

Abstract
The outbreak of a novel coronavirus associated with acute respiratory disease, called COVID‐19, marked the introduction of the third spillover of an animal coronavirus (CoV) to humans in the last two decades. The genome analysis with various bioinformatics tools revealed that the causative pathogen (SARS‐CoV‐2) belongs to the subgenus Sarbecovirus of the genus Betacoronavirus, with highly similar genome as bat coronavirus and receptor‐binding domain (RBD) of spike glycoprotein as Malayan pangolin coronavirus. Based on its genetic proximity, SARS‐CoV‐2 is likely to have originated from bat‐derived CoV and transmitted to humans via an unknown intermediate mammalian host, probably Malayan pangolin. Further, spike protein S1/S2 cleavage site of SARS‐CoV‐2 has acquired polybasic furin cleavage site which is absent in bat and pangolin suggesting natural selection either in an animal host before zoonotic transfer or in humans following zoonotic transfer. In the current review, we recapitulate a preliminary opinion about the disease, origin and life cycle of SARS‐CoV‐2, roles of virus proteins in pathogenesis, commonalities, and differences between different corona viruses. Moreover, the crystal structures of SARS‐CoV‐2 proteins with unique characteristics differentiating it from other CoVs are discussed. Our review also provides comprehensive information on the molecular aspects of SARS‐CoV‐2 including secondary structures in the genome and protein–protein interactions which can be useful to understand the aggressive spread of the SARS‐CoV‐2. The mutations and the haplotypes reported in the SARS‐CoV‐2 genome are summarized to understand the virus evolution.
Keywords: ACE2, angiotensin‐converting enzyme 2, pandemic coronavirus, SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
1. INTRODUCTION
Coronaviruses (CoVs) are the positive‐stranded RNA viruses which taxonomically come under the family Coronaviridae and subfamily Coronavirinae. Coronaviruses are a group of related RNA viruses. Coronaviruses constitute the subfamily Orthocoronavirinae, in the family Coronaviridae, order Nidovirales, and realm Riboviria. They are enveloped viruses with a positive‐sense single‐stranded RNA genome having spherical, oval or pleomorphic shape. The diameter ranges between 60 and 140 nm [1]. The subfamily can be divided into four genera: Alpha‐, Beta‐, Gamma‐, and Deltacoronavirus [2]. The CoVs are not new to human being and most of them produce mild respiratory diseases in human and are known to infect domesticated animals from decades [3]. But, since the beginning of 21st century, they emerged as a big threat to human population and warrant immediate and researchful remedies. There were six CoVs known, out of which severe acute respiratory syndrome CoV (SARS‐CoV) and Middle East respiratory syndrome CoV (MERS‐CoV) outbreak took a wide toll of human life in 2002 and 2012, respectively. In 2002, SARS‐CoV emerged in China and infected 8422 persons leading to the death of 916 individuals. Later, MERS‐CoV appeared in the Arabian countries and infected around 1800 humans. Recently in 2019, seventh CoV caused large‐scale epidemic affecting almost all countries across the globe. Being a close relative of SARS‐CoV the novel coronavirus was named as SARS‐CoV‐2 (details discussed in Section 1.1). As compared with SARS‐CoV and MERS‐CoV, SARS‐CoV‐2 is spreading faster and number of deaths are multifold higher [4].
1.1. Historical background
In December 2019, patients with pneumonia‐like symptoms were reported from several local health facilities in the Wuhan city of China. The cause was unknown and most of the patients were from sea/wet food market in Wuhan, China. The pathogen was confirmed as virus by polymerase chain reaction (PCR), real‐time polymerase chain reaction and sequenced rapidly by next‐generation sequencing. The virus was considered as novel because its genome did not completely match with any previously sequenced virus genome. Also, the clinical symptoms were distinguishable from that of the other known viral infections. Hence, the virus was named as 2019‐nCov where “n” stands for “novel” [4] and the disease caused by this virus was named as COVID‐19. On the basis of the highest conserved protein‐encoding open reading frame (ORF) 1a/1b sequence, the new virus clustered with SARS‐CoV under genus Betacoronavirus. Thus, the name was changed to SARS‐CoV‐2 by International Committee on Taxonomy of Viruses [5].
2. GENOMIC ORGANIZATION
Genome sequencing of the SARS‐CoV‐2 started at an early stage of the outbreak in Wuhan. The bronchoalveolar lavage fluid samples were collected from the initial patients. The quantitative PCR assays with pan‐CoV primers, including nonstructural protein (nsp) RNA‐dependent RNA polymerase (RdRP) primers confirmed CoV as a causative pathogen. Zhu et al. [4] reported the early genome sequences of SARS‐CoV‐2 of approximate size 29,891 bp using next‐generation sequencing technology [6]. After these initial submissions of SARS‐CoV‐2 genome sequences, multiple entries from different parts of world are continuously appearing in GISAID. By the cut‐off date of this article, more than 84,000 complete genome sequences (>29 kb) were submitted to GISAID. This large volume of sequencing data provides great opportunity to identify the variations in virus strain, the trend of virus evolution/mutations, and their impact on pathogenic potential of SARS‐CoV‐2.
The SARS‐CoV‐2 contains a positive‐sense single‐stranded RNA genome packed in the protein envelope. The spherical envelope possesses spike‐like projections of surface glycoprotein [6, 7]. The GC content of the genomic RNA (gRNA) is very low, that is, 38% as compared with other CoVs. The gRNA consists of 5ʹ‐cap and 3ʹ‐poly‐A tail structure. The number of ORFs varies across the CoVs. In SARS‐CoV‐2, ORF1a is the longest ORF and occupies almost two‐third portion of the genome. Further, ORF1b overlaps with ORF1a following which shorter sub‐gRNAs (sgRNA) encoding four structural proteins namely, spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins along with other accessory proteins encoding sequences are present. The SARS‐CoV‐2 genome (Figure 1a) displays the architectural feature of leader and transcription‐regulatory sequence (TRS) commonly possessed by the CoVs. The leader sequence of 70 bases is present at 5ʹ end of RNA of which 7–10 bases are transcription‐regulatory sequences known as TRS‐L. Similarly, adjacent to each ORF (body sequence) TRS‐B motifs are present. The TRS‐L and TRS‐B regulate discontinuous synthesis of intermediate negative strands of sgRNA [8]. The genome of SARS‐CoV‐2, like other CoVs, represents suppression of CpG islands. This is essential as the host vertebrates possess zinc‐finger antiviral protein (ZAP) which identifies the CpG motifs of the viral genome and process them for degradation. Across the genome, ORF encoding E protein displays the highest content of CpG island motifs [9].
Figure 1.
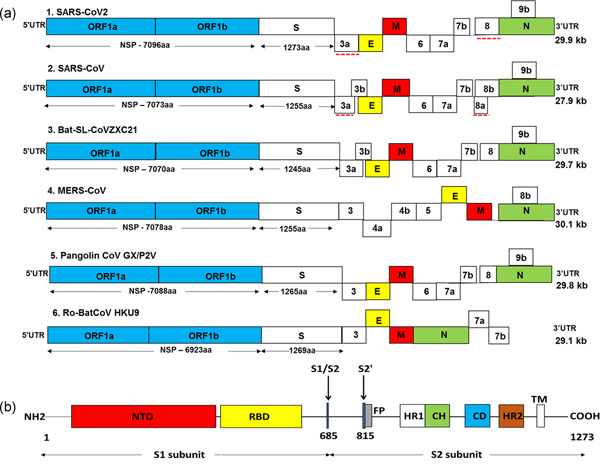
(a) Genome structure of SARS‐CoV‐2 and other coronaviruses. The genome of CoVs comprises of 5ʹ and 3ʹ untranslated region (UTR) and open reading frame (ORF) 1a/b (blue boxes). The structural genes present at 3ʹ terminus encodes for the structural proteins including spike (S; white boxes), envelope (E; yellow boxes), membrane (M; red boxes), and nucleocapsid (N; green boxes) which are common features to all CoVs. In addition, the accessory genes interspaced between the structural genes encodes for accessory proteins. The comparison of coding regions of SARS‐CoV‐2 with different CoVs showed a similar genome organization to SARS‐CoV, bat SL‐CoVZXC21, and pangolin CoV GX/P2V. There is no remarkable difference in the ORF1 of different CoVs but it encodes for NSPs of variable lengths and there is a distinction in the accessory genes. The red dotted line shows a notable variation between SARS‐CoV‐2 and SARS‐CoV. Red dotted line: notable variation between SARS‐CoV‐2 and SARS‐CoV. (b) SARS‐CoV‐2 spike (S) glycoprotein. The S1 region of spike protein contains a N‐terminal domain (NTD; red box) and a C‐domain or receptor‐binding domain (RBD; yellow box). The S2 subunit contains the fusion peptide (FP; gray box), heptad repeat 1 (HR1; white box), central helix (CH; green), connector domain (CD; blue box), heptad repeat 2 (HR2; brown box), and transmembrane domain (TM). Black arrows: cleavage sites at S1/S2 boundary (R685) and S2ʹ (R815)
2.1. Secondary structures (SS) of gRNA
The genomes of CoVs are known to form SS. They act as a regulatory elements, hence are essential for the virus life cycle. It has also been proposed that the evolution in RNA viruses is more aggressive in RNA structures than that of coding sequences [10]. Thus, it becomes equally essential to study the SS in SARS‐COV‐2 RNA genome.
Once the viral RNA is released in the host cell after N protein dissociation, RNA adopts various SS. Such SS includes hairpin‐forming inverted repeats (IR), quadruplex sites, and slippery sequence with downstream pseudoknot, the last one is extensively reported from different DNA/RNA viruses and is frequently observed across the CoVs. These SS are essential for virus replication and, hence, are potential therapeutic targets [11, 12]. The scanning of virus genome predicted higher number of genome‐scale ordered RNA structures (GORS) in SARS‐CoV‐2 than other viruses like hepatitis C virus. The GORS protects viral RNA genome from getting recognized by the host vigilance system. The contour plotting method has been used to analyze GORS in the viral genome [10]. This relatively new method is a promising technique for a deep study of the structural features and positions of the RNA SS.
The SARS‐CoV‐2 possesses the highest frequency of IR per 1000 nucleotides (nt) across the nidoviruses. The majority of the IRs in 5ʹ untranslated region (UTR) region are of length 12+ nucleotides while 3ʹ‐UTR IRs are <12 nt. This difference in length of IRs may indicate the differential regulatory roles of these two UTRs [13]. There are five stem–loop structures (SL) predicted in 5ʹ‐UTR region of SARS‐CoV‐2 and designated as SL1–SL5. The inline and RNase V1 probing has provided the experimental evidence for the stability of SS in 5ʹ region. These structures are found to be conserved across the variants of SARS‐CoV‐2 [14]. SL2 is essential for sgRNA synthesis. TRS‐L belongs to SL3 while SL5 contains the start codon for the ORF1a. A pseudoknot structure is predicted to be present in ORF1ab which controls the frame shifting during the overlapped translation of ORF1a and ORF1ab and known as programmed −1 ribosomal frameshift (−1 PRF) signal. Apart from the pseudoknot, the −1 PRF signal is also composed of slippery sequence and linker region [15]. At 3ʹ‐UTR SL2‐like motif is present, crystal structure of which shows homology with ribosomal RNA (rRNA) loop indicating its role in the initiation of protein translation [16]. Another important SS possessed by SARS‐CoV‐2 is G‐quadruplex. The four guanine bases forms a square planer structure known as guanine tetrad, stacking of which builds G‐quadruplex. In comparison to IR, occurrence of putative quadruplex sites like G‐quadruplex (G‐PQS) is meager, which is obvious as quadruplex sites in virus genome can easily expose the virus to the host immune system. But on the contrary, they are critical elements for virus replication, assembly, and for modulation of host immune response as well [17]. There are 25 G‐PQS in SARS‐CoV‐2 predicted specifically in ORF1ab, ORF3a, S, M, and N genes. Out of these in silico predicted G‐PQS, multiple in vitro spectroscopic assays have confirmed G‐PQS at positions 13385 and 24268. Their interaction with nsp13, a helicase, may provide efficient therapeutic target [18, 19]. The accessory protein‐encoding ORF8 is also predicted to possess SS, functions of which are yet to be elucidated [20].
3. VIRUS PROTEINS
Inside the host cell, ORF1a and ORF1ab translate into polyproteins pp1a and pp1a/b. From these polyproteins, 16 nsps are generated through proteolytic cleavage carried out by nsp3, that is, papain‐like protease (PLpro) and nsp5, that is, main protease (Mpro/3CLpro). PLpro, Mpro/3CLpro, and nsp12 (RdRP) with its cofactors nsp7 and nsp8 forms a replication–transcription complex (RTC) for further synthesis of genomic and sgRNA from which structural and accessory proteins are translated. Figure 1a represents the arrangement of RNA genome and proteins encoded by it. Details about viral nsps, structural, and accessory proteins are discussed below.
3.1. NSPS
The nsps performs multiple functions in the life cycle of virus. The section below provides the functional and structural details of the nsps. We have broadly grouped these nsps according to their functions, though many nsps perform multiple and overlapping roles.
3.1.1. The nsps modulating host immunity
The 5ʹ region of gRNA encodes the host immune response modulating proteins nsp1 and nsp2. The catRAPID (computational tool which calculates RNA‐binding capacity of the proteins) analysis of more than 10,000 interactions revealed that the 5ʹ region exhibits strong tendency to interact with human proteins correlated to the virus infection [21].
The nsp1 interacts with host ribosomal small subunit to stop the antiviral protein synthesis by switching off the host protein synthesis in infected cells [22]. The amino acid sequence of nsp1 of SARS‐CoV and that of SARS‐CoV‐2 is 84% identical which indicates common functions carried out by nsp1 in both the viruses. Crystal structure of SARS‐CoV‐2 nsp1 interacting with human 40S ribosomal subunits suggests that its monomer constitutes 180 amino acid residues comprising of first short α‐helix followed by a short loop and second larger α‐helix. The hydrophobic interaction between the two helices stabilizes the protein structure. The residues K164 and H165 belonging to short loop are essential for the functionality of the protein. The C‐terminal domain of nsp1 makes tight interactions with uS3, uS5, and rRNA helix 18 on the 40S ribosomal small subunit and blocks the messenger (mRNA) entry tunnel (Figure 2) [23]. The nsp2 interacts with signals involved in host cell cycle. In SARS‐CoV, nsp2 specifically interacts with human proteins prohibitin 1 and prohibitin 2 which are involved in mitochondrial function and morphology, and cell proliferation (Figure 2) [24].
Figure 2.
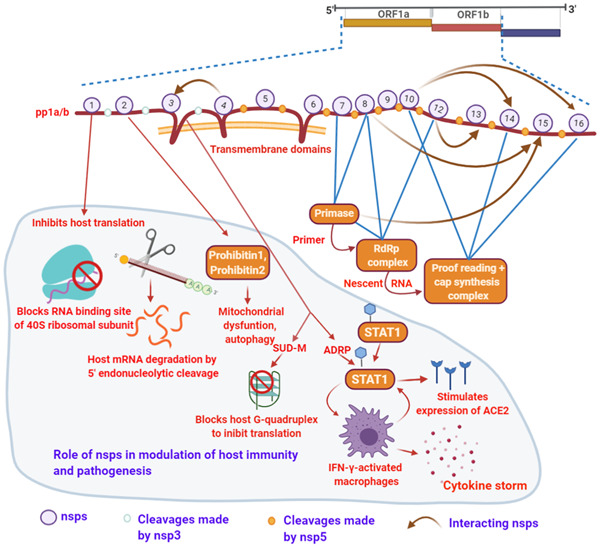
SARS‐CoV‐2 nonstructural proteins (nps), replication cycle, and host pathogenesis. The multiple interactions or the complexes which acts as activators are essential for the virus replication. Numbers on polypeptide pp1a/b labels the nsps; white dots: cleavage made by nsp3; orange dots: nsp5 (Mpro/3CLpro) cleave sites. Brown arrow: nsp–nsp interactions. The portion of the figure with faint blue background specifies the role of nsps in host pathogenesis. Figure created with BioRender.com
The nsp3 is a multifunctional largest protein encoded by SARS‐CoV‐2 genome. It shows multiple domains including Ubiquitin‐like domain 1 (Ubl1)—a RNA‐binding domain, highly variable N‐terminal, ADP‐ribose phosphatase domain (ADRP; also known as macrodomain and X domain), SARS‐unique domain (SUD), RNA‐binding domain, and transmembrane domain. ADRP possesses ADP‐ribose (ADPr) binding site which interacts with host immune signaling, specifically nuclear factor kappa B (NF‐κB) signaling and hence, plays a major role in the generation of cytokine storm in the host tissue [25, 26]. Very interestingly, the viral ADPr‐binding site shows significant identity only with human ADP‐ribose binding modules across the complete human proteome. This suggests that viral ADPr‐binding site can participate functionally in the host protein deMARylation/MARylation. ADRP deMARylates the transcription factor “signal transducer and activator of transcription 1” (STAT1). The STAT1 works as an important switch in innate immune signaling mediated through interferon receptors. The deMARylation of STAT1 produces IFN‐γ‐activated macrophages and expression of interferon‐stimulated genes. As a consequence, cytokine storm is generated which is the fundamental cause for the severity associated with SARS‐CoV‐2 infection. Moreover, angiotensin‐converting enzyme 2 (ACE2), a receptor of SARS‐CoV‐2, is also under the control of interferon and its promoter shows STAT1‐binding site, thus deMARylation of STAT1 probably sets a circuit which may also stimulate the expression of ACE2 accelerating the multiplication of SARS‐CoV‐2 inside the host (Figure 2).
Crystal structure of ADRP in apo form as well as with ADPr substrate has been solved. It comprises of central seven‐stranded β‐sheet, of which, both sides are occupied by α helices. The C‐terminal edges of the central β strands (β3, β5, β6, β7) forms the substrate‐binding pocket. In presence of ADPr, substrate‐binding pocket undergoes major conformational changes [27]. The SUD is observed in human CoVs only. The SUD constitutes three macrodomains; SUD‐N, SUD‐M, and SUD‐C [28]. SUD‐M interacts with genomic SS specifically G‐quadruplex in viral RNA and host RNA as well, with interacting residues K565, K568, and E571 [13]. As mentioned earlier, virus genome possesses much less numbers of G‐quadruplexes; hence, it is possible that major activity of domain M is associated with the host genome.
3.1.2. The nsps with proteolytic activity
Among these 16 nsps, PLpro, and Mpro/3CLpro received special attention as therapeutic targets as they perform the first protein cleavage inside the host cell. Crystal structure of Mpro has been revealed recently which is made up of protomers A and B; each protomer consists three domains designated as I, II, and III. These protomers dimerizes to form Mpro dimer. The substrate‐binding site is present between six‐stranded antiparallel beta barrels of domain I (chymotrypsin‐like domain) and II (picornavirus 3C protease‐like domain). Domain III is a globular cluster of five helices, regulating dimerization of the Mpro through the salt–bridge interaction between two protomers. The two protomers show tight binding with contact interface of 1394 Å2. Serine (S1) of protomer B and glutamine (Q166) of protomer A interacts to ascertain S1 pocket shape and confirmation for substrate binding. The SARS‐CoV‐2 Mpro is distinguishable from SARS‐CoV Mpro as it exhibits replacement of threonine by alanine at position 285 which enhances the catalytic activity of SARS‐CoV‐2 Mpro [29]. PLpro cleaves the nsps residing toward the N‐terminal of the polyprotein chain, that is, nsp1, nsp2, and itself from the long polypeptide chain. As nsp3 possesses transmembrane domain, it anchors the host membrane and provides stability for assembling of viron particles. Later, it forms double‐membrane vesicle in coordination with nsp4.
3.1.3. The nsps involved in RNA synthesis, proofreading, and modification
The nsp12 occupies the major portion of RTC. Cryo‐electron microscopy of nsp12 exhibits nidovirus RdRp‐associated nucleotidyltransferase (NiRAN), a nidoviral signature domain present at N‐terminal of the RdRp. The NiRAN is thought to contribute in nucleic acid ligation and mRNA capping [30]. An interface domain connects NiRAN with “right‐handed” RdRp domain. The RdRp domain constitutes finger, palm, and thumb domain. Notable feature of the SARS‐CoV‐2 polymerase is that it possesses additional N‐terminal β‐hairpin inserted into the groove clamped by the NiRAN and the palm subdomain stabilizing the overall polymerase structure. As seen in other COVs, the active site of RdRp is comprised of seven motifs (A–G) in palm subdomain. A heterodimer nsp7–nsp8 and individual nsp8 are bonded with RdRp similar to the RdRp complex observed in other CoVs [31].
Although nsp12 can synthesize the RNA independently, binding of nsp7 and nsp8 to nsp12 increases the efficiency of the enzyme. As revealed by crystal structure, two nsp7–nsp8 dimers further dimerizes in 2:2 proportion to form heterotetrameric primase complex. Dimerization (i.e., nsp7–nsp8) and tetramerization (i.e., nsp7–nsp8 × 2) occurs mainly via hydrophobic interaction and stabilized by a disulfide bridge between the symmetric cysteine residues of nsp7 [32, 33]. As proposed by Wang et al., primase complex dissociates into two equal units from which one unit further contributes to RdRp complex. Binding of nsp7–nsp8 dimer to the RdRp is mainly mediated by nsp7 and takes place above the thumb subdomain of RdRp. The nsp7–nsp8 heterodimer also sandwiches the finger extension loops of the RdRp to make it more stable, while the tip of the finger subdomain is clamped by another nsp8 co‐factor. Interestingly, both nsp8 cofactors show different confirmations in the complex. In presence of template RNA, RdRp and nsp8 exhibit conformational changes which are further discussed in Section 6.
Recently, the crystal structure of nsp15, a nidoviral RNA uridylate‐specific endoribonuclease (NendoU) has been solved. It plays an important role in protecting virus from the host immune response. It is composed of dimers of trimer forming double ring hexamer. Each monomer is composed of N‐terminal oligomerization domain, a middle domain, and catalytic C terminal domain. Middle domain of each monomer gives concave shape to the surface allowing multiple interactions with other proteins and RNA [34].
The nsp14 is another interesting nsp which performs two distinct roles. Its N‐terminal domain exhibits exoribonuclease activity, hence, named as ExoN while C‐terminal mediates N7 guanine‐methyl transferase activity in mRNA cap synthesis. The N‐ and C‐terminals are connected through hinge region made up of one loop and three strands providing flexibility to the protein essential to perform these two activities. The nsp10 acts as an activator of the nsp14 and forms dimer in 1:1 ratio [35]. The nsp13, a helicase of SARS‐CoV‐2, shows 100% similarity with that of SARS‐CoV. It performs helicase activity during synthesis of RNA and also contributes in 5ʹ‐RNA capping. It unwinds double‐stranded RNA in 5ʹ–3ʹ direction and presents single‐stranded template to polymerase for further elongation. It exhibits five domains forming pyramidal‐shaped structure. The zinc binding domain with two zinc fingers and one stalk domain are critical for the activity [36]. The nsp16, a 2ʹ‐O‐methyltransferase, is one of the conserved proteins among CoVs forming complex with nsp10. Crystal structure of nsp16 and nsp10 heterodimer suggests that catalytic core adopts typical Rossmann‐like β‐sheet fold which is composed of 11 α‐helices, 7 β‐strands, and loops. It uses S‐adenosyl‐l‐methionine (SAM) as the methyl group donor and shows two SAM‐binding sites, first near β1 and β2 strands of Rossmann‐like β‐sheet and second, a SAM‐binding pocket formed by three loops. The nsp10 displays central anti‐parallel pair of β‐strands which are surrounded by a large loop and two zinc fingers. The hydrophobic and positively charged surface of nsp10 stabilizes binding of nsp16 and SAM [37]. The nsp9 is one of the essential proteins for virus amplification. It is thought to be involved in RNA replication as single‐stranded RNA‐binding protein, although the exact mechanism is not clearly understood [38]. The apo structure of nsp9 consists of CoV‐specific unusual fold of β‐strand barrel. A series of extended loops projects from these strands. The N‐terminal β‐strand and C‐terminal α1 helix forms the dimer interface. The β‐strand 2, 3, and 4 protrudes two loops which are positively charged and proposed as a site of RNA binding [39].
The interactions between nsps are needed for efficiently carrying different molecular mechanisms. For example, binding of nsp12 to nsp13 increases helicase activity of nsp13. Figure 2 represents these nsp–nsp interactions, their independent functions and their mode of action in pathogenesis. Table 1 summarizes the roles of nsps in the life cycle of SARS‐CoV‐2 as well as in the host pathogenesis; either based on the reports from SARS‐CoV‐2 or from previously studied other CoVs.
Table 1.
Significance of the nonstructural proteins in virus replication cycle and host pathogenesis
| Protein | Role in virus life cycle | Role in host pathogenesis |
|---|---|---|
| nsp1 | Inhibits host protein translation by the interaction with 40S ribosomal subunit and host mRNA | |
| nsp2 | Disturbs cell cycle by binding to prohibitin 1 and prohibitin 2 proteins | |
| nsp3 (PLpro) | Protease, ssRNA binding | Interacts with host RNA G‐quadruplex to inhibit host translation, suppresses host innate immune responses by deubiquitination, deISGylation, and ADPr binding |
| nsp4 | Assembling the viral double‐membrane vesicles | |
| nsp5 (Mpro/3CLpro) | Protease | |
| nsp6 | Induction of autophagosomes | |
| nsp7 | Primer synthesis and RNA replication | |
| nsp8 | Primer synthesis and RNA replication | |
| nsp9 | Putative role as ssRNA binding | Interacts with DEAD‐box RNA helicase 5 (DDX5) cellular protein to facilitate virus replication |
| nsp10 | mRNA cap methylation | |
| nsp12 (RdRp) | RNA replication, mRNA capping | |
| nsp13 (Helicase) | Helicase activity during RNA replication, 5ʹ‐triphosphatase activity for mRNA capping | |
| nsp14 (ExoN) | Proof reading during RNA synthesis, N7‐methyltransferase during mRNA capping | |
| nsp15 (NendoU) | Endoribonuclease cleaves RNA at polyuridylate sites | |
| nsp16 | 2ʹ‐O‐ribose methyltransferase during mRNA capping |
Note: Information is based on SARS‐CoV‐2 nsps or identical nsps from other previously studied coronaviruses.
Abbreviations: mRNA, messenger RNA; ssRNA, single‐stranded RNA.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.2. Accessory proteins
Accessory protein coding genes are present in between the structural genes but dominantly clustered at 3ʹ end of the genome. They are thought to be replaceable but they must conduct essential role in virus life cycle as they have retained their position in the genome very well across the CoVs. Specific functions of some accessory proteins are experimentally reported and their possible role to counter attack host immune response is getting wider acceptance. Most of the CoVs contains eight accessory proteins out of which some accessory proteins are expressed selectively in few CoVs only [40]. There are at least six accessory protein‐encoding ORFs annotated in SARS‐CoV‐2 including 3a, 6, 7a, 7b, 8 (8b), and 9b [8]. The SARS‐CoV and SARS‐CoV‐2 shows variations in accessory proteins (Figure 1a). For example, 8a protein is absent in SARS‐CoV‐2 and 8b is 37 amino acid longer as compared with SARS‐CoV [1, 41]. The effect of these variations on the SARS‐CoV‐2 infectivity and pathogenicity have not been fully understood. Previous studies on other CoVs have identified the roles of accessories protein. For example, 3a and 7a are known to have functions like ion‐channel activity, upregulation of host inflammation regulators like NF‐κB and induction of host cell apoptosis [42]. Thus, further study may provide a connection between variations in accessory proteins and high degree of virulence shown by SARS‐CoV‐2. It may also highlight the ability of CoV to cross the species barriers [20, 43].
3.3. Structure proteins
3.3.1. S protein
The CoVs make entry in the host cell by engaging their S protein with host receptors. The S proteins are Class 1 transmembrane proteins which protrude extensively from the virus envelope. There are 15–30 freely rotating S proteins reported on the envelope of the virus [44, 45]. These trimeric proteins are composed of three regions, namely, ectodomain region, transmembrane region, and intracellular domain. Recently cryo‐electron microscopy revealed the structure of S protein suggesting it can make hinge‐like movement resulting into transitions between “up” and “down” confirmations [44, 46, 47]. The intracellular domain shows a short intracellular tail. The ectodomain region has S1 and S2 subdomains. The S1 domain of spike protein acts as a major surface antigen. It contains two subunits, N‐terminal domain (NTD) and C‐terminal domain (CTD) [47]. The S1‐CTD acts as a receptor‐binding domain (RBD). The RBD interacts with the 18 residues of ACE‐2 [48]. RBDs are shielded by glycosylation which is commonly observed in viral glycoproteins including S proteins from SARS‐CoV and HIV‐1. But glycosylation percentage of SARS‐CoV‐2 S protein is low as compared with HIV‐1 S protein [49]. The three RBDs form a trimer and rotate up to give receptor accessible confirmation [50]. The S2 domain is a membrane fusion subunit. It contains the fusion peptide (FP), heptad repeat 1 (HR1), central helix (CH), connector domain (CD), heptad repeat 2 (HR2), and transmembrane domain (TM). There are two cleavage sites, one at S1/S2 boundary (R685) and second at S2ʹ (R815; Figure 1b) [47, 51]. The HRs trimerises to form a coiled‐coil structure and drags virus envelope as well as the host cell bilayer to close proximity, facilitating their fusion [52]. At the boundary of S1 and S2 subunits, a furin cleavage site (RRAR) is present. This site distinguishes SARS‐CoV‐2 from SARS‐CoV and other CoVs. Another remarkable feature of SARS‐CoV‐2 is the addition of proline residue at the start of furin cleavage site (Figure 3) [53, 54]. This inserted proline creates a turn which is predicted to result into O‐linked glycosylation at positions S673, T678, and S686. Moreover, O‐linked glycan may contribute to strong shielding of SARS‐CoV‐2 epitopes [53, 55]. But, contradictory to this, recent liquid chromatography with tandem mass spectrometry (LC‐MS/MS) study reported the absence of such O‐linked glycosylation at the abovementioned position [56]. Instead of O‐linked glycosylation site, the role of N‐glycans neighboring (N61 and N603) to furin cleavage site, specifically in regulation of furin cleavage site accessibility has been strongly proposed. The S1/S2 protease cleavage site loop apex makes strong and stable interaction with N‐glycans at N61 and N603 making furin cleavage site more accessible [57]. Apart from the extensively studied furin cleavage site, GTNGTKR motif is also present in S1‐NTD domain which is thought to bind other receptors like protein or sugar receptor. A more thorough study is required to experimentally evidence the role of GTNGTKR motif in viral pathogenesis [58].
Figure 3.

The multiple sequence alignment of spike (S) glycoprotein along with S1/S2 and S2ʹ cleavage sites. The proprotein convertase (PPC) or furin motif RRAR with leading proline insertion is unique to SARS‐CoV‐2 (PRRA insertion highlighted in red box) although NL63 and MERS have proline without downstream additional basic residues. Such polybasic cleavage site is absent in other beta CoVs including bat, Chinese as well as Malayan pangolin, and even previous human SARS‐CoV. The S2ʹ cleavage site at R815 is conserved across all the sequences analyzed; however, SARS‐CoV‐2, bat, and pangolin has KPSKR and civet and hSARS‐CoV has KPTKR
Amino acid sequence of S protein of SARS‐CoV‐2 is 76% identical with SARS‐CoV while it shows more identity, that is, 97% with bat CoV RaTG13. The ACE2‐interacting region (460–520 amino acid) is highly conserved among CoVs [21]. Interestingly, identity between SARS‐CoV and SARS‐CoV‐2 decreases in the RBD region (Figure 4). Only 74% identical RBD possibly explains why they bind to two different receptors on the host cells [59]. In case of SARS‐CoV, it has been observed that mutations in RBD can occur to adopt with host cells during passage in cell culture [2, 60]. Thus, theoretically it is possible that SARS‐CoV‐2 gained the mutations in RBD as an adaptation during cross‐species transmission. Mutations in RBD not only enhance the structural stability of S protein but it can also weaken the binding of the antibody raised against the other strains [60, 61]. After the initial interaction between the S1 domain and the host receptor ACE2, S2 segment mediates membrane fusion of the host and the viral membrane that allows the virus RNA genome to enter inside the host cells [48]. Steps involved in virus entry are discussed in Section 6.
Figure 4.

Multiple sequence alignment of SARS‐CoV‐2 receptor‐binding domain (RBD) of spike glycoprotein (S). The contact amino acid residues of RBD that interacts with ACE2 receptor are marked with red boxes. All six amino acid residues exactly matches with Malayan pangolin CoV strains MP789 (NCBI acc no: MT084071) [62] and GD/P2S (GISAID acc no: EPI_ISL_410544) [63]; both the samples originated from the Guangdong Wildlife Rescue Center. These Malayan pangolins were rescued by the Anti‐Smuggling Customs Bureau in March 2019. This suggests that ancestral strain of SARS‐CoV‐2 might have infected Malayan pangolins
3.3.2. E protein
The E protein is a small polypeptide, ranging from 8.4 to 12 kDa. It comprises two distinct domains: the hydrophobic transmembrane domain and the charged cytoplasmic tail. Recently, topology of the E protein in eukaryotic membrane has been revealed. It represents E protein as single‐spanning membrane protein. Its N‐terminus being translocated across the membrane and the C‐terminus is exposed to the cytoplasmic side [64]. The E protein is the most conserved protein across the studied CoVs, and hence, displays common characteristic features and functions. For instance, SARS‐CoV E protein is identical to SARS‐CoV‐2 except for four variations (which are not expected to affect any feature or function of E protein). Thus, features shown by SARS‐CoV E protein including ion channel activity are also thought to be exhibited by SARS‐CoV‐2 E protein [65, 66]. E protein of CoV possesses another unique function of “oligomerization” resulting into formation of viroporin [67]. The viroporins are capable to selectively transport ions like Ca2+ and participate in assembling and release of virus particles from host cells [68, 69, 70]. The CoV E protein is also known to contribute in pathogenesis. It participates in increasing the protein folding load on endoplasmic reticulum (ER). This results in incorrect protein folding emerging into a condition known as unfolded‐protein response (UPR). UPR may ultimately lead to apoptosis [71]. Such pathogenesis by SARS‐CoV E protein is experimentally evidenced in cells infected with mutated strains rSARS‐CoV and rSARS‐CoVΔE, and can be explored for SARS‐CoV‐2 as well [72]. Further, E protein participates in the formation of specialized structure ER–Golgi intermediate compartment (ERGIC) facilitating release of matured virus [73].
3.3.3. M protein
The M glycoprotein is the most abundant constituent of the CoVs. The interaction of M protein with S and E reconciles the characteristic shape of the viron envelope. The M protein is a multispanning membrane protein which is characterized by three transmembrane domains having C‐terminal inside and N‐terminal outside. The third transmembrane domain contains amphipathic region at the end. This region is found to be highly conserved across Coronaviridae members. Apart from this region, other regions of M protein show variability in protein sequences, but interestingly, these variations does not impact SS of CoV M proteins [74]. When SARS‐CoV‐2 M protein sequence was compared with that of bat CoV RaTG13 and Malayan pangolin CoV MP789, unique insertion of a serine residue is observed at the N‐terminal. Moreover, alignment also showed substitutions at position 70, which is predicted to be a part of transmembrane domain. It has been proposed that such mutations in N‐terminal and transmembrane domain, which are exposed to the surface, may have contributed to cross‐species transfer of the SARS‐CoV‐2 [75]. Through various protein–protein interactions, M protein plays a major role in viral assembly and its internal homeostasis [48]. Transmembrane as well as endodomain of M protein participate in protein–protein interaction [76]. It has also been known that CoV M proteins can interact with RNAs which encodes information about genome‐packing signals [77]. These findings support their central role in the assembly of the virion particles. As one of the major proteins of the CoVs, it is hypothesized to be involved in the regulation of replication and packing of RNA into matured virus particles [78]. It has been evidenced that M proteins can endorse two structural confirmations, compact and elongated. Compact M proteins are frequently associated with low density of S proteins as compared with the elongated one [79]. Such confirmation needs to be studied in SARS‐CoV‐2. The M protein from SARS‐CoV is reported to interact with NF‐κB of host cell, lowering the gene expression of cyclooxygenase 2 (Cox 2). Moreover, M protein may contribute to pathogenesis by hijacking NF‐κB‐ and Cox‐2‐mediated host inflammatory response [80]. Being highly similar to that of SARS‐CoV, SARS‐CoV‐2 M protein may have a similar role in pathogenesis. In a very interesting report, in silico analyses showed that the M protein of SARS‐CoV‐2 is homologous to the prokaryotic sugar transport protein semiSWEET (sugar transporter); it is appealing to understand role of M protein in virus energy metabolism [81].
3.3.4. N protein
The N protein ranges from 43 to 50 kDa and binds to gRNA. In all, it is divided into three conserved domains, namely, N arm, central linker (CL), and C tail. The NTD and CTD are the important structural and functional domains. The function of the NTD is RNA binding and its major portion is occupied by positively charged amino acids. X‐ray crystallography of NTD displays four NTD monomers packed in asymmetric orthorhombic crystal. The complete structure of NTD can be divided into three regions: a protruded basic finger, a basic palm, and an acidic wrist. The monomers show right‐handed sandwich of loop–β‐sheet core and loop. β‐sheet core is made up of five antiparallel β‐strands with functionally essential β‐hairpin protruded in between β2 and β5 strand. Across the CoVs, most conserved residues are present in basic palm regions as compared with fingers and acidic wrist. Although the NTD of CoVs shows highly similar structure, the SARS‐CoV‐2 NTD possesses distinguished surface charge distributions, the significance of which is not yet understood, but probably adapted for more efficient binding to its RNA genome [82]. The CTD mediates dimerization of N protein by self‐association and contains nuclear localization signal. It plays important role in nucleocapsid protein oligomerization and N–M protein–protein interactions. The CL region is thought to interact specifically with M protein [83]. Amino acid sequence of SARS‐CoV‐2 N protein is approximately 90% identical with SARS‐CoV N protein [82]. The functions of N protein include replication and transcription of viral RNA, formation, and maintenance of the ribonucleoprotein (RNP) complex [48]. Moreover, it is also reported that N proteins are involved in host–virus interaction. They regulate host cell cycle including apoptosis to facilitate virus multiplication and spread [84]. Very recently, three nuclear localization signals (NLS1–NLS3) and two nuclear export signals (NES1 and NLS2) are reported in the N protein of SARS‐CoV‐2 which are supposed to play crucial role in viral protein assembling [85].
4. PHYLOGENETICS
To understand genome characteristics of SARS‐CoV‐2 structural, phylogenetic, and mutational studies are being carried out intensively [86]. As discussed in Section 2, RBD of S protein plays important role in the selection of the host for pathogenesis and variations in RBD distinguishes SARS‐CoV‐2 from other CoVs. Thus, for aforesaid reasons, we selected S protein from different hosts and performed multiple sequence alignment (MSA). The full‐length spike glycoprotein sequences were retrieved from UniProtKB, Genbank, and GISAID website.
MSA was performed using MUSCLE program and IQ‐Tree web server was used for tree building [87]. To understand the best fit model of spike glycoprotein evolution, Modelfinder tool was employed that evaluated more than 200 models. For the full‐length spike glycoprotein, model WAG + F + I+ G4 was found to be the best fit. We constructed the phylogenetic tree of spike glycoprotein sequences from various genera using maximum likelihood method with 1000 bootstraps. The consensus tree was visualized using FigTree software (http://tree.bio.ed.ac.uk/software/figtree/). As evident from the phylogenetic tree, spike glycoprotein of CoVs could cluster the various genera into Alpha‐, Beta‐, Gamma‐, and Deltacoronaviruses (Figure 5). SARS‐CoV‐2 is being clustered with betaCoV genera and having most similar taxa as CoV from pangolin isolate GX‐P5E, indicating possible intermediary host for SARS‐CoV‐2. Moreover, SARS‐CoV‐2 is forming separate clade from other CoVs having hosts such as bat, mouse, bovine, civet, porcine, and human including MERS‐CoVs with significant bootstrap value suggesting convergent evolution (Figure 5).
Figure 5.
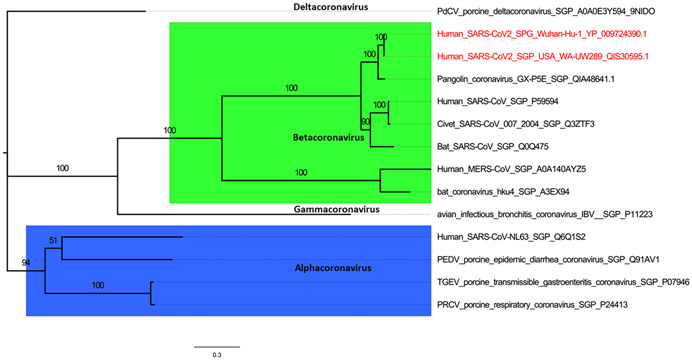
Phylogenetic relationship of various coronavirus spike (S) glycoproteins. The sequences downloaded from UniprotKB and GenBank website were clustered according to generas, namely, Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus. Multiple sequence alignment (MSA) was build using the MUSCLE tool of MEGAX software and phylogeny was inferred using maximum likelihood method with model of substitution: WAG + F + I+ G4 and 1000 bootstraps employing IQ‐Tree webserver (http://iqtree.cibiv.univie.ac.at/)
The SARS‐CoV‐2 spike glycoprotein consists of S1 and S2 subunits. RBD of approximately 230 amino acids recognizes the host ACE2 as its receptor. Therefore, RBD is the critical determinant of virus receptor interaction and reflects host selectivity, virus tropism, and infectivity [88, 89]. The RBD of S glycoprotein is responsible for initiating the viral attachment and viral entry and any mutation to RBD may have significant impact on receptor binding. Thus, it was earlier believed that the RBD should be highly conserved [89]. To investigate this hypothesis, we performed the MSA for the analysis of the mutational dynamics of RBD of SARS‐CoV‐2 against the RBD of most closely related CoVs using Clustal X2. Based on our results of MSA and as shown in recent reports [28, 53, 54], we confirmed that six amino acids of RBD those which are involved in interaction with ACE2, have been changed, possibly altering the host range. The SARS‐CoV‐2 S protein may bind to ACE2 through L455, F486, Q493, S494, N501, Y505 residues whereas in the case of SARS‐CoV Y422, L472, N479, D480, T487, and Y4911 are the interfacing positions for binding [55]. The red boxes in the Figure 4 indicate that five out of the six residues those which are crucial for interaction with human ACE2 differ between SARS‐CoV‐2 and SARS‐CoV. Interestingly, all these six residues are exactly the same in Malayan pangolin CoVs (MP789 and GD/P2S) and differ in Chinese pangolin CoV (GX/P5E) indicating Malayan pangolins as a possible intermediate host for SARS‐CoV‐2 (Figure 4). These mutations in RBD have altered the receptor binding affinities. Sequence and structural comparisons of RBD and ACE2 suggest that SARS‐CoV‐2 RBD is well suited for binding to ACE2 from humans and other species with high receptor homology [50, 62, 90].
5. RESERVOIR AND ZOONOTIC ORIGIN
As mentioned earlier in Section 1, CoVs are subdivided into four genera. Out of these, alpha CoVs and beta CoVs infect mammals while other two can infect birds mostly. The two alpha CoVs infecting humans (hCoVs) are hCoV‐NL63 and hCoV‐229E, and four beta hCoVs are hCoV‐OC43, hCoV‐HKU1, SARS‐CoV, and MERS‐CoV [55]. The SARS‐CoV‐2 is the fifth beta hCoV recently added to the list. As the initial pieces of evidence of SARS‐CoV‐2 infection were obtained from sea/wet food market, the link between seafood and the disease was hypothesized. Later, supportive pieces of evidence were lacking to link the origin of SARS‐CoV‐2 to seafood market as human‐to‐human spread of SARS‐CoV‐2 was substantiated [90, 91]. Meanwhile, genome sequence confirmed the virus as CoV, for which bats act as a major reservoir [8, 92]. The genome sequence of SARS‐CoV‐2 is found to be 96.1% identical with bat CoV (SARS‐CoV‐RaTG13). The CoV from Chinese pangolin (SARS‐CoV‐P4LGuangxi‐2017) was found to be 85.3% identical. The other CoVs were found to be similar at genome level in the range of 73.8%–78.6% with SARS‐CoV‐2 [93]. High similarity between bat CoV and SARS‐CoV‐2 indicates a common ancestor for them. Previously also, bats were extensively reported as major reservoirs of CoVs [2, 94, 95]. Thus, it is more likely that SARS‐CoV‐2 also originated from bats [96]. But interestingly, no bats were reported in the seafood market in Wuhan from where COVID‐19 emerged [92]. Hence, similar to the Himalayan palm civet and dromedary camel as intermediate hosts for SARS‐CoV and MERS‐CoV, respectively, a prima facie “unknown” intermediate host was considered for spreading SARS‐CoV‐2 from bats to human. The time to the most recent common ancestor (TMRCA) is estimated to be November 12, 2019 [97].
Another approach tried to understand the origin and intermediate host for SARS‐CoV‐2 was protein sequence alignment. Spike proteins of CoVs binds to receptors on host cells by their RBD. The ACE2 act as receptor for SARS‐CoV‐2 [98, 99]. By analyzing host receptor and viral spike proteins interaction, species which can act as host/intermediate host can be identified. Pangolin, turtle, and snakes were the species which also possess and express ACE2 receptors and hence, proposed as probable intermediate host for SARS‐CoV‐2 [56].
It has been known that viruses shows flexibility for codons according to their host genome to facilitate their interaction [100]. Relative synonymous codon usage (RSCU) provides possibility of viruses and their host interaction on the basis of “codon usage bias” shown by the viruses. By using RSCU, Ji et al. [99] reported that snake served as intermediate host for SARS‐CoV‐2. But later, Zhang et al. [101] proved that the findings of the experiment were inconsistent due to small size of sequence data analyzed and inclusion of outdated database. Their further study also provided strong pieces of evidence for Malayan pangolin as an intermediate host for SARS‐CoV‐2 through metagenomics. SARS‐CoV‐2‐like virus is also identified from Malayan pangolin which shows high similarity with SARS‐CoV‐2 at the amino acid level. The RBD of S protein from Malayan pangolin CoV showed single amino acid variation when compared with SARS‐CoV‐2 S protein, indicating Malayan pangolin as an intermediate host for SARS‐CoV‐2 [102]. Similar findings were also reported by Wahba et al. [103]. In a strong support of the Malayan pangolin as an intermediate host, they reported homology between the reads from lungs samples of dead pangolin and SARS‐CoV‐2. The RBD of Guangdong (Malayan origin) pangolin CoVs were closely related to SARS‐CoV‐2 RBD. Including present study and previous metagenomics analysis have consistently identified Malayan pangolin as an intermediate host for SARS‐CoV‐2 [63, 104, 105].
The RBD, which is important for binding with human ACE2 receptor, is an ancestral trait from bat viruses rather than recent recombination [106]. It has also been proposed that recombination in SARS‐CoV‐2 genome might have occurred in intermediate hosts. The genome of SARS‐CoV‐2 is 96.1% identical with bat CoV RaTG13. However, RBD domain of both viruses shows divergence. Strikingly, RBD residues of pangolin, specifically Malayan pangolin CoV and SARS‐CoV‐2 are 98% identical. Moreover, Malayan pangolin CoV RBD possessed all six key amino acids which are also present in SARS‐CoV‐2, whereas RaTG13 RBD could present only single key amino acid (Figure 4). These pieces of evidence advocated that recombination event between bat and pangolin CoV materialized in Malayan pangolin through which a new strain of virus might have emerged. But interestingly, insertion of polybasic furin cleavage motif (RRAR) at S1/S2 (Figure 3), which plays significant role in membrane fusion, is present only in SARS‐CoV‐2 and absent in other two CoVs. Thus, altogether, the study proposed that the recombination events occurred are complex and needs more detail experimentation to understand the intermediate host of SARS‐CoV‐2 [106].
6. VIRUS REPLICATION
6.1. Attachment and entry
Viral infections are initiated with the binding of viral particle, that is, glycoprotein spikes on the outer surface to the host surface receptor. The RBD domain of S1 region of the S protein interacts with the host receptor ACE2 [98]. The ACE2 receptor is present on the cell membranes of multiple organs including lungs, arteries, kidney, heart, and intestines. Cell types and the organs at risk of SARS‐CoV‐2 infection inside the human body can be predicted on the basis of ACE2 gene‐expressing cells. The expression of ACE2 is enhanced by interferons which are one of the body's main defenses when host detects the virus. The single‐cell RNA sequencing study revealed that Type II alveolar cells of lungs, myocardial cells, esophagus upper and stratified epithelial cells, and digestive system (specifically absorptive enterocytes from ileum and colon) shows high expression ACE2 mRNA [107, 108, 109]. Further, high expression of ACE2 in mucosa transcriptome of the oral cavity probably emphasizes the entry routes of SARS‐CoV‐2 [110]. Interestingly, 14 ACE2 orthologs from a broad range of animal species were predicted which may also be used by S protein of SARS‐CoV‐2 [111].
The SARS‐CoV‐2 RBD shows higher affinity to ACE2 compared with SARS‐CoV RBD. Apart from the amino acid sequence variations in RBD domain of these viruses, the presence of variability in glycosylation pattern may also have contributed to differential affinity shown by these two viruses [53]. The RBD can possess two confirmations, that is, “up” and “down.” However, RBD with up‐confirmation binds more efficiently compared with the other one.
In addition, the entry requires S protein activation mediated by host type II transmembrane serine protease 2 (TMPRSS2). Human TMPRSS2 protein is chymotrypsin family serine proteases (492 aa) possess three functional domains. It has been shown that TMPRSS2 is expressed in prostate, salivary gland, colon, and stomach [112]. It mediates first cleavage of S protein at the S1–S2 boundary (R685) and second cleavage at S2ʹ (R815) sites. The S1/S2 cleavage site of SARS‐CoV‐2 S protein contains repeated basic arginine residues generating high cleavability [113]. Essentialness of TMPRSS2 is evidenced by recent multiple experiments, thus, the co‐expression of ACE2 and TMPRESS2 protein is a prerequisite for the initiation of pathogenesis. The co‐expression analysis for ACE2 and TMPRSS2 proteins using single‐cell transcriptome analysis of various human cells has identified three cells types: nasal goblet epithelial cells, type II pneumocytes, and enterocytes, thus, possible host cells for SARS‐CoV‐2 [114]. Further polybasic furin cleavage site in S protein efficiently increases priming of the S protein. Recently, in a meticulous study, ACE2, TMPRSS2, and FURIN are shown to co‐express in human lung tissue probably due to which multiplication of SARS‐CoV‐2 is higher in the lungs [114, 115]. After the cleavage at S2ʹ site, the fusion peptide is inserted into the host membrane. The two HR regions, that is, HR1 and HR2 in S2 domain form antiparallel six‐helix bundles (6‐HB). The HR1 region of SARS‐CoV‐2 shows mutations when compared with SARS‐CoV. These variations are expected to provide stability to 6‐HB [116]. The 6‐HB bundle brings about the fusion of two membranes and releases viral genome in the host cell.
6.2. Genome multiplication
In many CoVs, it has been reported that 5ʹ and 3ʹ UTRs of viral gRNA possesses cis‐acting elements. Host factors interact with viral RNA at these sites and participates in viral RNA synthesis [117]. There are many host factors which includes heterogeneous nuclear ribonucleoprotein A1 and Q, polypyrimidine tract‐binding protein, and poly(A)‐binding protein, for which experimental pieces of evidence are available to confirm their role in CoV RNA synthesis [118]. Further, viral RNA being positive‐stranded is translated into a polypeptide chain by using host cell machinery. A programmed frameshift in translation of ORF1a synthesizes pp1a and pp1a/b from the 5ʹ end of ORF. Viral proteases, main protease (Mpro/3CLpro), and papain‐like protease cleave these pp1a/b chains to generate various nsps. Thus Mpro is one of the targets for drug discovery against SARS‐CoV‐2. These nsps then assemble to form RTC. The RdRP with its cofactors nsp7 and nsp8 forms the RNA replication unit of the RTC. The RNA template enters to motifs A and C though a groove fastens by motifs F and G, while the primer stand is supported by motif E and thumb subunit. Motif G initiates RNA synthesis by interacting with the primer strand. Additionally, 5ʹ RNA hook is required to bind motif F which activates RNA synthesis. The nucleotide triphosphate (NTP) substrate reaches the catalytic site through a channel formed at the rear side of RdRp palm subdomain. RNA exit channel is formed at the front side of RdRp through which product–template hybrids exits [119, 120]. Crystal structures of catalytic complex of nsp12–nsp7–nsp8 in the presence of template RNA, and nsp7–nsp8 primase complex have been recently explored. These structures provide in‐depth knowledge of the steps involved in viral RNA synthesis. A transition model has been proposed according to which RNA template first binds to primase complex to synthesize primer strand. Later, half portion of primase dissociates to form RdRp complex with nsp12 and nsp8. This hypothesis is also supported by the crystal structure of primase complex. In any primase, dense positively charged surface is required to facilitate RNA binding. Such positively charged surface is observed in heterotetramers and not in dimers of nsp7–nsp8 [32]. Thus, probably, primase synthesizes primer and transfer it to newly assembling RdRp complex.
Two states of RdRp catalytic complexes have been observed, called as pre‐translocation and post‐translocation states. In pre‐translocation complex, N‐terminal extension of nsp8 (binding independently to nsp12) shows either 45° bend (confirmation I) or the two copies of nsp8 interacts with exiting RNA from the opposite sides providing stable platform for the replication of large RNA genome (confirmation II). Confirmation II is thought to be more rigid and preferred by the virus. Template–product RNA hybrid and nsp12 active motif interacts in a non‐sequence‐specific manner. A loop region of the motif B (present in the palm subdomain) is pushed backward to accommodate this hybrid. Motif G of the finger also goes under conformational changes to interact with the template–product complex in such way that it controls the rate of translocation [119, 120]. The extension of nsp8 forms sliding pole along which exiting RNA slides which indeed prevents premature RdRp dissociation during replication of long RNA genome [119, 121]. Interestingly, the exiting RNA is double‐stranded (ds) hybrid of template–product RNA while for translation, single‐stranded RNA is required. It is thus proposed that dsRNA hybrid is processed by other nsp cofactors, for which studies are lacking at present [121].
Different sgRNAs as well as gRNAs are then synthesized by the RTC complex [8]. From the gRNA, intermediate negative‐strand RNAs are synthesized which are used to generate positive strands of genomic and sgRNAs. The synthesis of sgRNA is discontinuous. The polymerase pauses when it reaches TRS‐B and shifts the newly synthesized fragment to TRS‐L, fusing leader sequence to the body sequence. Apart from the earlier‐mentioned canonical RNAs, a population of noncanonical RNA has been observed in the transcriptome which indicates polymerase jumping events. The 5ʹ capping of mRNA, which is common in nidoviruses, plays an important role in protecting viral mRNA from degradation by host factors. It is made up of triphosphate bridge linking methylated GTP, that is, 7‐methylguanosine to the 5ʹ nucleoside. The nsp13 hydrolyzes the first NTP making it available for capping. The C‐terminal N7‐MTase domain of nsp14 methylates the N7 position of the guanosine. In the next step, nsp16 adds methyl group to the 2ʹ‐O‐ribose of the NTP and nsp10 act as an activator of the complete process. The crystal structure of heterodimer nsp16–nsp10 also has been recently revealed. Interestingly, this complex from SARS‐CoV‐2 is 98% identical with that of SARS‐CoV and all variations found outside of the catalytic site, substrate‐binding site, and interface between nsp10 and nsp16 [122].
Although, RNA modifications other than 5ʹ capping and 3ʹ tailing are reported from different viruses, their details in SARS‐CoV‐2 are not yet extensively reported. But, on the basis of difference in the ionic current between negative control and viral transcripts observed in nanopore DRS data, 41 potential RNA modification sites have been recently identified [8]. The CoVs recruits NendoU for cleavage of viral polyuridine sequences so as to hide virus from host immune sensing system. The role and importance of NendoU activity has been experimentally evidenced in SARS‐CoV. The SARS‐CoV NendoU mutant could activate strong MDA5‐dependent interferon response in the host [122]. In the presence of manganese ion, NendoU cuts the viral RNA at 3ʹ of the polyuridine site [34].
6.3. Viral assembly and release
In the next step, SARS‐CoV‐2 uses host transfer RNA for the translation of its own proteins. Thus, it is obligatory for the virus to adopt its codon usage according to host cells. The RSCU and CAI (codon adaptation index) profiles indicate that SARS‐CoV‐2 has adapted its codon usage and GC content according to the genes expressed in human lungs [123]. The codons ending with either A or U represents the major pool of the virus codons. Though the codon usage bias is very low for SARS‐CoV‐2, geolocation‐wise there are slight variations in codon usage pattern [124]. The high level of viral transcripts facilitates the virus translation and suppresses the host transcript translation [125]. The translated structural and accessory proteins are released in ER. A specialized smooth‐walled Golgi intermediate compartment (ERGIC) carries these viral particles across the secretary pathways. The ERGIC is a characteristic feature of CoVs [126, 127]. For assembling virion‐like particles, viral protein–protein interaction is required and mediated by M protein. One constrain in this assembly is that the membrane proteins through secretory pathway reaches plasma membrane but they are required to be retained near ERGIC for efficient assembling [77]. For this purpose, viral protein possesses intrinsic intracellular retention signals. One of such properly studied signal is ER retrieval signal retained by the cytoplasmic tail of S proteins [128, 129]. In between, gRNA translated earlier, forms RNP complex by interacting with N protein. The N protein forms a complex with RNA which resembles the “beads on string” structure. The bimolecular condensates of N protein and RNA phase separates in the host cell. The highly disordered N and C termini of the protein are rich in arginine and lysine which promotes the interaction with negatively charged RNA backbone while serine‐arginine‐rich motif probably facilitates the phase separation. This complex then attaches to M protein of the ERGIC [130]. The fully assembled virion is then released by exocytosis [92, 129, 131]. The protein encoded by ORF3a, a dimer with six transmembrane helices, forms ion channel which participates in virus release [132]. The schematic representation of pathogenesis is shown in Figure 6.
Figure 6.
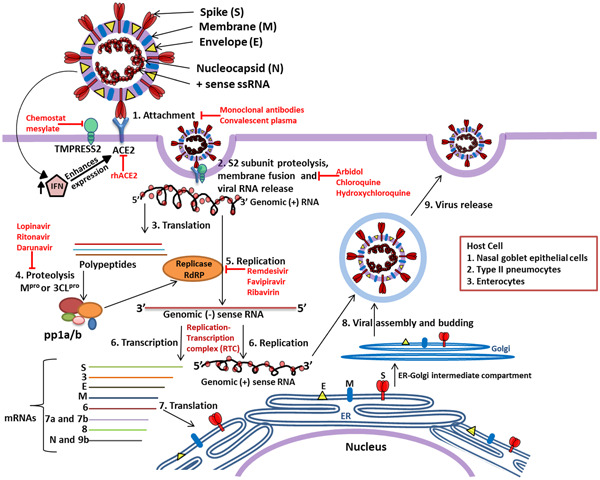
Replication cycle of SARS‐CoV‐2 and potential therapeutic target sites. The SARS‐CoV‐2 enters human body through nasal–oral route and in response to the virus, the body initiates innate response by producing interferons (IFNs); however, IFN activates expression of ACE2 protein which acts as receptor for virus attachment to host cell. Receptor‐binding domain (RBD) of S1 region of S protein interact with ACE2 which leads to proteolytic cleavage at the S1–S2 boundary and S2ʹ R815 site mediated by TMPRSS2 induces the viral and host cell plasma membrane fusion. The viral genomic single‐stranded RNA is translated by host machinery to produce viral polypeptide and these polypeptide undergo proteolytic cleavage by Mpro or 3CLpro synthesizing pp1a and pp1a/b. These polyproteins encode replication–transcription complex (RTC) which continuously replicates and produces a series of subgenomic messenger RNAs encoding the accessory and structural proteins. The viral genomic RNA and proteins are assembled to form the virus particles and buds in the ER and Golgi. Later, the virus containing vesicles fuse with plasma membrane of the host and release the viral particles out of the cell. The antiviral molecules with target sites are highlighted in red color
7. MUTATIONS AND HAPLOTYPE STUDIES
Viruses are prone to rapid mutations which enables them to evade host immunity [133]. Although Coronaviridae possesses unique (distinct from other viruses) proofreading activity for RNA synthesis, a function attributed to nsp14, however, they too mutate but at a low rate [134]. For instance, SARS‐CoV is reported to be mutated at the rate of 9.0 × 10−7 substitutions/nucleotide/replication cycle [135]. Genome sequence of the SARS‐CoV‐2 is now available from the different parts of the world. Several mutations in viral genome have been identified using these whole genome sequences. The phylogeny of SARS‐CoV‐2 genome sequences shows geographical structuring arisen due to local evolutions after the geographically long‐distance dispersal of the virus [136]. For example, 30% of proteomic sequence from Wuhan‐Hu‐1 does not show any similarity with that of Indian isolates [137]. There are no concrete correlations derived between the reported mutations and pathogenicity/spread of the virus yet. But the study of these mutations will definitely be helpful to predict the future path of virus outbreaks, and for the development of antiviral treatments and vaccines as well. At an early stage of the outbreak, the study of data set, including 32 genome assemblies, revealed incremental genetic divergence in SARS‐CoV‐2 genome [138]. On the basis of single‐nucleotide polymorphisms (SNPs) at nucleotide positions 8782 and 28144, Tang et al. divided the SARS‐CoV‐2 strains in two lineages, that is, S and L lineage. The S represents SNP at 28144 resulting into codon for serine, while L accommodates codon for leucine. They found that mutations at these two sites are closely linked. A haplotype network established using all SNPs in studied 103 genomes also supported the divergence of SARS‐CoV‐2 genome in these two strains. The L lineage was found to be more prone for mutation compared with the S strain [139].
The D614G mutation in S protein is the most dominating mutation reported increasingly with the spread of the virus and its significance is functionally characterized. On human lung epithelial calu‐3 cell line, this mutation increases the virus infectivity compared with non‐mutant type of the virus. In the golden Syrian hamster model, D614G mutant titer was higher in nasal wash and different lobes of the lung indicating increased efficiency for infection to the upper respiratory tract which ultimately enhances the viral replication. The mutation D614G do not affect the characteristic S1/S2 cleavage which is in favor of the virus again [140]. In Europe, among all variants of SARS‐CoV‐2, a variant with D614G mutation co‐exhibiting A222V substitution in S protein, has the fastest expansion [141]. Wang et al. [142] reported 13 mutations in SARS‐CoV‐2 genome from the genome sequences submitted till February 2020. In another study, 93 mutations were identified across the SARS‐CoV‐2 genome which includes three mutations in RBD of S protein demanding further study to understand the impact of these mutations on antigenicity of the SARS‐CoV‐2 [143]. Dorp et al. studied 7666 public genome assemblies of SARS‐CoV‐2 and identified invariant and diversified regions of the genome. They observed 198 recurrent mutations across the genome when compared with reference genomes Wuhan‐Hu‐1 (accession IDs NC_045512.2 and EPI_ISL_402125) [144]. These authors found that most (80%) of the mutations were non‐synonymous at the protein level. They also identified highly recurrent mutational sites in nsp6, nsp11, nsp13, and S proteins. Further, they noted that the number of mutations increased with the chronology of sampling dates, suggesting progressive status of mutations. The data set used by these authors also exhibited the highest homoplasies in ORF1a at position 11083 which resides in a region known for triggering CD4+ and CD8+ response in host cells. Interestingly, homoplasy detected in S protein across the data set was outside of the RBD and N‐terminal, thus not affecting virus–host cell receptor interaction [144]. The tendency to acquire mutations varies among the genomic region. The N protein displays the strongest propensity for mutations. Analysis of 61,485 genome sequences of the N protein encoding ORF revealed 1034 mutations resulting in 684 amino acid substitutions. The high‐frequency non‐synonymous substitutions in NTD and CTD are predicted to impact their SS. In‐frame deletions are also reported globally in N protein encoding ORF. Such deletions may impact the functionality of the protein [145]. Pachetti et al. [146] studied 220 genomic sequences across the world. They categorized the process of mutation, geographical region‐wise and observed the highest mutations in genome from North America while genomes sequenced from Asia were the most invariable. In addition to the previously reported mutations, they confirmed 12 most conserved mutations in nsp2, nsp3, RdRP, nsp13, nsp14, S, and ORF9a. They observed a silent and a missense mutation (at nucleotide position 14408) in RdRP from the United Kingdom and Italy, respectively. Interestingly, accumulation of this specific mutation in RdRP is followed by large numbers of mutations in Europe and America compared with genomic assemblies having non‐mutant RdRP from other geographical sites. Thus, the authors have proposed that the mutation in RdRP at nucleotide position 14408 might have affected its binding to nsp14 resulting into higher mutational rate in SARS‐CoV‐2 genomes from Europe and America [146]. In a different approach, based on the polymorphism displayed worldwide in nucleotides encoding NSPs (3, 4, 6, 12, 13, 14), S, N, and ORF8 the virus population is divided into 6 well‐defined and 10 poorly defined subtypes [147]. Very recently, mutations from the Indian isolates has been reported, few of which are supposed to induce conformational changes in viral proteins. In ORF1ab, substitution of C by T at position 1059 results in the replacement of polar uncharged amino acid threonine by hydrophobic isoleucine at the protein level, which may disturb the hydrogen bonds made between threonine and the surrounding amino acids. Another mutation in nsp12, substituting proline by threonine, may result in unusual turn in the alpha‐helix where the cofactors, nsp7 and nsp8 binds to form RdRP complex. Similarly, mutation replacing threonine by methonine at position 644 in nsp12 may affect protein folding and stability [148]. Apart from the impact on protein coding, mutations also exert effect on protein structure morphology, protein stability, and protein–protein interaction. Four such mutations (Q57H and G251V in ORF3a, R203K/G204R in N proteins) have been reported. Q57H increases the affinity of ORF3a for S and N proteins resulting in an impact on virus budding and release [149]. On the contrary, some mutations (like P323L in NSP12 and D614G in S protein) do not affect the protein biology of the virus but play a role in RNA structures [150]. However, to understand the impacts of observed mutations, more systematic studies and in vitro evidences are required.
To sum up, there are multiple studies identifying mutations in SARS‐CoV‐2 genome at the nucleotide and protein level as well. The sites, number, and type (synonymous, non‐synonynous) of mutation varies with the data set used. Most of the mutations are synonymous, which are also claimed as a responsible factor for the evolution of recent SARS‐CoV‐2 virus from its ancestor [151], and non‐synonymous mutations faces strong negative selection pressure. But the process of mutation or evolution of SARS‐CoV‐2 genome is in progress and a genome‐wide study indicates the haplotypes of viral strains are getting established [152, 153].
7.1. Mechanisms driving mutations
There are multiple drivers behind the mutations in virus genome which includes errors during replication, damage to nucleic acid, and host‐induced editing of virus genome as an antiviral defense [154]. These factors may work individually or in combination. Among the observed mutations in SARS‐CoV‐2, C to U/T (most of the literatures have used T in the nucleotide sequences instead of U for the convenience) pyrimidine transition outrightly dominates the other mutations [153]. Deamination of cytosine and 5‐methylcytocine brings about this transitional mutation. The transversion of G to T is also observed in ample data sets. This mutation may have arisen due to the action of reactive oxygen species altering guanine to 8‐oxo‐guanine ultimately replacing G by T. Very recently, probable contribution of host RNA editing mechanisms in SARS‐CoV‐2 genome mutation is also reported. There are two deaminase families, adenosine deaminase that acts on RNA and apolipoprotein B mRNA editing enzyme, catalytic polypeptide‐like (ADAR and APOBEC, respectively), which are involved in mammalian antiviral defense. ADAR edits double‐stranded RNA while APOBECs can edit single strands of RNA or DNA. ADAR‐ and APOBEC‐type RNA editing (i.e., A to I and C to U, respectively) has been observed in the sequences derived from bronchoalveolar lavage fluids obtained from COVID‐19 patients along with the detection of APOBEC in the transcriptome from the same samples. APOBEC is otherwise undetectable in healthy tissue; hence, its detection supports viral RNA editing mediated by host machinery. Although these two enzymes act for the defense of host cells, their activity possibly have contributed to the mutations and evolution of viral genome [151, 154, 155].
8. CONCLUSIONS
During the initial period of the outbreak of COVID‐19, several studies have been published highlighting the characterization, genetic evolution, receptor binding, pathogenesis, and clinical manifestation of SARS‐CoV‐2. Many research groups are working vigorously on the prevention and control of this novel coronavirus, as studies in this area are of high priority to reduce the impact of this outbreak. The sequence‐based analysis suggests horseshoe bat to be the natural reservoir and primary pieces of evidence prompt Malayan pangolin as an intermediate host. The spike protein plays a vital role in determining the host range and the analysis of RBD of spike protein established that SARS‐CoV‐2 and Malayan pangolin CoV share identical binding residues to ACE2. The RNA genome of SARS‐CoV‐2 acquires SS which are crucial signals for translation. Once inside the host cell, the nsp1 and nsp2 play important role in host immune response modulation. Apart from the nsp–nsp stable complexes, nsp–nsp interactions are essential for the life cycle of virus. The ADRP domain of nsp3 is the key regulator of cytokine storm in COVID‐19. Although SARS‐CoV‐2 possesses unique proofreading activity for nascent RNA, mutations in the viral genome are reported in multiple genomic regions. The human antiviral defense mechanisms such as ADAR and APOBEC are also suspected to contribute in SARS‐CoV‐2 mutations.
Due to the medical emergency raised globally, most of the initial review literatures have focused on the mode of transmission, primary/intermediate host and possible therapeutic targets, epidemiology and remedies for SARS‐CoV‐2 [1, 156, 157, 158]. Astuti et al. reviewed the available structure, origin and body response to SARS‐CoV‐2 infection based on the available literature about SARS‐CoV‐2, MERS‐related CoV, and SARS‐CoVs. They further explained possible reasons for the hyper‐immune response, that is, cytokine storm generated after the infection of SARS‐CoV‐2 [131]. Anderson et al. [55] reviewed the notable features of SARS‐CoV‐2 to identify the proximal origin of the virus. In a very interesting review, Mousavizadeh et al. [157] compared the genotype and phenotype of the novel virus with other CoVs in the family to shed light on the pathogenesis of SARS‐CoV‐2. Romano et al. thoroughly reviewed the structure of SARS‐CoV‐2 replication machinery. They further utilized the data from other CoVs to understand the protein–protein interactions essential for efficient working of SARS‐CoV‐2 RNA polymerase machinery [159].
As the SARS CoV‐2 is a novel virus, data from previously well studied CoVs like SARS‐CoV were optimally used in published reviews. These reviews have served as an excellent information platform to the scientific community and sufficed the need of situation. The present article reviews the updated information available about the molecular biology of this novel virus with major emphasis on molecular and structural aspects of the SARS‐CoV‐2 genome. The crystal structures of different proteins, their interaction and their proposed roles in pathogenesis are discussed with special attention on the distinguishable components of SARS‐CoV‐2 possibly responsible for the wide spread of the virus (Table 2). The reported mutations and haplotypes are discussed with putative factors driving these mutations to provide insights on the current status of the virus evolution.
Table 2.
SARS‐CoV‐2 proteins: Distinguishing features and significances
| Region | Distinguishing feature | Significance |
|---|---|---|
| RdRp complex | N‐terminal β‐hairpin inserted into the groove clamped by the NiRAN and the palm subdomain | Stabilizes the polymerase structure |
| Spike protein | Furin cleavage site (RRAR) at the boundary of S1 and S2 subunits | Priming of the S protein increases the affinity for receptor |
| Furin cleavage site | The insertion of proline residue at the start of furin cleavage site | O‐linked glycosylation at S673, T678, and S686 resulting in strong shielding of SARS‐CoV‐2 epitopes |
| S1‐NTD | GTNGTKR motif | May assist to bind multiple receptors |
| N protein | NTD possesses distinguished surface charge distributions | Efficient binding between N protein and RNA genome |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ACKNOWLEDGMENTS
We duly acknowledge Savitribai Phule Pune University, Pune, India, for all the support provided during the lockdown period. We also thank GISAID‐Initiative (www.gisaid.org/) for granting access to large‐scale SARS‐CoV‐2 genome sequencing data. Further, we would like to show our gratitude toward all the researchers, medical practitioners, and individuals working toward understanding and combating the COVID‐19 pandemic. We would like to thank anonymous reviewers for critical assessment and constructive suggestions leading to substantial improvements in the manuscript.
Kadam SB, Sukhramani GS, Bishnoi P, Pable AA, Barvkar VT. SARS‐CoV‐2, the pandemic coronavirus: Molecular and structural insights. J Basic Microbiol. 2021;61:180–202. 10.1002/jobm.202000537
REFERENCES
- 1. Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R. COVID‐19 infection: origin, transmission, and characteristics of human coronaviruses. J Adv Res. 2020;24:91‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corman VM, Lienau J, Witzenrath M. Coronaviruses as the cause of respiratory infections. Internist (Berl). 2019;60:1136‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Gulyaeva AA, Haagmans BL, et al. The species and its viruses—a statement of the coronavirus study group. bioRxiv. 2020. 10.1101/2020.02.07.937862 [DOI] [Google Scholar]
- 6. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, et al. Genome composition and divergence of the novel coronavirus (2019‐nCoV) originating in China. Cell Host Microbe. 2020;27:325‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuo L, Masters PS, Vennema H, Rottier PJM. Coronavirus particle assembly: primary structure requirements of the membrane protein. J Virol. 1998;72:6838‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Digard P, Lee HM, Sharp C, Grey F, Gaunt E. Intra‐genome variability in the dinucleotide composition of SARS‐CoV‐2. Virus Evol. 2020;6(2):veaa057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simmonds P. Pervasive RNA secondary structure in the genomes of SARS‐CoV‐2 and other coronaviruses. mBio. 2020;11(6):e01661–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim D, Lee J, Yang J, Kim JW, Kim VN, Chang H. The architecture of SARS‐CoV‐2 transcriptome. Cell. 2020;181:914‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bridges R, Correia S, Wegner F, Venturini C, Palser A, White RE, et al. Essential role of inverted repeat in Epstein‐Barr virus IR‐1 in B cell transformation; geographical variation of the viral genome. Philos Trans R Soc B Biol Sci. 2019;374:20180299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dutkiewicz M, Stachowiak A, Swiatkowska A, Ciesiołka J. Structure and function of RNA elements present in enteroviral genomes. Acta Biochim Pol. 2016;63:623‐30. [DOI] [PubMed] [Google Scholar]
- 14. Miao Z, Tidu A, Eriani G, Martin F. Secondary structure of the SARS‐CoV‐2 5ʹ‐UTR. RNA Biol. 2020. 1‐10. 10.1080/15476286.2020.1814556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bartas M, Brázda V, Bohálová N, Cantara A, Volná A, Stachurová T, et al. In‐depth bioinformatic analyses of Nidovirales including human viruses suggest important roles of non‐canonical nucleic acid structures in their lifecycles. Front Microbiol. 2020;11:1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kelly JA, Olson AN, Neupane K, Munshi S, San Emeterio J, Pollack L, et al. Structural and functional conservation of the programmed‐1 ribosomal frameshift signal of SARS coronavirus 2 (SARS‐CoV‐2). J Biol Chem. 2020;295:10741‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rangan R, Zheludev IN, Das R. RNA genome conservation and secondary structure in SARS‐CoV‐2 and SARS‐related viruses: a first look. RNA. 2020;26:937‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang R, Ke X, Gu Y, Liu H, Sun X. Whole genome identification of potential G‐quadruplexes and analysis of the G‐quadruplex binding domain for SARS‐CoV‐2. bioRxiv. 2020. 10.1101/2020.06.05.135749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ji D, Juhas M, Tsang CM, Kwok CK, Li JS, Zhang Y. Discovery of G‐quadruplex‐forming sequences in SARS‐CoV‐2. Brief Bioinform. 2020. 10.1093/bib/bbaa114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pereira F. Evolutionary dynamics of the SARS‐CoV‐2 ORF8 accessory gene. Infect Genet Evol. 2020;85:104525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vandelli A, Monti M, Milanetti E, Armaos A, Rupert J, Zacco E, et al. Structural analysis of SARS‐CoV‐2 genome and predictions of the human interactome. Nucleic Acids Res. 2020;48:11270‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schubert K, Karousis ED, Jomaa A, Scaiola A, Echeverria B, Gurzeler LA, et al. SARS‐CoV‐2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol. 2020;27:959‐66. [DOI] [PubMed] [Google Scholar]
- 23. Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS‐CoV‐2. Science. 2020;369:1249‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cornillez‐Ty CT, Liao L, Yates JR, Kuhn P, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol. 2009;83:10314‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Claverie JM. A putative role of de‐mono‐ADP‐ribosylation of STAT1 by the SARS‐CoV‐2 Nsp3 protein in the cytokine storm syndrome of COVID‐19. Viruses. 2020;12:646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alhammad YMO, Fehr AR. The viral macrodomain counters host antiviral ADP‐ribosylation. Viruses. 2020;12:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Michalska K, Kim Y, Jedrzejczak R, Maltseva NI, Stols L, Endres M, et al. Crystal structures of SARS‐CoV‐2 ADP‐ribose phosphatase (ADRP): from the apo form to ligand complexes. IUCrJ. 2020;7:814‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chang YS, Ko BH, Ju JC, Chang HH, Huang SH, Lin CW. SARS unique domain (SUD) of severe acute respiratory syndrome coronavirus induces NLRP3 inflammasome‐dependent CXCL10‐mediated pulmonary inflammation. Int J Mol Sci. 2020;21:3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved α‐ketoamide inhibitors. Science. 2020;368:409‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lehmann KC, Gulyaeva A, Zevenhoven‐dobbe JC, Janssen GMC, Ruben M, Overkleeft HS, et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase‐containing protein of all nidoviruses. Nucleic Acids Res. 2015;43:8416‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, et al. Structure of the RNA‐dependent RNA polymerase from COVID‐19 virus. Science. 2020;368:779‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Konkolova E, Klima M, Nencka R, Boura E. Structural analysis of the putative SARS‐CoV‐2 primase complex. J Struct Biol. 2020;211:107548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kirchdoerfe RN, Ward AB. Structure of the SARS‐CoV nsp12 polymerase bound to nsp7 and nsp8 co‐factors. Nat Commun. 2019;10:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim Y, Jedrzejczak R, Maltseva NI, Wilamowski M, Endres M, Godzik A, et al. Crystal structure of Nsp15 endoribonuclease NendoU from SARS‐CoV‐2. Protein Sci. 2020;29:1596‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma Y, Wu L, Shaw N, Gao Y, Wang J, Sun Y, et al. Structural basis and functional analysis of the SARS coronavirus nsp14‐nsp10 complex. Proc Natl Acad Sci U S A. 2015;112:9436‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jia Z, Yan L, Ren Z, Wu L, Wang J, Guo J, et al. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47:6538‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lemus MR, Minasov G, Shuvalova L, Inniss NL, Kiryukhina O, Wiersum G, et al. The crystal structure of nsp10‐nsp16 heterodimer from SARS CoV‐2 in complex with S‐adenosylmethionine. bioRxiv. 2020. 10.1101/2020.04.17.047498 [DOI] [Google Scholar]
- 38. Frieman M, Yount B, Agnihothram S, Page C, Donaldson E, Roberts A, et al. Molecular determinants of severe acute respiratory syndrome coronavirus pathogenesis and virulence in young and aged mouse models of human disease. J Virol. 2012;86:884‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Littler DR, Gully BS, Colson RN, Rossjohn J. Crystal structure of the SARS‐CoV‐2 non‐structural protein 9 Nsp9. IScience. 2020;23:101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lai MMC, Cavanaght D. The molecular biology of coronaviruses. In: Maramorosch K, Murphy FA, Shatkin AJ, editors. Advances in virus research. New York, NY: Academic Press; 1997. p. 1‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, et al. Genome composition and divergence of the novel coronavirus (2019‐nCoV) originating in China. Cell Host Microbe. 2020;27:325‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Narayanan K, Huang C, Makino S. SARS coronavirus accessory proteins. Virus Res. 2008;133:11321‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song HD, Tu CC, Zhang GW, Wang SY, Zheng K, Lei LC, et al. Cross‐host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci U S A. 2005;102:2430‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yuan M, Wu NC, Zhu X, Lee CCD, So RTY, Lv H, et al. A highly conserved cryptic epitope in the receptor‐binding domains of SARS‐CoV‐2 and SARS‐CoV. Science. 2020;368:630‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3:237‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yao H, Song Y, Chen Y, Wu N, Xu J, Sun C, et al. Molecular architecture of the SARS‐CoV‐2 virus. Cell. 2020;183:730‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ke Z, Oton J, Qu K, Cortese M, Zila V, McKeane L, et al. Structures and distributions of SARS‐CoV‐2 spike proteins on intact virions. Nature. 2020;588:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581:215‐20. [DOI] [PubMed] [Google Scholar]
- 49. Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M. Site‐specific analysis of the SARS‐CoV‐2 glycan shield. Science. 2020;369:330‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019‐nCoV contains a furin‐like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jahn R, Sudhof T. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777‐810. [DOI] [PubMed] [Google Scholar]
- 52. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell. 2020;181:281‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bagdonaite I, Wandall HH. Global aspects of viral glycosylation. Glycobiology. 2018;28:443‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367:1260‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS‐CoV‐2. Nat Med. 2020;26:450‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shajahan A, Supekar NT, Gleinich AS, Azadi P. Deducing the N‐and O‐glycosylation profile of the spike protein of novel coronavirus SARS‐CoV‐2. Glycobiology. 2020;30:981‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lemmin T, Kalbermatter D, Harder D, Plattet P, Fotiadis D. Structures and dynamics of the novel S1/S2 protease cleavage site loop of the SARS‐CoV‐2 spike glycoprotein. J Struct Biol: X. 2020;4:100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Behloul N, Baha S, Shi R, Meng J. Role of the GTNGTKR motif in the N‐terminal receptor‐binding domain of the SARS‐CoV‐2 spike protein. Virus Res. 2020;286:198058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ou J, Zhou Z, Zhang J, Lan W, Zhao S, Wu J, et al. RBD mutations from circulating SARS‐CoV‐2 strains enhance the structure stability and infectivity of the spike protein. bioRxiv. 2020. 10.1101/2020.03.15.991844 [DOI] [Google Scholar]
- 60. Sheahan T, Rockx B, Donaldson E, Sims A, Pickles R, Corti D, et al. Mechanisms of zoonotic severe acute respiratory syndrome coronavirus host range expansion in human airway epithelium. J Virol. 2008;82:2274‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li F. Receptor recognition and cross‐species infections of SARS coronavirus. Antiviral Res. 2013;100:246‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu Z, Xiao X, Wei X, Li J, Yang J, Tan H, et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS‐CoV‐2. J Med Virol. 2020;92:595‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lam TTY, Jia N, Zhang YW, Shum MHH, Jiang JF, Zhu HC, et al. Identification of 2019‐nCoV related coronaviruses in Malayan pangolins in southern China. Nature. 2020;583:282‐5. [DOI] [PubMed] [Google Scholar]
- 64. Alam I, Kamau AA, Kulmanov M, Jaremko Ł, Arold ST, Pain A, et al. Functional pangenome analysis suggests inhibition of the protein E as a readily available therapy for COVID‐2019. Front Cell Infect Microbiol. 2020;10:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wilson L, Mckinlay C, Gage P, Ewart G. SARS coronavirus E protein forms cation‐selective ion channels. Virology. 2004;330:322‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nieva JL, Madan V, Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol. 2012;10:563‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang R, Wang K, Lv W, Yu W, Xie S, Xu K, et al. The ORF4a protein of human coronavirus 229E functions as a viroporin that regulates viral production. Biochim Biophys Acta–Biomembr. 2014;1838:1088‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liao Y, Tam JP, Liu DX. Viroporin activity of SARS‐CoV E protein. Adv Exp Med Biol. 2006;581:199‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pham T, Perry JL, Dosey TL, Delcour AH, Hyser JM. The rotavirus NSP4 viroporin domain is a calcium‐conducting ion channel. Sci Rep. 2017;7:43487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Duart G, García‐Murria MJ, Grau B, Acosta‐Cáceres JM, Martínez‐Gil L, Mingarro I. SARS‐CoV‐2 envelope protein topology in eukaryotic membranes. Open Biol. 2020;10:200209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu Q, Zhang Y, Lü H, Wang J, He X, Liu Y, et al. The E protein is a multifunctional membrane protein of SARS‐CoV. J Genomics Proteomics. 2003;1:131‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fung TS, Liu DX. Coronavirus infection, ER stress, apoptosis and innate immunity. Front Microbiol. 2014;5:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. DeDiego ML, Nieto‐Torres JL, Jiménez‐Guardeño JM, Regla‐Nava JA, Álvarez E, Oliveros JC, et al. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLOS Pathog. 2011;7(10):e1002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jiang S, Hillyer C, Du L. Neutralizing antibodies against SARS‐CoV‐2 and other human coronaviruses. Trends Immunol. 2020;41:355‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Arndt AL, Larson BJ, Hogue BG. A conserved domain in the coronavirus membrane protein tail is important for virus assembly. J Virol. 2010;84:11418‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bianchi M, Benvenuto D, Giovanetti M, Angeletti S, Pascarella S. SARS‐CoV‐2 envelope and membrane proteins: differences from closely related proteins linked to cross‐species transmission? BioMed Res Int. 2020;2020:1‐6. 10.20944/preprints202004.0089.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. de Haan CAM, Vennema H, Rottier PJM. Assembly of the coronavirus envelope: homotypic interactions between the M proteins. J Virol. 2000;74:4967‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Narayanan K, Chen CJ, Maeda J, Makino S. Nucleocapsid‐independent specific viral RNA packaging via viral envelope protein and viral RNA signal. J Virol. 2003;77:2922‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hu Y, Wen J, Tang L, Zhang H, Zhang X, Li Y, et al. The M protein of SARS‐CoV: basic structural and immunological properties. Genomics Proteomics Bioinformatics. 2003;1:118‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fang X, Gao J, Zheng H, Li B, Kong L, Zhang Y, et al. The membrane protein of SARS‐CoV suppresses NF‐κB activation. Antivir Ther. 2007;79:1431‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Thomas S. The structure of the membrane protein of SARS‐CoV‐2 resembles the sugar transporter semiSWEET. Pathog Immunity. 2020;5:342‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kang S, Yang M, Hong Z, Zhang L, Huang Z, Chen X, et al. Crystal structure of SARS‐CoV‐2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm Sin B. 2020;10:1228‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Surjit M, Lal SK. The SARS‐CoV nucleocapsid protein: a protein with multifarious activities. Infect Genet Evol. 2008;8:397‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. McBride R, van Zyl M, Fielding BC. The coronavirus nucleocapsid is a multifunctional protein. Viruses. 2014;6:2991‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kumar A, Parveen A, Kumar N, Bairy S, Kaushik V, Chandola C, et al. Characterization of nucleocapsid (N) protein from novel coronavirus SARS‐CoV‐2. Preprints. 2020. 10.20944/preprints202005.0413.v1 [DOI] [Google Scholar]
- 86. Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol Biol Evol. 2015;32:268‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Su Q, Yi Y, Zou Y, Jia Z, Qiu F, Wang F, et al. The biological characteristics of SARS‐CoV‐2 spike protein Pro330‐Leu650. Vaccine. 2020;38:5071‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan. J Virol. 2020;94:e00127–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li X, Zai J, Zhao Q, Nie Q, Li Y, Foley BT, et al. Evolutionary history, potential intermediate animal host, and cross‐species analyses of SARS‐CoV‐2. J Med Virol. 2020;92:602‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID‐19) outbreak—an update on the status. Mil Med Res. 2020;7:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lau SKP, Luk HKH, Wong ACP, Li KSM, Zhu L, He Z, et al. Possible bat origin of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis. 2020;26:1542‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Poon LLM, Chu DKW, Chan KH, Wong OK, Ellis TM, Leung YHC, et al. Identification of a novel coronavirus in bats. J Virol. 2005;79:2001‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fan Y, Zhao K, Shi ZL, Zhou P. Bat coronaviruses in China. Viruses. 2019;11:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang YZ, Holmes EC. A genomic perspective on the origin and emergence of SARS‐CoV‐2. Cell. 2020;181:223‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nie Q, Li X, Chen W, Liu D, Chen Y, Li H, et al. Phylogenetic and phylodynamic analyses of SARS‐CoV‐2. Virus Res. 2020;287:198098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor‐binding domain complexed with receptor. Science. 2005;309:1864‐8. [DOI] [PubMed] [Google Scholar]
- 99. Ji W, Wang W, Zhao X, Zai J, Li X. Cross‐species transmission of the newly identified coronavirus 2019‐nCoV. J Med Virol. 2020;92:433‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bahir I, Fromer M, Prat Y, Linial M. Viral adaptation to host: a proteome‐based analysis of codon usage and amino acid preferences. Mol Syst Biol. 2009;5:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang C, Zheng W, Huang X, Bell EW, Zhou X, Zhang Y. Protein structure and sequence reanalysis of 2019‐nCoV genome refutes snakes as its intermediate host and the unique similarity between its spike protein insertions and HIV‐1. J Proteome Res. 2020;19:1351‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Xiao K, Zhai J, Feng Y, Zhou N, Zhang X, Zou JJ, et al. Isolation and characterization of 2019‐nCoV‐like coronavirus from Malayan pangolins. bioRxiv. 2020. 10.1101/2020.02.17.951335 [DOI] [Google Scholar]
- 103. Wahba L, Jain N, Fire AZ, Shoura MJ, Artiles KL, McCoy MJ, et al. An extensive meta‐metagenomic search identifies SARS‐CoV‐2‐homologous sequences in pangolin lung viromes. mSphere. 2020;5(3):e00160–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS‐CoV‐2 associated with the COVID‐19 outbreak. Curr Biol. 2020;30:1346‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wong MC, Cregeen SJJ, Ajami NJ, Petrosino JF. Evidence of recombination in coronaviruses implicating pangolin origins of nCoV‐2019. bioRxiv. 2020. 10.1101/2020.02.07.939207 [DOI] [Google Scholar]
- 106. Boni MF, Lemey P, Jiang X, Lam TTY, Perry BW, Castoe TA, et al. Evolutionary origins of the SARS‐CoV‐2 sarbecovirus lineage responsible for the COVID‐19 pandemic. Nat Microbiol. 2020;5:1408‐17. [DOI] [PubMed] [Google Scholar]
- 107. Zhang H, Kang Z, Gong H, Xu D, Wang J, Li Z, et al. The digestive system is a potential route of 2019‐nCov infection: a bioinformatics analysis based on single‐cell transcriptomes. bioRxiv. 2020. 10.1101/2020.01.30.927806 [DOI] [Google Scholar]
- 108. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single‐cell RNA expression profiling of ACE2, the receptor of SARS‐CoV‐2. Am J Respir Crit Care Med. 2020;202:756‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single‐cell RNA‐seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019‐nCoV infection. Front Med. 2020;14:185‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Xu H, Zhong L, Deng J, Peng J, Dan H, Zeng X, et al. High expression of ACE2 receptor of 2019‐nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhao X, Chen D, Szabla R, Zheng M, Li G, Du P, et al. Broad and differential animal angiotensin‐converting enzyme. J Virol. 2020;94:e00940–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hussain M, Jabeen N, Amanullah A, Ashraf Baig A, Aziz B, Shabbir S, et al. Structural basis of SARS‐CoV‐2 spike protein priming by TMPRSS2. AIMS Microbiol. 2020;6:350‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ziegler C, Allon SJ, Nyquist SK, Mbano I, Miao VN, Cao Y, et al. SARS‐CoV‐2 Receptor ACE2 is an interferon‐stimulated gene in human airway epithelial cells and is enriched in specific cell subsets across tissues. SSRN Electron J. 2020;181:1016‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Lukassen S, Chua RL, Trefzer T, Kahn NC, Schneider MA, Muley T, et al. SARS‐CoV‐2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020;18:e105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S, et al. Inhibition of SARS‐CoV‐2 (previously 2019‐nCoV) infection by a highly potent pan‐coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Chen SC, Olsthoorn RCL. Group‐specific structural features of the 5′‐proximal sequences of coronavirus genomic RNAs. Virology. 2010;401:29‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Nakagawa K, Lokugamage KG, Makino S. Viral and cellular mRNA translation in coronavirus‐infected cells. Adv Virus Res. 2016;96:165‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Peng Q, Peng R, Yuan B, Zhao J, Wang M, Wang X, et al. Structural and biochemical characterization of the nsp12‐nsp7‐nsp8 core polymerase complex from SARS‐CoV‐2. Cell Rep. 2020;31:107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wang Q, Wu J, Wang H, Gao Y, Liu Q, Mu A, et al. Structural basis for RNA replication by the SARS‐CoV‐2 Polymerase. Cell. 2020;182:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, Cramer P. Structure of replicating SARS‐CoV‐2 polymerase. Nature. 2020;584:154‐6. [DOI] [PubMed] [Google Scholar]
- 122. Hackbart M, Deng X, Baker SC. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc Natl Acad Sci U S A. 2020;117:8094‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Li Y, Yang X, Wang N, Wang H, Yin B, Yang X, et al. GC usage of SARS‐CoV‐2 genes might adapt to the environment of human lung expressed genes. Mol Genet Genomics. 2020;295:1537‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hou W. Characterization of codon usage pattern in SARS‐CoV‐2. Virol J. 2020;17:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Finkel Y, Mizrahi O, Nachshon A, Weingarten‐Gabbay S, Morgenstern D, Yahalom‐Ronen Y, et al. The coding capacity of SARS‐CoV‐2. Nature. 2020;589:125‐30. 10.1038/s41586-020-2739-1 [DOI] [PubMed] [Google Scholar]
- 126. Krijnse‐Locker J, Ericsson M, Rottier PJM, Griffiths G. Characterization of the budding compartment of mouse hepatitis virus: evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J Cell Biol. 1994;124:55‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kuo L, Masters PS. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J Virol. 2002;76:4987‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ujike M, Huang C, Shirato K, Makino S, Taguchi F. The contribution of the cytoplasmic retrieval signal of severe acute respiratory syndrome coronavirus to intracellular accumulation of S proteins and incorporation of S protein into virus‐like particle. J Gen Virol. 2016;97:1853‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92:418‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Jack A, Ferro LS, Trnka MJ, Wehri E, Nadgir A, Costa K, et al. SARS CoV‐2 nucleocapsid protein forms condensates with viral genomic RNA. bioRxiv. 2020. 10.1101/2020.09.14.295824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Astuti I, Ysrafil. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2): an overview of viral structure and host response. Diabetes Metab Syndr. 2020;14:407‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Marquez‐Miranda V, Rojas M, Duarte Y, Diaz‐Franulic I, Holmgren M, Cachau RE, et al. Analysis of SARS‐CoV‐2 ORF3a structure reveals chloride binding sites. bioRxiv. 2020. 10.1101/2020.10.22.349522 [DOI] [Google Scholar]
- 133. Stern A, Andino R. Viral evolution: it is all about mutations. In: Katze M, Korth M, Law GL, Nathanson N, editors. Viral pathogenesis. New York, NY: Academic Press; 2016. p. 233‐40. [Google Scholar]
- 134. Minskaia E, Hertzig T, Gorbalenya AE, Campanacci V, Cambillau C, Canard B, et al. Discovery of an RNA virus 3′ → 5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc Natl Acad Sci U S A. 2006;103:5108‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Eckerle LD, Becker MM, Halpin RA, Li K, Venter E, Lu X, et al. Infidelity of SARS‐CoV Nsp14‐exonuclease mutant virus replication is revealed by complete genome sequencing. PLOS Pathog. 2010;6:e1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Jones LR, Manrique JM. Quantitative phylogenomic evidence reveals a spatially structured SARS‐CoV‐2 diversity. Virology. 2020;550:70‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Prathiviraj R, Kiran GS, Selvin J. Phylogenomic proximity and comparative proteomic analysis of SARS‐CoV‐2. Gene Reports. 2020;20:100777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Li X, Wang W, Zhao X, Zai J, Zhao Q, Li Y, et al. Transmission dynamics and evolutionary history of 2019‐nCoV. J Med Virol. 2020;92:501‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the origin and continuing evolution of SARS‐CoV‐2. Natl Sci Rev. 2020;7:1012‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, et al. Spike mutation D614G alters SARS‐CoV‐2 fitness. Nature. 2020. 10.1038/s41586-020-2895-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hodcroft EB, Zuber M, Nadeau S, Comas I, Candelas FG, Stadler T, et al. Emergence and spread of a SARS‐CoV‐2 variant through Europe in the summer of 2020. medRxiv. 2020. 10.1101/2020.10.25.20219063 [DOI] [Google Scholar]
- 142. Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, et al. The establishment of reference sequence for SARS‐CoV‐2 and variation analysis. J Med Virol. 2020;92:667‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Phan T. Genetic diversity and evolution of SARS‐CoV‐2. Infect Genet Evol. 2020;81:104260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. van Dorp L, Acman M, Richard D, Shaw LP, Ford CE, Ormond L, et al. Emergence of genomic diversity and recurrent mutations in SARS‐CoV‐2. Infect Genet Evol. 2020;83:104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rahman MS, Islam MR, Alam ASMRU, Islam I, Hoque MN, Akter S, et al. Evolutionary dynamics of SARS‐CoV‐2 nucleocapsid (N) protein and its consequences. J Med Virol. 2020. jmv.26626. 10.1002/jmv.26626 [DOI] [PubMed] [Google Scholar]
- 146. Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al. Emerging SARS‐CoV‐2 mutation hot spots include a novel RNA‐dependent‐RNA polymerase variant. J Transl Med. 2020;18:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Morais IJ, Polveiro RC, Souza GM, Bortolin DI, Sassaki FT, Lima ATM. The global population of SARS‐CoV‐2 is composed of six major subtypes. Sci Rep. 2020;10:18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Gaurav S, Pandey S, Puvar A, Shah T, Joshi M, Joshi C, et al. Identification of unique mutations in SARS‐CoV‐2 strains isolated from India suggests its attenuated pathotype. bioRxiv. 2020. 10.1101/2020.06.06.137604 [DOI] [Google Scholar]
- 149. Wu S, Tian C, Liu P, Guo D, Zheng W, Huang X, et al. Effects of SARS‐CoV‐2 mutations on protein structures and intraviral protein–protein interactions. J Med Virol. 2020. 1‐9. 10.1002/jmv.26597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Rad AH, McLellan AD. Implications of SARS‐CoV‐2 mutations for genomic RNA structure and host microRNA targeting. Int J Mol Sci. 2020;21:4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Simmonds P. Rampant C → U hypermutation in the genomes of SARS‐CoV‐2 and other coronaviruses—causes and consequences for their short and long evolutionary trajectories. mSphere. 2020;5:e00408–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Chan JFW, Kok KH, Zhu Z, Chu H, To KKW, Yuan S, et al. Genomic characterization of the 2019 novel human‐pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Wang H, Pipes L, Nielsen R. Synonymous mutations and the molecular evolution of SARS‐Cov‐2 origins. Virus Evolution. 2020;6:veaa098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Di Giorgio S, Martignano F, Torcia MG, Mattiuz G, Conticello SG. Evidence for host‐dependent RNA editing in the transcriptome of SARS‐CoV‐2. Sci Adv. 2020;6:eabb5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Sanjuán R, Domingo‐Calap P. Mechanisms of viral mutation. Cell Mol Life Sci. 2016;73:4433‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Li H, Zhou Y, Zhang M, Wang H, Zhao Q, Liu J. Updated approaches against SARS‐CoV‐2. Antimicrob Agents Chemother. 2020;64:e00483–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID‐19: their roles in pathogenesis. J Microbiol Immunol Infect. 2020. 10.1016/j.jmii.2020.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and coronavirus disease‐2019 (COVID‐19): the epidemic and the challenges. Int J Antimicrob Agents. 2020;55:105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R. A Structural view of SARS‐CoV‐2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells. 2020;9:1267. [DOI] [PMC free article] [PubMed] [Google Scholar]