

Abstract
We investigate binding of linoleate and other potential ligands to the recently discovered fatty acid binding site in the SARS‐CoV‐2 spike protein, using docking and molecular dynamics simulations. Simulations suggest that linoleate and dexamethasone stabilize the locked spike conformation, thus reducing the opportunity for ACE2 interaction. In contrast, cholesterol may expose the receptor‐binding domain by destabilizing the closed structure, preferentially binding to a different site in the hinge region of the open structure. We docked a library of FDA‐approved drugs to the fatty acid site using an approach that reproduces the structure of the linoleate complex. Docking identifies steroids (including dexamethasone and vitamin D); retinoids (some known to be active in vitro, and vitamin A); and vitamin K as potential ligands that may stabilize the closed conformation. The SARS‐CoV‐2 spike fatty acid site may bind a diverse array of ligands, including dietary components, and therefore provides a promising target for therapeutics or prophylaxis.
Keywords: molecular modeling, retinoids, SARS-CoV-2 spike fatty-acid-site, steroids, vitamins
Vitamin K2 docked at the fatty acid site in the closed SARS‐CoV‐2 spike protein. Simulations indicate that ligands binding at this site stabilize the closed spike conformation.
Introduction
The global COVID‐19 pandemic is caused by the SARS‐CoV‐2 coronavirus. Eleven months since the first infections were reported, over 72 million people worldwide have become infected, with more than 1.6 million deaths in confirmed cases. [1] Initially thought to be a predominantly respiratory disease, its effects are not limited to the respiratory tract, and in severe cases extensive organ damage and death can result from for example, a virus‐induced cytokine storm, [2] blood clots or organ failure. Even in less severe cases of COVID‐19, there is now evidence that infection can cause damage to heart muscle: for example, in a recent study, [3] MRI scans of the heart tissue of 100 recovered COVID‐19 subjects found that 78 people, including those who had not been admitted to hospital, had sustained persistent heart damage, irrespective of the severity of their respiratory symptoms. Notwithstanding the small size of that study, and the as yet unknown extent of any tissue recovery, these findings suggest implications for reduced longevity in a “COVID‐recovered” population. Quality of life has also been adversely affected in the growing numbers of people described as suffering from “long COVID”, with some suffering neurological symptoms including fibromyalgia and profound fatigue, lasting for months after their initial illness. [4] Indeed, a recent longitudinal study of COVID‐recovered, symptomatic individuals, published by the COVERSCAN study investigators, reveals that in this relatively young, low risk population 70 % had impaired organ function 4 months later. [5] Treatment regimens to attenuate initial infection, to reduce the severity of disease, and prevent its residual effects, are an urgent and growing medical need.
The surface of SARS‐CoV‐2 virus particles is decorated with a characteristic “crown” of spikes, formed of trimers of the spike (S) protein, which enables viral entry into human cells via receptor‐mediated plasma membrane fusion, after priming by the host proteases furin and TMPRSS2.[ 6 , 7 ] The binding interaction occurs between the spike receptor binding domain (RBD) and the cell surface angiotensin‐converting enzyme‐2 (ACE2) receptor. A furin protease cleavage site potentiates the infectivity of SARS‐CoV‐2, [8] but mutations in and around the SARS‐CoV‐2 furin site are sometimes found in clinical isolates.[ 9 , 10 ] Laboratory passaged SARS‐CoV‐2 lacking a functional furin cleavage site fails to gain entry via membrane fusion via ACE2, and is instead capable of infection via ACE2‐mediated endocytosis, albeit less efficiently, involving proteins such as NPC intracellular cholesterol transporter 1 (NPC1) and 2 that, are involved in endosomal trafficking and membrane fusion. [11] Structural studies have revealed that the spike adopts a mixed population of conformational states.[ 12 , 13 , 14 , 15 ]
We recently [12] showed that the spike trimer contains a fatty acid (FA) binding site situated at each of the three interfaces between two RBDs from adjacent subunits: in the resulting “locked” conformers, inter‐subunit contacts between RBDs are maximized (Figure 1) such that a molecule of linoleate (LA−), a nutritionally essential dietary FA, is bound in each of three binding sites of the spike trimer. The K d of LA− for the isolated SARS‐CoV‐2 spike RBD has been determined by surface plasmon resonance as ≈41 nM. [12] The affinity of LA− for the (physiological) spike trimer is likely to be higher than this, given that the interactions with the residues R408, Q409 and K417 from the adjacent chain in the trimer (see below) are not available to the isolated RBD.
Figure 1.
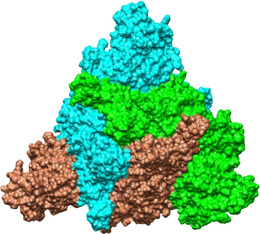
Inter‐subunit contacts between RBDs are maximized in the locked spike. This top view of the SARS‐CoV‐2 spike trimer with three linoleate molecules bound following 200 ns of MD simulation of the 6ZB5 PDB structure, illustrates the close contacts between the subunits. Individual chains are shown in surface representation and are colored cyan, green and brown.
In the “open” state of the spike trimer, one of the ACE2‐binding domains (RBD) adopts a more extended, solvent exposed conformation. Crucially, the ACE2 interaction surfaces of the RBDs are occluded in the locked state and only available for receptor interaction in the open states (see Figure 1).
The spike protein FA binding site [12] lies adjacent to a flexible hinge region beneath each of the RBDs and at its interface with neighboring subunits. This hinge is believed to allow the large movement required to extend the RBD into an open conformation capable of binding ACE2. LA− is bound with its hydrophobic tail nestled in a hydrophobic groove on the underside of the RBD from one chain, while the carboxylate head group is within salt‐bridging distance of two charged residues, R408 and K417, and hydrogen‐bonding distance of Q409 on the adjacent RBD chain. The extensive LA− interactions, involving two RBDs, in these locked structures support the conclusion that LA− binding stabilizes the locked state of the spike trimer. This proposal is supported by our observations of EM grids, where images suggest that ≈70 % of spike trimers on the EM grid adopt the locked form if exposed to LA−, whereas only 30 % are closed (all RBDs down) if fatty acids are lacking. [12] N.B. the possibility that linoleate (and possibly other endogenous ligands) binds at the fatty acid site during folding/assembly of the spike cannot be ruled out.
These observations led us to hypothesize that stabilizing the locked (ACE2‐interaction‐face occluded) conformation of the spike protein reduces the opportunity for receptor‐mediated cell entry via interaction with ACE2. [12] The spike protein FA pocket represents a potentially druggable site: this stabilization might therefore also be achieved by binding of high‐affinity small molecule ligands: such ligands might potentially reduce cellular ingress by maintaining the spike protein in the locked conformation incompetent for ACE2 interaction. This proposal is supported by our finding that prior exposure to linoleate reduces SARS‐CoV‐2 replication in human epithelial cells. [12] Also we note that a recent study seeking biomarkers for COVID‐19 found that disease severity was associated with lower circulating levels of polyunsaturated fatty acids, such as linoleic acid. [16]
Various drug targeting strategies, including extensive computational efforts, sometimes as part of extensive, open science collaborative programs (e.g. the COVID Moonshot project) have been employed to design both competitive and covalent inhibitors for the SARS‐CoV‐2 targets, notably the main protease. [17] While there have been intensive efforts to target several SARS‐CoV‐2 proteins,[ 18 , 19 , 20 , 21 ] efforts to modulate the function of the spike protein are to our knowledge more limited, although covalent stabilisation of the closed form of the spike for example, by introduction of disulphide bonds [22] is relevant for vaccine development as well as, potentially, small molecule therapeutic development. Molecular simulations have also been applied to study the behavior of the spike protein in both its glycosylated,[ 22 , 23 , 24 ] and unglycosylated states,[ 12 , 25 , 26 ] exploring for example its interactions with receptors in addition to ACE2, including neuropilin [25] and potentially, nicotinic acetylcholine receptors. [26]
In the light of these findings, we here investigate binding of LA− and other potential ligands at the SARS‐CoV‐2 spike protein FA binding site, using molecular dynamics (MD) simulations to explore the dynamic properties of the spike trimer in the “apo” (unliganded) state and in the LA−‐bound (“holo”) form in the locked and open conformations. Motivated by reports that the SARS‐CoV‐2 spike protein binds cholesterol, [27] we also investigate the interactions of cholesterol with the FA binding site. Furthermore, as cholesterol and the anti‐inflammatory steroid dexamethasone (an agent demonstrated to improve outcomes for severely ill SARS‐CoV‐2 patients) [28] are chemically and structurally similar, we introduced dexamethasone into the FA binding site to establish whether it too can be accommodated and retained there. Lastly, we used the Bristol University Docking Engine (BUDE) [29] to screen an approved drug library for candidate small molecule ligands that may bind to the spike protein LA− binding site in the locked conformation, and thus potentially stabilize the spike in a form that is less competent for ACE2 interaction. Virtual screening using BUDE identifies multiple compound classes, including naturally occurring and synthetic retinoids and steroids and the fat‐soluble vitamins D, K and A, as candidate ligands for the spike FA binding site. Our data suggest that a range of small molecule ligands, including pharmaceuticals and dietary components, and some compounds with known activity against SARS‐CoV‐2 replication in vitro, may bind to the SARS‐CoV‐2 spike protein at this site, and may thereby be capable of influencing the conformational changes required for receptor binding, potentially reducing viral infectivity.
Results
Molecular Dynamics Simulations of Ligand Complexes of the Locked Form of the Spike
To investigate interactions of the FA binding site with small molecule ligands, and the effects of the ligands on the spike and its dynamics, we performed MD simulations of the apo spike protein (i.e. no ligand bound at the FA site) and of its complexes with the deprotonated, charged form of linoleic acid (as it would predominantly be at physiological pH) linoleate (LA−), cholesterol (previously proposed to bind to the SARS‐CoV‐2 spike [27] ), and the chemically and structurally similar anti‐inflammatory agent dexamethasone (Figure 2). Dexamethasone is increasingly widely used as a therapeutic intervention for SARS‐CoV‐2‐infected patients. [28] This work builds on our previously reported MD studies of LA− binding: [12] here, we significantly extend the duration of simulations of the spike LA− complex and also expand our investigations to include other possible ligands.
Figure 2.
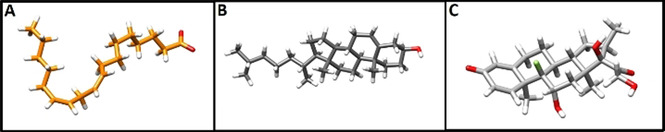
Structures of A) linoleate; B) cholesterol; and C) dexamethasone from equilibrated MD simulations of complexes with the locked SARS‐CoV‐2 spike. Oxygen atoms are shown in red, dexamethasone fluorine atom in green.
In 200 ns MD simulations of the locked structures, starting from the cryo‐EM structure (PDB code 6ZB5 [12] ), the surrounding residues are ordered around the bound LA− ligand (Figure 3 A). The LA− molecules themselves (Figure 4 A) move very little within the FA binding site (RMSD ≤3 Å). Hydrogen bonds of the LA− carboxylate head group with Q409, and salt bridge contacts with R408, are largely maintained; when the latter salt bridge is lost it is replaced by interactions with K417 (Figure S1). Comparison of overall Cα RMSD and by‐residue RMSF plots of the uncomplexed and LA−‐bound locked forms of the spike show that, while, on the timescale of these simulations, the overall dynamic properties of the spike trimer as a whole are little affected by LA− binding (Figure 4 D, E), the presence of ligand rigidifies the FA binding site (residues 330–480) as evidenced by the reduced fluctuations of residues surrounding the ligand (Figure 4 H, I) and has an effect that extends towards the N‐terminus as far as residue 310. In contrast, in simulations of the uncomplexed apo spike protein in the locked form, the FA site exhibits larger fluctuations (Figure 4 H) that lead to its eventual collapse.
Figure 3.
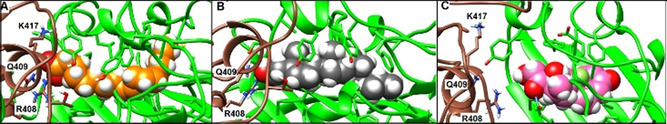
Representative structures of bound linoleate, cholesterol and dexamethasone after 200 ns of MD simulations of complexes with the locked spike trimer. Brown and green ribbons denote different chains of the spike trimer (same color coding used throughout). A) Representative structure of bound linoleate (spheres, carbon atoms orange) after 200 ns MD simulation starting from the cryo‐EM structure (PDB code 6ZB5). Note the interactions of the linoleate head group with R408, Q409 and K417. B) Representative structure of cholesterol (spheres, carbon atoms grey) from MD simulation. C) Representative structure of dexamethasone (spheres, carbon atoms pink) from MD simulation in the locked spike trimer.
Figure 4.
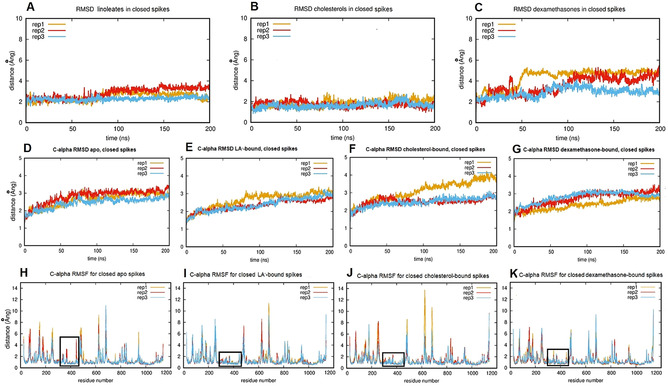
Ligand and protein dynamics during MD simulations of the locked spike trimer in apo and complexed forms. A, B, C) RMSD plots showing averaged ligand movement over time within binding sites for repeat simulations of (A) linoleate (LA−) (B) cholesterol and (C) dexamethasone. Note the greater relative motion of dexamethasone. D, E, F, G) Cα RMSDs averaged over time for repeat simulations of the apo (unliganded) locked spike trimer (D) and of its complexes with LA− (E), cholesterol (F) and dexamethasone (G). H, I, J, K) Cα RMSFs for repeat simulations of the apo (unliganded) locked spike trimer (H) and of its complexes with LA− (I), cholesterol (J) and dexamethasone (K). Black rectangles indicate the FA pocket and adjacent regions (residues 310–480) that are rigidified (lower fluctuations than in the apo form) in MD simulations of the ligand complexes.
Taking into account reports that the SARS‐CoV‐2 spike protein binds cholesterol, [27] we next investigated the interactions of cholesterol within the FA site. Cholesterol was manually positioned in the FA binding site by superposition onto the position of bound LA− in the cryo‐EM structure (PDB ID 6ZB5) after energy minimization, and oriented such that the polar end of cholesterol aligned with the LA− carboxylate head group and could replicate the inter‐chain interactions made by LA− bound to the spike trimer. All three cholesterol molecules remain bound to the closed spike trimer throughout three replicate 200 ns MD simulations (Figure 4 B) with their hydroxyl moieties maintaining interactions with R408, Q409 and K417 in the FA binding site, as well as making occasional contacts with the threonine residues T415 and T373 (Figure 3 B). Notably, the (ligand) RMSD values for bound cholesterol appear lower than those for either LA− (above) or dexamethasone (below), indicating that this ligand fits tightly into the spike FA site. It should however be noted that while these MD simulations were repeated three times to test significance, they are of relatively limited duration (200 ns). In consequence, while they do characterize the conformational sampling of the complexes on this timescale, they do not conclusively determine binding affinity or stability.
In order to explore further the potential for other ligands to bind in the FA site, the synthetic corticosteroid dexamethasone, a compound structurally and chemically similar to cholesterol, was oriented in the FA site as described above for cholesterol, and the complex subjected to 200 ns MD simulations. During the three replicate simulations, all the dexamethasone molecules (three per spike trimer) remain bound. RMSD plots (Figure 4 C) reveal that dexamethasone moves more freely within the FA site than either LA− or cholesterol, and samples a wider range of orientations due to its smaller size (Figure 2 C, Figure 3 C).
Comparison of Cα RMSD plots from MD simulations of the respective ligand complexes with the apo form indicates that binding of LA− (Figure 4 E), and, to a lesser extent, dexamethasone (Figure 4 G), has a stabilizing effect on the locked conformation of the spike protein, as shown by reduced average Cα RMSDs. In contrast, one of the three simulations of the cholesterol complex (rep1, Figure 4 F), appears to be destabilized, as shown by an increase in RMSD. As a larger, rigid ligand, cholesterol is more constrained in the FA binding site (Figure 3 B), which may favor opening of the pocket, disrupting more distant structural elements, via neighboring secondary structure elements. In this simulation, residues 480–500 in chain B (directly above the FA binding site), residues 620–630 in chains A and B, and residues 680–700 in chain B (containing the furin cleavage site) are disrupted (Figure S2). Notably, the apparently globally destabilizing effect observed in this simulation of the cholesterol complex (Figure 4 F) is not evident in simulations of the spike trimer in the apo, LA− or dexamethasone‐bound forms (Figure 4 D, E, G).
Comparison of RMSF plots for the four sets of simulations (Figure 4 I–K) shows that the presence of any of the bound ligands reduces fluctuations in the region 330–480 (residues surrounding the FA binding site) compared to the unliganded spike (Figure 4 H). This rigidifying effect extends towards the N‐terminus, as far as residue 310. Despite the relative freedom of movement of bound dexamethasone, its presence rigidified the FA binding site and its surroundings similarly to LA− and cholesterol (Figure 4 K).
Molecular Dynamics Simulations of Ligand Complexes of the Open Form of the Spike
In contrast to the locked form, cryo‐EM structures of the spike protein in the partially open form are less well ordered, and bound LA− was not readily resolved. To explore possible interactions of LA− with the spike protein in the open form, the locked spike trimer (PDB code 6ZB5 [12] ) was aligned with the open form and LA− (positioned as in the locked structure) was placed into the open structure (Toelzer et al. Electron Microscopy Databank EMD‐11146) such that interactions between the carboxylate head group and residues R408, K417 and Q409 at the subunit interface were maintained. Where this was not possible, the pose of bound LA− was adjusted slightly to allow alternative hydrogen bonds or salt‐bridge interactions to be made. The open conformation (EMD‐11146), contains three distinct environments for the three bound LA− molecules, that are referred to here as sites 1, 2 and 3. Site 1 is analogous to the LA− binding site in the locked conformation, and retains the salt‐bridge and hydrogen‐bond interactions of the carboxylate head group with R408, Q409 and K417 on the adjacent chain at the interface between RBDs. However, in the open form of the spike protein the presence of the RBD of one chain in a raised, open conformation disrupts the other two FA binding sites. Thus, in site 2, the LA− carboxylate head group which, in the locked state, would associate with R408 and Q408 on the subunit opposite, instead contacts residues K386 and N388 from the same spike subunit, and, unlike site 1, does not make contacts that bridge the subunit interface. In site 3 bound LA− associates with the underside of the RBD that is raised in the open conformation. LA− binding at site 3 allows the linoleate head group to interact across the domain interface with residues R408, Q409 and Q414 on the opposite chain and with K386 on the same chain, while residues F392, F515, L387, V382 and the C391‐C525 disulphide interact with the hydrophobic LA− tail.
This LA−‐bound open spike complex was then subjected to 200 ns MD simulations, in triplicate. LA− molecules bound in site 1 (equivalent to its binding site in the locked structure) maintain their original orientation and contacts during all repeat simulations. LA− bound in site 2 retains contacts with K386 and N388 at the rim of the binding site but does not make any interdomain connections. The behavior of LA− in site 3 is more dynamic. In two of the three replicate simulations, LA− in site 3 migrates further from its starting position but revisits the site regularly. In all three replicates, the RMSD of LA− at site 3 is higher than that for sites 1 and 2. However, in one of the repeat simulations, the LA− molecule originally bound in site 3, makes little contact with what would be the hydrophobic floor of the closed conformation site, and begins to dissociate after 150 ns (Figure S3). These results show weaker binding of LA− to the SARS‐CoV‐2 spike protein in the open conformation, specifically at the binding site (site 3) that differs most from its equivalent in the locked form, and indicate a correlation between tight LA−‐binding and closure of the RBD.
Complexes of the open form of the spike with cholesterol were generated as above and subjected to three replicate 200 ns MD simulations. During these simulations, all three cholesterol molecules remained bound to the spike trimer. The behavior of cholesterol bound at sites 1 and 2 was similar to that observed for LA−. However, the behavior of cholesterol bound in site 3 was noticeably different. In two of the three replica simulations, cholesterol at site 3 (i.e., associated with the open chain of the spike trimer) migrates to an alternative binding site (Figure 5 B, circled in black) within the first 5 ns, and remains there for the rest of the simulation. This alternative site is composed of prolines 330, 579 and 521 at the far end and is lined with hydrophobic residues including the C391–C525 disulphide, F329 and V327 (Figure 5 B, D). Residues 318 to 326 and 588 to 595 have been identified as responsible for the rigid body rotation [30] of the RBD, and 330 to 335 and 527 to 531 for the lift [30] required to enable the RBD to rise into a position that presents it for interaction with ACE2 (as highlighted in Figure S4). Cholesterol binding at this alternative site would be likely to restrict the motion required for full closure. In the third replicate simulation, cholesterol at site 3 also moves into this site, but binds in the opposite orientation with its more polar end towards the hydrophobic end of the binding site and is less stable. Altogether, these results indicate that cholesterol binds less tightly than LA− at site 3, and that other sites in the open spike conformation may have higher affinity for this ligand.
Figure 5.
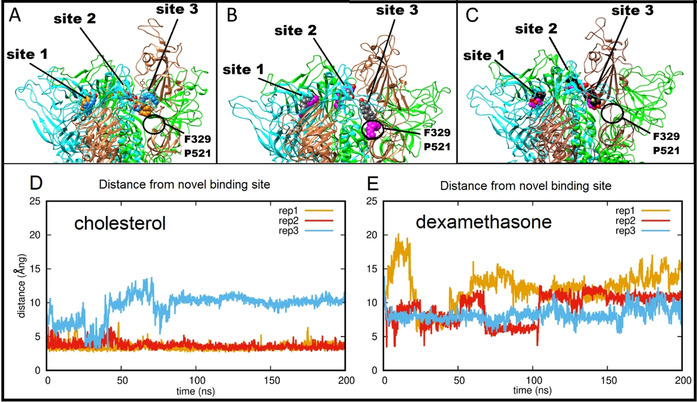
Ligand interactions of the SARS‐CoV‐2 spike protein in the open form. A) Starting (carbon atoms shown in orange) and final (carbon atoms blue) positions of three LA− molecules from one 200 ns MD simulation of their complex with the SARS‐CoV‐2 spike in the open conformation. B) Starting (carbon atoms grey) and final (carbon atoms magenta) positions of three cholesterol molecules from an equivalent 200 ns MD trajectory. C) Starting (carbon atoms black) and final (carbon atoms purple) positions of three dexamethasone molecules in the last frame of an equivalent 200 ns MD trajectory. A black circle (panels A, B, C) denotes the alternative binding site 3 sampled by the site 3 cholesterol (see main text). D, E) Time dependence of distance between the site 3 cholesterol (D) or dexamethasone (E) and residues F329 and P521 of the alternative site 3 favored by cholesterol in the hinge region.
We also performed MD simulations of dexamethasone bound to the open form, with three molecules of dexamethasone modelled into the FA binding sites as described above. As for simulations with cholesterol (above), the behavior of dexamethasone bound in sites 1 and 2 was analogous to that observed for LA. Dexamethasone bound in site 1 retained contact with R408, Q409 and K417, while dexamethasone bound in site 2 retained contact with N388 and the hydrophobic residues F377, F374 and Y365. However, dexamethasone bound in site 3 was not stably bound, and drifted further from its starting position (Figure S5) towards, but not remaining near, the alternative site found by cholesterol (Figure 5 C). Thus, in these MD simulations, dexamethasone did not locate a high affinity binding site close to the spike RBD in the subunit with the open conformation. These data indicate that dexamethasone does not have a favored binding site around site 3 of the open spike and does not make persistent interactions with the alternative cholesterol binding site (above) in the spike protein hinge region, though it may remain bound at the other FA binding sites in the trimer.
Virtual Ligand Screening at the Locked SARS‐CoV‐2 Spike Protein Fatty Acid Binding Site
The identification of a binding pocket for free fatty acids in the SARS‐CoV‐2 spike protein [12] led us to explore the possibility of this site as a target for binding of approved drugs (Figure 6 A–D). We used the Bristol University Docking Engine (BUDE [29] ) to screen in silico a library of approved drugs that are potential candidates for repurposing against COVID‐19. As the starting structure for docking, we chose an equilibrated frame from the MD simulations of the spike trimer in the locked, LA−‐bound form described above (hereafter cryo‐EMeq), because this structure had sidechains optimally oriented to form salt‐bridges and hydrogen bonds with the carboxylate head group (LA− is treated throughout as linoleate, as above). Docking results were filtered on the basis of ligand size, such that compounds having <15 or >45 heavy atoms were eliminated. The remaining compounds were then ranked based on their predicted binding affinity (BUDE energy) and the single best conformer for compounds scoring ≤ −100 kJ mol−1 was selected for more detailed analysis. The list of the top 100 compounds ranked in this way is given in Supporting Information (Table S1). The utility of BUDE here is supported by the similarity of the position and interactions of docked LA− to experimental and MD structures (see Figure 6).
Figure 6.
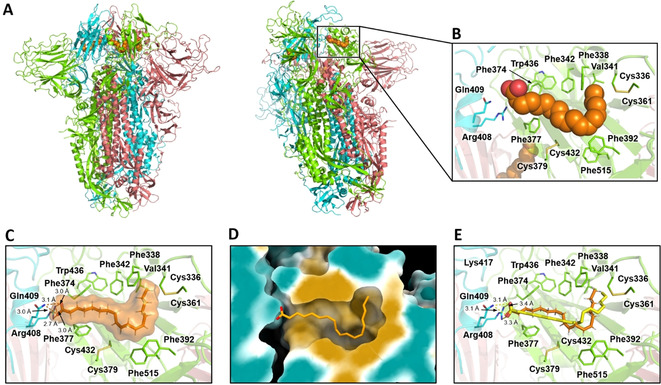
BUDE docks linoleate into the locked SARS‐CoV‐2 spike trimer in a conformation consistent with cryo‐EM structures. A) Cryo‐EM structure of the LA−‐bound spike trimer in the locked conformation. B) LA− binding site in the same structure. C) LA− binding pose in the structure used for docking (i.e. the cryo‐EM structure after MD equilibration). D) Surface representation of the LA− binding pocket in this structure, colored according to hydrophobicity: cyan for most hydrophilic through white to gold for most hydrophobic. The linoleate binding mode after equilibration by MD is shown as sticks. E) Superimposition of the BUDE predicted linoleate binding pose (yellow) and MD equilibrated structure (orange); the RMSD between the two poses is 1.6 Å.
The fat‐soluble vitamins D (vitamin D3 metabolite calcitriol), K and A ranked more highly than LA− by BUDE binding energy. Vitamin K2 is the highest ranked compound overall; vitamin A acetate is ranked 17th; vitamin D (calcitriol) 24th; vitamin A 38th and vitamin K1 70th. The lowest energy docked poses of these vitamins align very well with that of LA− bound to the locked spike trimer after 200 ns of MD simulation (Figure 7): the double bonds in the sidechains of calcitriol (Figure 7 A, D) and vitamin K2 (Figure 7 B, E) align closely with the hydrophobic LA− tail, and so these ligands are predicted to fit well into the FA binding site.
Figure 7.
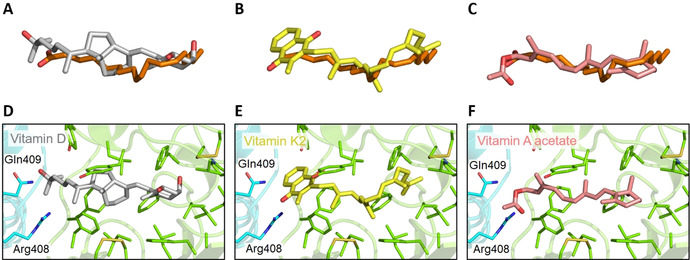
Docked poses of vitamins in the FA binding site of the SARS‐CoV‐2 spike. Overlays are shown of LA− (carbon atoms in orange, structure after MD simulation of the locked spike complex) with the lowest energy conformations from BUDE docking of: A) calcitriol (vitamin D, carbon atoms grey); B) vitamin K2 (carbon atoms yellow); and C) vitamin A acetate (carbon atoms salmon). Panels (D), (E) and (F) show the docked positions in the FA site of: D) vitamin D; E) vitamin K2; and F) vitamin A acetate.
The retinoids, tretinoin, acitretin and tazarotene identified by Riva et al. [31] to inhibit SARS‐CoV‐2 replication in Vero E6 cells are prominent in the ordered list of compounds identified by BUDE, ranking 5th, 8th, and 214th, respectively by BUDE energy. The predicted binding poses for these compounds involve salt‐bridge and hydrogen‐bonding interactions with R408 and Q409, and hydrophobic interactions with the remainder of the pocket (Figure 8 A, C, D). These data suggest that these compounds may act on viral replication by binding to this free FA site, similarly to LA−. Consistent with the MD simulations above, the dexamethasone binding mode predicted by BUDE (Figure 8 B) does not involve interactions with R408 or Q409 (unlike LA− and the retinoic acid receptor agonists) and instead is dominated by hydrophobic interactions with residues on the opposite side of the binding site. Other steroids were also highly ranked by BUDE (see SI).
Figure 8.
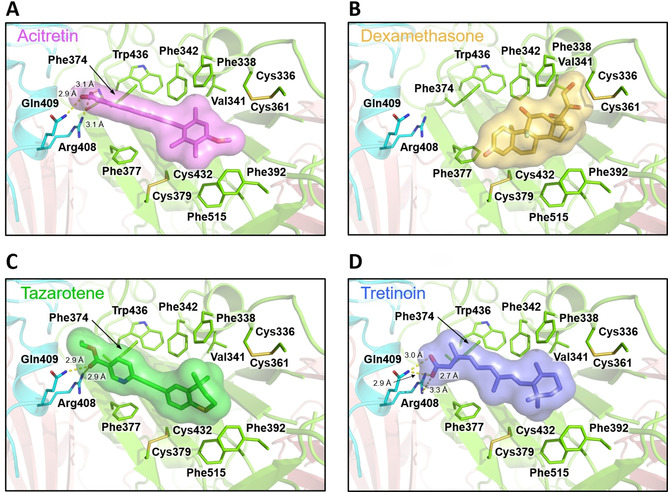
Docked poses of FDA‐approved drugs in the free FA binding pocket. In all cases, the highest scoring pose is shown. A) Acitretin (purple). B) Dexamethasone (orange). C) Tazarotene (green). D) Tretinoin (blue). Electrostatic interactions are shown as dashed yellow lines.
This virtual screening provides evidence that the SARS‐CoV‐2 spike FA site may bind physiological and synthetic ligands, including fat‐soluble vitamins, retinoids and steroids. It appears to be a promising, potentially druggable site, and ligand binding at this site may be responsible for previous experimentally observed activity.
Discussion
The unexpected discovery of a FA binding site in locked structures of the SARS‐CoV‐2 spike protein trimer, and the subsequent inference that LA− binding at this site stabilizes a locked conformation of the spike that is less competent for ACE2 receptor binding, raises the possibility that small molecule ligands binding at this site might modulate the conformation of the spike and disrupt SARS‐CoV‐2 infection. Here, we have explored these hypotheses, investigating the dynamic properties of the spike trimer in locked and open conformations, in complex with LA− and other potential ligands. Virtual screening suggests additional small molecule ligands from an approved drug library.
LA− makes persistent and stable interactions with the SARS‐CoV‐2 spike in its locked form. The locked spike has reduced mobility when LA− is bound (Figure 4). The conclusion that the FA binding site is rigidified by bound ligand is corroborated by the observations that the FA site collapses in MD simulations of the unliganded (apo) structure; it is also collapsed in cryo‐EM structures of the apo form. [12] Our results provide further evidence that the locked form of the spike is stabilized by LA− binding and demonstrate that it affects the conformational dynamics of the spike. The region stabilized by ligand binding extends beyond the immediate FA site, at least as far as residue 310, and contacts portions of the spike protein known to influence infectivity. Specifically, these include position 614 (the site of the D614G mutation that increasingly predominates in circulating SARS‐CoV‐2 strains [32] ) and the furin cleavage site (residues 680‐6) important in activation of the spike to enable interaction with ACE2. [33] Our simulations of the open spike show that LA− in the FA binding site that involves the repositioned subunit of the spike trimer (site 3) is more mobile and may dissociate, indicating a connection between LA− binding at the FA site and the open/closed conformation of the RBD.
We also investigated whether cholesterol may bind at the FA site, given that the spike protein has been demonstrated to bind cholesterol [27] and accumulating evidence associating individuals with prior elevated cholesterol levels with enhanced viral infectivity and adverse patient outcomes.[ 34 , 35 ] We also investigated dexamethasone as a possible ligand for the FA site. Both cholesterol and dexamethasone can be readily accommodated in the locked LA− site and remain bound to the closed spike during MD simulations on the timescales investigated here (200 ns; longer simulations would be required to investigate binding and conformational effects in more detail). Unlike LA− or dexamethasone, which apparently stabilize the closed structure, the structural changes in one of the replicate MD simulations indicate that cholesterol binding has a destabilizing effect on the locked spike, and suggest it has a weaker binding affinity at this site. Moreover, in two of the three replicate simulations of cholesterol bound to the open spike, cholesterol in the site formed by the open subunit (site 3) migrates to an alternative site in the open structure. The proximity of this alternative site to the hinge region, which is important for conformational changes of the spike between the locked and open forms,[ 30 , 36 ] suggests a possible mechanism by which cholesterol binding at this position may affect the dynamics and preferred conformation of the spike RBD, potentially stabilizing a form which presents the RBD for ACE2 binding.
Increasing evidence connects cholesterol levels with COVID‐19 disease risk and outcomes. For example, the apolipoprotein (apo)E4 genotype is associated with increased risk for severe COVID‐19 [37] and with increased circulating cholesterol. [38] Cholesterol‐lowering statins can reduce the risk of developing severe COVID‐19 and reduce recovery time in less severe cases. [39] Cholesterol has been proposed to promote SARS‐CoV‐2 infection by multiple mechanisms, including promoting ACE2 and furin trafficking to viral entry points, [34] but plasma levels are reduced in patients with established disease and correlate inversely with disease severity.[ 16 , 27 , 40 , 41 ] When considered together with previous findings of cholesterol binding to the spike protein, our results suggest possible additional involvement of cholesterol in infection, i.e., through binding to the spike. Cholesterol binding by the spike protein could constitute a sequestration mechanism that maintains local, cellular cholesterol at a level that supports continued infectivity in the face of a reduction in blood levels. Alternatively, it is possible that cholesterol bound to the spike protein may be more directly involved in the infection process, potentially through effects on the spike conformation and presentation of the RBD for interaction with ACE2. Involvement of cholesterol in virus entry is also likely in the case of spike variants lacking a functional furin cleavage site,[ 10 , 11 ] that may utilize an endosomal entry mechanism, as evidenced by identification of genes such as the host cholesterol transporter NPC1 as essential to cell killing by such strains. [11]
In silico screening of a library of approved drugs against the locked form of the SARS‐CoV‐2 spike protein yielded numerous candidates for ligands that may bind to the FA pocket. Our chosen docking tool, BUDE, identified LA− as a high‐scoring ligand and predicted a conformation similar to that observed experimentally, providing validation of our approach. Comparison with previous experimental screening of approved drugs suggests that some compounds known to be active against SARS‐CoV‐2 replication in cultured cells may bind to this site. Docking also identified the fat‐soluble vitamins D, K and A as potential ligands for the FA site. Notably, the vitamin D3 metabolite calcitriol reduces viral titers in monkey and human nasal epithelial cell lines. [42] Alongside socio‐economic deprivation, [43] evidence is accumulating that deficiencies in at least two of these vitamins (D and K) are associated with increased risk of developing severe COVID‐19.[ 44 , 45 ] Obesity is a known risk factor for poor COVID‐19 outcome, with the risk of death higher by 40 % for individuals with a body‐mass‐index (BMI) between 35–40, rising to 90 % for a BMI over 40. [46] It is worth noting that vitamin D becomes sequestered in adipose tissue, increasing the incidence of deficiency in obese individuals. [47] Although we are unaware of any demonstrated link between population vitamin A levels and COVID‐19 risk, we note the heavy COVID‐19 burden in some regions where vitamin A deficiency may be prevalent. [48] Aside from the role of vitamin D in modulating the host immune response to respiratory infections, [49] our data suggest that, as for cholesterol (above), sequestration through binding to the SARS‐CoV‐2 spike protein is a possible mechanism by which vitamin levels may be reduced in COVID‐19 disease. Furthermore, the implication that, by binding to the spike FA site, fat‐soluble vitamins may stabilize the locked form of the spike and inhibit viral entry suggests that these compounds could also play a more direct role in protecting host cells from viral infection. This possible involvement of micronutrients in countering SARS‐CoV‐2 highlights the potential for food poverty as a risk factor for COVID‐19 disease. These possibilities are worthy of further investigation.
In addition to vitamin A, retinoids [50] are highly represented among the docking hits, including three retinoids (acitretin, tazarotene and tretinoin) that have been shown to inhibit SARS‐CoV‐2 replication in Vero E6 cells. [31] Four more (adapalene, fenretinide, etretinate and isotretinoin (13‐cis retinoic acid)) are also prominent hits. Retinoids have been proposed for inclusion in COVID‐19 treatment regimes, particularly in combination with type 1 interferons [51] (IFN‐I), based upon their ability to upregulate host antiviral responses, rather than any hypothesized effect upon viral entry or replication. Coronaviruses such as SARS‐CoV and MERS‐CoV are known for their ability to disrupt IFN‐I signaling. [52] In vitro, SARS‐CoV‐2 is more susceptible than SARS‐CoV to the IFN‐I response,[ 53 , 54 ] although results from the WHO SOLIDARITY trial [55] did not show any benefits from subcutaneous interferon‐β1a for hospitalized COVID‐19 patients. It has been suggested that IFN‐I administration may be most effective as a prophylactic or early treatment option; thus agents such as retinoids that may stimulate secretion of, and potentiate response to, IFN‐I are worthy of investigation. Our data provide additional motivation for this by suggesting that, through binding to the FA pocket and keeping the spike protein in the locked conformation, retinoids may affect viral entry, particularly early in the infection process, as well as via upregulation of the host antiviral response. We note a clinical trial (NCT04353180) of a retinoid (isotretinoin/13‐cis retinoic acid) as a treatment for COVID‐19, but this remains at an early stage. [56]
Virtual screening also identifies several approved steroid drugs as potential candidate ligands for the FA site. This is of particular interest because recent meta‐analysis [57] showed that corticosteroids other than dexamethasone (methylprednisolone and hydrocortisone) are also beneficial to critically ill COVID‐19 patients, as measured by 28‐day all‐cause mortality, in randomized clinical trials. [55] Although steroids are not prominent in compound lists from cell‐based screens of approved drugs, and compounds for which clinical trial data are available (dexamethasone, methylprednisolone and hydrocortisone) are not the highest‐scoring hits (respectively 273rd, 47th (hemisuccinate), 266th and 97th in BUDE energy rankings), they feature highly in the virtual screening output. Together with our MD simulations which support binding of dexamethasone at the FA site, these results suggest that corticosteroids known to be effective against COVID‐19 may have an additional mode of action involving direct interaction with the spike, as well as their effects on the host response.
The interactions that we identify suggest that some compounds known to be active against SARS‐CoV‐2 replication in cultured cells may bind at the spike FA site, and this interaction may be responsible for some of their observed antiviral activity. It is important to emphasize that we make no claims regarding the possible clinical efficacy of compounds identified in the virtual screening described here. No recommendation on treatment should ever be based on the results of molecular simulations alone.
Conclusion
MD simulations indicate that the physiological ligand LA− stabilizes the locked form of the spike. The results are consistent with our findings that LA− stabilizes the locked spike, thus reducing infectivity. It binds more weakly to the exposed FA site of the open form. Cholesterol, which is also retained by the locked spike in MD simulations on the timescales explored here, is not retained by the FA site of the open spike. Indeed, the propensity of cholesterol to migrate to a distinct site, situated in the hinge region that connects the spike RBD to the remainder of the protein, suggests the possibility that interactions with cholesterol may modulate conformational changes in the SARS‐CoV‐2 spike and, potentially, may promote infection. Despite the similarity between dexamethasone and cholesterol, they behave differently in simulations. Our MD simulations and docking indicate that dexamethasone may bind to the FA site and may stabilize the locked conformation. Dexamethasone binding to the SARS‐CoV‐2 spike may represent an additional contribution to its efficacy in treating COVID‐19.
Docking suggests that some compounds that are known to be active against SARS‐CoV‐2 may bind at the FA site of the spike. Docking also identifies vitamins A, D and K as potential ligands for the spike FA site. Further study is warranted to explore whether fat soluble vitamins play a direct role in protecting against SARS‐CoV‐2 infection, for example, by stabilizing the locked spike conformation. In addition to vitamin A, three retinoids that are known inhibitors of SARS‐CoV‐2 replication are predicted to bind at the FA site. Docking also suggests that the FA pocket may bind a range of natural and synthetic steroids. These findings suggest that, in addition to their effects on host response, certain vitamins, retinoids and steroids may also affect SARS‐CoV‐2 by binding to the spike protein. This is worthy of further investigation.
Abbreviations
LA− linoleate; FA fatty acid; ACE2 angiotensin converting enzyme 2; MD molecular dynamics.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank BrisSynBio, a BBSRC/EPSRC Synthetic Biology Research Centre (Grant Number: BB/L01386X/1) for funding DKS and the HPC BlueGem and EPSRC vis HECBIOSIM (hecbiosim.ac.uk) for providing ARCHER time through a COVID‐19 rapid response call. A.J.M., J.S. and C.K.C. thank the British Society for Antimicrobial Chemotherapy (grant number BSAC‐COVID‐30). A.J.M. thanks EPSRC (grant number EP/M022609/1, CCP‐BioSim) for support. C.S. and I.B. are supported by the Wellcome Trust (210701/Z/18/Z; 106115/Z/14/Z) and the BBSRC (BB/P000940/1). This work used computational facilities of the Advanced Computing Research Centre, University of Bristol, http://www.bris.ac.uk/acrc/. We also thank Oracle Research for Oracle Public Cloud Infrastructure (https://cloud.oracle.com/en_US/iaas) time under an award for COVID‐19 research and the Elizabeth Blackwell Institute, University of Bristol, for their support.
D. K. Shoemark, C. K. Colenso, C. Toelzer, K. Gupta, R. B. Sessions, A. D. Davidson, I. Berger, C. Schaffitzel, J. Spencer, A. J. Mulholland, Angew. Chem. Int. Ed. 2021, 60, 7098.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.13143761.v1).
Contributor Information
James Spencer, Email: Jim.Spencer@bristol.ac.uk.
Adrian J. Mulholland, Email: Adrian.Mulholland@bristol.ac.uk.
References
- 1.Johns-Hopkins, pp. COVID-19 dashboard https://coronavirus.jhu.edu/map.html.
- 2. Mehta P., McAuley D. F., Brown M., Sanchez E., Tattersall R. S., Manson J. J., Collaboration U.K. HLH Across Speciality, Lancet 2020, 395, 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Puntmann V. O., Carerj M. L., Wieters I., Fahim M., Arendt C., Hoffmann J., Shchendrygina A., Escher F., Vasa-Nicotera M., Zeiher A. M., Vehreschild M., Nagel E., JAMA Cardiol. 2020, 5, 1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yelin D., Wirtheim E., Vetter P., Kalil A., Bruchfeld J., Runold M., Guaraldi G., Mussini C., Gudiol C., Pujol M., Bandera A., Scudeller L., Paul M., Kaiser L., Leibovici L., Lancet Infect. Dis. 2020, 20,1115–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.A. Dennis, M. Wamil, S. Kapur, J. Alberts, A. D. Badley, G. A. Decker, S. A. Rizza, R. Banerjee, A. Banerjee, medRxiv 2020, 10.1101/2020.10.14.20212555. [DOI]
- 6. Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F., Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walls A. C., Park Y. J., Tortorici M. A., Wall A., McGuire A. T., Veesler D., Cell 2020, 183, 1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xia S., Lan Q., Su S., Wang X., Xu W., Liu Z., Zhu Y., Wang Q., Lu L., Jiang S., Signal Transduction Targeted Ther. 2020, 5, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xing Y., Li X., Gao X., Dong Q., Front. Genet. 2020, 11, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Z., Zheng H., Lin H., Li M., Yuan R., Peng J., Xiong Q., Sun J., Li B., Wu J., Yi L., Peng X., Zhang H., Zhang W., Hulswit R. J. G., Loman N., Rambaut A., Ke C., Bowden T. A., Pybus O. G., Lu J., J. Virol. 2020, 94, https://jvi.asm.org/content/94/17/e00790-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Y. Zhu, F. Feng, G. Hu, Y. Wang, Y. Yu, Y. Zhu, W. Xu, X. Cai, Z. Sun, W. Han, R. Ye, H. Chen, Q. Ding, Q. Cai, D. Qu, Y. Xie, Z. Yuan, R. Zhang, bioRxiv 2020, 10.1101/2020.08.25.266775. [DOI]
- 12. Toelzer C., Gupta K., Yadav S. K. N., Borucu U., Davidson A. D., Kavanagh Williamson M., Shoemark D. K., Garzoni F., Staufer O., Milligan R., Capin J., Mulholland A. J., Spatz J., Fitzgerald D., Berger I., Schaffitzel C., Science 2020, 370, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wrapp D., Wang N., Corbett K. S., Goldsmith J. A., Hsieh C. L., Abiona O., Graham B. S., McLellan J. S., Science 2020, 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ke Z., Oton J., Qu K., Cortese M., Zila V., McKeane L., Nakane T., Zivanov J., Neufeldt C. J., Cerikan B., Lu J. M., Peukes J., Xiong X., Krausslich H. G., Scheres S. H. W., Bartenschlager R., Briggs J. A. G., Nature 2020, 588, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan Y., Cao D., Zhang Y., Ma J., Qi J., Wang Q., Lu G., Wu Y., Yan J., Shi Y., Zhang X., Gao G. F., Nat. Commun. 2017, 8, 15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.T. Dierckx, J. van Elslande, H. Salmela, B. Decru, E. Wauters, J. Gunst, Y. Van Herck, J. Wauters, B. Stessel, P. Vermeersch, medRxiv 2020, 10.1101/2020.11.09.20228221 . [DOI]
- 17.H. Achdout, A. Aimon, E. Bar-David, H. Barr, A. Ben-Shmuel, J. Bennett, M. L. Bobby, J. Brun, B.V.N.B.S. Sarma, M. Calmiano, A. Carbery, E. Cattermole, J.D. Chodera, A. Clyde, J. E. Coffland, G. Cohen, J. Cole, A. Contini, L. Cox, M. Cvitkovic, A. Dias, A. Douangamath, S. Duberstein, T. Dudgeon, L. Dunnett, P. K. Eastman, N. Erez, M. Fairhead, D. Fearon, O. Fedorov, M. Ferla, H. Foster, R. Foster, R. Gabizon, P. Gehrtz, C. Gileadi, C. Giroud, W. G. Glass, R. Glen, I. Glinert, M. Gorichko, T. Gorrie-Stone, E. J. Griffen, J. Heer, M. Hill, S. Horrell, M. F. D. Hurley, T. Israely, A. Jajack, E. Jnoff, T. John, A. L. Kantsadi, P. W. Kenny, J. L. Kiappes, L. Koekemoer, B. Kovar, T. Krojer, A. A. Lee, B. A. Lefker, H. Levy, N. London, P. Lukacik, H. Bruce Macdonald, B. MacLean, T. R. Malla, T. Matviiuk, W. McCorkindale, S. Melamed, O. Michurin, H. Mikolajek, A. Morris, G. M. Morris, M. J. Morwitzer, D. Moustakas, J. B. Neto, V. Oleinikovas, G. J. Overheul, D. Owen, R. Pai, J. Pan, N. Paran, B. Perry, M. Pingle, J. Pinjari, B. Politi, A. Powell, V. Psenak, R. Puni, V. L. Rangel, R. N. Reddi, S. P. Reid, E. Resnick, M. C. Robinson, R. P. Robinson, D. Rufa, C. Schofield, A. Shaikh, J. Shi, K. Shurrush, A. Sittner, R. Skyner, A. Smalley, M. D. Smilova, J. Spencer, C. Strain-Damerell, V. Swamy, H. Tamir, R. Tennant, A. Thompson, W. Thompson, S. Tomasio, A. Tumber, I. Vakonakis, R. P. van Rij, F. S. Varghese, M. Vaschetto, E. B. Vitner, V. Voelz, A. von Delft, F. von Delft, M. Walsh, W. Ward, C. Weatherall, S. Weiss, C. F. Wild, M. Wittmann, N. Wright, Y. Yahalom-Ronen, D. Zaidmann, H. Zidane, N. Zitzmann, ChemRxiv 2020, 10.26434/chemrxiv.13158218.v1. [DOI]
- 18. Amaro R. E., Mulholland A. J., Comput. Sci. Eng. 2020, 22, 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.A. Acharya, R. Agarwal, M. Baker, J. Baudry, D. Bhowmik, S. Boehm, K. G. Byler, L. Coates, S. Y. Chen, C. J. Cooper, O. Demerdash, I. Daidone, J. D. Eblen, S. Ellingson, S. Forli, J. Glaser, J. C. Gumbart, J. Gunnels, O. Hernandez, S. Irle, J. Larkin, T. J. Lawrence, S. LeGrand, S. H. Liu, J. C. Mitchell, G. Park, J. M. Parks, A. Pavlova, L. Petridis, D. Poole, L. Pouchard, A. Ramanathan, D. Rogers, D. Santos-Martins, A. Scheinberg, A. Sedova, S. Shen, J. C. Smith, M. D. Smith, C. Soto, A. Tsaris, M. Thavappiragasam, A. F. Tillack, J. V. Vermaas, V. Q. Vuong, J. Yin, S. Yoo, M. Zahran, L. Zanetti-Polzi, ChemRxiv 2020, 10.26434/chemrxiv.12725465. [DOI]
- 20. Francés-Monerris A., Hognon C., Miclot T., García-Iriepa C., Iriepa I., Terenzi A., Grandemange S., Barone G., Marazzi M., Monari A., J. Proteome Res. 2020, 19, 4291–4315. [DOI] [PubMed] [Google Scholar]
- 21. Yadav M., Dhagat S., Eswari J. S., Eur. J. Pharm. Sci. 2020, 155, 105522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCallum M., Walls A. C., Bowen J. E., Corti D., Veesler D., Nat. Struct. Mol. Biol. 2020, 27, 942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Turoňová B., Sikora M., Schürmann C., Hagen W. J. H., Welsch S., Blanc F. E. C., von Bülow S., Gecht M., Bagola K., Horner C., van Zandbergen G., Landry J., de Azevedo N. T. D., Mosalaganti S., Schwarz A., Covino R., Mühlebach M. D., Hummer G., Krijnse Locker J., Beck M., Science 2020, 370, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Casalino L., Gaieb Z., Goldsmith J. A., Hjorth C. K., Dommer A. C., Harbison A. M., Fogarty C. A., Barros E. P., Taylor B. C., McLellan J. S., Fadda E., Amaro R. E., ACS Cent. Sci. 2020, 6, 1722–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Daly J. L., Simonetti B., Klein K., Chen K. E., Williamson M. K., Anton-Plagaro C., Shoemark D. K., Simon-Gracia L., Bauer M., Hollandi R., Greber U. F., Horvath P., Sessions R. B., Helenius A., Hiscox J. A., Teesalu T., Matthews D. A., Davidson A. D., Collins B. M., Cullen P. J., Yamauchi Y., Science 2020, 370, 861–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.A. S. F. Oliveira, A. A. Ibarra, I. Bermudez, L. Casalino, Z. Gaieb, D. K. Shoemark, T. Gallagher, R. B. Sessions, R. E. Amaro, A. J. Mulholland, bioRxiv 2020, 10.1101/2020.07.16.206680. [DOI]
- 27.Y. Peng, L. Wan, C. Fan, P. Zhang, X. Wang, J. Sun, Y. Zhang, Q. Yan, J. Gong, H. Yang, X. Yang, H. Li, Y. Wang, Y. Zong, F. Yin, X. Yang, H. Zhong, Y. Cao, C. Wei, medRxiv 2020, 10.1101/2020.04.16.20068528. [DOI]
- 28. Group R. C., Horby P., Lim W. S., Emberson J. R., Mafham M., Bell J. L., Linsell L., Staplin N., Brightling C., Ustianowski A., Elmahi E., Prudon B., Green C., Felton T., Chadwick D., Rege K., Fegan C., Chappell L. C., Faust S. N., Jaki T., Jeffery K., Montgomery A., Rowan K., Juszczak E., Baillie J. K., Haynes R., Landray M. J., N. Engl. J. Med. 2020, 10.1056/NEJMoa2021436. [DOI] [Google Scholar]
- 29. McIntosh-Smith S., Price J., Sessions R. B., Ibarra A. A., Int. J. High Perform. Comput. Appl. 2015, 29, 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Melero R., Sorzano C. O. S., Foster B., Vilas J. L., Martinez M., Marabini R., Ramirez-Aportela E., Sanchez-Garcia R., Herreros D., Del Cano L., Losana P., Fonseca-Reyna Y. C., Conesa P., Wrapp D., Chacon P., McLellan J. S., Tagare H. D., Carazo J. M., IUCrJ 2020, 7, 1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riva L., Yuan S., Yin X., Martin-Sancho L., Matsunaga N., Pache L., Burgstaller-Muehlbacher S., De Jesus P. D., Teriete P., Hull M. V., Chang M. W., Chan J. F., Cao J., Poon V. K., Herbert K. M., Cheng K., Nguyen T. H., Rubanov A., Pu Y., Nguyen C., Choi A., Rathnasinghe R., Schotsaert M., Miorin L., Dejosez M., Zwaka T. P., Sit K. Y., Martinez-Sobrido L., Liu W. C., White K. M., Chapman M. E., Lendy E. K., Glynne R. J., Albrecht R., Ruppin E., Mesecar A. D., Johnson J. R., Benner C., Sun R., Schultz P. G., Su A. I., Garcia-Sastre A., Chatterjee A. K., Yuen K. Y., Chanda S. K., Nature 2020, 586, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korber B., Fischer W. M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E. E., Bhattacharya T., Foley B., Hastie K. M., Parker M. D., Partridge D. G., Evans C. M., Freeman T. M., de Silva T. I., Sheffield C.-G. G., McDanal C., Perez L. G., Tang H., Moon-Walker A., Whelan S. P., LaBranche C. C., Saphire E. O., Montefiori D. C., Cell 2020, 182, 812–827 e819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hoffmann M., Kleine-Weber H., Pohlmann S., Mol. Cell 2020, 78, 779–784 e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.H. Wang, Z. Yuan, M. A. Pavel, S. B. Hansen, bioRxiv 2020, 10.1101/2020.05.09.086249. [DOI]
- 35. Zaki N., Alashwal H., Ibrahim S., Diabetes Metab. Syndr. 2020, 14, 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Henderson R., Edwards R. J., Mansouri K., Janowska K., Stalls V., Gobeil S. M. C., Kopp M., Li D., Parks R., Hsu A. L., Borgnia M. J., Haynes B. F., Acharya P., Nat. Struct. Mol. Biol. 2020, 27, 925-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuo C. L., Pilling L. C., Atkins J. L., Masoli J. A. H., Delgado J., Kuchel G. A., Melzer D., J. Gerontol. Ser. A 2020, 75, 2231–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saito M., Eto M., Nitta H., Kanda Y., Shigeto M., Nakayama K., Tawaramoto K., Kawasaki F., Kamei S., Kohara K., Matsuda M., Matsuki M., Kaku K., Diabetes Care 2004, 27, 1276–1280. [DOI] [PubMed] [Google Scholar]
- 39. Daniels L. B., Sitapati A. M., Zhang J., Zou J., Bui Q. M., Ren J., Longhurst C. A., Criqui M. H., Messer K., Am. J. Cardiol. 2020, 136, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wei X., Zeng W., Su J., Wan H., Yu X., Cao X., Tan W., Wang H., J. Clin. Lipidol. 2020, 14, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.X. Hu, C. Dong, L. Wu, W. Lianpeng, G. He, W. Ye, Lancet SSRN. 2020, 10.2139/ssrn.3544826. [DOI]
- 42.C. K. Mok, Y. L. Ng, B. A. Ahidjo, R. C. Hua Lee, M. W. Choy Loe, J. Liu, K. S. Tan, P. Kaur, W. J. Chng, J. E.-L. Wong, D. Y. Wang, E. Hao, X. Hou, Y. W. Tan, T. M. Mak, C. Lin, R. Lin, P. Tambyah, J. Deng, J. J. Hann Chu, bioRxiv 2020, DOI: 10.1101/2020.06.21.162396. [DOI]
- 43. Patel J. A., Nielsen F. B. H., Badiani A. A., Assi S., Unadkat V. A., Patel B., Ravindrane R., Wardle H., Public Health 2020, 183, 110–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Benskin L. L., Front. Public Health 2020, 8, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dofferhoff A. S. M., Piscaer I., Schurgers L. J., Visser M. P. J., van den Ouweland J. M. W., de Jong P. A., Gosens R., Hackeng T. M., van Daal H., Lux P., Maassen C., Karssemeijer E. G. A., Vermeer C., Wouters E. F. M., Kistemaker L. E. M., Walk J., Janssen R., Clin. Infect. Dis. 2020, 10.1093/cid/ciaa1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mahase E., BMJ 2020, 370, m2994. [DOI] [PubMed] [Google Scholar]
- 47. Di Nisio A., De Toni L., Sabovic I., Rocca M. S., De Filippis V., Opocher G., Azzena B., Vettor R., Plebani M., Foresta C., J. Clin. Endocrinol. Metab. 2017, 102, 2564–2574. [DOI] [PubMed] [Google Scholar]
- 48. Akhtar S., Ahmed A., Randhawa M. A., Atukorala S., Arlappa N., Ismail T., Ali Z., J. Health Popul. Nutr. 2013, 31, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Greiller C. L., Martineau A. R., Nutrients 2015, 7, 4240–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thompson B., Katsanis N., Apostolopoulos N., Thompson D. C., Nebert D. W., Vasiliou V., Hum. Genomics 2019, 13, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Trasino S. E., Clin. Exp. Pharmacol. Physiol. 2020, 47, 1765–1767. [DOI] [PubMed] [Google Scholar]
- 52. Park A., Iwasaki A., Cell Host Microbe 2020, 27, 870–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lokugamage K. G., Hage A., de Vries M., Valero-Jimenez A. M., Schindewolf C., Dittmann M., Rajsbaum R., Menachery V. D., J. Virol. 2020, 94, 10.1128/JVI.01410-20 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mantlo E., Bukreyeva N., Maruyama J., Paessler S., Huang C., Antiviral Res. 2020, 179, 104811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Solidarity Trial Consortium W. H. O., Pan H., Peto R., Henao-Restrepo A. M., Preziosi M. P., Sathiyamoorthy V., Abdool Karim Q., Alejandria M. M., Hernandez Garcia C., Kieny M. P., Malekzadeh R., Murthy S., Reddy K. S., Roses Periago M., Abi Hanna P., Ader F., Al-Bader A. M., Alhasawi A., Allum E., Alotaibi A., Alvarez-Moreno C. A., Appadoo S., Asiri A., Aukrust P., Barratt-Due A., Bellani S., Branca M., Cappel-Porter H. B. C., Cerrato N., Chow T. S., Como N., Eustace J., Garcia P. J., Godbole S., Gotuzzo E., Griskevicius L., Hamra R., Hassan M., Hassany M., Hutton D., Irmansyah I., Jancoriene L., Kirwan J., Kumar S., Lennon P., Lopardo G., Lydon P., Magrini N., Maguire T., Manevska S., Manuel O., McGinty S., Medina M. T., Mesa Rubio M. L., Miranda-Montoya M. C., Nel J., Nunes E. P., Perola M., Portoles A., Rasmin M. R., Raza A., Rees H., Reges P. P. S., Rogers C. A., Salami K., Salvadori M. I., Sinani N., Sterne J. A. C., Stevanovikj M., Tacconelli E., Tikkinen K. A. O., Trelle S., Zaid H., Rottingen J. A., Swaminathan S., N. Engl. J. Med. 2020, 10.1056/NEJMoa2023184. [DOI] [Google Scholar]
- 56. https://clinicaltrials.gov/ct2/show/NCT04353180 [Accessed 22 September 2020] 2020.
- 57. Sterne J. A. C., Murthy S., Diaz J. V., Slutsky A. S., Villar J., Angus D. C., Annane D., Azevedo L. C. P., Berwanger O., Cavalcanti A. B., Dequin P. F., Du B., Emberson J., Fisher D., Giraudeau B., Gordon A. C., Granholm A., Green C., Haynes R., Heming N., Higgins J. P. T., Horby P., Juni P., Landray M. J., Le Gouge A., Leclerc M., Lim W. S., Machado F. R., McArthur C., Meziani F., Moller M. H., Perner A., Petersen M. W., Savovic J., Tomazini B., Veiga V. C., Webb S., Marshall J. C., JAMA J. Am. Med. Assoc. 2020, 324, 1330–1341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary