Hyp), and PIP for 48 h. Note HIF1α expression in Cos7 and PGCs in hypoxia and in PIP-supplemented cultures. Scale bars correspond to 25 μm.
Figure 6.
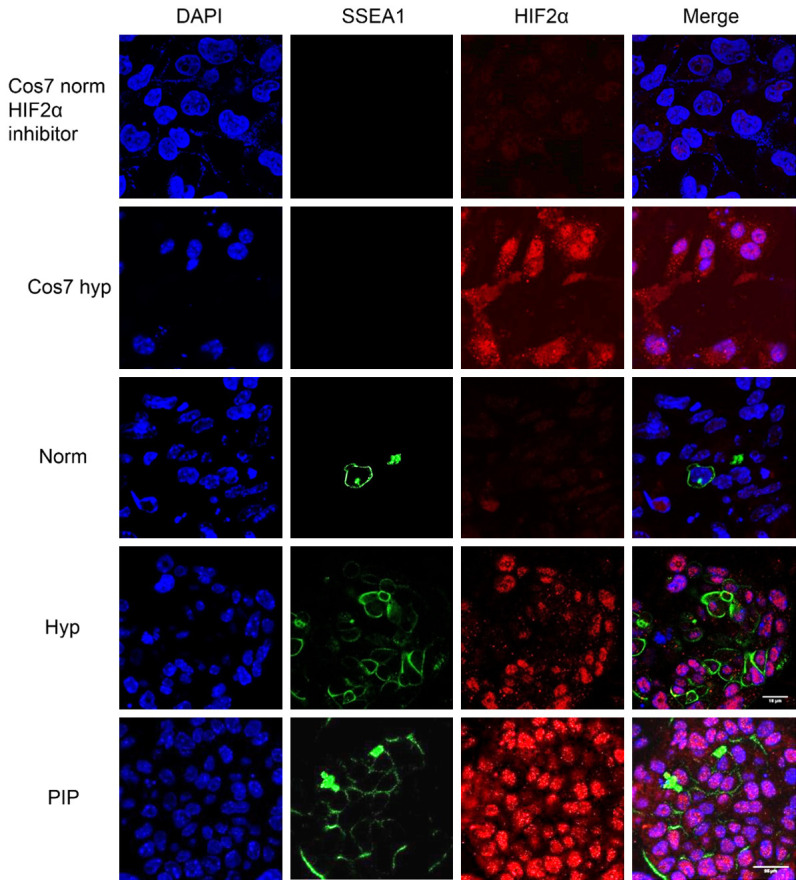
Confocal microscopy images for immunofluorescence against SSEA1 and HIF2α in PGC cultures. Samples include Cos7 cells cultured in normoxic conditions as a negative control and in hypoxia as a positive control. Images also show PGCs cultured in normoxia (norm), hypoxia (Hyp), and PIP for 48 h. Note HIF2 expression in Cos7 and PGCs in hypoxia and in PIP-supplemented cultures. Scale bars correspond to 25 μm.
PKCε inhibition did not change the classical pluripotency gene expression of PGCs
Nanog expression was used as a pluripotency marker. PGCs naturally express Nanog, and in the case of differentiation, this expression would be lost. Immunofluorescence studies were performed in cultures in normoxia, PIP, and hypoxia. The nuclear expression of Nanog was maintained in all cases, as expected, adding to the evidence that reprogramming toward pluripotency had been achieved (Figure S4).
In PGC reprogramming conditions exerted by hypoxia cultures, HIF1 stabilization promotes deregulation of Oct4 levels, which in PGCs are already high, lowering them to promote reprogramming [2]. To ascertain whether reprogramming in normoxia by PIP uses the same mechanism, we performed flow cytometry for Oct4-GFP in such cultures. Intriguingly, although a medium Oct-4 expression level population arose in PIP supplemented cultures, the change was not statistically significant (Figure S5).
Similarly, no expression of c-Myc (Figure 7A) or Klf4 (Figure 7B) was detected in PIP-supplemented cultures, indicating that PKCε inhibition uses a different reprogramming set of factors to achieve pluripotency.
Figure 7.
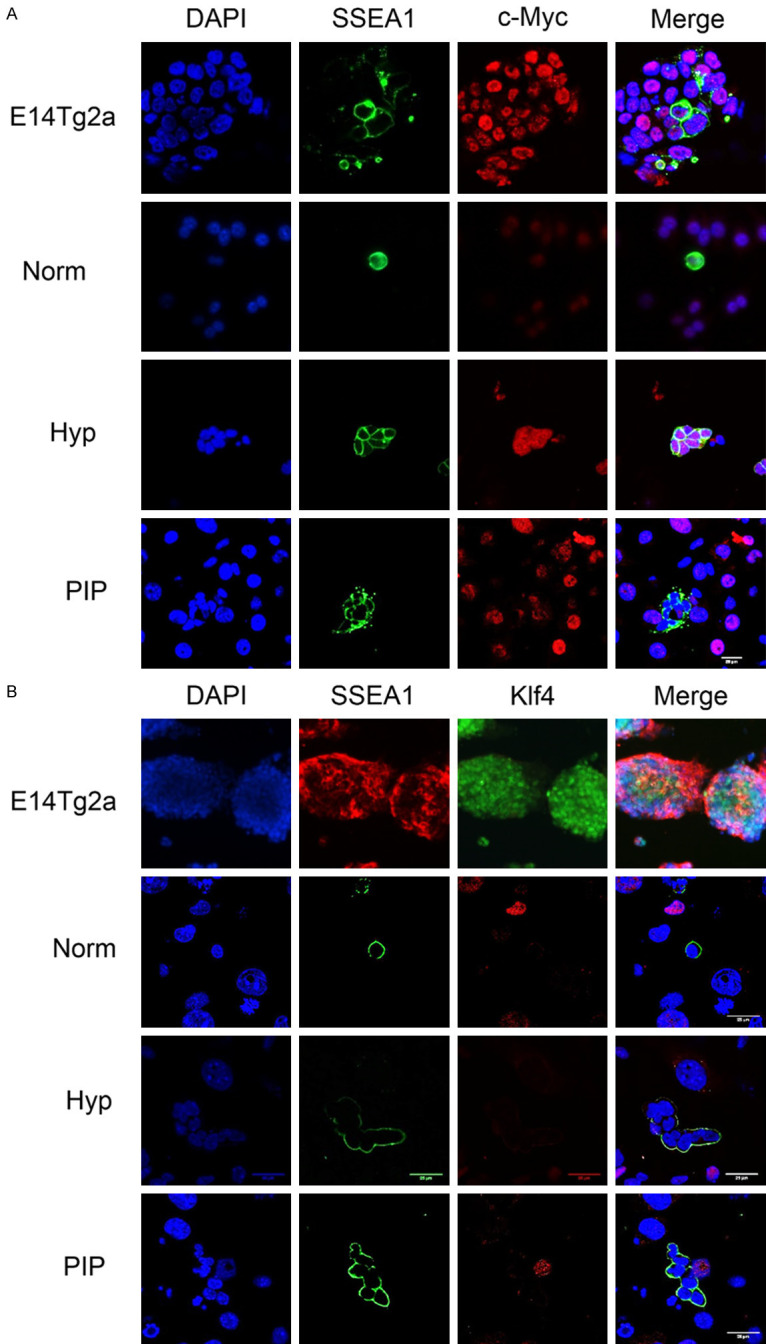
Immunofluorescence microscopy against SSEA1 and c-Myc (A) and Klf4 (B) in PGC cultures, showing a negative reaction in SSEA1+ cell (PGCs) nuclei in PIP-supplemented cultures. Images also show PGCs cultured in normoxia (norm) and hypoxia (Hyp) for 48 h. Positive controls are the ESC line E14Tg2 and also hypoxic PGC cultures are positive for cMyc. Scale bars correspond to 25 µm.
PKCε inhibition changed the histone acetylation of PGCs
Given PKC activation increases the activity of histone acetyltransferases [14], we aimed to study the histone acetylation state of reprogrammed PGCs by exposure to PIP. Actual histone acetylation was evaluated by immunocytochemistry against H2BacK20, H3acK9, and H4acK5K8, K12, K16. Our results showed that whereas histones H2B, H3, and H4 are nonacetylated in normoxic unipotent PGCs, they all become acetylated in PIP-supplemented cultures within as little as 48 h of treatment, similarly to positive controls subjected to hypoxia (Figure 8), suggesting that reprogramming by PKCε inhibition is mediated by histone acetylation.
Figure 8.
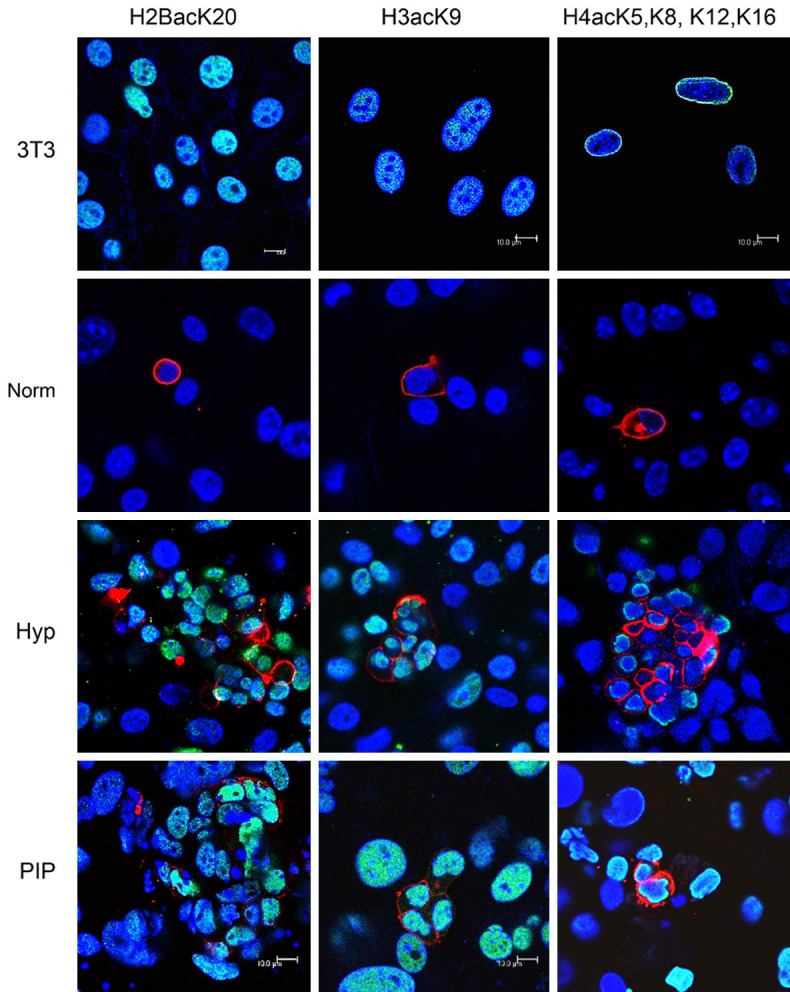
Confocal merged images of H2BacK20, H3acK9, and H4acK5K8, K12, K16 immunofluorescence (in green in the nuclei) of PGC cultures under normoxic, hypoxic, and PIP conditions, showing a positive reaction in SSEA1+ cells (PGCs, membranes in red). Blue shows nuclei with DAPI stain. Colocalization is seen as light blue. Positive controls are an NIH3T3 mouse cell line subjected to the histone deacetylase inhibitor Na+ butyrate at 0.25 μM for 16 hours. Note histone acetylation in PGCs under reprogramming conditions only, both in hypoxia and PIP-supplemented cultures. Scale bars correspond to 100 μm in every photograph.
Discussion
Ca2+ acts as a versatile messenger, effecting the extracellular, cellular, and subcellular levels at a wide dynamic time range. The extracellular matrix is the largest source of free Ca2+ ions in a multicellular organism, at a concentration of approximately 1.2 mM, a 10,000-fold higher concentration than cytosol. Inside the cell, the Ca2+ concentration is maintained at 50-100 nM by a wide signaling toolkit coordinating diverse subcellular compartments (nuclei, lysosomes, mitochondria, endoplasmic reticulum) and proteins (receptors, channels, Ca2+-transporting ATPases, Na+/Ca2+ exchangers, and Ca2+ binding proteins). Every cell expresses a different set of proteins of the Ca2+ signaling toolkit, creating systems with widely different spatial and temporal properties [15].
Our results showed that PGC survival and reprogramming requires a normal extracellular Ca2+ concentration range. This finding is interesting, given neoplastic or transformed cells can continue to proliferate in Ca2+-deficient media [16,17], differentiating normal reprogramming from neoplastic transformation.
Our results, showing that high intracellular Ca2+ concentrations are toxic for PGCs, are not surprising. The accumulation of Ca2+ inside the cells mediates a cascade of destructive events, including alterations in the critical functions of enzymes and organelles and consequent apoptotic cell death (for a concise review see [18]). This situation probably also accounts for the toxic effects at high concentrations of both ionomycin and thapsigargin. At lower concentrations, Ionomycin prevented reprogramming, whereas inconsistent effects of thapsigargin were found in our cultures. Other studies have shown discrepancies in the effects of thapsigargin and ionomycin [19-21]. The fact that thapsigargin increases AKT and PAX3-FOXO1 phosphorylation, could also be aiding in the discrepancy, as AKT supports reprogramming (see below).
Our results showed that a spike in Ca2+ extracellular concentration of 1-3 mM is able to reprogram PGCs in the absence of FGF2. Although we have not explored which calcium channels are active in PGC cultures, it is obvious that a high Ca2+ extracellular concentration exerts changes in the concentration of free intracellular calcium. These changes have been shown to initiate specific courses of cell behavior in many circumstances. For example, one of the first events after fertilization, leading to physiological reprogramming of germ cells, is an alteration of Ca2+ levels (for a review see [22]).
Calmodulin (CaM) is one of the most important transducers of Ca2+ signaling, and one of its many effectors is Ca2+/calmodulin-dependent protein kinase (CaMK), which is involved in phosphorylation of transcription factors involved in the cell cycle [23]. Not surprisingly, CaM, CAMKII, and calcineurin inhibitors did not affect PGC reprogramming in our cultures, probably due to the fact that both CAMKII and Ppp3cc (the catalytic subunit of protein phosphatase 2B holoenzyme [aka calcineurin]) are upregulated in reprogrammed PGC by classical methods, suggesting that activation but not inhibition of this pathway might be involved in reprogramming.
An activation of this pathway also correlates with the increased expression of Atp2a2 (the gene encoding for sarcoendoplasmic reticulum Ca2+ ATPase isoform 2), which transports Ca2+ from the cytosol to the endoplasmic reticulum [ER] lumen [24] in reprogrammed EGCs [2], and also correlates with the inconsistent results obtained with thapsigargin and the lack of effect of BAPTA, all of which suggest that very narrow ER Ca2+-released concentrations must be exquisitely fine-tuned for reprogramming.
Minuscule changes in intracellular free Ca2+ are quickly converted into changes in the activity of several kinases, including the PKC family [14]. Inhibition of all 3 families of PKCs with staurosporine resulted in PGC reprogramming, as occurred in the case of iPSC reprogramming with other broad PKC inhibitors, such as Gö6983 [25] and GF109203X [26]. By microarray comparison between unipotent PGCs and pluripotent EGCs, we had previously identified differential expression of several PKC genes encompassing all 3 types of PKCs: β, ε, and ζ. Accordingly, our results also showed that a mere soluble factor inhibiting classical PKCs (Ca2+ and DAG-dependent) and further inhibiting only a novel PKCε (only DAG-dependent) induced acquisition of pluripotency in PGCs, as we and others had previously shown using only hypoxia [2] and other compounds related to glycolysis switch [13] or histone acetylation [11,13].
The fact that a Ca2+-independent PKC isoform inhibition was able to reprogram PGCs was very intriguing and prompted us to deepen the analysis of how reprogramming was achieved. Among PKCε isoform functions, the regulation of Nanog by phosphorylating and stabilizing it in a nuclear location stands out [27]. Our results showed that the inhibition of PKCε translocation promotes PGC reprogramming, whereas its activation with FR236924 does not have any effect. This result is striking, given the inhibition of PKCε by interfering in its DAG binding should in turn lower Nanog stability; however, Nanog remained unaltered in our PGC cultures. A recent study has suggested that PKC does not phosphorylate mouse Nanog as it does human Nanog [28]. Nanog activation by phosphorylation could therefore have occurred via a PKC-independent pathway in our study. Alternatively, given Nanog is already expressed in unipotent PGCs, it is possible that this PKC function is not operating in PGCs.
Other groundbreaking studies have shown several PKCs to be involved in early events of mammalian fertilization, such as meiotic resumption and egg activation [29]. PKCδ translocates to the meiotic spindle after fertilization [30], correlating and outlasting Ca2+ oscillations [31]. Representatives from all three PKC categories (conventional PKCα and γ, atypical PKCζ and λ, and novel PKCµ and δ), are present in the unfertilized egg until the 8-cell stage embryo at the protein level [32]. All 11 PKC isotypes are expressed in undifferentiated ESC [33], persisting in differentiating embryoid bodies, and 6 of them (α, β, δ, θ, ι and ζ were downregulated or suppressed during cardiomyocyte differentiation [33]). However, PKCζ inhibition is not sufficient to maintain pluripotency when ESC are given differentiation cues [34]. Interestingly, PKCε was upregulated in cardiomyocyte beating areas of ESC differentiation [33]. Similarly, in our case, PKCε inhibition might be working along with other isoforms (probably classical PKCβ and atypical PKCζ) in maintaining pluripotency. Further studies inhibiting individual isoforms would be necessary to clarify the role each one plays in the transduction pathway of PGC reprogramming.
Inhibition of another isoform, PKCλ/ι, has been shown to boost the reprogramming efficiency in iPSCs [35]. Low levels of PKCλ/ι could increase metabolic reprogramming toward glycolysis, HIF1 stabilization, and a pluripotent state [36]altered mitochondrial function and promotion of aerobic glycolysis are key to maintain and induce pluripotency. However, signaling mechanisms that regulate mitochondrial function and reprogram metabolic preferences in self-renewing versus differentiated PSC populations are poorly understood. Here, using murine embryonic stem cells (ESCs), similar to our findings for PKCε in PGC reprogramming. In fact, many studies have shown that PKC isoforms can regulate HIF1α in some types of cancer: PKC-ζ transactivates HIF1α by promoting its association with p300 in renal cancer [37], and PKCs play an important role in increasing HIF1α gene transcription [38]. Our results show that PKCε inhibition is accompanied by both HIF1α and HIF2α upregulation and an increase in inactive mitochondria and thus an increase in glycolytic metabolism.
When HlFs proteins are stabilized, they assemble into a heterodimer that recognizes and binds hypoxia-responsive elements (HREs) in DNA, triggering the expression of hypoxia response genes [39]. HREs contain the consensus sequence RCGTG, found in hundreds of genes. Among those, many are related to glucose metabolism, TCA cycle, energy and amino-acid metabolism, primarily aiding in the shift of cell metabolism from oxidative phosphorylation towards glycolysis (for a review see [40]). This switch in the cell metabolism is required for reprogramming of somatic cells to iPSCs [41], and is an active process occurring at the beginning of the reprogramming process. We have previously demonstrated that in PGCs it is the metabolic shift that activates the stem cell program [2,13]; for a review see [1].
HIF1 induces the glucose transporter, pyruvate dehydrogenase kinase 1, lactate dehydrogenase A, and hexokinase expression needed for high glycolytic activity. Also, HIF stabilization through prolyl hydroxylase inhibition has been shown to induce PGC reprogramming [2]. Our reprogrammed cells in the presence of PIP showed HIF1α and HIF2α upregulated expression levels similar to those of hypoxia-reprogrammed EGCs (hiEGLs), with the same temporal pattern. This result could mean that the reprogramming mechanism of PIP is similar to that occurring in hypoxia, this is, metabolic reprogramming and cellular rearrangement through autophagy by HIF1 inducing Bnip3 expression, which in turn may inhibit mTORC1 binding to Rheb, an mTOR activator [13]. It is remarkable that distinct pathways leading to reprogramming share HIF1α expression as a common feature. In previous studies, we had shown that different ways of PGC reprogramming show HIF1α upregulation and protein stabilization after a 48-h culture, turning HIF1α-negative at approximately the sixth day of culture. Thus, HIF1α appears to be essential for PGC reprogramming. Similarly, iPSC classical derivation is impaired if HIF1α is absent [42]. Also, ectopic expression of HIF1α during the reprogramming process promotes the emergence of iPSC colonies [43].
In addition to the metabolic switch, a previous study has shown that over 2% of all human genome is regulated by HlF1 in endothelial cells [44]. HlFs regulate the expression of those hundreds of genes heavily dependent on cell type and cell context [40]. HIFs enable the expression of genes involved in a great variety of processes other than glucose and cell metabolism, such as cell proliferation (for example CyclinD2, IGF2), apoptosis (for example Bnip3, Bcl-2), differentiation, autophagy, and transcriptional regulation (for example DEC1, ETS-1) and many others (see Table 1 in [40]). Among genes directly regulated by HIFs are also pluripotency genes such as Oct4 and c-Myc, regulated by HIF2, or Notch and ETS-1, regulated by HIF1 [45]. HIF1 also acts through Activin/Nodal signaling to maintain pluripotency [46]. Moreover, very recently HIF1 has been acknowledged to increase Act16a expression and thus histone acetylation and reprogramming in iPSC [47]. Similar data were shown for ESCs, in which Act16a inhibition led to primitive endoderm differentiation [48]. In our cultures, PKCε inhibition by PIP increases HIF1α, which might account for the appearance of H3K9ac markers.
Within intracellular signaling cascades, 3 proteins have been positively identified as PKCε substrates: protein kinases B (AKT), protein kinase D, and the transcription factor signal transducer and activator of transcription 3 [49]. Interestingly, HIF1α stabilization also increases AKT phosphorylation ([50], for a review see [40]). In previous studies, we showed that AKT signaling pathway activation increases PGC proliferation [51]. In fact, PTEN-/- mice (thus, constitutive AKT pathway activation) show increased PGC reprogramming ability and teratoma formation [52].
Interestingly, AKT has been implicated in metabolic reprogramming and histone acetylation in tumor cells [53]. AKT pathway activation increases the generation of glucose-derived citrate, and then ATP-citrate-lyase cleaves it into oxaloacetate and acetyl-CoA, which in turn can be transferred to histones by histone acetyltransferases [54]. Thus, in our cultures, PKCε inhibition by PIP could also activate the AKT pathway, which in turn might facilitate histone acetylation-mediated reprogramming.
The glycolytic switch has also been shown to increase acetyl-CoA production and elevate histone acetylation in transformed cells [55]. Another product of glycolysis, pyruvate, acts as an inhibitor of histone deacetylases (HDACs) 1 and 3 and thus promotes histone acetylation [56]. Lactate, the end product of aerobic glycolysis, has also been reported to inhibit the activity of HDACs [56]. In our cultures, PKCε inhibition by PIP increased inactive mitochondria, which would increase glycolysis and consequently acetyl-CoA availability for histone acetylation.
We have previously shown that histone acetylation was in fact capable of direct reprogramming. We made use of valproic acid (VPA), a wide histone deacetylase inhibitor, demonstrating that VPA is capable of PGC reprogramming in normoxia [13]. Interestingly, VPA-treated cultures were positive for cMyc. Another HDAC inhibitor, trichostatin A, can reprogram PGCs, also in the absence of FGF2 [1,11]. HDAC inhibitors have been widely used in the field of cell reprogramming, given their addition to culture increases reprogramming efficiency. Specifically, VPA has been shown to exert this improvement in the rise of iPSCs, even making Klf4 and cMyc dispensable from the reprogramming cocktail [57]. This result agrees with our finding a lack of Klf4 and cMyc expression in our PIP-reprogrammed PGC cultures.
The exact manner by which histone acetylation reprograms PGCs is poorly understood. However, it is not surprising that epigenetic modifications relate to potency. PSCs display an open chromatin conformation and active chromatin markers, such as H3K4me and H3K9ac [45,46]. Conversely, differentiated cells show repressed chromatin markers, such as H3K27me. Partially differentiated cells show a bivalent chromatin, with both active and repressing markers. AKT, one of the primary factors related to PGC reprogramming as noted earlier, also promotes a more active chromatin, mainly by inhibition of Mbd3, a component of the nucleosome remodeling deacetylase complex. Mbd3 is important in heterochromatin formation, and its inhibition promotes reprogramming in both EGC and iPSC derivation [60]. Interestingly, PKC signaling is also involved in the regulation of promoter activity via detachment of HDAC1 from the promoter region and by the reduction of DNA methyltransferase activity [61].
In our cultures, inhibition of PKC induced HIF-1α, an increase in inactive mitochondria, and a glycolytic switch, while maintaining Oct4 and Nanog expression, resulting in histone acetylation and thus a more open-state chromatin-promoting reprogramming.
Acknowledgements
Authors thank funding from Roche Farma SA, Foundation Domingo Martínez and Foundation Jesus Antolín Garciarena. A Moratilla and M Cadenas Martin are financed by Consejería de Educación, Juventud y Deporte of Comunidad de Madrid and by Fondo Social Europeo (Programa Operativo de Empleo Juvenil, and Iniciativa de Empleo Juvenil (YEI)), (PEJD-2018-PRE/BMD-8878, PEJ15/BIO/AI/0093 and PEJD-2018-PRE/BMD-9040). Authors thank Morote Traducciones for English editing, the Statistics Facility of La Paz Hospital for statistical advice, and Victor Toledano for flow-cytometry.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.De Miguel MP, Alcaina Y, de la Maza DS. Primordial germ cell reprogramming. In: Germ Cell. IntechOpen; 2018. pp. 43–62. [Google Scholar]
- 2.López-Iglesias P, Alcaina Y, Tapia N, Sabour D, Arauzo-Bravo MJ, Sainz De La Maza D, Berra E, O’Mara AN, Nistal M, Ortega S, Donovan PJ, Schöler HR, De Miguel MP. Hypoxia induces pluripotency in primordial germ cells by HIF1α stabilization and Oct4 deregulation. Antioxid Redox Signal. 2015;22:205–223. doi: 10.1089/ars.2014.5871. [DOI] [PubMed] [Google Scholar]
- 3.Leitch HG, Nichols J, Humphreys P, Mulas C, Martello G, Lee C, Jones K, Surani MA, Smith A. Rebuilding pluripotency from primordial germ cells. Stem Cell Reports. 2013;1:66–78. doi: 10.1016/j.stemcr.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Resnick JL, Bixler LS, Cheng L, Donovan PJ. Long-term proliferation of mouse primordial germ cells in culture. Nature. 1992;359:550–551. doi: 10.1038/359550a0. [DOI] [PubMed] [Google Scholar]
- 5.Matsui Y, Zsebo K, Hogan BL. Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell. 1992;70:841–847. doi: 10.1016/0092-8674(92)90317-6. [DOI] [PubMed] [Google Scholar]
- 6.Donovan PJ, De Miguel MP. Turning germ cells into stem cells. Curr Opin Genet Dev. 2003;13:463–471. doi: 10.1016/j.gde.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 7.Koshimizu U, Taga T, Watanabe M, Saito M, Shirayoshi Y, Kishimoto T, Nakatsuji N. Functional requirement of gp130-mediated signaling for growth and survival of mouse primordial germ cells in vitro and derivation of embryonic germ (EG) cells. Development. 1996;122:1235–1242. doi: 10.1242/dev.122.4.1235. [DOI] [PubMed] [Google Scholar]
- 8.Felici M, Farini D, Dolci S. In or out stemness: comparing growth factor signalling in mouse embryonic stem cells and primordial germ cells. Curr Stem Cell Res Ther. 2009;4:87–97. doi: 10.2174/157488809788167391. [DOI] [PubMed] [Google Scholar]
- 9.Surani MA, Hayashi K, Hajkova P. Genetic and epigenetic regulators of pluripotency. Cell. 2007;128:747–762. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Leitch HG, Blair K, Mansfield W, Ayetey H, Humphreys P, Nichols J, Surani MA, Smith A. Embryonic germ cells from mice and rats exhibit properties consistent with a generic pluripotent ground state. Development. 2010;137:2279–2287. doi: 10.1242/dev.050427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durcova-Hills G, Tang F, Doody G, Tooze R, Surani MA. Reprogramming primordial germ cells into pluripotent stem cells. PLoS One. 2008;3:e3531. doi: 10.1371/journal.pone.0003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo H, Hu B, Yan L, Yong J, Wu Y, Gao Y, Guo F, Hou Y, Fan X, Dong J, Wang X, Zhu X, Yan J, Wei Y, Jin H, Zhang W, Wen L, Tang F, Qiao J. DNA methylation and chromatin accessibility profiling of mouse and human fetal germ cells. Cell Res. 2017;27:165–183. doi: 10.1038/cr.2016.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sainz De La Maza D, Moratilla A, Aparicio V, Lorca C, Alcaina Y, Martín D, De Miguel MP. Metabolic reprogramming, autophagy, and reactive oxygen species are necessary for primordial germ cell reprogramming into pluripotency. Oxid Med Cell Longev. 2017;2017:4745252. doi: 10.1155/2017/4745252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mellström B, Naranjo JR. Mechanisms of Ca2+-dependent transcription. Curr Opin Neurobiol. 2001;11:312–319. doi: 10.1016/s0959-4388(00)00213-0. [DOI] [PubMed] [Google Scholar]
- 15.Gerbino A, Colella M. The different facets of extracellular calcium sensors: old and new concepts in calcium-sensing receptor signalling and pharmacology. Int J Mol Sci. 2018;19:999. doi: 10.3390/ijms19040999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitfield JF. Calcium signals and cancer. Crit Rev Oncog. 1992;3:55–90. [PubMed] [Google Scholar]
- 17.Kahl CR, Means AR. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr Rev. 2003;24:719–736. doi: 10.1210/er.2003-0008. [DOI] [PubMed] [Google Scholar]
- 18.O’Reilly D, Buchanan P. Calcium channels and cancer stem cells. Cell Calcium. 2019;81:21–28. doi: 10.1016/j.ceca.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Lew PD, Wollheim CB, Waldvogel FA, Pozzan T. Modulation of cytosolic-free calcium transients by changes in intracellular calcium-buffering capacity: correlation with exocytosis and O2-production in human neutrophils. J Cell Biol. 1984;99:1212–1220. doi: 10.1083/jcb.99.4.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nüße O, Serrander L, Foyouzi-Youssefi R, Monod A, Lew DP, Krause KH. Store-operated Ca2+ influx and stimulation of exocytosis in HL-60 granulocytes. J Biol Chem. 1997;272:28360–28367. doi: 10.1074/jbc.272.45.28360. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y, Putney JW. Relationship between intracellular calcium store depletion and calcium release-activated calcium current in a mast cell line (RBL-1) J Biol Chem. 1998;273:19554–19559. doi: 10.1074/jbc.273.31.19554. [DOI] [PubMed] [Google Scholar]
- 22.Whitaker M. Calcium at fertilization and in early development. Physiol Rev. 2006;86:25–88. doi: 10.1152/physrev.00023.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swulius MT, Waxham MN. Ca2+/calmodulin-dependent protein kinases. Cell Mol Life Sci. 2008;65:2637–2657. doi: 10.1007/s00018-008-8086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu LH, Boivin GP, Prasad V, Periasamy M, Shull GE. Squamous cell tumors in mice heterozygous for a null allele of Atp2a2, encoding the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump. J Biol Chem. 2001;276:26737–26740. doi: 10.1074/jbc.C100275200. [DOI] [PubMed] [Google Scholar]
- 25.Lee J, Park YJ, Jung H. Protein kinases and their inhibitors in pluripotent stem cell fate regulation. Stem Cells Int. 2019;2019:1569740. doi: 10.1155/2019/1569740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinehara M, Kawamura S, Tateyama D, Suga M, Matsumura H, Mimura S, Hirayama N, Hirata M, Uchio-Yamada K, Kohara A, Yanagihara K, Furue MK. Protein kinase C regulates human pluripotent stem cell self-renewal. PLoS One. 2013;8:e54122. doi: 10.1371/journal.pone.0054122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang M, Li K, Xie M, Ding S. Chemical approaches to controlling cell fate. Princ Dev Genet Second Ed. 2015;28:59–76. [Google Scholar]
- 28.Saunders A, Li D, Faiola F, Huang X, Fidalgo M, Guallar D, Ding J, Yang F, Xu Y, Zhou H, Wang J. Context-dependent functions of NANOG phosphorylation in pluripotency and reprogramming. Stem Cell Reports. 2017;8:1115–1123. doi: 10.1016/j.stemcr.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallicano GI, Schwarz SM, McGaughey RW, Capco DG. Protein kinase C, a pivotal regulator of hamster egg activation, functions after elevation of intracellular free calcium. Dev Biol. 1993;156:94–106. doi: 10.1006/dbio.1993.1061. [DOI] [PubMed] [Google Scholar]
- 30.Tatone C, Delle Monache S, Francione A, Gioia L, Barboni B, Colonna R. Ca2+-independent protein kinase C signalling in mouse eggs during the early phases of fertilization. Int J Dev Biol. 2003;47:327–333. [PubMed] [Google Scholar]
- 31.Gonzalez-Garcia JR, Machaty Z, Lai FA, Swann K. The dynamics of PKC-induced phosphorylation triggered by Ca2+ oscillations in mouse eggs. J Cell Physiol. 2013;228:110–119. doi: 10.1002/jcp.24110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pauken CM, Capco DG. The expression and stage-specific localization of protein kinase C isotypes during mouse preimplantation development. Dev Biol. 2000;223:411–421. doi: 10.1006/dbio.2000.9763. [DOI] [PubMed] [Google Scholar]
- 33.Zhou X, Quann E, Gallicano GI. Differentiation of nonbeating embryonic stem cells into beating cardiomyocytes is dependent on downregulation of PKCβ and ζ in concert with upregulation of PKCε. Dev Biol. 2003;255:407–422. doi: 10.1016/s0012-1606(02)00080-5. [DOI] [PubMed] [Google Scholar]
- 34.Dutta D, Ray S, Home P, Larson M, Wolfe MW, Paul S. Self-renewal versus lineage commitment of embryonic stem cells: protein kinase C signaling shifts the balance. Stem Cells. 2011;29:618–628. doi: 10.1002/stem.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mah IK, Soloff R, Hedrick SM, Mariani FV. Atypical PKC-iota controls stem cell expansion via regulation of the Notch pathway. Stem Cell Reports. 2015;5:866–880. doi: 10.1016/j.stemcr.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahato B, Home P, Rajendran G, Paul A, Saha B, Ganguly A, Ray S, Roy N, Swerdlow RH, Paul S. Regulation of mitochondrial function and cellular energy metabolism by protein kinase C-λ/ι: a novel mode of balancing pluripotency. Stem Cells. 2014;32:2880–2892. doi: 10.1002/stem.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Datta K, Li J, Bhattacharya R, Gasparian L, Wang E, Mukhopadhyay D. Protein kinase Cζ transactivates hypoxia-inducible factor α by promoting its association with p300 in renal cancer. Cancer Res. 2004;64:456–462. doi: 10.1158/0008-5472.can-03-2706. [DOI] [PubMed] [Google Scholar]
- 38.Pagé EL, Robitaille GA, Pouysségur J, Richard DE. Induction of hypoxia-inducible factor-1α by transcriptional and translational mechanisms. J Biol Chem. 2002;277:48403–48409. doi: 10.1074/jbc.M209114200. [DOI] [PubMed] [Google Scholar]
- 39.Aprelikova O, Wood M, Tackett S, Chandramouli GV, Barrett JC. Role of ETS transcription factors in the hypoxia-inducible factor-2 target gene selection. Cancer Res. 2006;66:5641–5647. doi: 10.1158/0008-5472.CAN-05-3345. [DOI] [PubMed] [Google Scholar]
- 40.Miguel MP, Alcaina Y, de la Maza D, Lopez-Iglesias P. Cell metabolism under microenvironmental low oxygen tension levels in stemness, proliferation and pluripotency. Curr Mol Med. 2015;15:343–359. doi: 10.2174/1566524015666150505160406. [DOI] [PubMed] [Google Scholar]
- 41.Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell. 2009;5:237–241. doi: 10.1016/j.stem.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 42.Prigione A, Rohwer N, Hoffmann S, Mlody B, Drews K, Bukowiecki R, Blümlein K, Wanker EE, Ralser M, Cramer T, Adjaye J. HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells. 2014;32:364–376. doi: 10.1002/stem.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathieu J, Zhou W, Xing Y, Sperber H, Ferreccio A, Agoston Z, Kuppusamy KT, Moon RT, Ruohola-Baker H. Hypoxia-inducible factors have distinct and stage-specific roles during reprogramming of human cells to pluripotency. Cell Stem Cell. 2014;14:592–605. doi: 10.1016/j.stem.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 45.Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol. 2008;9:285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, Ruohola-Baker H. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012;31:2103–2116. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cui P, Zhang P, Zhang Y, Sun L, Cui G, Guo X, Wang H, Zhang X, Shi Y, Yu Z. HIF-1α/Actl6a/H3K9ac axis is critical for pluripotency and lineage differentiation of human induced pluripotent stem cells. FASEB J. 2020;34:5740–5753. doi: 10.1096/fj.201902829RR. [DOI] [PubMed] [Google Scholar]
- 48.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 49.Newton PM, Messing RO. The substrates and binding partners of protein kinase Cε. Biochem J. 2010;427:189–196. doi: 10.1042/BJ20091302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerrada I, Ruiz-Saurí A, Carrero R, Trigueros C, Dorronsoro A, Sanchez-Puelles JM, Diez-Juan A, Montero JA, Sepúlveda P. Hypoxia-inducible factor 1 alpha contributes to cardiac healing in mesenchymal stem cells-mediated cardiac repair. Stem Cells Dev. 2013;22:501–511. doi: 10.1089/scd.2012.0340. [DOI] [PubMed] [Google Scholar]
- 51.De Miguel MP, Cheng L, Holland EC, Federspiel MJ, Donovan PJ. Dissection of the c-Kit signaling pathway in mouse primordial germ cells by retroviral-mediated gene transfer. Proc Natl Acad Sci U S A. 2002;99:10458–10463. doi: 10.1073/pnas.122249399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimura T, Suzuki A, Fujita Y, Yomogida K, Lomeli H, Asada N, Ikeuchi M, Nagy A, Mak TW, Nakano T. Conditional loss of PTEN leads to testicular teratoma and enhances embryonic germ cell production. Development. 2003;130:1691–1700. doi: 10.1242/dev.00392. [DOI] [PubMed] [Google Scholar]
- 53.Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, Jackson E, Aiello NM, Haas NB, Rebbeck TR, Judkins A, Won KJ, Chodosh LA, Garcia BA, Stanger BZ, Feldman MD, Blair IA, Wellen KE. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20:306–319. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Intlekofer AM, Finley LWS. Metabolic signatures of cancer cells and stem cells. Nat Metab. 2019;1:177–188. doi: 10.1038/s42255-019-0032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clementino M, Xie J, Yang P, Li Y, Lin HP, Fenske WK, Tao H, Kondo K, Yang C, Wang Z. A positive feedback loop between c-Myc up-regulation, glycolytic shift and histone acetylation enhances cancer stem cell-like property and tumorigenicity of Cr(VI)-transformed cells. Toxicol Sci. 2020;177:71–83. doi: 10.1093/toxsci/kfaa086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu X, Ma R, Wu Y, Zha Y, Li S. Reciprocal regulation of metabolic reprogramming and epigenetic modifications in cancer. Front Genet. 2018;9:394. doi: 10.3389/fgene.2018.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen X, Zhai Y, Yu D, Cui J, Hu JF, Li W. Valproic acid enhances iPSC induction from human bone marrow-derived cells through the suppression of reprogramming-induced senescence. J Cell Physiol. 2016;231:1719–1727. doi: 10.1002/jcp.25270. [DOI] [PubMed] [Google Scholar]
- 58.Gaspar-Maia A, Alajem A, Meshorer E, Ramalho-Santos M. Open chromatin in pluripotency and reprogramming. Nat Rev Mol Cell Biol. 2011;12:36–47. doi: 10.1038/nrm3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hou P, Li Y, Zhang X, Liu C, Guan J, Li H, Zhao T, Ye J, Yang W, Liu K, Ge J, Xu J, Zhang Q, Zhao Y, Deng H. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341:651–654. doi: 10.1126/science.1239278. [DOI] [PubMed] [Google Scholar]
- 60.Sekita Y, Nakamura T, Kimura T. Reprogramming of germ cells into pluripotency. World J Stem Cells. 2016;8:251. doi: 10.4252/wjsc.v8.i8.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ryu JM, Lee SH, Seong JK, Han HJ. Glutamine contributes to maintenance of mouse embryonic stem cell self-renewal through PKC-dependent downregulation of HDAC1 and DNMT1/3a. Cell Cycle. 2015;14:3292–3305. doi: 10.1080/15384101.2015.1087620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.