

Abstract
The compartmentalization of eukaryotic cells, which is essential for their viability and functions, is ensured by single or double bilayer membranes that separate the cell from the exterior and form boundaries between the cell’s organelles and the cytosol. Nascent nuclear envelopes and autophagosomes, which both are enveloped by double membranes, need to be sealed during the late stage of their biogenesis. On the other hand, the integrity of cellular membranes such as the plasma membrane, lysosomes and the nuclear envelope can be compromised by pathogens, chemicals, radiation, inflammatory responses and mechanical stress. There are cellular programmes that restore membrane integrity after injury. Here, we review cellular mechanisms that have evolved to maintain membrane integrity during organelle biogenesis and after injury, including membrane scission mediated by the endosomal sorting complex required for transport (ESCRT), vesicle patching and endocytosis.
Keywords: autophagy, endocytosis, ESCRT, lysosome, membrane repair
Subject Categories: Membrane & Intracellular Transport
This review provides an overview of the cellular mechanisms that have evolved to maintain membrane integrity during organelle biogenesis and after injury in eukaryotic cells.

Introduction
The bilayered membranes of eukaryotic cells are vital to their very existence, with the plasma membrane separating the cells from their surroundings, and the endomembranes enclosing the various organelles. It is crucial that these membranes are intact so that only gated transport of molecules and ions across them can occur. Defective membrane sealing is indeed associated with a large number of diseases, including myopathies, central and peripheral neurological disorders, and coronary diseases (Cooper & McNeil, 2015). Sealing of holes in membranes is therefore of great importance in biology, both during biogenesis of double‐membrane organelles and as response to membrane damage. For instance, all newly formed nuclei contain transient holes (Anderson & Hetzer, 2008), and it has been estimated that as many as 20–30% of cardiac and skeletal muscle cells and 6% of skin cells have transient openings in their plasma membrane (McNeil & Steinhardt, 1997). Whereas membrane openings during organelle biogenesis have a defined size of < 100 nm (Olmos et al, 2015; Denais et al, 2016), holes in membranes due to damage may range from a few nm to several µm (Cooper & McNeil, 2015). The molecular and cellular mechanisms that have evolved to seal such holes will be discussed in this review.
Mechanisms of membrane sealing
Holes in bilayer membranes can be sealed by several alternative mechanisms (Fig 1), most of which require specialized protein machineries. Below is a brief overview of different mechanisms that have been proposed.
Figure 1. Mechanisms of membrane sealing.
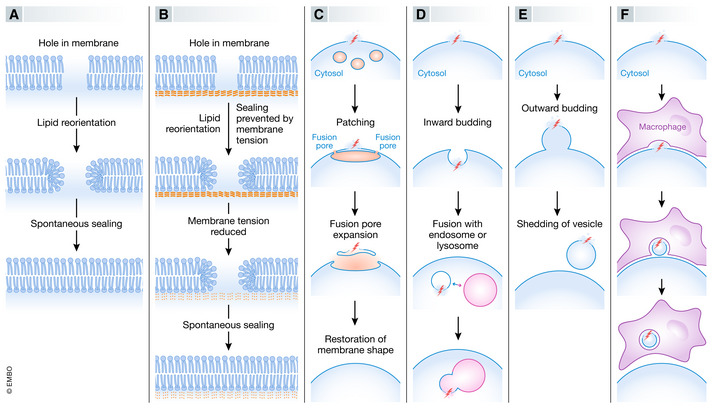
Holes in lipid bilayers can be sealed by several alternative mechanisms, including membrane self‐sealing (A), reduction in membrane tension to promote self‐sealing (B), patching of hole by vertex fusion of large intracellular vesicles (C), inward budding of hole‐containing membrane area (D), outward budding of hole‐containing membrane area (E), or removal of hole‐containing membrane area by adjacent cell (F).
Self‐sealing of membranes
Studies of artificial membranes have shown that ruptured bilayer membranes can seal spontaneously without the need for auxiliary proteins (Gozen & Dommersnes, 2014) (Fig 1A). When a hole is formed in a lipid bilayer, lipid disorder present on the curved edges of the hole provides a driving force for resealing so that edge tension can be minimized. On the other hand, resealing is counteracted by membrane tension, which is low in most artificial membranes but is high in a cellular context where the membrane is attached to rigid components such as the cytoskeleton or the nuclear lamina. The high tension of cellular membranes thus disfavours self‐sealing of holes of more than a few nm (McNeil & Kirchhausen, 2005).
Reduction in membrane tension
Because membrane tension limits the ability of cellular membranes to self‐seal, cellular mechanisms that reduce membrane tension can promote membrane sealing. One such mechanism is addition of more membrane via vesicle fusion (Fig 1B). A second mechanism for reducing membrane tension is to alter membrane curvature, whereas a third mechanism is to disrupt membrane‐associated protein scaffolds.
Patching by fusion of intracellular membranes
Early work on membrane repair revealed numerous small cytoplasmic vesicles close to the site of injury, that formed larger vesicles via homotypic fusion (Terasaki et al, 1997) (Fig 1C). This generated the hypothesis that large cytoplasmic vesicles can patch up large (up to several µm) damaged membrane areas. It has been proposed that the patch vesicle makes fusion pores with the plasma membrane at several discrete sites around the damaged area and that these pores expand to form a continuous union of patch vesicle and the plasma membrane. In this way, the damaged plasma membrane will be replaced by the new membrane (McNeil & Kirchhausen, 2005). This mechanism is still debated as it would require stabilization and lateral expansion of the fusion pore to form a ring around the site of damage that is shed when the fusion pores meets. An alternative mechanism for how vesicle fusion might mediate membrane repair is the concept that such fusion causes release of an enzyme that promotes membrane sealing (Tam et al, 2010).
Inward budding and scission of damaged membrane area
One of the proposed mechanisms for membrane repair entails inward budding (i.e., in the direction towards the cytosol) of a membrane area that contains the damaged zone (Idone et al, 2008) (Fig 1D). The resulting cytoplasmic vesicle, which will contain a damaged membrane area, can then fuse with an endosome or lysosome, thus effectively transferring the damage from the original membrane to the endosome or lysosome membrane, from where it can be internalized into the lumen in the form of an intraluminal vesicle (ILV) (Andrews et al, 2014).
Outward budding and scission of damaged membrane area
Outward vesicle budding (i.e. in the direction away from the cytosol) ensures that damaged membrane areas are removed from their site of origin and shed either as extracellular vesicles or ILVs that form inside organelles such as endosomes or lysosomes (Fig 1E). This type of vesicle budding shares topology with closure of nascent double‐membrane organelles such as autophagosomes and the nuclear envelope and is mediated by the same molecular machinery.
Removal of damaged membrane area by adjacent cells
Even though membrane repair has mostly been described as a cell autonomous mechanism, muscle cells, which are particularly prone to damage of their plasma membrane, can also engage neighbouring macrophages to pinch off damaged portions of the muscle cell plasma membrane (Middel et al, 2016) (Fig 1F). How macrophage‐mediated removal of the damaged plasma membrane can proceed in a way that preserves membrane integrity still remains to be resolved.
Proteins that promote membrane sealing
Proteins that promote membrane sealing can roughly be subdivided into detectors of membrane damage, fusion regulators and fission regulators (see Table 1).
Table 1.
Proteins involved in membrane sealing. See text for references.
| General function in membrane sealing | Protein name | Specific function | Site of action | |
|---|---|---|---|---|
| Sensing of membrane integrity | Sensing of Ca2+ influx into cytosol | PDCD6 | Binds to ALIX to promote ESCRT recruitment | Plasma membrane, endolysosomes |
| ALIX | Promotes ESCRT recruitment | Plasma membrane, endolysosomes | ||
| Annexins | Together with Ca2+ binding proteins of the S100 family, involved in membrane fusion, shaping, sealing and tension reduction | Plasma membrane, endolysosomes | ||
| SYT7 | Endolysosomal protein that activates SNAREs in membrane fusion | Plasma membrane | ||
| Dysferlin | Accumulates phosphatidylserine at the site of membrane damage, as an “eat‐me” signal for macrophages. Interacts with some annexins | Plasma membrane | ||
| TMEM16F | Lipid scramblase that causes exposure of phosphatidylserine at the extracytosolic leaflet | Plasma membrane | ||
| Sensing of β‐galactoside sugars on membrane proteins not normally exposed to cytosol | GAL3 | Interacts with ALIX and the autophagy‐associated ubiquitin E3 ligase TRIM16 | Endolysosomes | |
| GAL8 | Inhibits mTORC1 and interacts with the autophagy receptor NDP52 | Endolysosomes | ||
| GAL9 | Activates AMPK | Endolysosomes of macrophages and hematopoietic cells | ||
| Sensing of nuclear envelope integrity | CHMP7 | Binds to the inner nuclear membrane proteins LEMD2 and LEMD3 and recruits ESCRT‐III proteins | Nuclear envelope, micronuclear envelope | |
| Membrane fusion | SNARE proteins (VAMP, syntaxin and SNAP families) | Mediate fusion of specific membranes through formation of tetrahelical trans‐SNARE complexes | Multiple | |
| Membrane scission towards cytosol | Acidic sphingomyelinase | Lysosomal enzyme that is shed by Ca2+‐dependent exocytosis in response to wounding. Converts sphingomyelin into ceramide, which triggers caveolae endocytosis | Plasma membrane | |
| GRAF1 | GTPase‐activating protein for the small GTPase Cdc42. Promotes clathrin‐independent endocytosis | Plasma membrane, endosomes | ||
| Membrane scission away from cytosol | ESCRT‐I | Recruits ESCRT‐III | Multiple | |
| ESCRT‐III | Filamentous complex that mediates scission of narrow membrane necks filled with cytosol, to achieve membrane sealing | Multiple | ||
| VPS4 | AAA ATPase that regulates ESCRT‐III assembly and disassembly | Multiple | ||
Ca2+‐binding proteins as sensors of membrane damage
Whereas cytosolic levels of Ca2+ are low (about 100 nM), the Ca2+ concentrations in extracellular fluids and organelles such as the endoplasmic reticulum (ER) and lysosomes can reach several mM (Kass & Orrenius, 1999). The steep Ca2+ gradients across cellular membranes indeed form the basis of a simple yet powerful mechanism for detection of membrane integrity. Leakage of Ca2+ into cytosol is detected by several Ca2+–binding proteins that promote membrane sealing by membrane fusion, fission or tension reduction.
Annexins are abundant Ca2+‐binding cytosolic proteins that tend to assemble on membranes in response to rises in cytosolic Ca2+ levels (Gerke & Moss, 2002). Several annexins bind to small Ca2+‐binding proteins of the S100 family, which contribute to their functions as Ca2+ effectors. Evidence from both lower organisms such as Paramecium and mammalian cells has shown that annexins play roles in membrane fusion, shaping and sealing although it has been difficult to pinpoint their exact functional mechanisms (Koerdt et al, 2019; Bendix et al, 2020). One of the functions of annexins in membrane sealing may be to reduce membrane tension so that membrane self‐sealing is promoted (Bouter et al, 2011). Annexins have also been proposed to assemble into multimeric structures that physically cap the hole in the membrane (Demonbreun et al, 2016; Robinson et al, 2020).
Synaptotagmins are integral membrane proteins that function as Ca2+‐sensors that mediate membrane fusion. Among these, Synaptotagmin‐7 (SYT7) has been found to be particularly important for membrane repair. The cytosolic part of SYT7 contains two Ca2+‐ and phospholipid‐binding C2 domains that sense cytosolic Ca2+and mediate interactions with membranes. Like other members of the Synaptotagmin family, SYT7 regulates formation of SNARE complexes that drive membrane fusion and thus transduces Ca2+ sensing into membrane fusion (see below).
Whereas SYT7 couples Ca2+ sensing to membrane fusion in membrane repair, another Ca2+ sensor, PDCD6 (also known as ALG‐2), connects with a membrane fission machinery that mediates repair. PDCD6 is a small penta‐EF‐hand protein that contains a binding site for Ca2+ and Mg2+ and interacts with other proteins in a Ca2+‐dependent manner. One of the interacting partners of PDCD6 is ALIX (PDCD6IP), a scaffolding protein involved in diverse cellular functions. ALIX also binds Ca2+, albeit with lower affinity than PDCD6. Injury‐triggered influx of Ca2+ causes accumulation of PDCD6 at the site of injury, and PDCD6 in turn recruits ALIX. ALIX then recruits components of the ESCRT machinery that mediates membrane repair by outward budding and fission of the damaged membrane area (see below).
Muscle cells contain an additional Ca2+‐sensing membrane protein, Dysferlin (DYSF). The cytosolic region of DYSF harbours seven C2 domains, and Ca2+ sensing by DYSF plays a key role in repair of damaged plasma membranes of muscle cells as illustrated by the involvement of DYSF mutations in genetic myopathies (Cardenas et al, 2016). DYSF has both cell autonomous and non‐autonomous functions in membrane sealing. Via an arginine‐rich motif, DYSF mediates accumulation of phosphatidylserine at the site of plasma membrane damage, and this triggers removal of the damaged membrane region by neighbouring macrophages (Middel et al, 2016). Translocation of phosphatidylserine to the extracytoplasmic leaflet can be mediated by a Ca2+ –activated lipid scramblase, TMEM16F, which promotes repair of the plasma membrane after injury by pore‐forming toxins by reducing membrane tension and facilitating release of extracellular vesicles containing damaged membranes (Wu et al, 2020).
Galectins as sensors of endomembrane damage
Galectins are a family of cytosolic lectins that bind specifically to β‐galactoside carbohydrates, which are linked to the extracytosolic domains of a large number of integral membrane proteins (Vasta, 2009). They function as pattern recognition receptors and can serve as sensors of membrane integrity by binding to sugars that are not normally exposed to cytosol. This has been found to be the case with the ubiquitously expressed Galectin‐3 and Galectin‐8, and also with Galectin‐9, which is mainly expressed in macrophages and cells of the haematopoietic system. Galectins can transmit signals about compromised membrane integrity in several different ways. For example, upon lysosome damage, Galectin‐3 interacts with ALIX and the autophagy‐associated E3 ubiquitin ligase TRIM16 (Chauhan et al, 2016; Jia et al, 2020b), Galectin‐8 inhibits the mTORC1 signalling complex and interacts with the autophagy receptor NDP52 (Jia et al, 2018; Weng et al, 2018), and Galectin‐9 activates the low energy‐sensing kinase AMPK (Jia et al, 2020a). By these mechanisms, galectins can couple sensing of membrane damage to membrane repair but also to metabolic control and autophagy of damaged organelles.
SNARE proteins in membrane fusion during membrane sealing
Soluble NSF attachment protein receptors (SNAREs) are a large group of small proteins that mediate membrane fusion (Sudhof & Rothman, 2009). They are frequently subdivided into vesicle (v)‐SNAREs (synaptobrevin/VAMP family) and target membrane (t)‐SNAREs (Syntaxin and SNAP families). Most SNAREs are integral membrane proteins that are tail‐anchored to specific membranes, but there are also examples of soluble SNAREs and SNAREs that are membrane‐bound via cysteine‐linked palmitoyl chains. All SNAREs are characterized by their content of a 60–70 amino acid region called the SNARE motif, which contains heptad repeats. The well‐characterized core trans‐SNARE complex in fusion of synaptic vesicles with the presynaptic plasma membrane consists of a four‐α‐helix bundle with one α‐helix contributed by the synaptic vesicle protein VAMP2, one helix by the plasma membrane protein STX1A, and two α‐helices by the plasma membrane protein SNAP‐25 (Poirier et al, 1998). SNARE assembly into stable four‐helix bundles not only bridges membranes but also provides the energy for initiating membrane fusion.
ESCRT proteins in membrane scission and sealing
Originally identified for its function in endosomal sorting of ubiquitinated membrane proteins into ILVs of endosomes (Katzmann et al, 2002), the endosomal sorting complex required for transport (ESCRT) machinery is now recognized as the major catalyst of scission and sealing reactions at diverse cellular locations that share the same topology (Schoneberg et al, 2017; Vietri et al, 2020a). The machinery consists of three subcomplexes, ESCRT‐I, ‐II and –III. ESCRT‐I is an elongated helical heterotetramer with the ubiquitin‐binding protein TSG101 as a core component. ESCRT‐II is a heterotetramer of three winged‐helix (WH) domain proteins (EAP30, EAP45 and two subunits of EAP20), whereas ESCRT‐III is a biochemically more heterogenous complex that forms helical filaments consisting of small highly charged α‐helical proteins of the CHMP family. The membrane scission/sealing activity of ESCRT is mediated by the ESCRT‐III filaments, whose activity is controlled and modulated by the AAA ATPase VPS4. ESCRT‐III can be recruited to membranes by several alternative mechanisms, including direct interactions with ESCRT‐I, ESCRT‐II, the ESCRT‐II/‐III like protein CHMP7 or the ESCRT‐I‐binding protein ALIX.
During sealing of damaged plasma membrane and lysosome membranes, both the ESCRT‐I subunit TSG101 and ALIX have been assigned important functions in ESCRT‐III recruitment (Jimenez et al, 2014; Scheffer et al, 2014; Radulovic et al, 2018; Skowyra et al, 2018), whereas the Ca2+ sensor PDCD6 and the carbohydrate sensor GAL3 have been implicated in ALIX recruitment (Scheffer et al, 2014; Skowyra et al, 2018; Jia et al, 2020b). ESCRT‐I also plays a pivotal role in ESCRT‐III recruitment for closure of nascent autophagosomes (Takahashi et al, 2019). During reformation or repair of nuclear envelopes, CHMP7, a protein with both ESCRT‐II‐ and ESCRT‐III‐like domains, functions as a sensor for openings in the nuclear envelope and mediates recruitment of ESCRT‐III (Vietri et al, 2015; Denais et al, 2016; Olmos et al, 2016; Raab et al, 2016; Vietri et al, 2020b). Available data suggest that the ESCRT machinery is suited for sealing small holes in membranes, with a diameter < 100 nm (Jimenez et al, 2014; Olmos et al, 2015; Denais et al, 2016; Bohannon & Hanson, 2020), whereas other mechanisms probably account for sealing of larger holes. It is interesting to note that low membrane tension promotes ESCRT‐III recruitment, and reduction in membrane tension after membrane injury could thus by itself contribute to ESCRT‐III recruitment (Mercier et al, 2020).
Membrane sealing during organelle biogenesis
Most of the cell’s organelles form by membrane budding and fusion reactions that do not involve membrane sealing. However, two organelles enclosed by double membranes, the nucleus and the autophagosome, contain holes that need to be closed during the end of their biogenesis.
Sealing of the nascent nuclear envelope
During mitosis in mammalian cells, duplicated chromosomes condense during prophase, and the nuclear envelope is disassembled. The chromosomes align at the equator of the dividing cell in metaphase and are then pulled by microtubule bundles to each pole in anaphase. At the end of anaphase, new nuclear envelopes assemble around the two separated chromosome clusters to form daughter nuclei, which are completed during telophase. This is followed by separation of the two daughter cells at the end of cytokinesis. The nascent nuclear envelopes are formed from membranes mobilized from the endoplasmic reticulum, which fuse to form a continuum. However, the completion of the new nuclear envelopes requires both the removal of the microtubule bundles used for chromosome separation and the closure of holes remaining after the microtubules have been removed (De Magistris & Antonin, 2018). Both these events are accomplished by the ESCRT machinery (Fig 2A).
Figure 2. Sealing of holes during biogenesis of double‐membrane organelles.
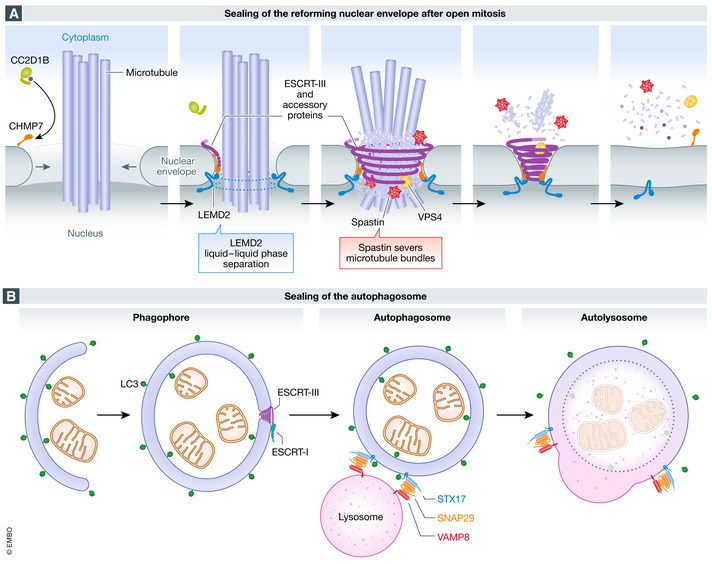
(A) sealing of the reforming nuclear envelope after open mitosis. During anaphase, when the reforming nuclear envelope meets microtubule bundles still connected to chromatin, the inner nuclear membrane protein LEMD2 undergoes liquid‐phase separation and activates the ESCRT‐III specific nuclear envelope recruitment factor CHMP7, which drives ESCRT‐III polymerization. Timing of ESCRT‐III recruitment is also regulated by CC2D1B, which prevents its premature localization to the reforming membrane. Spastin recruitment by ESCRT‐III is required for severing of mitotic spindle microtubules, while VPS4 modelling of ESCRT‐III filaments promotes membrane constriction and sealing. (B) sealing of the autophagosome. During autophagy, the double‐membrane phagophore expands to sequester cytoplasmic material for degradation. When the resulting LC3‐positive autophagosome is complete, a small hole remains. This hole is sealed by ESCRT‐III, which is recruited by ESCRT‐I. Sealing is followed by recruitment to the autophagosome membrane of the SNARE protein STX17, which forms a complex with the cytosolic SNARE SNAP29 and the lysosomal SNARE VAMP8, and this mediates fusion of the autophagosome with the lysosome to form an autolysosome.
Firstly, ESCRT‐III is recruited and its polymerization triggered to the microtubule‐traversed holes. This requires the interaction of endoplasmic reticulum‐associated CHMP7 with the integral inner nuclear membrane protein LEMD2 (Vietri et al, 2015; Olmos et al, 2016; Webster et al, 2016; Gu et al, 2017). ESCRT‐III subsequently recruits the microtubule‐severing AAA ATPase Spastin via the ESCRT‐III associated protein IST1, and this causes removal of the spindle microtubules (Vietri et al, 2015). The remaining holes are then closed by ESCRT‐III (Olmos et al, 2015; Vietri et al, 2015).
Regulation of these events in time and space requires the interaction of CHMP7 with LEMD2. LEMD2 bridges the nuclear envelope with the underlying chromatin through an N‐terminal Lap2, Emerin, Man1 (LEM)‐domain. LEMD2 also contains a C‐terminal WH domain, responsible for interaction and activation of CHMP7. At the sites where the membrane is intersected by microtubule bundles, LEMD2 accumulates and undergoes liquid‐phase separation, thereby triggering CHMP7 activation and ESCRT‐III assembly (von Appen et al, 2020). Furthermore, ESCRT‐III recruitment and activity are regulated by additional factors, including CC2D1B, which prevents premature ESCRT‐III polymerization (Ventimiglia et al, 2018), and UFD1, a co‐factor of the AAA ATPase p97 that has been shown to regulate ESCRT‐III subunit CHMP2A into ESCRT‐III filaments (Olmos et al, 2015).
Because the nuclear envelope is a double membrane, this ESCRT‐mediated closure is topologically equivalent to ESCRT‐III‐dependent fission reactions such as shedding of extracellular vesicles from the plasma membrane or formation of intraluminal vesicles in endosomes (Vietri et al, 2020a). ESCRT‐III driven nuclear envelope sealing after mitosis is evolutionarily conserved and has even been described in lower eukaryotes. In semi‐open mitosis of Schizosaccharomyces japonicus, similar events leading to re‐establishment of nucleocytoplasmic compartmentalization in mitotic exit have been described, including the accumulation of a LEMD2 orthologue at intersections between the nuclear envelope and the mitotic spindle, as well as the importance of orthologues of CHMP7, CHMP4B and VPS4 (Yam et al, 2011; Pieper et al, 2020).
Sealing of the nascent autophagosome
Autophagy is a vital cellular process for lysosomal degradation of cytoplasmic content, which ensures cell survival under starvation conditions and removal of potentially harmful cytoplasmic objects such as microorganisms, protein aggregates and damaged organelles (Mizushima et al, 2008; Mizushima et al, 2011). The process starts with a double‐membrane phagophore membrane that forms around bulk cytoplasm or specific cargo, and the phagophore eventually closes to form a complete autophagosome. When the autophagosome fuses with a lysosome to form an autolysosome, the sequestered cargo becomes degraded by lysosomal hydrolases. Even though contributions to the phagophore membrane may come from the plasma membrane, endosomes, mitochondria or de novo synthesis, most studies to date indicate the endoplasmic reticulum as a major membrane source (Melia et al, 2020). When the phagophore membrane has grown around cytoplasmic content and shaped to form a complete autophagosome, it contains a small hole that needs to be closed (Knorr et al, 2015). This is achieved by ESCRT‐III by a mechanism that is topologically similar to other ESCRT‐dependent fission reactions (Takahashi et al, 2018; Zhen et al, 2020) (Fig 2B). Recruitment of ESCRT‐III to the nascent autophagosome requires ESCRT‐I, and a putative ubiquitin E2 variant (UEV) domain of the ESCRT‐I subunit VPS37A is important for autophagosome closure (Takahashi et al, 2019). How ESCRT‐I is recruited to the phagophore still remains to be understood, but studies of budding yeast mutants have suggested the involvement of the small endosomal GTPase Rab5 and Atg17, a subunit of the Atg1 autophagic kinase complex, as upstream regulators (Zhou et al, 2017; Zhou et al, 2019). Because autophagosomes are known to fuse with endosomes (Berg et al, 1998), it still remains to be established whether Rab5 and Atg17 (or its mammalian homologue, FIP200) are directly involved in ESCRT recruitment to nascent autophagosomes. The importance of ESCRT‐mediated autophagosome sealing is illustrated by the fact that ESCRT depletion causes accumulation of autophagosomes that are incapable of fusing with lysosomes (Filimonenko et al, 2007; Rusten et al, 2007). This phenomenon can probably be explained by the failure of unsealed autophagosomes to recruit Syntaxin 17, a SNARE protein required for autophagosome‐lysosome fusion (Itakura et al, 2012).
Membrane repair
Most cellular membranes are exposed to damage, and repair machineries promote cell survival by closing the holes. Below, we will discuss those membranes that have been most studied in the context of damage and repair.
Repair of the damaged plasma membrane
Because the plasma membrane faces the exterior of the cell, it is particularly prone to rupture by pathogens, chemicals and mechanical forces. In addition, holes in the plasma membrane are also employed in inflammatory cell death pathways, including complement‐induced necrosis, inflammasome‐induced pyroptosis and tumour necrosis factor α‐induced necroptosis (Liu & Lieberman, 2020). Several mechanisms have been described for repair of damaged plasma membranes, including patching by intracellular membranes or removal of damaged area by endocytosis, outward budding or pinching‐off by neighbouring macrophages (Fig 3).
Figure 3. Plasma membrane repair.
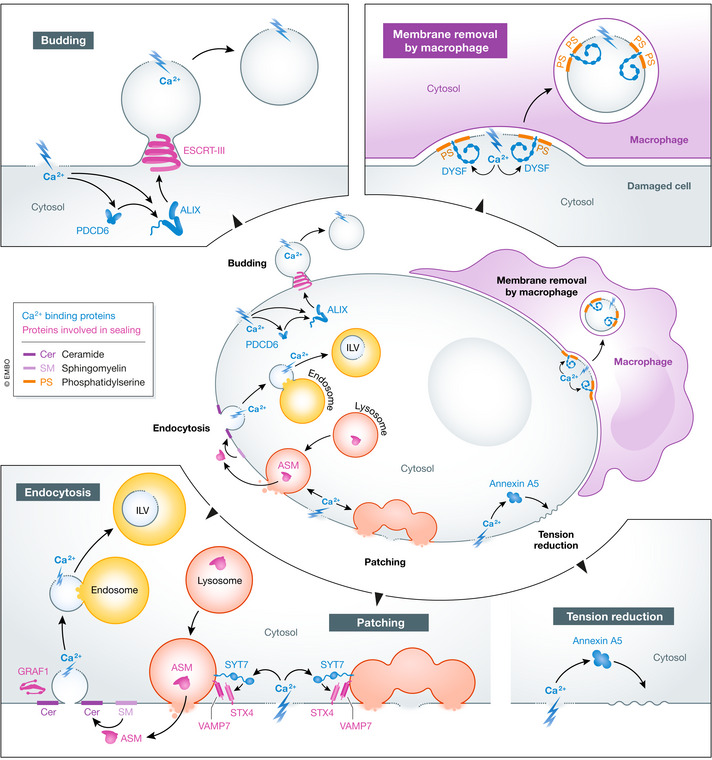
Holes in the plasma membrane can be sealed by patching, endocytosis, budding, macrophage‐mediated membrane removal or reduction in membrane tension. Ca2+ influx triggers all these processes. Ca2+ binding proteins are in green font, other proteins involved in sealing are in red font. Cer, ceramide; ILV, intraluminal vesicle; PS, phosphatidylserine.
The involvement of vesicle fusion in plasma membrane repair originally led to the hypothesis that large intracellular vesicles, formed by homotypic fusion of smaller vesicles, patch up the damaged area of the plasma membrane (Terasaki et al, 1997; McNeil et al, 2000). Indeed, exocytosis of lysosomes plays a well‐documented role in plasma membrane repair after mechanical or bacterial toxin‐induced injury. Ca2+ influx through holes in the plasma membrane activates SYT7 to promote fusion between peripheral lysosomes and the plasma membrane, mediated by a trans‐SNARE complex consisting of the lysomal v‐SNARE VAMP7 and the plasma membrane t‐SNAREs STX4 and SNAP23 (Reddy et al, 2001; Rao et al, 2004). Interference with SYT7 or SNARE functions prevents plasma membrane repair, demonstrating that fusion between lysosomes and the plasma membrane is essential. It has been difficult to explain how addition of more membrane to the plasma membrane would seal large holes, and also how this would serve to close long‐lasting pores formed by bacterial toxins. However, a mechanism that involves formation of expanding fusion pores around the damaged area, resulting in replacement of this area with a membrane patch from a large intracellular vesicle (such as a lysosome) has gained support (see Fig 1) (McNeil & Kirchhausen, 2005).
Later studies have shown that injury‐dependent lysosome exocytosis after plasma membrane damage with Streptolysin O is followed by a burst of endocytosis of the damaged membrane area via flask‐shaped invaginations known as caveolae (Idone et al, 2008) and that the pores end up in ILVs of late endosomes and lysosomes where they are degraded. A lysosomal enzyme, acid sphingomyelinase (ASM), provides the link between damage‐induced lysosome exocytosis and caveolar endocytosis. When ASM is released from cells as lysosomes fuse with the plasma membrane, sphingomyelin in the extracellular leaflet of the plasma membrane is converted into ceramide, and this triggers rapid endocytosis via caveolae (Tam et al, 2010). There is also evidence that decreased plasma membrane tension triggers another type of endocytosis mediated by GRAF1 (Holst et al, 2017), a protein with a known involvement in muscle cell plasma membrane repair (Lenhart et al, 2015).
Plasma membrane damage induced by mechanical force, laser radiation, detergents or pore‐forming toxins causes a rapid redistribution of ESCRT‐I and ‐III proteins to the site of damage (Jimenez et al, 2014). This is accompanied by the accumulation of extracellular buds that are shed from the membrane. Depletion of ESCRT‐III subunits or VPS4B prevents plasma membrane repair, and it is reasonable to conclude that ESCRT‐III and VPS4B cooperate to mediate plasma membrane repair by promoting extracellular budding and shedding of plasma membrane domains containing the site of damage. Damage‐induced ESCRT recruitment to the plasma membrane is Ca2+‐dependent, and the requirement of PDCD6, ALIX and Annexin A7 for ESCRT‐III recruitment and plasma membrane repair suggests that these proteins could function as Ca2+ sensors that trigger ESCRT recruitment (Scheffer et al, 2014; Sonder et al, 2019). It still remains to be understood how ESCRT‐III is recruited around the site of damage so that the damaged area becomes incorporated into the ESCRT‐dependent bud.
Mouse perivascular cells devoid of Annexin A5 exhibit a severe defect in repair of their plasma membrane (Bouter et al, 2011), indicating a role for this annexin in plasma membrane repair. Annexin A5 assembles into local two‐dimensional arrays at the plasma membrane in response to Ca2+ influx, and it has been proposed that these arrays prevent wound expansion and promote resealing by reducing cytoskeleton‐induced membrane tension (Bouter et al, 2011).
Perhaps owing to their particular exposure to mechanical damage, muscle cells rely on an additional mechanism for repair of their plasma membrane, namely heterologous repair mediated by macrophages. Damage‐induced influx of Ca2+ causes recruitment of the membrane protein Dysferlin (DYSF), whose cytosolic Ca2+ binding domain ensures clustering at the site of damage. A phosphatidylserine‐binding motif in DYSF promotes accumulation of phosphatidylserine in the extracellular leaflet of the plasma membrane around the site of injury, marked by a “repair patch” consisting of proteins and lipids, and phosphatidylserine then causes recruitment of macrophages in an analogous way to apoptotic cells. However, whereas apoptotic cells, which contain phosphatidylserine all over their plasma membrane, are fully engulfed by macrophages, the macrophages only pinch off the damaged areas of the muscle cell plasma membrane (Middel et al, 2016). A potential source of DYSF and PS might be caveolae, which unfold on membrane stress and thus rapidly increase membrane surface (Sinha et al, 2011). It is interesting to note that DYSF interacts with MG53/TRIM72, another phosphatidylserine‐binding protein involved in repair of the muscle cell plasma membrane (Cai et al, 2009). The proposed mechanism of TRIM72 is in fusion‐mediated patching of holes in the plasma membrane, but its involvement in macrophage‐mediated repair has not been clarified.
Repair of the damaged nuclear envelope
Because of its rigidity and large size, the nucleus is particularly prone to membrane damage when the cell moves through a confined space. In addition, reversible nuclear envelope ruptures are frequently detected in genetic disorders caused by mutations in proteins of the nuclear lamina, known as laminopathies, or in cancer cells (De Vos et al, 2011; Vargas et al, 2012; Denais et al, 2016; Raab et al, 2016; Earle et al, 2020). Upon rupture, uncoordinated mixing of nucleocytoplasmic components such as transcription factors, mRNA processing proteins, DNA damage repair factors and mitochondria and PML bodies occur and may affect a number of cellular functions (De Vos et al, 2011; Vargas et al, 2012).
The exposure of genomic DNA to the cytosolic nuclease TREX1 causes accumulation of DNA damage and leads to reduced cell viability in laminopathies and senescence in non‐transformed cell lines (Earle et al, 2020). Nuclear envelope ruptures at chromatin bridges and micronuclei lead to extensive DNA damage, kataegis, chromosome shattering and chromothripsis, supporting the generation of complex genomes in cancer cells (Maciejowski et al, 2015; Zhang et al, 2015; Umbreit et al, 2020). Along with these effects, prolonged exposure of DNA to the cytoplasm activates innate immunity and inflammatory responses dependent on the cytosolic DNA sensor, cyclic GMP‐AMP synthase (cGAS) (Harding et al, 2017; Mackenzie et al, 2017).
In mammalian cells, repair of large ruptures first requires the mobilization of nuclear envelope membranes to coat the exposed chromatin. This is achieved through the action of barrier‐to‐autoantigen factor (BAF), a protein that exists in the cell as an unphosphorylated cytosolic pool, and as a phosphorylated nucleoplasmic pool which bridges the chromatin to the nuclear envelope. Upon nuclear envelope rupture, cytosolic BAF readily binds the exposed DNA and recruits membrane through its interaction with nuclear transmembrane proteins containing a LEM domain, such as LEMD2 (Halfmann et al, 2019; Young et al, 2020). BAF may in addition have a role in keeping the exposed chromatin compacted so that membrane coating and resealing are facilitated (Samwer et al, 2017; Penfield et al, 2018; Robijns et al, 2018). Subsequently, sealing depends on ESCRT‐III (Denais et al, 2016; Raab et al, 2016), similarly to sealing of nascent nuclear envelopes during mitotic exit (Olmos et al, 2015; Vietri et al, 2015). The overall mechanism of sealing appears to be the same as with nascent nuclear envelopes, with CHMP7 playing an essential role as nuclear envelope integrity sensor and ESCRT‐III recruiter (Fig 4). CHMP7 contains a nuclear export signal which ensures that CHMP7 is kept out from the nucleus at steady state (Thaller et al, 2019; Vietri et al, 2020b). However, whenever the integrity of the nuclear envelope is compromised, CHMP7 is recruited to the inner nuclear membrane by interacting with the inner nuclear membrane proteins LEMD2 and LEMD3. This translocation of CHMP7 is succeeded by ESCRT‐III recruitment and membrane repair (Vietri et al, 2020b).
Figure 4. Nuclear envelope repair.
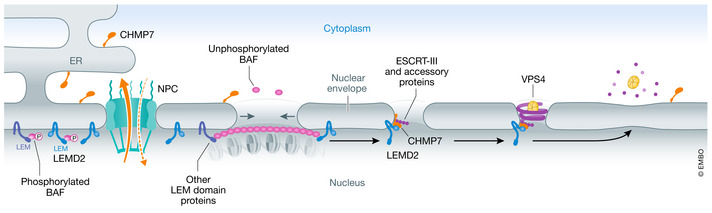
Under unperturbed conditions, the inner nuclear membrane protein LEMD2 and the endoplasmic reticulum‐associated protein CHMP7 are spatially separated because CHMP7 is actively exported out of the nucleus through nuclear pore complexes (NPCs). Following rupture, unphosphorylated cytosolic BAF coats the exposed chromatin. Interaction of BAF with integral membrane LEM‐domain proteins facilitates recruitment of nuclear membrane and decreases the size of the rupture. Subsequently, the CHMP7 and LEMD2 interaction promotes nucleation and polymerization of ESCRT‐III, which together with the regulatory ATPase VPS4 further constricts the rupture and promotes sealing.
It is interesting to note that excessive CHMP7 and ESCRT‐III recruitment, rather than promoting repair, causes aberrant folding of the nuclear envelope, accompanied by chromosome damage due to torsional stress. While such excessive ESCRT recruitment to the primary nucleus does not normally occur, it happens frequently to damaged micronuclei—single chromosomes enclosed by a micronuclear envelope. Thus, injury to the micronuclear envelope does not lead to repair but rather to catastrophic membrane rearrangements and chromosome damage (Vietri et al, 2020b).
Repair of the damaged lysosome membrane
Molecules, particles and microorganisms that are internalized into cells are harboured in organelles of the endocytic pathway, including endosomes, phagosomes, macropinosomes and lysosomes. All these organelles are exposed to membrane damage, and this is particularly the case with lysosomes, since these represent the terminal stations for many internalized cargoes and since their low intraluminal pH favours accumulation of weak bases that may cause membrane damage due to osmotic swelling.
Severely damaged lysosomes are removed by a selective autophagy process known as lysophagy, which entails sequestration of the damaged lysosome into an autophagosome, followed by fusion with a healthy lysosome in order to degrade the sequestered content (Hung et al, 2013; Maejima et al, 2013). However, more limited damage to the lysosome membrane can be repaired, and here, the ESCRT machinery plays a key role (Radulovic et al, 2018; Skowyra et al, 2018). Current evidence suggests that both Ca2+ flow from the damaged lysosome and cytosolic access to intraluminal β‐galactoside sugars play roles in recruitment of the ESCRT machinery (Skowyra et al, 2018; Jia et al, 2020b) (Fig 5).
Figure 5. Lysosome repair.
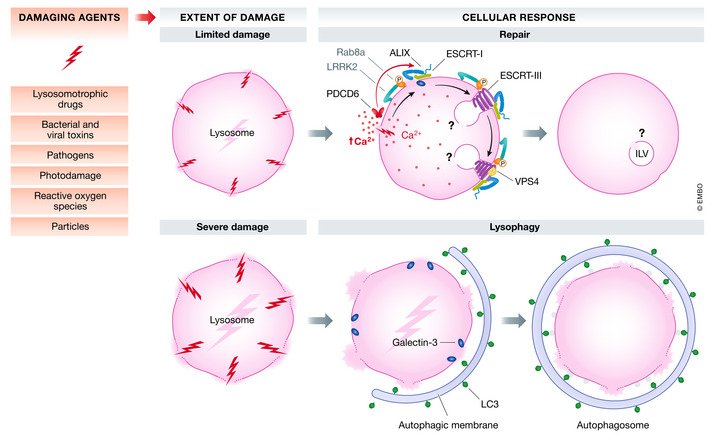
Damaging agents such as lysosomotropic drugs, photodamage, pathogens etc. may inflict varying extent of lysosomal damage, leading to Ca2+ efflux from damaged lysosomes. The ESCRT machinery orchestrates repair of the limited lysosomal damage through multiple mechanisms in a coordinated manner. The increase in cytosolic Ca2+ is probably sensed by PDCD6 which in turn recruits the ESCRT‐III binding protein ALIX. The ESCRT‐I protein TSG101 and ALIX further recruit the ESCRT‐III machinery together with VPS4 to seal the membrane lesions. Although Ca2+ efflux might provide an immediate signal for ESCRT‐III recruitment, the β‐galactoside sensor GAL3, which interacts with ALIX, is required for efficient recruitment of ALIX and ESCRT‐III to the damaged lysosomes. This sealing might be accompanied by formation of ILVs containing the damage, similar to the process of endosomal ILV biogenesis. In macrophages, phagolysosome or lysosome damage triggers activation of the kinase LRRK2. Once activated, LRRK2 phosphorylates the small GTPase Rab8A, and ESCRT‐III is recruited to mediate endolysosomal membrane repair in Ca2+‐dependent fashion. Severely damaged lysosomes are engulfed and degraded via lysophagy, initiated by β‐galactoside sensors such as GAL3, which recruits LC3‐containing autophagic membranes.
Like with damage to the plasma membrane, Ca2+ efflux from damaged lysosomes is accompanied by recruitment of the Ca2+ sensor PDCD6 (Skowyra et al, 2018), and several laboratories have reported an inhibitory effect of Ca2+ chelators on ESCRT recruitment to damaged lysosomes (Skowyra et al, 2018; Jia et al, 2020b). Indeed, the Ca2+‐ and PDCD6‐binding protein ALIX plays an important role in ESCRT‐III recruitment to damaged lysosomes. Surprisingly, however, depletion of ALIX alone has minimal effect on ESCRT‐III recruitment whereas its co‐depletion with the ESCRT‐I subunit TSG101 (which has a moderate effect on ESCRT‐III recruitment by itself) strongly prevents ESCRT‐III recruitment (Radulovic et al, 2018; Skowyra et al, 2018). This indicates that the mechanism of ESCRT‐III recruitment is more complex than that to the damaged plasma membrane and may involve additional factors. One of these is GAL3, which interacts with ALIX and is required for efficient recruitment of ALIX and ESCRT‐III to damaged lysosomes. Whereas Ca2+ efflux might provide an immediate signal for ESCRT‐III recruitment, GAL3 could provide a later and more sustained signal, and GAL3 has the additional function of promoting lysophagy in case of more severe lysosome damage (Jia et al, 2020b).
An additional factor that determines the fate of damaged phagolysosomes and lysosomes in macrophages is the Parkinson’s disease‐related leucine‐rich repeat kinase 2 (LRRK2). LRRK2 is activated upon lysosome damage, which then triggers further recruitment of the small GTPase Rab8A and subsequently the ESCRT‐III protein CHMP4B to damaged organelles (Herbst et al, 2020). Whether this cascade also mediates repair in other cell types or is specific for macrophages requires further investigation.
It is not yet known how the ESCRT machinery functions to seal holes in the lysosome membrane, but from the known topologies of other ESCRT‐dependent processes (Vietri et al, 2020a) it is tempting to speculate that it functions by shedding of vesicles containing membrane damage into the lumen of the lysosome.
Even though leakiness of lysosomes is generally thought to be harmful to the cell, a spatially and temporally controlled leakage of lysosomes is actually required for accurate chromosome segregation in normal mammalian cell division (Hamalisto et al, 2020). During metaphase, cathepsin B leaks out of chromosome‐proximal lysosomes to cleave a small subset of histone H3 in order to maintain chromosome fidelity in mitosis. It will be interesting to understand how this selective lysosome permeabilization is initiated and terminated.
Conclusions and perspectives
There is strong evidence that multiple mechanisms have evolved to seal holes in membranes, and there may be good reasons why separate mechanisms exist. Firstly, because membrane integrity is so crucial for cellular viability and functions, the existence of multiple sealing mechanisms could ensure successful sealing even if one mechanism fails. Secondly, the different mechanisms are optimized for sealing of different types of holes. The smallest holes with a diameter of a few nm may be self‐sealed, or self‐sealing could be promoted by proteins that reduce membrane tension, such as annexins. Holes with a diameter of 50–100 nm are typically sealed by endocytosis or ESCRT‐mediated outward scission, whereas the largest holes of more than 100 nm could require patching by intracellular membranes, or engulfment by neighbouring cells. It is also plausible that additional mechanisms of membrane sealing exist, which have not been characterized yet. For instance, regulated lipid synthesis has been proposed as a mechanism (Penfield et al, 2020), and it is also possible to envisage intermembrane lipid transfer to seal holes, possibly mediated via membrane contact sites (Elbaz & Schuldiner, 2011; Raiborg et al, 2015).
Although we here have focused on sealing of the plasma membrane, the nuclear envelope, lysosomes and autophagosomes, the integrity of other cellular membranes is certainly also critical in biology and health. For example, permeabilization of mitochondrial membranes is known as a major trigger of cell death (Kroemer & Reed, 2000). It will therefore be exciting to learn whether there are cellular mechanisms that seal such membranes as well.
The importance of cellular membrane integrity is underscored by the many genetic diseases associated with compromised integrity of the plasma membrane and the nuclear envelope in particular. Mutations that either compromise membrane integrity as such or affect membrane repair systems are typically associated with diseases that involve reduced functions of cells exposed to mechanical damage, including skeletal and cardiac muscle cells. Long‐lived cells with very limited capacity for self‐renewal, such as neurons, are also vulnerable to membrane damage, and this is reflected by the number of diseases of the peripheral and central nervous system associated with compromised membrane integrity (Chi et al, 2009).
While repair of damaged membranes provides crucial cellular defence against viruses and microorganisms and their pathogenicity factors (Etxaniz et al, 2018), some microorganisms can subvert cellular membrane repair mechanisms for their entry or replication. For instance, Trypanozoma cruzi hijacks the ASM‐mediated membrane repair pathway for its invasion into cells (Fernandes et al, 2011), whereas Coxiella burnetii takes advantage of ESCRT‐mediated repair of the bacterial vacuole for its intravacuolar replication (Radulovic et al, 2018). This means that, under some conditions, membrane sealing can be a disadvantage for the host.
The importance of membrane sealing for cell viability and organismal health, and its exploitation by certain pathogens, makes it relevant to ask whether manipulation of membrane sealing could have therapeutic applications. Selective prevention of membrane sealing could be beneficial for killing cancer cells or preventing intracellular replication of pathogens that rely on an intact vacuole. Conversely, stimulating membrane sealing could ameliorate symptoms in patients with genetic diseases associated with membrane fragility of dysfunctional repair. While prevention of membrane sealing could potentially be achieved with small‐molecule inhibitors of sealing factors, stimulating sealing would probably require gene therapeutic approaches. A major obstacle to these approaches is the complexity of cellular membrane sealing reactions, and their reliance on proteins that often have multiple cellular functions. Nevertheless, as we learn more and more about the mechanisms of cellular membrane sealing, therapies directed against membrane sealing could soon become a reality.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
HS was supported by grants from the Research Council of Norway (302994), South‐Eastern Norway Regional Health Authority (2016087), the Norwegian Cancer Society (182698) and the European Research Council (788954). MV was supported by a researcher grant from the South‐Eastern Norway Regional Health Authority (2018043). This work was in part supported by the Research Council of Norway through its Centres of Excellence funding scheme (262652).
The EMBO Journal (2021) 40: e106922.
References
- Anderson DJ, Hetzer MW (2008) The life cycle of the metazoan nuclear envelope. Curr Opin Cell Biol 20: 386–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews NW, Almeida PE, Corrotte M (2014) Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol 24: 734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Appen A, LaJoie D, Johnson IE, Trnka MJ, Pick SM, Burlingame AL, Ullman KS, Frost A (2020) LEM2 phase separation promotes ESCRT‐mediated nuclear envelope reformation. Nature 582: 115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendix PM, Simonsen AC, Florentsen CD, Hager SC, Mularski A, Zanjani AAH, Moreno‐Pescador G, Klenow MB, Sonder SL, Danielsen HM et al (2020) Interdisciplinary synergy to reveal mechanisms of annexin‐mediated plasma membrane shaping and repair. Cells 9: 1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO (1998) Isolation and characterization of rat liver amphisomes ‐ Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem 273: 21883–21892 [DOI] [PubMed] [Google Scholar]
- Bohannon KP, Hanson PI (2020) ESCRT puts its thumb on the nanoscale: fixing tiny holes in endolysosomes. Curr Opin Cell Biol 65: 122–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouter A, Gounou C, Berat R, Tan S, Gallois B, Granier T, d'Estaintot BL, Poschl E, Brachvogel B, Brisson AR (2011) Annexin‐A5 assembled into two‐dimensional arrays promotes cell membrane repair. Nat Commun 2: 270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, Ko JK, Lin P, Thornton A, Zhao X et al (2009) MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol 11: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas AM, Gonzalez‐Jamett AM, Cea LA, Bevilacqua JA, Caviedes P (2016) Dysferlin function in skeletal muscle: possible pathological mechanisms and therapeutical targets in dysferlinopathies. Exp Neurol 283: 246–254 [DOI] [PubMed] [Google Scholar]
- Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun JA et al (2016) TRIMs and galectins globally cooperate and TRIM16 and galectin‐3 Co‐direct autophagy in endomembrane damage homeostasis. Dev Cell 39: 13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi YH, Chen ZJ, Jeang KT (2009) The nuclear envelopathies and human diseases. J Biomed Sci 16: 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ST, McNeil PL (2015) Membrane repair: mechanisms and pathophysiology. Physiol Rev 95: 1205–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Magistris P, Antonin W (2018) The dynamic nature of the nuclear envelope. Curr Biol 28: R487–R497 [DOI] [PubMed] [Google Scholar]
- De Vos WH, Houben F, Kamps M, Malhas A, Verheyen F, Cox J, Manders EM, Verstraeten VL, van Steensel MA, Marcelis CL et al (2011) Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum Mol Genet 20: 4175–4186 [DOI] [PubMed] [Google Scholar]
- Demonbreun AR, Quattrocelli M, Barefield DY, Allen MV, Swanson KE, McNally EM (2016) An actin‐dependent annexin complex mediates plasma membrane repair in muscle. J Cell Biol 213: 705–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J (2016) Nuclear envelope rupture and repair during cancer cell migration. Science 352: 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earle AJ, Kirby TJ, Fedorchak GR, Isermann P, Patel J, Iruvanti S, Moore SA, Bonne G, Wallrath LL, Lammerding J (2020) Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat Mater 19: 464–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz Y, Schuldiner M (2011) Staying in touch: the molecular era of organelle contact sites. Trends Biochem Sci 36: 616–623 [DOI] [PubMed] [Google Scholar]
- Etxaniz A, Gonzalez‐Bullon D, Martin C, Ostolaza H (2018) Membrane repair mechanisms against permeabilization by pore‐forming toxins. Toxins 10: 234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes MC, Cortez M, Flannery AR, Tam C, Mortara RA, Andrews NW (2011) Trypanosoma cruzi subverts the sphingomyelinase‐mediated plasma membrane repair pathway for cell invasion. J Exp Med 208: 909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179: 485–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerke V, Moss SE (2002) Annexins: from structure to function. Physiol Rev 82: 331–371 [DOI] [PubMed] [Google Scholar]
- Gozen I, Dommersnes P (2014) Pore dynamics in lipid membranes. EurPhysJ Special Topics 223: 1813–1829 [Google Scholar]
- Gu M, LaJoie D, Chen OS, von Appen A, Ladinsky MS, Redd MJ, Nikolova L, Bjorkman PJ, Sundquist WI, Ullman KS et al (2017) LEM2 recruits CHMP7 for ESCRT‐mediated nuclear envelope closure in fission yeast and human cells. Proc Natl Acad Sci USA 114: E2166–E2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfmann CT, Sears RM, Katiyar A, Busselman BW, Aman LK, Zhang Q, O'Bryan CS, Angelini TE, Lele TP, Roux KJ (2019) Repair of nuclear ruptures requires barrier‐to‐autointegration factor. J Cell Biol 218: 2136–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamalisto S, Stahl JL, Favaro E, Yang Q, Liu B, Christoffersen L, Loos B, Guasch Boldu C, Joyce JA, Reinheckel T et al (2020) Spatially and temporally defined lysosomal leakage facilitates mitotic chromosome segregation. Nat Commun 11: 229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548: 466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst S, Campbell P, Harvey J, Bernard EM, Papayannopoulos V, Wood NW, Morris HR, Gutierrez MG (2020) LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J 39: e104494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst MR, Vidal‐Quadras M, Larsson E, Song J, Hubert M, Blomberg J, Lundborg M, Landstrom M, Lundmark R (2017) Clathrin‐independent endocytosis suppresses cancer cell blebbing and invasion. Cell Rep 20: 1893–1905 [DOI] [PubMed] [Google Scholar]
- Hung YH, Chen LM, Yang JY, Yang WY (2013) Spatiotemporally controlled induction of autophagy‐mediated lysosome turnover. Nat Commun 4: 2111 [DOI] [PubMed] [Google Scholar]
- Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW (2008) Repair of injured plasma membrane by rapid Ca2+‐dependent endocytosis. J Cell Biol 180: 905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi‐Itakura C, Mizushima N (2012) The hairpin‐type tail‐anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151: 1256–1269 [DOI] [PubMed] [Google Scholar]
- Jia J, Abudu YP, Claude‐Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M et al (2018) Galectins control mTOR in response to endomembrane damage. Mol Cell 70: 120–135.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Bissa B, Brecht L, Allers L, Choi SW, Gu Y, Zbinden M, Burge MR, Timmins G, Hallows K et al (2020a) AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin‐directed ubiquitin signal transduction system. Mol Cell 77: 951–969.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Claude‐Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA et al (2020b) Galectin‐3 coordinates a cellular system for lysosomal repair and removal. Dev Cell 52: 69–87.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez AJ, Maiuri P, Lafaurie‐Janvore J, Divoux S, Piel M, Perez F (2014) ESCRT machinery is required for plasma membrane repair. Science 343: 1247136 [DOI] [PubMed] [Google Scholar]
- Kass GE, Orrenius S (1999) Calcium signaling and cytotoxicity. Environ Health Perspect 107(Suppl 1): 25–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD (2002) Receptor downregulation and multivesicular‐body sorting. Nat Rev Mol Cell Biol 3: 893–905 [DOI] [PubMed] [Google Scholar]
- Knorr RL, Lipowsky R, Dimova R (2015) Autophagosome closure requires membrane scission. Autophagy 11: 2134–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerdt SN, Ashraf APK, Gerke V (2019) Annexins and plasma membrane repair. Curr Top Membr 84: 43–65 [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC (2000) Mitochondrial control of cell death. Nat Med 6: 513–519 [DOI] [PubMed] [Google Scholar]
- Lenhart KC, O'Neill TJT, Cheng Z, Dee R, Demonbreun AR, Li J, Xiao X, McNally EM, Mack CP, Taylor JM (2015) GRAF1 deficiency blunts sarcolemmal injury repair and exacerbates cardiac and skeletal muscle pathology in dystrophin‐deficient mice. Skelet Muscle 5: 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lieberman J (2020) Knocking 'em dead: pore‐forming proteins in immune defense. Annu Rev Immunol 38: 455–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T (2015) Chromothripsis and kataegis induced by telomere crisis. Cell 163: 1641–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A et al (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548: 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y et al (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32: 2336–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Kirchhausen T (2005) An emergency response team for membrane repair. Nat Rev Mol Cell Biol 6: 499–505 [DOI] [PubMed] [Google Scholar]
- McNeil PL, Steinhardt RA (1997) Loss, restoration, and maintenance of plasma membrane integrity. J Cell Biol 137: 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Vogel SS, Miyake K, Terasaki M (2000) Patching plasma membrane disruptions with cytoplasmic membrane. J Cell Sci 113(Pt 11): 1891–1902 [DOI] [PubMed] [Google Scholar]
- Melia TJ, Lystad AH, Simonsen A (2020) Autophagosome biogenesis: from membrane growth to closure. J Cell Biol 219: e202002085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier V, Larios J, Molinard G, Goujon A, Matile S, Gruenberg J, Roux A (2020) Endosomal membrane tension regulates ESCRT‐III‐dependent intra‐lumenal vesicle formation. Nat Cell Biol 22: 947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middel V, Zhou L, Takamiya M, Beil T, Shahid M, Roostalu U, Grabher C, Rastegar S, Reischl M, Nienhaus GU et al (2016) Dysferlin‐mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nat Commun 7: 12875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self‐digestion. Nature 451: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- Olmos Y, Hodgson L, Mantell J, Verkade P, Carlton JG (2015) ESCRT‐III controls nuclear envelope reformation. Nature 522: 236–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos Y, Perdrix‐Rosell A, Carlton JG (2016) Membrane binding by CHMP7 coordinates ESCRT‐III‐dependent nuclear envelope reformation. Curr Biol 26: 2635–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfield L, Shankar R, Szentgyorgyi E, Laffitte A, Mauro MS, Audhya A, Muller‐Reichert T, Bahmanyar S (2020) Regulated lipid synthesis and LEM2/CHMP7 jointly control nuclear envelope closure. J Cell Biol 219: e201908179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfield L, Wysolmerski B, Mauro M, Farhadifar R, Martinez MA, Biggs R, Wu HY, Broberg C, Needleman D, Bahmanyar S (2018) Dynein‐pulling forces counteract lamin‐mediated nuclear stability during nuclear envelope repair. Mol Biol Cell 29: 852–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper GH, Sprenger S, Teis D, Oliferenko S (2020) ESCRT‐III/Vps4 controls heterochromatin‐nuclear envelope attachments. Dev Cell 53: 27–41.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier MA, Xiao WZ, Macosko JC, Chan C, Shin YK, Bennett MK (1998) The synaptic SNARE complex is a parallel four‐stranded helical bundle. Nature Struct Biol 5: 765–769 [DOI] [PubMed] [Google Scholar]
- Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon‐Dumenil AM, Manel N et al (2016) ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352: 359–362 [DOI] [PubMed] [Google Scholar]
- Radulovic M, Schink KO, Wenzel EM, Nahse V, Bongiovanni A, Lafont F, Stenmark H (2018) ESCRT‐mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 37: e106162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Wenzel EM, Stenmark H (2015) ER‐endosome contact sites: molecular compositions and functions. EMBO J 34: 1848–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SK, Huynh C, Proux‐Gillardeaux V, Galli T, Andrews NW (2004) Identification of SNAREs involved in synaptotagmin VII‐regulated lysosomal exocytosis. J Biol Chem 279: 20471–20479 [DOI] [PubMed] [Google Scholar]
- Reddy A, Caler EV, Andrews NW (2001) Plasma membrane repair is mediated by Ca(2+)‐regulated exocytosis of lysosomes. Cell 106: 157–169 [DOI] [PubMed] [Google Scholar]
- Robijns J, Houthaeve G, Braeckmans K, De Vos WH (2018) Loss of nuclear envelope integrity in aging and disease. Int Rev Cell Mol Biol 336: 205–222 [DOI] [PubMed] [Google Scholar]
- Robinson J, Berselli GB, Ryadnov MG, Keyes TE (2020) Annexin V drives stabilization of damaged asymmetric phospholipid bilayers. Langmuir 36: 5454–5465 [DOI] [PubMed] [Google Scholar]
- Rusten TE, Vaccari T, Lindmo K, Rodahl LM, Nezis IP, Sem‐Jacobsen C, Wendler F, Vincent JP, Brech A, Bilder D et al (2007) ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol 17: 1817–1825 [DOI] [PubMed] [Google Scholar]
- Samwer M, Schneider MWG, Hoefler R, Schmalhorst PS, Jude JG, Zuber J, Gerlich DW (2017) DNA cross‐bridging shapes a single nucleus from a set of mitotic chromosomes. Cell 170: 956–972.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, Jaiswal JK (2014) Mechanism of Ca(2)(+)‐triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun 5: 5646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoneberg J, Lee IH, Iwasa JH, Hurley JH (2017) Reverse‐topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol 18: 5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, Stan RV, Butler‐Browne G, Vedie B, Johannes L et al (2011) Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144: 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI (2018) Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360: eaar5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonder SL, Boye TL, Tolle R, Dengjel J, Maeda K, Jaattela M, Simonsen AC, Jaiswal JK, Nylandsted J (2019) Annexin A7 is required for ESCRT III‐mediated plasma membrane repair. Sci Rep 9: 6726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323: 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, He H, Tang Z, Hattori T, Liu Y, Young MM, Serfass JM, Chen L, Gebru M, Chen C et al (2018) An autophagy assay reveals the ESCRT‐III component CHMP2A as a regulator of phagophore closure. Nat Commun 9: 2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Liang X, Hattori T, Tang Z, He H, Chen H, Liu X, Abraham T, Imamura‐Kawasawa Y, Buchkovich NJ et al (2019) VPS37A directs ESCRT recruitment for phagophore closure. J Cell Biol 218: 3336–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, Schuchman E, Tabas I, Andrews NW (2010) Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol 189: 1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M, Miyake K, McNeil PL (1997) Large plasma membrane disruptions are rapidly resealed by Ca2+‐dependent vesicle‐vesicle fusion events. J Cell Biol 139: 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaller DJ, Allegretti M, Borah S, Ronchi P, Beck M, Lusk CP (2019) An ESCRT‐LEM protein surveillance system is poised to directly monitor the nuclear envelope and nuclear transport system. Elife 8: e45284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbreit NT, Zhang CZ, Lynch LD, Blaine LJ, Cheng AM, Tourdot R, Sun L, Almubarak HF, Judge K, Mitchell TJ et al (2020) Mechanisms generating cancer genome complexity from a single cell division error. Science 368: eaba0712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JD, Hatch EM, Anderson DJ, Hetzer MW (2012) Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 3: 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasta GR (2009) Roles of galectins in infection. Nat Rev Microbiol 7: 424–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventimiglia LN, Cuesta‐Geijo MA, Martinelli N, Caballe A, Macheboeuf P, Miguet N, Parnham IP, Olmos Y, Carlton JG, Weissenhorn W et al (2018) CC2D1B coordinates ESCRT‐III activity during the mitotic reformation of the nuclear envelope. Dev Cell 47: 547–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietri M, Radulovic M, Stenmark H (2020a) The many functions of ESCRTs. Nat Rev Mol Cell Biol 21: 25–42 [DOI] [PubMed] [Google Scholar]
- Vietri M, Schink KO, Campsteijn C, Wegner CS, Schultz SW, Christ L, Thoresen SB, Brech A, Raiborg C, Stenmark H (2015) Spastin and ESCRT‐III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 522: 231–235 [DOI] [PubMed] [Google Scholar]
- Vietri M, Schultz SW, Bellanger A, Jones CM, Petersen LI, Raiborg C, Skarpen E, Pedurupillay CRJ, Kjos I, Kip E et al (2020b) Unrestrained ESCRT‐III drives micronuclear catastrophe and chromosome fragmentation. Nat Cell Biol 22: 856–867 [DOI] [PubMed] [Google Scholar]
- Webster BM, Thaller DJ, Jager J, Ochmann SE, Borah S, Lusk CP (2016) Chm7 and Heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. EMBO J 35: 2447–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng IC, Chen HL, Lo TH, Lin WH, Chen HY, Hsu DK, Liu FT (2018) Cytosolic galectin‐3 and ‐8 regulate antibacterial autophagy through differential recognition of host glycans on damaged phagosomes. Glycobiology 28: 392–405 [DOI] [PubMed] [Google Scholar]
- Wu N, Cernysiov V, Davidson D, Song H, Tang J, Luo S, Lu Y, Qian J, Gyurova IE, Waggoner SN et al (2020) Critical role of lipid scramblase TMEM16F in phosphatidylserine exposure and repair of plasma membrane after pore formation. Cell Rep 30: 1129–1140.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam C, He Y, Zhang D, Chiam KH, Oliferenko S (2011) Divergent strategies for controlling the nuclear membrane satisfy geometric constraints during nuclear division. Curr Biol 21: 1314–1319 [DOI] [PubMed] [Google Scholar]
- Young AM, Gunn AL, Hatch EM (2020) BAF facilitates interphase nuclear membrane repair through recruitment of nuclear transmembrane proteins. Mol Biol Cell 31: 1551–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D (2015) Chromothripsis from DNA damage in micronuclei. Nature 522: 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen Y, Spangenberg H, Munson MJ, Brech A, Schink KO, Tan KW, Sorensen V, Wenzel EM, Radulovic M, Engedal N et al (2020) ESCRT‐mediated phagophore sealing during mitophagy. Autophagy 16: 826–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Wu Z, Zhao M, Murtazina R, Cai J, Zhang A, Li R, Sun D, Li W, Zhao L et al (2019) Rab5‐dependent autophagosome closure by ESCRT. J Cell Biol 218: 1908–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Zou S, Chen Y, Lipatova Z, Sun D, Zhu X, Li R, Wu Z, You W, Cong X et al (2017) A Rab5 GTPase module is important for autophagosome closure. PLoS Genet 13: e1007020 [DOI] [PMC free article] [PubMed] [Google Scholar]