

Abstract
Polymerase chain reaction (PCR), a central technology for molecular diagnostics, is highly sensitive but susceptible to the risk of false positives caused by aerosol contamination, especially when an end‐point detection mode is applied. Here, we proposed a solution by designing a clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 eraser strategy for eliminating potential contamination amplification. The CRISPR/Cas9 engineered eraser is firstly adopted into artpcr reverse‐transcription PCR (RT‐PCR) system to achieve contamination‐free RNA detection. Subsequently, we extended this CRISPR/Cas9 eraser to the PCR system. We engineered conventional PCR primers to enable the amplified products to contain an implanted NGG (protospacer adjacent motif, PAM) site, which is used as a code for specific CRISPR/Cas9 recognition. Pre‐incubation of Cas9/sgRNA with PCR mix leads to a selective cleavage of contamination amplicons, thus only the template DNA is amplified. The developed CRISPR/Cas9 eraser, adopted by both RT‐PCR and PCR systems, showed high‐fidelity detection of SARS‐CoV‐2 and African swine fever virus with a convenient strip test.
Keywords: contamination‐free, CRISPR/Cas9, end‐point detection, PCR, RT‐PCR
A novel strategy combining CRISPR/Cas9 with conventional RT‐PCR and PCR was developed to construct a contamination‐free amplification, termed CRISPR/Cas9 eraser. Wei Lin and coworkers used a combination of RT‐PCR/PCR system, CRISPR recognition, and lateral flow test strip strategy to detect clinical SARS‐CoV‐2 samples and pig blood samples from suspected ASFV infection. They demonstrated the reliability, accuracy and convenience of this method and its potential as a more convenient molecular diagnostic technology without worrying the contamination and expensive detection device.
1. INTRODUCTION
Polymerase chain reaction (PCR) technology is the most widely used technique in molecular diagnostics for detecting small amounts of target DNA and RNA (Park et al., 2011). In a typical PCR reaction, up to 109‐fold replication efficiency can be achieved due to its exponential amplification ability (You et al., 2020). After completing a PCR process, aerosol cross‐contamination from product exposure easily occurs when downstream operations, such as end‐point detection, are performed (Aslanzadeh, 2004; Borst et al., 2004; Cimino et al., 1990; Mens et al., 2012; Salter et al., 2014; Xu et al., 2014; C. Zhang et al., 2018; S. Zhang et al., 2020). These aerosol particles, containing up to 106 amplicons, will contaminate laboratory reagents and equipment in a short period of time. In such a contaminated situation, negative or control PCR tests often present the same amplification results as the positive reactions, which is termed false‐positive amplification. This is a tricky problem encountered in molecular diagnostics (Aslanzadeh, 2004; Borst et al., 2004; Cimino et al., 1990; Salter et al., 2014).
Now, PCR can be executed by a closed detection mode. For example, in fluorescent quantitative PCR (qPCR), both amplification and detection are completed in a closed‐tube mode, which greatly reduces the possibility of PCR contamination (though not completely) (Corless et al., 2000). Nevertheless, there is still a market for open‐tube PCR detection for downstream applications. For example, PCR combined with end‐point clustered regularly interspaced short palindromic repeat (CRISPR) detection can further improve detection sensitivity and specificity. And PCR combined with test strip detection can avoid the use of an expensive fluorescence detection device, therefore it can reduce the cost of testing (Mens et al., 2012; Xu et al., 2014; C. Zhang et al., 2018; S. Zhang et al., 2020). In addition, some PCR applications, such as digital PCR, cannot use the closed‐tube mode because it involves an essential droplet transfer process (Chen et al., 2017; Liao et al., 2020).
For end‐point PCR detection, once PCR contamination occurs, the equipment involved in the PCR process needs to be cleaned and all PCR reagents need to be replaced. To prevent such cross‐contamination, the physical space isolation method is currently widely adopted by molecular diagnostic laboratories (Aslanzadeh, 2004; Borst et al., 2004; Genzen, 2020; Salter et al., 2014). This method requires reagent preparation, sample preparation, amplification, and detection steps to be performed in separate spaces. Space isolation is effective in preventing cross‐contamination, but it also makes the construction of such a PCR laboratory expensive. Other methods, such as irradiation and enzymatic hydrolysis have been used to nonselectively degrade contaminated amplicons. However, these nonselective degradation methods are often incompatible with single‐tube operations, thus increasing the complexity of the procedure (Deragon et al., 1990; Kwok, 1990; Sarkar & Sommer, 1991; Walder et al., 1993).
Therefore, an ideal method should be able to selectively degrade contaminated amplicons without affecting the integrity of primers and DNA templates. It has been reported that by designing restriction enzyme recognition sites on PCR primers, the contaminated amplicons can be degraded before a new round of amplification. However, this method is not universal due to the limitation of the sequence specificity of the restriction enzyme (Dougherty et al., 1993). Another currently widely used method is the uracil‐N‐glycosylase (UNG) assay (Pang et al., 1992; Pruvost et al., 2005; Tetzner et al., 2007; Udaykumar et al., 1993). UNG is a DNA repair enzyme that recognizes and removes uracil from DNA. When thymine in the PCR reagent is replaced with uracil, the contaminated amplicons would contain a large number of uracil bases, which can be digested by UNG. the DNA template is not affected because it does not contain uracil. The UNG assay works well for amplifying DNA templates. However, the UNG assay cannot be directly applied to the one‐step reverse‐transcription PCR (RT‐PCR) system because UNG also degrades the uracil embedded in reverse transcription c‐DNA product, which results in the break of the c‐DNA template. In addition, uracil‐containing amplification products also have obstacles for downstream operations, such as cloning, southern blot hybridization, and restriction enzyme recognition.
Here, we proposed a solution by designing a CRISPR/Cas9 eraser strategy for eliminating contamination amplicons. The CRISPR/Cas9 engineered eraser is firstly adopted into RT‐PCR and RT‐qPCR system for achieving high‐fidelity RNA amplification. Furthermore, we also prove that this CRISPR/Cas9 eraser strategy can be extended to the PCR system. For DNA amplification system, engineering of conventional PCR primers enables the amplified products to contain an implanted NGG (protospacer adjacent motif, PAM) site, which is used as a code for specific CRISPR/Cas9 recognition and cleavage. We also combined this contamination‐free RT‐PCR and PCR system, CRISPR recognition, and lateral flow test strip strategy to construct a convenient end‐point method for detecting RNA (severe acute respiratory syndrome coronavirus 2 [SARS‐CoV‐2]) and DNA (African swine fever virus, ASFV) virus. We proved the reliability and accuracy of the test results and the convenience of the method by analyzing clinical SARS‐CoV‐2 samples and pig blood samples from a suspected ASFV infection. The developed CRISPR/Cas9 eraser, adopted by both RT‐PCR and PCR system, showing high‐fidelity detection of SARS‐CoV‐2 and ASFV, proved that PCR can be developed as a more convenient molecular diagnostic technology without the need for an expensive detection device.
2. MATERIALS AND METHODS
2.1. Materials
All the DNA and RNA sequences listed in Table S1 were supplied by Sangon Biotech. Citric acid, sodium bicarbonate, sodium carbonate, sodium hydrogen phosphate, sodium citrate dihydrate, sodium dihydrogen phosphate, streptavidin, sucrose, Tween‐20, Triton X‐100, Tris–HCl, 20 × sodium chloride–sodium citrate (SSC) buffer, 20 × phosphate‐buffered saline (PBS), sodium chloride were purchased from Sangon Biotech. HAuCl4 solution was purchased from Aladdin. Bovine serum albumin (BSA) and isopropyl β‐d‐thiogalactopyranoside (IPTG) were purchased from Sigma‐Aldrich. The enzymes, buffers, and NTPs used for RNA transcription were obtained from Bio‐Life Science. Evo M‐MLV One‐Step RT‐PCR Kit was purchased from Accurate Biotechnology. Uracil‐N‐glycosylase, dUTP mixture, Premix Taq (Takara Taq Version 2.0), and TaKaRa Taq™ HS PCR Kit, UNG plus were purchased from Takara. Bacterial genomic DNA was extracted by the TIANamp Bacteria DNA Kit. Reagents used for protein expression and purification were purchased from Abiotech. The pNS20‐SpCas9‐SNAP plasmid used to prepare S. pyogenes Cas9 was a gift from Feng Zhang (Addgene plasmid #11371; http://n2t.net/addgene:113717; RRID: Addgene_113717).
2.2. Apparatus
The XYZ three‐dimensional film and spray gold instrument used for preparing test strips were purchased from Shanghai Jinbiao Biotechnology Co., Ltd. High‐speed centrifuge and PCR instruments were purchased from Eppendorf Co. Polyacrylamide gel electrophoresis (PAGE) and agarose electrophoresis experiments were performed using an electrophoresis apparatus (Liuyi Corporation) and imaged by a Gel Imaging System (Beijing).
2.3. Cas9 protein expression and purification
The Cas9 protein was expressed in Escherichia coli Rosetta2 (DE3) cells and cultured in Terrific Broth (TB) containing 34 μg/ml chloramphenicol and 50 μg/ml ampicillin. The cells were cultured overnight at 18°C until an OD600 = 0.6 was reached, after that, 0.1 mM IPTG was added and cells were cultured at 26°C for 4 h to induce Cas9 protein expression. The collected bacterial solution was centrifuged and the supernatant was then removed. The precipitate was ultrasonically lysed in lysis buffer (20 mM Tris–HCl, pH 7.5, 1 M NaCl, and 10% glycerol). After centrifugation at 4°C, the supernatant was purified on a nickel column (Abiotech), and washed by washing buffer (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 20 mM imidazole, and 10% glycerol) to remove the heterologous proteins, and finally eluted with elution buffer (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 500 mM imidazole, and 10% glycerol). The eluted Cas9 proteins were confirmed by PAGE and Coomassie Blue staining (Figure S1). Then, the eluted Cas9 proteins were collected and dialyzed with dialysis solution (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, and 50% glycerol), finally, it was stored at −20°C for future use.
2.4. Synthesis and DNA modification of 13 nm AuNPs
AuNPs were prepared using the sodium citrate reduction method. In brief, all the glassware were soaked in aqua regia (HNO3:HCl = 1:3) for more than 30 min, followed by rinsing with ultrapure water. After that, 100 ml of 1 mM HAuCl4 solution was added into a 250 ml flask and then heated to boiling. Subsequently, 10 ml of 38.8 mM sodium citrate solution was added quickly with stirring into the flask, which resulted in a color change of the solution from pale yellow to wine red in 20 min. Then, we stopped heating, but continued stirring until the solution dropped to room temperature. The prepared solution was stored at 4°C in the dark.
For DNA modification, 500 µl prepared AuNPs was mixed with poly A‐tagged DNA probes (100 μM, 25 μl) and frozen at −20°C for at least 2 h. After thawing, the solution was centrifuged at 13000 rpm for 30 min at 4°C. Then, the supernatant was discarded and a suspension buffer was added to suspend the solution. The centrifugation step needs to be repeated four times. The suspension buffer used for the first two centrifugation was 0.1 M NaCl, 0.01 M PB, and the suspension buffer used for the third centrifugation was ultrapure water (>18.25 MΩ cm). Finally, the supernatant was carefully aspirated and the pellet was suspended with 300 μl buffer (20 mM Na3PO4, 5% BSA, 0.25% Tween‐20, and 10% sucrose). The resulting AuNP‐DNA probes were stored at 4°C in the dark until further use.
2.5. Preparation of the lateral flow device
The lateral flow device consists of a sample pad, a conjugate pad, an NC membrane, an absorbent pad, and a bottom plate. The sample pad, which was composed of glass fibers, was first thoroughly soaked with a buffer (pH 8.0) containing 0.25% Triton X‐100, 0.05 M Tris–HCl, and 0.15 M NaCl, and then dried at 37°C. The material used for the conjugate pad was also glass fibers which had been completely embedded with the AuNP‐DNA probe solution and dried at 37°C. The test line on the NC membrane was sprayed with streptavidin solution and the control line was sprayed with a streptavidin‐biotinylated DNA probe solution. Then the NC membrane was dried at 37°C. Finally, the sample pad, the conjugate pad, the NC membrane, and the absorption pad were attached to the bottom plate and stored at 4°C for future use.
2.6. Synthesis of single guide RNA (sgRNA)
All sgRNAs described in this assay were transcribed in vitro from DNA templates using T7 RNA polymerase. The DNA template was obtained by a bridge‐fill‐in PCR method. And in a bridge‐fill‐in PCR, the forward primer comprises a T7 promoter sequence and a 20‐nt targeting sequence, while the reverse primer consists primarily of sequences encoding the 3′ end of the sgRNA scaffold. PCR was performed using Premix Taq under the following thermal cycling program: 95°C for 2 min, followed by 35 cycles at 95°C for 20 s, 63°C for 10 s, and 72°C for 45 s, a final extension at 72°C for 10 min and 10°C for hold. The PCR product served as a template for transcription. The transcription system contained 0.5 mM NTPs, 250 U T7 RNA polymerase, 50 U recombination RNase inhibitor, and 200 ng of DNA template, then it was incubated at 37°C for 4 h. The obtained sgRNA was used immediately or stored at −80°C.
2.7. Viral RNA and genomic DNA extraction
SARS‐CoV‐2 RNA standards were obtained from the National Institute of Metrology. Nasopharyngeal swab samples were collected from the suspected COVID‐19 patients in Wuhan, China (during March 2020). The RNA extraction of these swab samples was executed by the automatic extraction platform (Qiagen, EZ1) in the P3 biosafety laboratory of Hubei Provincial Center for Disease Control and Prevention and this study is approved by the Ethics Committee of Hubei Provincial Center for Disease Control and Prevention/Academy of Preventive Medicine (No. 2020‐061‐01)
The Neisseria meningitidis genomic DNA used in this assay was extracted by the TIANamp Bacteria DNA Kit. The extracted DNA was stored at −20°C until use. Viral DNA templates from ASFV were obtained from the African Swine Fever Regional Laboratory of China (Guangzhou). Swine serum samples were collected from Huangpu District, Guangzhou City, Guangdong Province, China (OIE Surveillance Data Archive, https://www.oie.int/wahis_2/public/wahid.php/Reviewreport/Review?page_refer=MapEventSummary%26reportid=29047). It should be noted that all the swine serum samples have been inactivated at 60°C for 30 min before use. A heating and chemical reduction method was employed to prepare virus DNA for PCR amplification. In this assay, tris(2‐carboxyethyl)phosphine (TCEP) and EDTA were added to swine serum samples at final concentrations of 100 and 1 mM, respectively. Then, the swine serum samples were inactivated at 50°C for 5 min followed by heating at 64°C for another 5 min. The supernatant obtained by brief centrifugation (5000 rpm/min) can be directly used for the subsequent PCR reaction.
2.8. Conventional RT‐PCR and PCR conditions
The conventional RT‐PCR was performed in a 50 μl system. RT‐PCR primers of SARS‐CoV‐2 were designed according to the recommendations of the Chinese Center for Disease Control and Prevention and are shown in Table S1. The 50 μl RT‐PCR reaction system was conducted by Evo M‐MLV One‐Step RT‐PCR Kit. The reaction system contains 1 × One‐Step Reaction Solution A, 2 uL One Step Enzyme Mix, 0.4 μM forward primer, 0.4 μM reverse primer, and 2 μl RNA template. The temperature procedure of RT‐PCR was 37°C for 30 min, then 95°C for 2 min, 35 circles at 94°C for 30 s, 60°C for 30 s and 72°C for 1 min, finally extension at 72°C for 5 min and 10°C for hold. In the end, the RT‐PCR products were stored at −20°C for further use.
PCR primers used for conventional PCR were shown in Table S1. A total of 50 μl PCR reaction system contained 300 nM forward and reverse primers, 25 μl 2 × Premix Taq, 2 μl DNA templates, and deionized water. The PCR thermal cycling program of ctrA from Neisseria meningitidis was 94°C for 5 min, 33 cycles at 95°C for 30 s, 54°C for 35 s, 72°C for 35 s, then extension at 72°C for 10 min and 10°C for maintenance. And the PCR thermal cycling program of the P72 gene from ASFV was 95°C for 15 min, followed by 40 cycles of 95°C for 15 s, 51°C for 30 s, 72°C for 45 s, and a final extension at 72°C for 10 min, then 10°C for hold. Finally, the PCR products were stored at –20°C for further use.
2.9. UNG assay in RT‐PCR system
The UNG assay in the RT‐PCR system was carried out in a 50 μl solution containing 1 U of UNG and 0.6 mM dUTP, as well as other reagents used in conventional RT‐PCR. And the reaction was executed using the same condition as the conventional RT‐PCR described above.
2.10. CRISPR/Cas9 eraser‐assisted RT‐PCR and RT‐qPCR
The RT‐PCR with Cas9/sgRNA reaction was performed in a 50 μl system containing 20 nM Cas9, 20 nM sgRNA, and other reagents used in conventional RT‐PCR. The RT‐qPCR with Cas9/sgRNA reaction was executed in a 20 μl system containing 20 nM Cas9, 20 nM sgRNA, 1 × SYBR Green I, and other reagents used in conventional RT‐PCR. Both the reactions were carried out with the same condition as the conventional RT‐PCR described above.
2.11. Cas9‐mediated lateral flow nucleic acids assay (CASLFA) detection of SARS‐CoV‐2
The CASLFA detection system includes the Cas9 reaction and lateral flow detection. First, 20 μl of the Cas9 reaction contains 100 nM Cas9, 100 nM sgRNA, 5 mM MgCl2 and biotinylated amplicons of SARS‐CoV‐2 from CRISPR/Cas9 eraser‐assisted RT‐PCR. Then, it was incubated at 37°C for 5 min to obtain the Cas9‐sgRNA‐biotinylated amplicon complex. Then, a 30 μl running buffer (components: 4 × SSC, 0.05% Tween‐20 (v/v), 1 × PBS, and 1% BSA) with 20 μl Cas9 reaction solution was added to the sample pad of the lateral flow device. 2 min later, 50 μl of running buffer were added. Then, the bands in the test line and control line appeared within 1 min. After finishing the test, a photograph of the lateral flow device was captured.
2.12. In vitro CRISPR/Cas9 cleavage experiments
A total of 10 µl reaction solution included 5 mM MgCl2, 100 nM Cas9, 100 nM sgRNA, and 2 µl target DNA. Then it was incubated at 37°C for 10 min. Reactions were stopped by heating the samples to 95°C for 5 min. The cleaved products were analyzed by PAGE electrophoresis (Figures S3 and S5).
2.13. CRISPR/Cas9 eraser‐assisted PAM‐implanted PCR
PCR primers with one or two C base modifications used for PAM‐implanted PCR are shown in Table S1. The total of the 50 μl PAM‐implanted PCR reaction system contained 300 nM forward and reverse primers, 25 μl 2 × Premix Taq, 20 nM sgRNA, 20 nM Cas9, 2 μl DNA templates, and deionized water. Before the PCR program, the reaction solution was first incubated at 37°C for 10 min for cleavage. Then, PCR thermal cycling programs of ctrA from Neisseria meningitides or P72 gene from ASFV were the same as the conventional PCR amplifications described above, respectively. The PCR products were stored at −20°C for further use.
2.14. CASLFA detection of ASFV
The CASLFA detection of ASFV was the same as the SARS‐CoV‐2 described above. PAM‐implanted PCR products of ASFV were served as target DNA in Cas9 reaction.
3. RESULTS
3.1. Design of the CRISPR/Cas9 eraser strategy for RT‐PCR
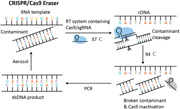
RT‐PCR, including the reverse transcription (RT) and PCR amplification step, is used to amplify and detect RNA sequences, such as mRNA, noncoding RNA, 16sRNA, and virus RNA. To simplify the procedure, RT and PCR procedures can be integrated into a one‐tube operation. Here, we experimentally confirmed that the UNG assay, a widely used contamination removal strategy in PCR, is not suitable for one‐tube homogeneous RT‐PCR. As shown in Figure 1a, in the UNG assay, dUTP and UNG enzymes must be included. In the process of RT, dUTPs are embedded into the newly synthesized cDNA strands. However, the cDNA strands embedded with dUTPs are easily degraded by the UNG enzyme. RT‐PCR and subsequent electrophoresis analysis showed that adding both dUTP and UNG enzymes for single‐tube RT‐PCR reaction lead to amplification failure, while adding only dUTP or UNG enzymes alone did not affect RT‐PCR (Figure 1a).
Figure 1.
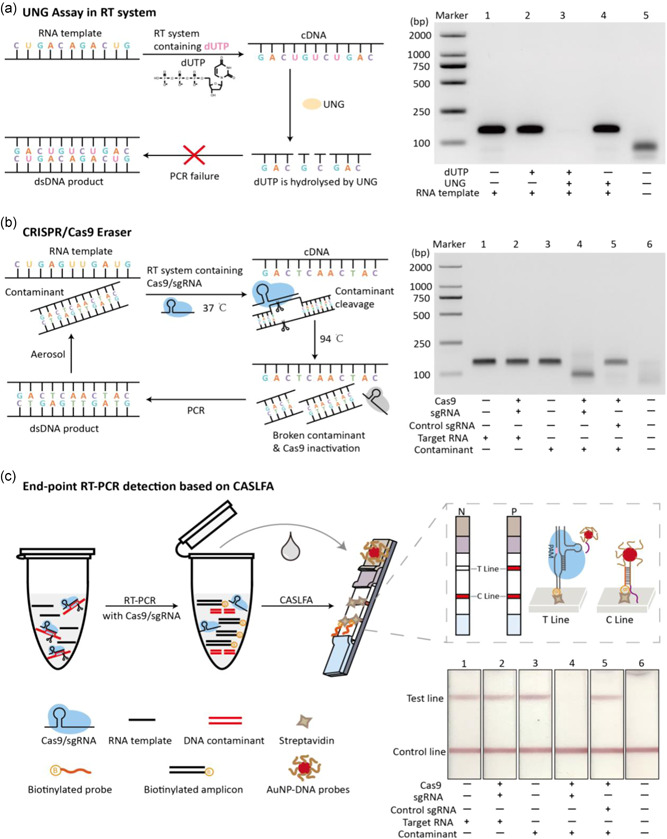
Development of CRISPR/Cas9 eraser for building contamination‐free RT‐PCR system. (a) UNG assay cannot be applied to one‐tube RT‐PCR system. Scheme presentation of UNG assay cannot be applied to RT‐PCR (left), and experimental confirmation of UNG assay cannot be applied to one‐step RT‐PCR (right). (b) Design of CRISPR/Cas9 eraser for RT‐PCR. Flow chart of CRISPR/Cas9 eraser working principle in RT‐PCR (left). Experimental confirmation of CRISPR/Cas9 eraser‐assisted contamination‐free RT‐PCR (right). (c) End‐point detection of RT‐PCR products by combining with CASLFA. The bottom right is the test strips analysis of the RT‐PCR products corresponding to (b). CASLFA, Cas9‐mediated lateral flow nucleic acids assay; cDNA, complementary DNA; CRISPR, clustered regularly interspaced short palindromic repeat; RT‐PCR, reverse‐transcription polymerase chain reaction; sgRNA, single guide RNA; UNG, uracil‐N‐glycosylase [Color figure can be viewed at wileyonlinelibrary.com]
Cas9 is an enzyme that can recognize specific DNA sequence and cut the DNA at an adjacent PAM site (NGG) under sgRNA programmability (Jiang et al., 2015; Jinek et al., 2012). The NGG PAM sequence occurs once on average every 8 bps of random DNA that gives CRISPR/Cas9 the versatility of DNA sequence cleavage and downstream bioanalytical applications (Bao et al., 2020; Gu et al., 2016; Hajian et al., 2019; Huang et al., 2018; S. H. Lee et al., 2017; H. Lee et al., 2018; Liu et al., 2019; Quan et al., 2019; T. Wang et al., 2019; Yang et al., 2018; W. Zhou, Hu, et al., 2018). Based on this feature, here we designed a CRISPR/Cas9 eraser strategy for building a contamination‐free RT‐PCR system (Figure 1b). In this strategy, a sgRNA was designed to target the double‐stranded DNA (dsDNA) amplification products. In the one‐tube RT‐PCR process, the designed Cas9/sgRNA is added and the potential dsDNA contamination amplicons can be selectively cleaved at 37°C RT step. However, the RNA template, single strand cDNA and DNA primers in the system can be free of Cas9/sgRNA cleavage. After the RT process completed, the PCR program is switched to the PCR thermal cycling process. At this time point Cas9/sgRNA is inactivated due to its instability at high temperature. Therefore, only the cDNA obtained from RT rather than potential DNA contamination is amplified, ensuring the fidelity of the RT‐PCR.
This concept was identified using a simulated RT‐PCR contamination system. As shown in Figure 1b, adding trace amounts of DNA contaminant amplicons to an RNA template omitted RT‐PCR system caused a false positive amplification (Figure 1b, line 3). Electrophoresis analysis shows that false positive amplification band is indistinguishable from real RNA amplification reactions (Figure 1b, line 1). Applying a designed Cas9/sgRNA in the RT‐PCR system, the DNA contaminants that caused false positive amplification disappeared (Figure 1b, line 4) but the RNA template mediated amplification was not affected (Figure 1b, line 2). By employing nontargeted sgRNA, the CRISPR/Cas9 eraser strategy becomes invalid, proving its dependence on sgRNA programmability and specificity (Figure 1b, line 5).
It seems that CRISPR/Cas9 eraser strategy may be particularly useful for end‐point RT‐PCR detection. For example, using test strips instead of expensive fluorescence device for detecting RT‐PCR products can reduce instrument costs and provide more convenient readout. Conventionally, these end‐point detection methods require the PCR test tube to be opened, which increases the contamination risk. We therefore evaluated the applicability of this CRISPR/Cas9 eraser strategy in RT‐PCR by adopting a CASLFA detection strategy that we previously developed. CASLFA is a nucleic acid strip detection method through engineering a CRISPR/Cas9 system to enhance detection specificity but avoid the conventional probe hybridization process (X. Wang, Xiong, et al., 2020). As shown in Figure 1c, by employing the CRISPR/Cas9 eraser strategy, the false positives caused by this contamination amplification disappeared. It is therefore expected that this high‐fidelity strategy will significantly improve the applicability of the CASLFA method. At the same time, we proved that CRISPR/Cas9 eraser was also compatible with RT‐qPCR system (Figure S2). we found that adding Cas9/sgRNA to the RT‐qPCR system does not affect RT‐qPCR efficiency. And only when the correctly matched sgRNA is present can the contamination be removed. Therefore, the CRISPR/Cas9 eraser can also be applied to RT‐qPCR to prevent contamination.
Encouraged by experimental verification of a high‐fidelity RT‐PCR end‐point detection method based on CASLFA, we moved to further identify its applicability for the development of a low‐cost, easy‐to‐use SARS‐CoV‐2 detection technology. Currently, there are almost 50 million individuals infected with SARS‐CoV‐2 worldwide (Sethuraman et al., 2020; Xia, 2020). Large‐scale population diagnosis is one of the key measures to control the spread of SARS‐CoV‐2 (da Silva et al., 2020; Feng et al., 2020; Qin et al., 2020). RT‐qPCR, due to its high sensitivity and specificity, simplicity of primer design, and easy availability of amplification enzymes, is the current gold standard technology for SARS‐CoV‐2 diagnosis (Y. Wang, Kang, et al., 2020). However, two issues may hinder its adoption in developing countries and regions. On the one hand, the RT‐qPCR instrument needs to be equipped with an expensive fluorescent detection device. For example, an RT‐qPCR instrument is usually ten times more expensive than an ordinary PCR instrument. On the other hand, it is also expensive to equip a PCR laboratory with an air control system and physical space separation for eliminating cross‐contamination.
CASLFA, combined with a CRISPR/Cas9 eraser‐assisted RT‐PCR, is developed for detecting clinical SARS‐CoV‐2 samples (Figure 2a). This method retains the advantages of PCR, such as high sensitivity and specificity, simplicity of primer design, and easy availability of amplification enzymes, but reduces the cost of equipment and laboratory construction. The SARS‐CoV‐2 RNA genome is about 30,000 bases in length (Kim et al., 2020). Nucleocapsid (N), envelope (E), and open reading frame 1ab (ORF1ab) genes, due to their high sequence conservation, are usually used as target genes to detect SARS‐CoV‐2 (Broughton et al., 2020; Patchsung et al., 2020). Here, we choose the E gene as the target gene to evaluate the clinical applicability of this method. A sgRNA, used for both CRISPR/Cas9 eraser and CASLFA, is designed to target the middle region of the forward primer and reverse primer (Figures 2b and S3). Ten clinical SARS‐CoV‐2 RNA samples, firstly diagnosed with a clinically approved RT‐PCR Kit, were used for CRISPR/Cas9 eraser‐assisted RT‐PCR and CASLFA analysis. Results indicated that CASLFA shows consistency with RT‐PCR, and the primer dimer amplification did not cause interferences in CASLFA (Figure 2c).
Figure 2.
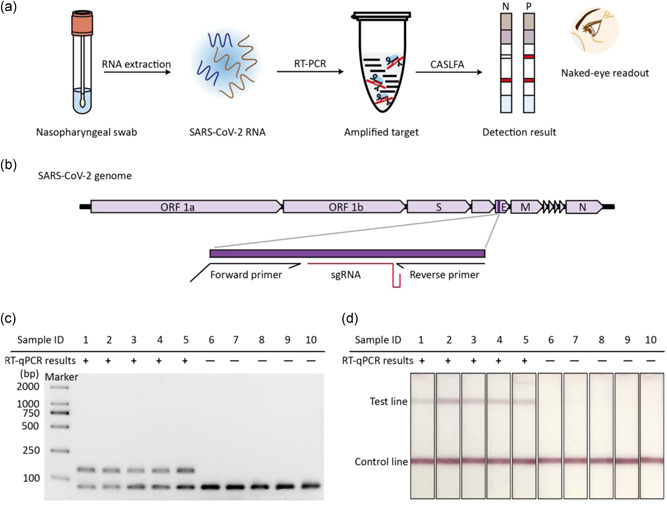
High‐fidelity detection of clinical SARS‐CoV‐2 samples. (a) Scheme of CASLFA detection of CRISPR/Cas9 eraser‐assisted RT‐PCR products for clinical SARS‐CoV‐2 diagnosis. (b) The genetic map of SARS‐CoV‐2, in which the amplified gene locations and target regions are marked in detail. (c) Electrophoresis analysis of 10 SARS‐CoV‐2 clinical samples based on RT‐PCR combined with CRISPR/Cas9 eraser strategy. (d) The corresponding CASLFA detection result of the clinical samples in (c). CASLFA, Cas9‐mediated lateral flow nucleic acids assay; CRISPR, clustered regularly interspaced short palindromic repeat; RT‐PCR, reverse‐transcription polymerase chain reaction; RT‐qPCR, quantitative RT‐PCR; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2 [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Design of the CRISPR/Cas9 eraser strategy for PCR
After proving that CRISPR/Cas9 eraser can be used in high‐fidelity RT‐PCR detection, we tried to extend its application to PCR amplification. However, direct transplantation of the CRISPR/Cas9 eraser strategy to PCR system is not feasible due to both DNA contaminants and the DNA template exhibiting the same target sequence that can be cleaved by designed Cas9/sgRNA. To successfully construct a CRISPR/Cas9 eraser for PCR, the designed CRISPR/Cas9 system should selectively cut the DNA contaminants but not the DNA template.
Considering that CRISPR/Cas9 requires a target sequence adjacent PAM site (NGG) for DNA recognition and subsequent cleavage, we decided to implant an artificial PAM site into the PCR products through primer engineering. Here, we proposed a strategy termed PAM‐implanted PCR to achieve this goal. For PAM‐implanted PCR, we attempted to engineer a conventional PCR primer to contain two extra CC bases at the near 3′ terminal region so that the amplified products will contain a new NGG PAM site (Figure 3a). If the 3′ terminal region of a primer happens to contain a C base, then we only need to add an additional C base to obtain such a PAM site (Figure 3a). We assumed that a minor modification of the 3′ terminal region of a PCR primer only has faint influence on its function. Using this two C bases or single C base implanted primer strategy, the amplified products will contain an artificial NGG PAM site at the opposed DNA strand. A corresponding 20 bp sgRNA with complementary pairing with the amplified PCR products can be designed for matching this artificial PAM site for Cas9/sgRNA recognition and cleavage. The designed Cas9/sgRNA was expected to cleave the site that is 3 bp away from 5′ of PAM site of the PCR products. Accordingly, in PAM‐implanted PCR, once PCR cross‐contamination occurs, the designed CRISPR/Cas9 eraser can selectively cut off the contaminated amplicons but not the template target DNA so that false positive amplification is abolished (Figure 3b).
Figure 3.
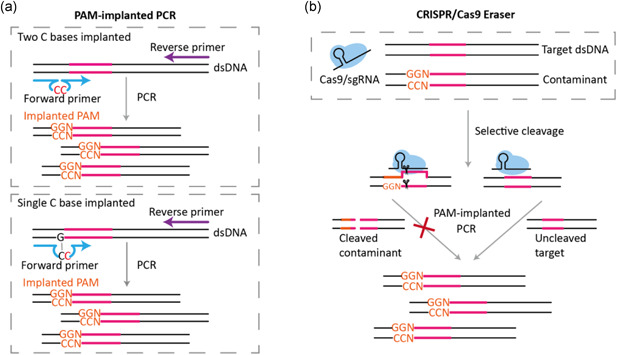
The design principle of applying CRISPR/Cas9 eraser to PCR. (a) The schematic of PAM‐implanted PCR. PAM‐implanted PCR can be achieved through a two C bases implanted primer engineering strategy (up) or a single C base implanted primer engineering strategy (blow), respectively. (b) The schematic of applying a Cas9/sgRNA eraser in PAM‐implanted PCR. Compared with template DNA, contaminated amplicons originated from the previous amplification containing additional PAM sites can be selectively excised. protospacer adjacent motif; PCR, polymerase chain reaction; sgRNA, single guide RNA [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Verification of the PAM‐implanted PCR
One of the most critical prerequisites to determine the feasibility of PAM‐implanted PCR is primer engineering. We firstly used two pairs of reported PCR primers to support this concept. These two reported primer pairs have been employed for PCR amplification of the ctrA gene of Neisseria meningitides (Hughes, 2017). In a conventional forward primer (5′‐GTCGCGGTGATGTGGTTA‐3′) there is no C base in its 3′ terminal region. Accordingly, we inserted two C bases at its 3′ terminal region and termed this modified primer as PAM‐implanted primer (5′‐GTCGCGGTGATGTGCCGTTA‐3′). Using this PAM‐implanted primer, we subsequently evaluated whether it still works well for PCR amplification. Comparisons showed that both the conventional primers and the PAM‐implanted primers exhibited similar amplification specificity and efficiency (Figure 4a, left). We further used DNA sequencing to verify whether the modified primers resulted in the insertion of two new CC bases in the amplified product. The sequencing of reverse DNA sequence showed that an additional GG site was obtained (Figure 4a, right).
Figure 4.
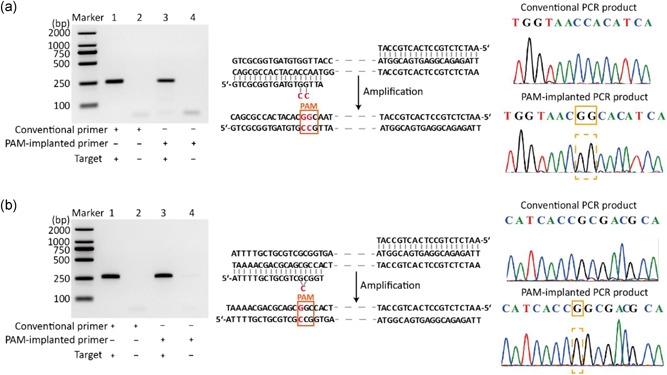
Verification of PAM‐implanted PCR. (a) On the left is the agarose gel analysis of products from conventional PCR and PAM‐implanted PCR which contain primers implanted by two C bases. In the middle is the schematic diagram of primer modification and amplification products. On the right is the sequencing result of products from conventional PCR (top) and PAM‐implanted PCR (bottom). (b) On the left is the agarose gel analysis of products from conventional PCR and PAM‐implanted PCR which contain primers implanted by single C base. In the middle is the schematic diagram of primer modification and amplification products. On the right is the sequencing result of products from conventional PCR (top) and PAM‐implanted PCR (bottom). PAM, protospacer adjacent motif; PCR, polymerase chain reaction [Color figure can be viewed at wileyonlinelibrary.com]
If the 3′ terminal region of a PCR primer contains a C base, then in the PAM‐implanted primer strategy only an additional C base needs to be inserted. We identified this idea by modifying another PCR primer (5′‐ATTTTGCTGCGTCGCGGT‐3′) for ctrA gene amplification. As shown in electrophoresis analysis of Figure 4b, the engineered PAM‐implanted primer (5′‐ATTTTGCTGCGTCGCCGGT‐3′) also exhibited similar amplification specificity and efficiency when compared to the conventional forward primer. Sequencing results also indicated that new PAM loci were obtained (Figure 4b, right).
3.4. Verification of the selective cleavage of PCR amplicons by CRISPR/Cas9 eraser
In the PAM‐implanted PCR, a new PAM site is obtained. Based on this PAM site, we designed a matched sgRNA and a scrambled sgRNA (control), respectively, to verify the selective recognition and cleavage for PAM‐implanted PCR products. We added the designed Cas9/sgRNA complex to the conventional PCR and PAM‐implanted PCR products to execute a cleavage reaction for verifying both two C bases and single C base implanted PCR products. Gel electrophoresis results showed that conventional PCR products are still intact regardless of using matched sgRNA or scrambled sgRNA (Figure 5a, left and right). This result indicated that the PAM site is essential for Cas9/sgRNA to perform the cleavage function. As expected, gel electrophoresis results showed that PAM‐implanted PCR products were cleaved into smaller fragments with matched sgRNA but not by control sgRNA (Figure 5a, left and right).
Figure 5.
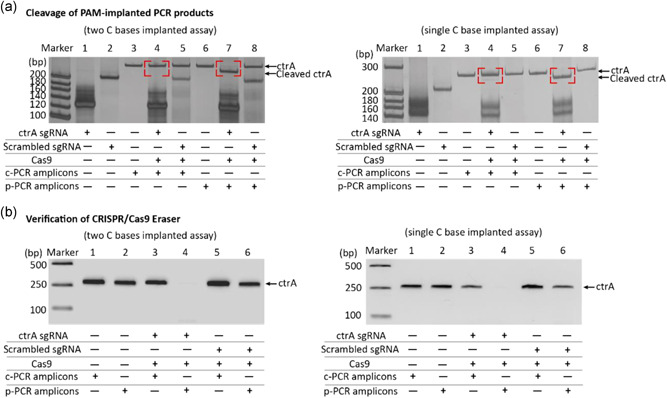
Verify the function of CRISPR/Cas9 eraser. (a) PAGE analysis of Cas9/sgRNA cleavage of conventional PCR products and PAM‐implanted PCR products which were amplified by primers with a two C bases implanted strategy (left) and with a single C base implanted strategy (right). (b) Agarose gel electrophoresis analysis of products from PAM‐implanted PCR in which the CRISPR/Cas9 eraser was used with two C bases implanted strategy (left) and with single C base implanted strategy (right). c‐PCR represents the conventional PCR, and p‐PCR represents the PAM‐implanted PCR. CRISPR, clustered regularly interspaced short palindromic repeat; PAGE, polyacrylamide gel electrophoresis; PAM, protospacer adjacent motif; PCR, polymerase chain reaction; sgRNA, single guide RNA [Color figure can be viewed at wileyonlinelibrary.com]
Next, we designed a simulated contaminated PCR system to prove the applicability of the developed CRISPR/Cas9 eraser strategy in high‐fidelity PCR amplification. Introduction of CRISPR/Cas9 eraser strategy to the PCR procedure will includes an additional short pre‐incubation cleavage step (10 min). After completing CRISPR/Cas9 cleavage reaction, this procedure was then switched to high‐temperature PCR amplification and Cas9/sgRNA system becomes inactive. We used both conventional PCR amplicons and PAM‐implanted PCR amplicons as model DNA contaminants for verifying this strategy. As illustrated in Figure 5b, when conventional PCR amplicons are used as a template, the contaminant caused PCR reaction cannot be inhibited by the CRISRP/Cas9 eraser. In contrast, the contaminant‐caused PCR reaction using PAM‐implanted PCR amplicons as a template was completely suppressed. Both PCR reactions are not affected when the control sgRNA is used. Both two C bases and single C base implanted strategies exhibit similar CRISPR/Cas9 eraser effect (Figure 5b, right).
3.5. CRISPR/Cas9 eraser for high‐fidelity PCR detection of ASFV
ASFV is a nucleocytoplasmic large DNA virus, which is highly infectious and pathogenic (Garigliany et al., 2019). Until now, there is no available vaccine or antiviral drug for ASFV. The fatality rate of pigs infected with ASFV is as high as 100% (Carlson et al., 2018; Kolbasov et al., 2018; X. Zhou, Li, et al., 2018). Establishing a rapid diagnostic method for the detection of ASFV is an effective means to effectively control the spread of African swine fever (ASF) (Yuan et al., 2020). The huge testing demand makes it urgent to develop low‐cost testing technology. Based on the CRISPR/Cas9 eraser strategy for PCR system, here, we established a PAM‐implanted PCR end‐point detection method for visual end‐point detection of ASFV (Figure 6a). Based on the amplification of the P72 gene, we obtained PAM‐implanted primers by modifying a pair of PCR primers used in conventional PCR (Figure 6b). This designed PAM‐implanted PCR system and CRISPR/Cas9 eraser have demonstrated good amplification and cleavage efficiency (Figures S4 and S5).
Figure 6.
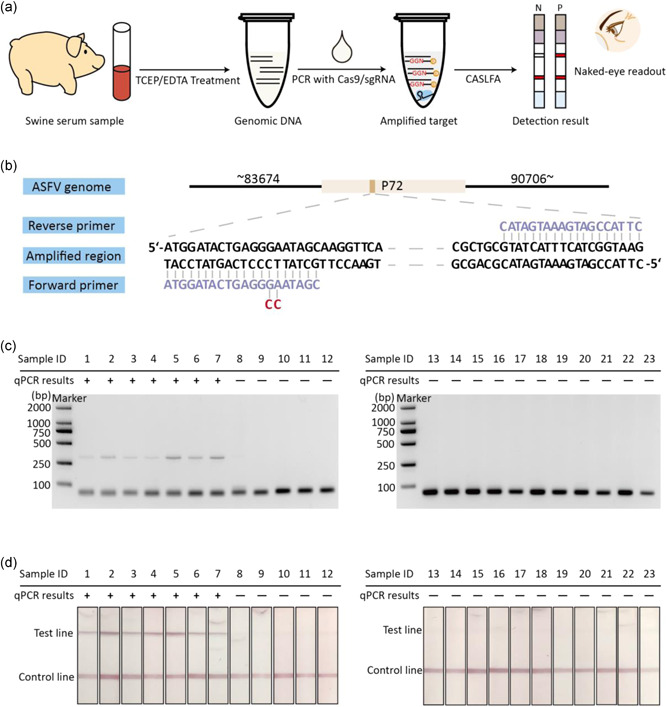
Detection of pig serum samples which are suspected to be infected with ASFV. (a) The flow chart of CASLFA detection of pig serum samples by combining PAM‐implanted PCR with CRISPR/Cas9 eraser. (b) The schematic diagram of ASFV genome and primers used for P72 gene amplification in this design. (c) The agarose gel electrophoresis analysis of 23 pig serum samples based on PAM‐implanted PCR with CRISPR/Cas9 eraser. (d) The corresponding CASLFA detection results of the samples in (c). ASFV, African swine fever virus; CASLFA, Cas9‐mediated lateral flow nucleic acids assay; CRISPR, clustered regularly interspaced short palindromic repeat; PAM, protospacer adjacent motif; PCR, polymerase chain reaction; qPCR, quantitative PCR; sgRNA, single guide RNA; TCEP, tris(2‐carboxyethyl)phosphine [Color figure can be viewed at wileyonlinelibrary.com]
WithIn the purpose of proving the ability of this method to tolerate cross‐contamination, we analyzed 23 pig serum samples. Seven of 23 pig serum samples were tested positive for ASFV by q‐PCR. These pig serum samples were first treated with a previously reported TCEP/EDTA method to release DNA and inactivate the enzymes in the blood sample. We then employed CRISPR/Cas9 eraser to perform PCR on these samples and subsequent CASLFA end‐point detection. In a contaminated PCR system, false‐positive amplification makes the test strip results difficult to distinguish from real positive samples (Figure S6). However, in the CRISPR/Cas9 eraser‐assisted PCR system, the electrophoresis and strip results show that all sample tests are consistent with the qPCR method, and there is no false positive caused by a cross‐contamination reaction under open‐tube detection (Figures 6c,d and S6).
4. DISCUSSION
The recent outbreaks of ASFV and SARS‐CoV‐2 pushed the importance of nucleic acid testing to the center stage. The huge demand for nucleic acid testing makes routine laboratory‐based methods, which require professional and harsh experimental conditions, difficult to deal with. The development of nucleic acid testing technology compatible with low‐complexity laboratories will make it easier for non‐professionals to operate, thereby expanding its accessibility for screening and controlling of infectious diseases.
PCR‐based methods, including fluorescence q‐PCR, are currently the gold standard method for nucleic acid testing. However, we did verify that the CRISPR/Cas9 eraser is also compatible with the RT‐qPCR system. But, q‐PCR instruments are expensive due to the need for fluorescence emission and detection devices. For example, q‐PCR instruments are about 10‐fold more expensive when compared with conventional PCR instruments. Thus, the development of nucleic acid testing by combining conventional PCR procedures with highly specific end‐point detection methods, such as strip tests, will be promising due to its simplicity of operation, low cost, and compatibility with low‐complexity laboratories.
Combining conventional PCR procedure and end‐point detection, like CASLFA, requires the amplification tube opened, thus increasing the risk of aerosol contamination. PCR amplicons diffused in the aerosol can be used as a template for the next round of amplification. As PCR is extremely sensitive, even trace amounts of aerosol contamination may cause false‐positive signals, which will be unacceptable in clinical testing applications. Strategy for selective destruction of contaminated amplicons in the PCR system but without damaging the primers and DNA templates will provide a solution to this problem. However, currently, there is no ideal solution that can be applied to both RT‐PCR and PCR. For example, a widely used strategy, the UNG assay, has been experimentally proven to be infeasible for one‐step RT‐PCR.
Here, we proposed a novel strategy to solve contamination amplification by designing a CRISPR/Cas9 based cleavage system. The mechanism of CRISPR/Cas9 cleaving the dsDNA sequence relies on base pairing between the dsDNA and the 20‐nt guide RNA sequence, as well as the presence of a PAM site proximal to the target region. These characteristics prompted us to develop a CRISPR/Cas9 eraser strategy to be applied to the RT‐PCR system for selective degradation of DNA contamination amplicons. As experimentally proved in a one‐tube RT‐PCR system, potential DNA contamination amplicons from the previous round amplification can be eliminated by the designed CRISPR/Cas9 eraser at the RT step. The target RNA and single‐stranded DNA primers are not affected because they are not the substrate of the CRISPR/Cas9 eraser. CRISPR/Cas9 eraser does not affect the subsequent PCR process because it is inactivated at high temperatures. The introduction of CRISPR/Cas9 eraser to the RT‐PCR is very convenient because all PCR reagents and the RT‐PCR procedure do not need to be changed. We applied this method to the detection of an RNA virus, SARS‐CoV‐2, and proved that it's a high‐fidelity end‐point detection method with no need to worry about false amplification risk. Detection of SARS‐CoV‐2 using this method only requires a thermal cycler and a preassembled test strip, thus giving it the potential to be used in low‐complexity laboratories.
Success in applying the CRISPR/Cas9 eraser into RT‐PCR drove us to develop a similar strategy suitable for the PCR method. It is not feasible to use the CRISPR/Cas9 eraser strategy directly into the PCR procedure, due to both DNA template and DNA contamination amplicons exhibit the same sequence characteristics. We solved this problem by engineering conventional PCR primers by inserting a single C base or two C bases proximal to the 3′ terminal. This primer engineering strategy enables the amplified products to contain an implanted NGG sites, which can be used as a code for specific CRISPR/Cas9 recognition. Therefore, in such a PAM‐implanted PCR system, only the contaminating amplicons can be cleaved by CRISPR/Cas9 eraser but the template DNA will be intact. We also proved that this PAM‐implanted PCR system is comparable to conventional PCR amplification in terms of amplification efficiency and specificity. This novel PAM‐implanted method, combined with CASLFA, has shown promising applications for high‐fidelity end‐point detection of 23 serum samples that are suspected of being infected with a DNA virus, ASFV.
A potential limitation of this CRISPR/Cas9 eraser strategy is that there is still a possibility of cross‐contamination between amplification of two closely related sequences, especially when PCR is applied to complex biological samples. In this case, the contamination may not be eradicated, and the amplified products caused by such contamination cannot be distinguished from electrophoresis or nonspecific SYBR‐Green RT‐qPCR detection. But the subsequent specific CASLFA test should be able to detect, which are the real amplified products. However, if there is contamination caused by an unknown gene sequence that is extremely homologous to the target gene, it may not be possible to rule out false positives even with a highly specific CASLFA test (due to the lack of corresponding knowledge for specific probe design). In this situation, gene sequencing must be employed to understand the source of contamination. Once the source of contamination is known, new primers and sgRNA can be designed to implement the CRISPR eraser strategy.
5. CONCLUSIONS
In conclusion, the developed CRISPR/Cas9 eraser strategy, compatible with RT‐PCR and PCR systems, solves a false‐positive obstacle in the PCR end‐point detection method. CRISPR/Cas9 eraser‐assisted PCR combined with the strip test technique provides a cheaper nucleic acid testing solution when compared to the fluorescence q‐PCR method. We expect this advancement will bring about end‐point PCR testing as a widely available screening strategy for infected disease and other diagnosis applications.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Wei Lin and Xiaoming Zhou designed the research; Wei Lin performed the research; Debin Zhu and Yongzhong Jiang contributed new reagents; Tian Tian and Erhu Xiong analyzed data; Wei Lin and Xiaoming Zhou wrote the paper.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (Grants 91959128; 21874049; 21904042; 81772246), the Special Project of Science and Technology Development of Guangdong Province (2017B020207011); the Special Support Program of Guangdong Province (Grant 2016TQ03R749), the Opening Project of State Key Laboratory of Chemo/Biosensing and Chemometrics of Hunan University (Grant 2019004) and the Open Funds of the State Key Laboratory of Electroanalytical Chemistry (SKLEAC202001). We thank Professor Jian Sun from South China Agricultural University for kindly providing inactivated African swine fever DNA samples.
Lin, W. , Tian, T. , Jiang, Y. , Xiong, E. , Zhu, D. , & Zhou, X . A CRISPR/Cas9 eraser strategy for contamination‐free PCR end‐point detection. Biotechnology and Bioengineering. 2021;118:2053–2066. 10.1002/bit.27718
Contributor Information
Debin Zhu, Email: zhudb@scnu.edu.cn.
Xiaoming Zhou, Email: zhouxm@scnu.edu.cn.
REFERENCES
- Aslanzadeh, J. (2004). Preventing PCR amplification carryover contamination in a clinical laboratory. Annals of Clinical & Laboratory Science, 34(4), 389–396. [PubMed] [Google Scholar]
- Bao, Y. , Jiang, Y. , Xiong, E. , Tian, T. , Zhang, Z. , Lv, J. , & Zhou, X. (2020). CUT‐LAMP: Contamination‐free loop‐mediated isothermal amplification based on the CRISPR/Cas9 Cleavage. ACS Sensors, 5(4), 1082–1091. 10.1021/acssensors.0c00034 [DOI] [PubMed] [Google Scholar]
- Borst, A. , Box, A. T. , & Fluit, A. C. (2004). False‐positive results and contamination in nucleic acid amplification assays: Suggestions for a prevent and destroy strategy. European Journal of Clinical Microbiology and Infectious Diseases, 23(4), 289–299. 10.1007/s10096-004-1100-1 [DOI] [PubMed] [Google Scholar]
- Broughton, J. P. , Deng, X. , Yu, G. , Fasching, C. L. , Servellita, V. , Singh, J. , & Chiu, C. Y. (2020). CRISPR‐Cas12‐based detection of SARS‐CoV‐2. Nature Biotechnology, 38(7), 870–874. 10.1038/s41587-020-0513-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, J. , Zani, L. , Schwaiger, T. , Nurmoja, I. , Viltrop, A. , Vilem, A. , & Blome, S. (2018). Simplifying sampling for African swine fever surveillance: Assessment of antibody and pathogen detection from blood swabs. Transboundary and Emerging Diseases, 65(1), e165–e172. 10.1111/tbed.12706 [DOI] [PubMed] [Google Scholar]
- Chen, Z. , Liao, P. , Zhang, F. , Jiang, M. , Zhu, Y. , & Huang, Y. (2017). Centrifugal micro‐channel array droplet generation for highly parallel digital PCR. Lab on a Chip, 17(2), 235–240. 10.1039/c6lc01305h [DOI] [PubMed] [Google Scholar]
- Cimino, G. D. , Metchette, K. C. , Tessman, J. W. , Hearst, J. E. , & Isaacs, S. T. (1990). Post PCR sterilization a method to control carryover contamination for the polymerase chain reaction. Nucleic Acids Research, 19(1), 99–107. 10.1093/nar/19.1.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corless, C. E. , Guiver, M. , Borrow, R. , Edwards‐Jones, V. , Kaczmarski, E. B. , & Fox, A. J. (2000). Contamination and sensitivity issues with a real‐time universal 16S rRNA PCR. Journal of Clinical Microbiology, 38(5), 1747–1752. 10.1128/JCM.38.5.1747-1752.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deragon, J. M. , Sinnett, D. , Mitchell, G. , Potier, M. , & Labuda, D. (1990). Use of γ irradiation to eliminate DNA contamination for PCR. Nucleic Acids Research, 18(20), 6149. 10.1093/nar/18.20.6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty, R. M. , Phillips, P. E. , Gibson, S. , & Young, L. (1993). Restriction endonuclease digestion eliminates product contamination in reverse transcribed polymerase chain reaction. Journal of Virological Methods, 41(2), 235–238. 10.1016/0166-0934(93)90130-J [DOI] [PubMed] [Google Scholar]
- Feng, W. , Newbigging, A. M. , Le, C. , Pang, B. , Peng, H. , Cao, Y. , & Le, X. C. (2020). Molecular diagnosis of COVID‐19: Challenges and research needs. Analytical Chemistry, 92(15), 10196–10209. 10.1021/acs.analchem.0c02060 [DOI] [PubMed] [Google Scholar]
- Garigliany, M. , Desmecht, D. , Tignon, M. , Cassart, D. , Lesenfant, C. , Paternostre, J. , & Linden, A. (2019). Phylogeographic analysis of African swine fever virus, Western Europe, 2018. Emerging Infectious Diseases, 25(1), 184–186. 10.3201/eid2501.181535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genzen, J. R. (2020). Wiping the slate clean‐assessing clinical laboratory contamination risk. Clinical Chemistry, 66(9), 1128–1130. 10.1093/clinchem/hvaa161 [DOI] [PubMed] [Google Scholar]
- Gu, W. , Crawford, E. D. , O'Donovan, B. D. , Wilson, M. R. , Chow, E. D. , Retallack, H. , & DeRisi, J. L. (2016). Depletion of abundant sequences by hybridization (DASH): Using Cas9 to remove unwanted high‐abundance species in sequencing libraries and molecular counting applications. Genome Biology, 17, 41. 10.1186/s13059-016-0904-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajian, R. , Balderston, S. , Tran, T. , deBoer, T. , Etienne, J. , Sandhu, M. , & Aran, K. (2019). Detection of unamplified target genes via CRISPR‐Cas9 immobilized on a graphene field‐effect transistor. Nature Biomedical Engineering, 3(6), 427–437. 10.1038/s41551-019-0371-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, M. , Zhou, X. , Wang, H. , & Xing, D. (2018). Clustered regularly interspaced short palindromic repeats/Cas9 triggered isothermal amplification for site‐specific nucleic acid detection. Analytical Chemistry, 90(3), 2193–2200. 10.1021/acs.analchem.7b04542 [DOI] [PubMed] [Google Scholar]
- Hughes, S. (2017). Neisseria lactamica in biofilm (Master of Philosophy thesis). University of Southampton.
- Jiang, F. , Zhou, K. , Ma, L. , Gressel, S. , & Doudna, J. A. (2015). A Cas9‐guide RNA complex preorganized for target DNA recognition. Science, 348, 1477–1481. 10.1126/science.aab1452 [DOI] [PubMed] [Google Scholar]
- Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J. A. , & Charpentier, E. (2012). A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Lee, J. Y. , Yang, J. S. , Kim, J. W. , Kim, V. N. , & Chang, H. (2020). The architecture of SARS‐CoV‐2 transcriptome. Cell, 181(4), 914–921 e910. 10.1016/j.cell.2020.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbasov, D. , Titov, I. , Tsybanov, S. , Gogin, A. , & Malogolovkin, A. (2018). African swine fever virus, Siber Russia, 2017. Emerging Infectious Diseases, 24, 796–798. 10.3201/eid2404.171238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok, S. (1990). Procedures to minimize PCR‐product carry‐over. PCR protocols: A guide to methods and applications, 142–145. [Google Scholar]
- Lee, H. , Choi, J. , Jeong, E. , Baek, S. , Kim, H. C. , Chae, J. H. , & Kim, S. J. (2018). dCas9‐mediated nanoelectrokinetic direct detection of target gene for liquid biopsy. Nano Letters, 18(12), 7642–7650. 10.1021/acs.nanolett.8b03224 [DOI] [PubMed] [Google Scholar]
- Lee, S. H. , Yu, J. , Hwang, G. H. , Kim, S. , Kim, H. S. , Ye, S. , & Bae, S. (2017). CUT‐PCR: CRISPR‐mediated, ultrasensitive detection of target DNA using PCR. Oncogene, 36(49), 6823–6829. 10.1038/onc.2017.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, P. , Jiang, M. , Chen, Z. , Zhang, F. , Sun, Y. , Nie, J. , Du, M. , Wang, J. , Fei, P. , & Huang, Y. (2020). Three‐dimensional digital PCR through light‐sheet imaging of optically cleared emulsion. Proceedings of the National Academy of Sciences, 117(41), 25628–25633. 10.1073/pnas.2002448117/-/DCSupplemental [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. J. , Orlova, N. , Oakes, B. L. , Ma, E. , Spinner, H. B. , Baney, K. L. M. , & Doudna, J. A. (2019). CasX enzymes comprise a distinct family of RNA‐guided genome editors. Nature, 566(7743), 218–223. 10.1038/s41586-019-0908-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mens, P. F. , de Bes, H. M. , Sondo, P. , Laochan, N. , Keereecharoen, L. , van Amerongen, A. , & Schallig, H. D. (2012). Direct blood PCR in combination with nucleic acid lateral flow immunoassay for detection of Plasmodium species in settings where malaria is endemic. Journal of Clinical Microbiology, 50(11), 3520–3525. 10.1128/JCM.01426-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, J. , Modlin, J. , & Yolken, R. (1992). Use of modified nucleotides and uracil‐DNA glycosylase (UNG) for the control of contamination in the PCR‐based amplification of RNA. Molecular and Cellular Probes, 6(3), 251–256. 10.1016/0890-8508(92)90024-R [DOI] [PubMed] [Google Scholar]
- Park, S. , Zhang, Y. , Lin, S. , Wang, T. H. , & Yang, S. (2011). Advances in microfluidic PCR for point‐of‐care infectious disease diagnostics. Biotechnology Advances, 29(6), 830–839. 10.1016/j.biotechadv.2011.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patchsung, M. , Jantarug, K. , Pattama, A. , Aphicho, K. , Suraritdechachai, S. , Meesawat, P. , & Uttamapinant, C. (2020). Clinical validation of a Cas13‐based assay for the detection of SARS‐CoV‐2 RNA. Nature Biomedical Engineering, 4, 1140–1149. 10.1038/s41551-020-00603-x [DOI] [PubMed] [Google Scholar]
- Pruvost, M. , Grange, T. , & Geigl, E. M. (2005). Minimizing DNA contamination by using UNG‐coupled quantitative real‐time PCR on degraded DNA samples application to ancient DNA studies. Biotechniques, 38(4), 569–575. 10.2144/05384ST03 [DOI] [PubMed] [Google Scholar]
- Qin, Z. , Peng, R. , Baravik, I. K. , & Liu, X. (2020). Fighting COVID‐19: Integrated micro‐ and nanosystems for viral infection diagnostics. Matter, 3(3), 628–651. 10.1016/j.matt.2020.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan, J. , Langelier, C. , Kuchta, A. , Batson, J. , Teyssier, N. , Lyden, A. , & Crawford, E. D. (2019). FLASH: A next‐generation CRISPR diagnostic for multiplexed detection of antimicrobial resistance sequences. Nucleic Acids Research, 47(14), e83. 10.1093/nar/gkz418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter, S. J. , Cox, M. J. , Turek, E. M. , Calus, S. T. , Cookson, W. O. , Moffatt, M. F. , Turner, P. , Parkhill, J. , Loman, N. J. , & Walker, A. W. (2014). Reagent and laboratory contamination can critically impact sequence‐based microbiome analyses. BMC Biology, 12(1), 87. 10.1186/s12915-014-0087-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar, G. , & Sommer, S. S. (1991). Parameters affecting susceptibility of PCR contamination to UV inactivation. Biotechniques, 10(5), 590–594. [PubMed] [Google Scholar]
- Sethuraman, N. , Jeremiah, S. S. , & Ryo, A. (2020). Interpreting diagnostic tests for SARS‐CoV‐2. Journal of the American Medical Association, 323(22), 2249–2251. 10.1001/jama.2020.8259 [DOI] [PubMed] [Google Scholar]
- da Silva, S. J. R. , Silva, C. , Guarines, K. M. , Mendes, R. P. G. , Pardee, K. , Kohl, A. , & Pena, L. (2020). Clinical and laboratory diagnosis of SARS‐CoV‐2, the virus causing COVID‐19. ACS Infectious Diseases, 6(9), 2319–2336. 10.1021/acsinfecdis.0c00274 [DOI] [PubMed] [Google Scholar]
- Tetzner, R. , Dietrich, D. , & Distler, J. (2007). Control of carry‐over contamination for PCR‐based DNA methylation quantification using bisulfite treated DNA. Nucleic Acids Research, 35(1), e4. 10.1093/nar/gkl955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udaykumar, Epstein, J. S. , & Hewlett, I. K. (1993). A novel method employing UNG to avoid carry‐over contamination in RNA‐PCR. Nucleic Acids Research, 21(16), 3917–3918. 10.1093/nar/21.16.3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walder, R. Y. , Hayes, J. R. , & Walder, J. A. (1993). Use of PCR primers containing a 3′terminal ribose residuce to prevent cross‐contamination of amplified sequences. Nucleic Acids Research, 21(18), 4339–4343. 10.1093/nar/21.18.4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T. , Liu, Y. , Sun, H. H. , Yin, B. C. , & Ye, B. C. (2019). An RNA‐guided Cas9 nickase‐based method for universal isothermal DNA amplification. Angewandte Chemie—International Edition In English, 58(16), 5382–5386. 10.1002/anie.201901292 [DOI] [PubMed] [Google Scholar]
- Wang, X. , Xiong, E. , Tian, T. , Cheng, M. , Lin, W. , Wang, H. , & Zhou, X. (2020). Clustered regularly interspaced short palindromic repeats/Cas9‐mediated lateral flow nucleic acid assay. ACS Nano, 14(2), 2497–2508. 10.1021/acsnano.0c00022 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Kang, H. , Liu, X. , & Tong, Z. (2020). Combination of RT‐qPCR testing and clinical features for diagnosis of COVID‐19 facilitates management of SARS‐CoV‐2 outbreak. Journal of Medical Virology, 92(6), 538–539. 10.1002/jmv.25721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, L. (2020). Research progress on new coronavirus SARS‐CoV‐2. Acta Laser Biology Sinica, 29, 1–10. 10.3969/j.issn.1007-7146.2020.01.001 [DOI] [Google Scholar]
- Xu, Y. , Liu, Y. , Wu, Y. , Xia, X. , Liao, Y. , & Li, Q. (2014). Fluorescent probe‐based lateral flow assay for multiplex nucleic acid detection. Analytical Chemistry, 86(12), 5611–5614. 10.1021/ac5010458 [DOI] [PubMed] [Google Scholar]
- Yang, W. , Restrepo‐Perez, L. , Bengtson, M. , Heerema, S. J. , Birnie, A. , van der Torre, J. , & Dekker, C. (2018). Detection of CRISPR‐dCas9 on DNA with solid‐state nanopores. Nano Letters, 18(10), 6469–6474. 10.1021/acs.nanolett.8b02968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You, M. , Li, Z. , Feng, S. , Gao, B. , Yao, C. , Hu, J. , & Xu, F. (2020). Ultrafast photonic PCR based on photothermal nanomaterials. Trends in Biotechnology, 38(6), 637–649. 10.1016/j.tibtech.2019.12.006 [DOI] [PubMed] [Google Scholar]
- Yuan, C. , Tian, T. , Sun, J. , Hu, M. , Wang, X. , Xiong, E. , & Zhou, X. (2020). Universal and naked‐eye gene detection platform based on the clustered regularly interspaced short palindromic repeats/Cas12a/13a System. Analytical Chemistry, 92(5), 4029–4037. 10.1021/acs.analchem.9b05597 [DOI] [PubMed] [Google Scholar]
- Zhang, C. , Liu, X. , Yao, Y. , Liu, K. , Hui, W. , Zhu, J. , & Cui, Y. (2018). Genotyping of multiple clinical samples with a combined direct PCR and magnetic lateral flow assay. iScience, 7, 170–179. 10.1016/j.isci.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Cai, Y. , Zhang, J. , Liu, X. , He, L. , Cheng, L. , Hua, K. , Hui, W. , Zhu, J. , Wan, Y. , & Cui, Y. (2020). Tetra‐primer ARMS‐PCR combined with GoldMag lateral flow assay for genotyping simultaneous visual detection of both alleles. Nanoscale, 12(18), 10098–10105. 10.1039/D0NR00360C [DOI] [PubMed] [Google Scholar]
- Zhou, W. , Hu, L. , Ying, L. , Zhao, Z. , Chu, P. K. , & Yu, X. F. (2018). A CRISPR‐Cas9‐triggered strand displacement amplification method for ultrasensitive DNA detection. Nature Communications, 9(1), 5012. 10.1038/s41467-018-07324-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Li, N. , Luo, Y. , Liu, Y. , Miao, F. , Chen, T. , & Hu, R. (2018). Emergence of African Swine Fever in China, 2018. Transboundary and Emerging Diseases, 65(6), 1482–1484. 10.1111/tbed.12989 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.