

Abstract
Partitioning of the genome in meiosis occurs through two highly specialized cell divisions, named meiosis I and meiosis II. Step‐wise cohesin removal is required for chromosome segregation in meiosis I, and sister chromatid segregation in meiosis II. In meiosis I, mono‐oriented sister kinetochores appear as fused together when examined by high‐resolution confocal microscopy, whereas they are clearly separated in meiosis II, when attachments are bipolar. It has been proposed that bipolar tension applied by the spindle is responsible for the physical separation of sister kinetochores, removal of cohesin protection, and chromatid separation in meiosis II. We show here that this is not the case, and initial separation of sister kinetochores occurs already in anaphase I independently of bipolar spindle forces applied on sister kinetochores, in mouse oocytes. This kinetochore individualization depends on separase cleavage activity. Crucially, without kinetochore individualization in meiosis I, bivalents when present in meiosis II oocytes separate into chromosomes and not sister chromatids. This shows that whether centromeric cohesin is removed or not is determined by the kinetochore structure prior to meiosis II.
Keywords: cohesin protection, meiosis, oocytes, Rec8, separase
Subject Categories: Cell Cycle
Surprisingly, bipolar tension across kinetochores is not required for sister chromatid separation in meiosis II, while separase activity in meiosis I is a prerequisite for centromeric cohesin deprotection.
Introduction
In meiosis I, chromosomes of different parental origin (“homologues”) are separated, and in meiosis II, sister chromatids. Because there is no S‐phase between the two meiotic divisions, the genome is thus halved and haploid gametes are generated. The meiosis‐specific segregation pattern requires the co‐orientation of the two sister kinetochores of one homologue to the same spindle pole in meiosis I. Hence, sister kinetochores are attached in a monopolar fashion—this is called mono‐orientation; however, in meiosis II, sister kinetochores are attached in a bipolar manner, they are bi‐oriented (Petronczki et al, 2003; Duro & Marston, 2015). Consequently, the direction of the tension forces applied by the bipolar spindle on sister kinetochore pairs is distinct in meiosis I and meiosis II. Sister kinetochores in meiosis I appear fused together when examined by confocal microscopy (Gomez et al, 2007; Lee et al, 2008; Chambon et al, 2013b), whereas they are clearly separated in meiosis II. It was proposed that this separation is due to the application of bipolar tension forces (Gomez et al, 2007; Lee et al, 2008), but it has never been addressed whether sister kinetochores also separate (“individualize”) without bipolar tension, and if they really separate only in meiosis II and not earlier.
To correctly execute both meiotic divisions, the physical connections between chromosomes and sister chromatids are removed in a step‐wise manner, from arms in meiosis I and the centromere region in meiosis II. Homologous chromosomes in meiosis I are maintained together by chiasmata, which form on chromosome arms at sites where a meiotic recombination event has taken place and was resolved by a cross‐over. Sister chromatids are held together through cohesion, brought about by the cohesin complex containing a kleisin subunit that is cleaved by the protease separase at metaphase‐to‐anaphase transition. The separation of chromosomes in meiosis I requires the resolution of chiasmata and the removal of cohesion from chromosome arms because genetic material has been exchanged between chromatids of different parental origin. Importantly, cohesin in the centromere region (where no recombination takes place) has to be preserved, to keep sister chromatids together until anaphase onset in meiosis II (Petronczki et al, 2003; Duro & Marston, 2015; Marston & Wassmann, 2017).
It was shown that protection of centromeric cohesin on homologous chromosome pairs (“bivalents”) in meiosis I depends on recruitment of the phosphatase PP2A‐B56 by a Shugoshin (Sgo) family protein, to the centromere region. There, PP2A‐B56 counteracts phosphorylation of the meiotic kleisin Rec8, preventing its cleavage by separase (Clift & Marston, 2011; Keating et al, 2020). As a caveat, it is important to mention that for up to now Rec8 phosphorylation as a requirement for cleavage by separase in vivo has only been demonstrated in yeast and Caenorhabditis elegans (Rogers et al, 2002; Ishiguro et al, 2010; Katis et al, 2010; Rumpf et al, 2010). In agreement with this model also applying in higher eukaryotes, inhibition of PP2A‐B56 activity, or loss of Sgo2 (which is required for PP2A‐B56 recruitment in higher eukaryotes) during the first meiotic division lead to cleavage of Rec8 not only on chromosome arms but also in the centromere region and, hence, precocious sister chromatid separation already in meiosis I (Mailhes et al, 2003; Lee et al, 2008; Llano et al, 2008; Chang et al, 2011; Rattani et al, 2013). Nevertheless, it is still unknown how protection of centromeric cohesin is removed in meiosis II only (a process also called “deprotection” (Chambon et al, 2013b; Wassmann, 2013; Arguello‐Miranda et al, 2017; Jonak et al, 2017)), to allow separation of sister chromatids.
Sgo2 fulfills several interwoven roles in meiosis I, ranging from its contribution to spindle assembly checkpoint (SAC) silencing, delaying tension establishment by the bipolar spindle, Aurora B kinase recruitment, and centromeric cohesin protection (Rattani et al, 2013). Together with the SAC protein Mad2, Sgo2 was shown very recently to additionally function as a separase inhibitor in mitotic cells, even though it is currently unknown whether this also applies to the meiotic divisions (Hellmuth et al, 2020). Not surprisingly, in oocytes, distinct pools of endogenous Sgo2 are recruited to the centromeric region (El Yakoubi et al, 2017), supposedly occupying different roles. We have shown that Sgo2 co‐localizing with centromere markers contributes to cohesin protection in meiosis I. The kinases Mps1 and Bub1 are involved in this recruitment process, and even though both proteins are required for Sgo2 localization, only the kinase activity of Mps1 is necessary for implementing its protective role (El Yakoubi et al, 2017).
When analyzing the localization of endogenous Sgo2 in mouse oocytes, we and others have found that Sgo2 is localized to the centromere region of paired sister chromatids (“dyads”) also in meiosis II, where centromeric cohesin has to be cleaved and Sgo2's protective role is not required (Lee et al, 2008; Chambon et al, 2013b). This is not surprising as such, because apart from preventing Rec8 cleavage, Sgo2's other roles are most likely also required for correct execution of meiosis II. PP2A‐B56 is recruited to centromeres and within the pericentromeric region at high levels, too (Lee et al, 2008; Chambon et al, 2013b), suggesting that there must be some kind of mechanism preventing PP2A‐B56 action on Rec8, to allow its cleavage by separase.
Several, not necessarily mutually exclusive, models have been proposed for deprotection of centromeric cohesin specifically in meiosis II. First, bipolar tension applied on sister kinetochores in meiosis II, but not meiosis I, was suggested to move Sgo2‐PP2A‐B56 far enough away from Rec8 at the pericentromere holding sister chromatids together to allow its phosphorylation (Gomez et al, 2007; Lee et al, 2008). Second, degradation of Sgo1 and Mps1 at anaphase II onset in an APC/C‐dependent manner was proposed to ensure deprotection in budding yeast (Arguello‐Miranda et al, 2017; Jonak et al, 2017). Third, using a Morpholino‐knockdown approach, we proposed that I2PP2A/Set, a histone chaperone and potential PP2A inhibitor, counteracts protection of Rec8 specifically in meiosis II in a tension‐independent manner (Chambon et al, 2013b). But apart from the first model, none allows us to understand the key event leading to the deprotection of centromeric cohesin for chromatid separation in meiosis II and not meiosis I.
Here, we revisit the question of which upstream event is required for deprotection of centromeric cohesin in mouse oocyte meiosis II, focusing on bipolar tension and Sgo2 displacement. We find that bipolar tension is dispensable for step‐wise cohesin removal, as deprotection of centromeric cohesin takes place also on tension‐less monopolar spindles in meiosis II. Centromeric cohesin cleavage does not require removal of Mps1 or Sgo2 at centromeres. Crucially, we find that fusion of sister kinetochores is resolved in a separase‐dependent manner already in anaphase I, hence independent of the bipolar tension established later. We show that this kinetochore individualization before entering meiosis II is the key event for allowing centromeric cohesin removal and sister chromatid separation in meiosis II. Our rescue experiments of oocytes devoid of separase (Kudo et al, 2006) demonstrate that separase activity before entry into meiosis II is required for kinetochore individualization and sister separation in meiosis II. Absence of separase in meiosis I and its presence only in meiosis II leads to removal of arm cohesin and segregation of bivalents into dyads instead of sister chromatids. In contrast, bivalents with individualized sister kinetochores separate sister chromatids in meiosis II. Hence, the key event for deprotection of centromeric cohesin for sister chromatid segregation in meiosis II takes place already at the final stages of meiosis I in mammalian oocytes. Our data show that centromeric cohesin removal is not determined solely by the cell cycle stage (meiosis I or meiosis II), but foremost the chromosome itself.
Results
Sister kinetochores individualize already in anaphase I
Inner kinetochores and CenPA, which substitutes for Histone H3 at the centromere, can be stained with anti‐CREST serum. Monopolar‐oriented sister kinetochores seem fused together in meiosis I and appear as two separated dots in metaphase of meiosis II upon CREST staining in oocytes (Lee et al, 2008; Chambon et al, 2013b; Kim et al, 2015). This sister kinetochore separation was thought to be due to the bipolar tension applied on dyads as they align at the metaphase plate and are oriented toward the opposite poles of the bipolar spindle in meiosis II (Lee et al, 2008). However, when we performed chromosome spreads or stained whole‐mount oocytes at the metaphase‐to‐anaphase transition of meiosis I, we observed that already in anaphase I sister kinetochores became visible as two separate dots (Fig 1A and B), even though they are still paired and attached to the same pole on the anaphase I spindle (Fig 1B) (Kitajima et al, 2011). We decided to refer to this first visible separation of the sister kinetochore signals in anaphase I, which was visible on both chromosome spreads and whole‐mount oocytes, as "kinetochore individualization". It is also visible in a previous study, but not mentioned as appearing in anaphase I (Kim et al, 2015). It is distinct from the previously described age‐related separation of sister kinetochores that occurs already in early meiosis I in mouse and human oocytes, which is due to loss of cohesin with maternal age (Chiang et al, 2010; Lister et al, 2010; Zielinska et al, 2015).
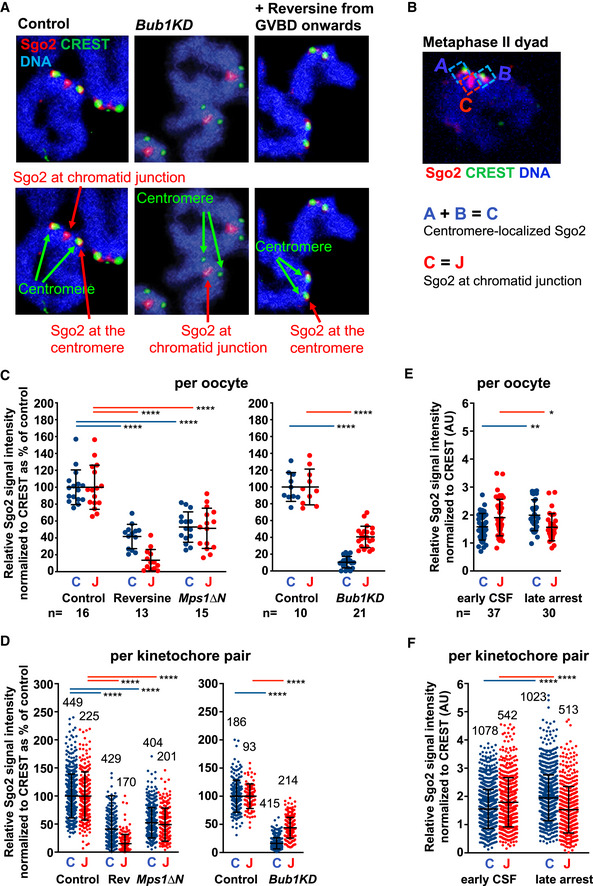
Figure 1. Sister kinetochores individualize already in anaphase I.
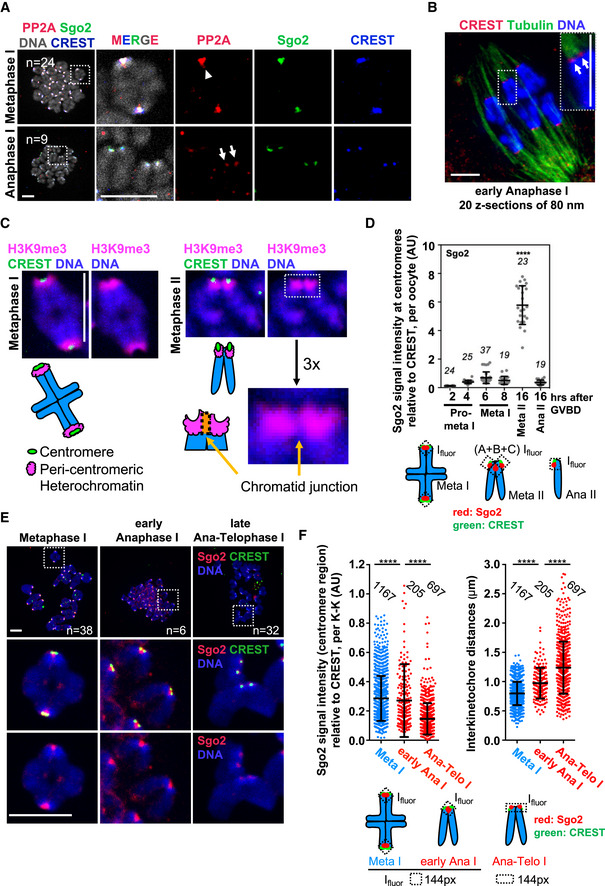
- Oocytes were fixed for chromosome spreads 8 h after GVBD and stained for PP2A‐c (red), Sgo2 (green), CREST (blue), and DNA (Hoechst, shown in gray). Spreads were classified into metaphase I and anaphase I, depending on whether chromosome segregation had taken place, or not. Arrowhead indicates fused sister kinetochores, and arrows indicate separated sister kinetochores.
- Example of a whole‐mount oocyte staining, in early anaphase I after cold treatment to visualize only cold‐stable microtubule fibers, stained with anti‐tubulin antibody (green). Kinetochores are stained with CREST serum (red) and chromosomes with Hoechst (blue). Shown is an overlay of 20 z‐sections. Arrows indicate separated sister kinetochores.
- Metaphase I (6 h after GVBD) and metaphase II (16–20 h after GVBD) chromosome spreads were stained with H3K9me3 antibody and CREST serum to reveal pericentromeric heterochromatin (pink) and the centromere (green), respectively. Chromosomes were stained with Hoechst (blue). Below, schematic representation of a bivalent in metaphase I and a dyad in metaphase II. Enlargement shows the centromere region and a region that we call “chromatid junction” with the corresponding scheme.
- Quantification of total Sgo2 signal at the centromere and the chromatid junction, normalized to CREST at centromeres, on chromosome spreads from oocytes at the indicated stages of meiotic maturation. Each dot shows mean per oocyte; the number of oocytes analyzed is indicated. Below: scheme illustrating how Sgo2 signals (in red) were measured at the indicated meiotic stages. Ifluor stands for a mean fluorescence intensity of the area within the box (dotted lines, see Materials and Methods for quantification approach).
- Chromosome spreads as in (A), stained for Sgo2 (red), CREST (green), and DNA (Hoechst, in blue). Spreads in early anaphase I and late anaphase I–telophase I were classified, depending on whether dyads of two chromosome sets had been found close to each other as one group, or were scattered apart into two groups. In early anaphase I, Sgo2 is found inbetween sister kinetochores that are already separated.
- Quantification of (E) showing total Sgo2 signal in the centromere region relative to CREST per kinetochore pair (dot plot on the left), and distance between sister kinetochores (dot plot on the right) at the indicated stages of meiosis I. Meta: metaphase, ana: anaphase, and telo: telophase. Each dot represents one kinetochore pair; the number of kinetochore pairs analyzed is indicated. Below: scheme of Sgo2 signal (in red) measurements.
Data information: On each graph mean is shown, error bars are ± SD, asterisks indicate significant difference (****P < 0.0001) according to Mann–Whitney U‐test. AU, arbitrary units. n indicates number of analyzed kinetochores, kinetochore pairs or oocytes, as indicated. Scale bars, 10 μm. See also Fig EV1.
Rec8 holding sister chromatids together in metaphase II is localized at the junction of the two sister chromatids between the two CREST dots (see also below and (Lee et al, 2006; Lee et al, 2008; Chambon et al, 2013b; preprint: Ogushi et al, 2020)). It was proposed that bipolar tension moves Sgo2 together with PP2A away from pericentromeric Rec8 for deprotection in metaphase II (Gomez et al, 2007; Lee et al, 2008). To determine whether bipolar tension in meiosis II was indeed the trigger to remove protection, we asked at what time during the transition from meiosis I to meiosis II Sgo2 and PP2A were removed from the region where sister chromatids are connected and which we call “chromatid junction” (Figs 1C and EV1A). The chromatid junction is significantly less dense in pericentromeric heterochromatin when stained for H3K9me3 (Figs 1C and EV1A), and also less dense when stained with Hoechst or propidium iodide (Fig EV1A and B). Unexpectedly, both Sgo2 and PP2A were removed from this region inbetween sister chromatids in anaphase I, whereas Sgo2 and PP2A co‐localizing with CREST at the centromere persisted in anaphase I (Fig 1A). (The CREST serum we were using gives a similar staining to CenPA staining, which defines chromatin at the centromere, Fig EV1B.) Hence, bipolar tension that is only established in metaphase II seems neither required for the first separation of sister kinetochores, nor the removal of Sgo2 co‐localizing with Rec8.
Figure EV1. Pericentromeric heterochromatin staining of mouse oocyte chromosomes in meiosis I and meiosis II (related to Fig 1C).
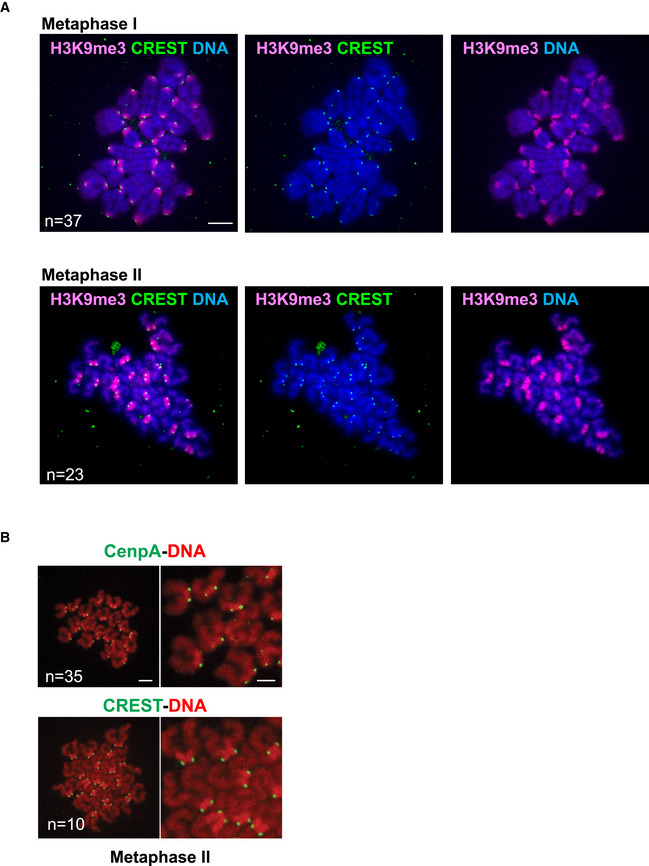
- Metaphase I (6 h after GVBD) and metaphase II chromosome spreads were stained with H3K9me3 antibody and CREST serum to reveal pericentromeric heterochromatin (pink) and the centromere (green), respectively. Chromosomes were stained with Hoechst (blue).
- Schematic representation of the centromere, pericentromeric heterochromatin and chromatid junction in a metaphase II dyad, for quantifications.
- Metaphase II spreads were stained with either CenPA antibody (green), or CREST serum (green), DNA was visualized with Propidium Iodide (red).
Data information: n indicates the number of analyzed oocytes. Scale bars are 10 µm in (A) and 5 µm in (B).
Removal of Sgo2 from the chromatid junction is not required for initial kinetochore individualization
To better understand how kinetochore individualization and Sgo2 localization are related, we re‐analyzed endogenous centromeric localization of Sgo2 in synchronized mouse oocytes cultured in vitro and fixed at key stages of meiosis I and meiosis II. Prophase I arrested oocytes were released to enter meiosis I and allowed to progress into meiosis II, until cell cycle arrest in metaphase II, where they await fertilization. Examination of endogenous Sgo2 localization on chromosome spreads showed that Sgo2 is recruited to the centromere region throughout meiosis I, as shown before (Fig 1D) (Lee et al, 2008; Lister et al, 2010; Rattani et al, 2013; El Yakoubi et al, 2017). We prepared chromosome spreads at the metaphase‐to‐anaphase transition of meiosis I and classified spreads into metaphase I, early anaphase, and late anaphase–telophase I, depending on whether separating dyads were found right next to each other, or dyads had moved away from each other into two independent pools. Sgo2 at the centromere can be detected throughout anaphase I, whereas Sgo2 between sister chromatids was present in early anaphase, and disappeared in late anaphase–telophase (Fig 1E). Kinetochore individualization however started in early anaphase I. At this stage, sister kinetochores that have separated, but still harbor Sgo2 at the chromatid junction, were detected (Fig 1F). Hence, Sgo2 inbetween sister chromatids is removed too late for being the initial trigger for sister kinetochore individualization in meiosis I.
Sgo2 re‐localizes to the sister chromatid junction in meiosis II
Strikingly, staining of endogenous Sgo2 showed an approximately 5‐fold increase of Sgo2 levels in metaphase II, compared to metaphase I (Fig 1D). The fraction of Sgo2 at the chromatid junction was removed in anaphase I, but re‐appeared there in metaphase II. Therefore, removal of this fraction of Sgo2 in metaphase II may still be essential for cohesin deprotection in anaphase II, prompting us to further analyze endogenous Sgo2 localization in meiosis II. Anaphase II takes place as soon as fertilization occurs, or alternatively, after chemical activation to mimic fertilization in oocytes cultured in vitro. We found that Sgo2 protein levels decreased again in anaphase II (Fig 1D), but importantly, Sgo2 was present only in the centromere region, co‐localizing with the CREST signal (Fig 2A). The fraction of Sgo2 that disappeared was indeed the one in between the two kinetochore signals of the sister chromatids, at the chromatid junction (Figs 1C and 2A). Our data indicate that there are at least three pools of Sgo2 as oocytes progress from meiosis I into meiosis II: one that remains mostly associated with centromeres, one that is removed from the chromatid junction after kinetochore individualization when oocytes exit meiosis I, and a fraction in meiosis II that is again localized to the chromatid junction and removed in anaphase II.
Figure 2. Sgo2 localized by Mps1 to the chromatid junction is dispensable for sister chromatid segregation.
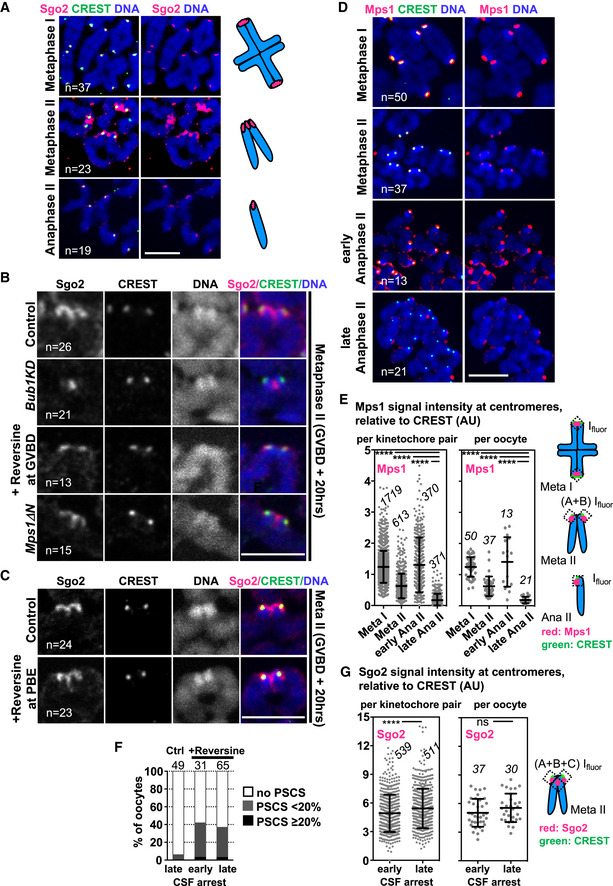
- Chromosome spreads at the indicated stages of meiosis, stained for endogenous Sgo2 (pink), centromeres with CREST serum (green) and DNA with Hoechst (blue). On the right, schemes illustrate the chromosome figures observed, and the corresponding Sgo2 staining (in pink).
- Dyads of metaphase II chromosome spreads of control, Bub1KD, Mps1ΔN and Reversine‐treated oocytes (from GVBD onwards) stained with Sgo2 antibody (pink), CREST serum (green) and Hoechst (blue).
- Dyads of metaphase II chromosome spreads of oocytes treated with Reversine from GVBD + 8 h onwards, corresponding to the time of anaphase I onset. PBE: polar body extrusion, stainings as in (B).
- Chromosome spreads at the indicated stages of meiosis, stained for endogenous Mps1 (pink), centromeres with CREST serum (green) and DNA with Hoechst (blue). For early and late anaphase II spreads, oocytes were chemically activated with Strontium and fixed 25 min or 1 h later, respectively.
- Quantification of (D) showing Mps1 signal relative to CREST per kinetochore pair (per single kinetochore in anaphase II) (left dot plot), and Mps1 signal averaged per oocyte (right dot plot) at the indicated timepoints. The number of kinetochore pairs (kinetochores in anaphase II) and oocytes analyzed is indicated. The scheme of measurements is shown on the right.
- Percentage of oocytes harboring one or more single sister chromatids when analyzed by chromosome spreads stained with CREST and Hoechst. Where indicated, oocytes were treated with Reversine from GVBD onwards, and treatment was renewed every 8 h. Early CSF corresponds to 8 h after GVBD (Reversine‐treated oocytes undergo anaphase I on average 4–5 h after GVBD), and late CSF corresponds to 36 h after GVBD (> 29 h in CSF arrest). PSCS, precocious sister chromatid segregation.
- Quantification of total Sgo2 signal on spreads in early or late CSF arrest (16 h or 30 h after GVBD, respectively) relative to CREST per kinetochore pair (per single kinetochore in anaphase II) (left dot plot), and Sgo2 signal averaged per oocyte (right dot plot) at the indicated stages. The number of kinetochore pairs and oocytes analyzed is indicated. The scheme of measurements is shown on the right. Meta: metaphase and ana: anaphase.
Data information: On each graph mean is indicated, error bars are ± SD, asterisks indicate significant difference (****P < 0.0001; ns = not significant) according to Mann–Whitney U‐test. AU, arbitrary units, n indicates number of analyzed kinetochores, kinetochore pairs or oocytes, as indicated. Scale bars, 10 μm. See also Fig EV2.
Sgo2 localized by Mps1 kinase to the chromatid junction is removed at anaphase II onset
In meiosis I, the different pools of endogenous Sgo2 can be distinguished within the centromere region. We have shown previously that Sgo2 brought to the centromere by Mps1 kinase and Bub1 protein (but not kinase activity) is required for centromeric cohesin protection in meiosis I (El Yakoubi et al, 2017). There is another pool of Sgo2 localized in a Bub1 kinase activity‐dependent manner within the pericentromere, but this one is not required for cohesin protection in meiosis I (El Yakoubi et al, 2017). It was attractive to speculate that the pool of Sgo2 that is removed in meiosis II is the one equally localized there by Mps1, hence potentially protecting centromeric cohesin until anaphase II onset. To address this issue, we asked first whether also in metaphase II, distinct pools of Sgo2 are localized by Mps1 and Bub1 kinase activities, such as in meiosis I. As previously done for meiosis I, we made use of two mouse models, one harboring a kinase‐dead version of Bub1 (Bub1KD) instead of wild‐type Bub1 (Ricke et al, 2012; El Yakoubi et al, 2017), and the other one expressing only a truncated version of Mps1 (Mps1ΔN) that cannot localize to kinetochores, but still harbors kinase activity (Hached et al, 2011). Additionally, we added the Mps1 inhibitor Reversine to wild‐type oocytes during the entire meiotic maturation from germinal vesicle breakdown (GVBD, corresponding to nuclear envelope breakdown in mitosis) onwards, to inhibit Mps1 kinase activity, but not its localization (El Yakoubi et al, 2017). We found that inhibition of either kinase leads to reduction of overall Sgo2 levels in the centromere region. Additionally and similar to meiosis I, Bub1 and Mps1 kinases preferentially localize distinct pools of Sgo2, but this time to the centromere and the chromatid junction, respectively (Figs 2B and EV2A–D). Whereas Bub1 kinase activity is not essential for localization of Sgo2 inbetween sister chromatids, Mps1 kinase activity promotes Sgo2's localization there. Upon inhibition of Mps1, Sgo2 at the centromere is significantly reduced, and hardly any Sgo2 at the chromatid junction was detected. Hence, the fraction of Sgo2 that is removed in anaphase II corresponds foremost to Sgo2 localized there by Mps1 kinase activity, suggesting that this pool, in analogy to meiosis I, may confer protection until anaphase II onset. Kinetochore localization of Mps1 is not specifically required for localizing Sgo2 to the chromatid junction, as Mps1ΔN oocytes do not preferentially lose Sgo2 from this location. Mps1ΔN oocytes do not show loss of centromeric cohesin protection in meiosis I or meiosis II (El Yakoubi et al, 2017), further indicating that kinetochore localization of Mps1 is not required for protection. Taken together, our data indicate that the Mps1 kinase activity‐dependent fraction of Sgo2 localized to the chromatid junction is removed for sister separation in meiosis II.
Figure EV2. Visualization and quantification of centromeric Sgo2 and Sgo2 at the chromatid junction in meiosis II (related to Fig 2).
-
A3D‐rendering with Arivis Vision 4‐D software of representative stainings of chromosome spreads in Fig 2B. (Chromosomes appear in blue, Sgo2 in red, and CREST in green). In the lower panels Sgo2 staining at centromeres and the chromatid junction are indicated.
-
BA representative wild‐type control chromosome spread of a dyad in metaphase II from Fig 2B is shown to indicate how the regions for quantifications were defined.
-
C, DThe graphs show the corresponding quantifications of the centromere signal (blue: overlapping with CREST) and the signal at the chromatid junction (red: not overlapping with CREST), for Fig 2B, per oocyte (C) and per kinetochore pair (D).
-
E, FQuantifications from Fig 2G, distinguishing centromere and chromatid junction pools of Sgo2, per oocyte (E), and per kinetochore pair (F).
Data information: In panels (C and D), Sgo2/CREST intensities at the centromere versus the chromatid junction in Bub1KD, Reversine‐treated and Mps1∆N oocytes were normalized to the mean of Sgo2/CREST intensities in wild‐type control oocytes. On each graph, mean is indicated, error bars are ± SD, asterisks indicate significant difference according to Mann–Whitney U‐test (*P < 0.1; **P < 0.01; ****P < 0.0001). n indicates the number of kinetochore pairs and number of oocytes analyzed.
Mps1 activity in meiosis I promotes Sgo2 localization to the chromatid junction in meiosis II
We were wondering at what stage of meiotic maturation Mps1 kinase activity was necessary to obtain Sgo2 at the chromatid junction in meiosis II. When we inhibited Mps1 only during meiosis II by adding Reversine at the time of meiosis I polar body extrusion (PBE, corresponds to exit from meiosis I), we did not lose this fraction of Sgo2 (Fig 2C), unlike what we observed upon inhibition of Mps1 from GVBD onwards (Fig 2B). This indicates that Mps1 activity in meiosis I and not meiosis II is required for proper localization of Sgo2 in metaphase II at the chromatid junction. As kinetochore localization of Mps1 was not required (Mps1ΔN, Fig 2B), and taking into account the overall increase of Sgo2 levels in the centromere region in meiosis II (Fig 1D), we conclude that cytoplasmic activity of Mps1 in meiosis I but not meiosis II is necessary for recruiting Sgo2 from the cytoplasm to the chromatid junction. Alternatively, Sgo2 brought to the centromere in meiosis I by Mps1 may move to the chromatid junction in meiosis II in an Mps1‐independent manner. However, we think this is unlikely, given the increase of the amount of Sgo2 in the centromere region in meiosis II, compared to meiosis I.
We had previously shown that Mps1 is co‐localizing with CREST in metaphase II (El Yakoubi et al, 2017); hence, removal of Mps1 may be required for deprotection in oocyte meiosis II. Indeed, in S. cerevisiae, degradation of Sgo1 and Mps1 at anaphase II onset was proposed to contribute to the deprotection of centromeric cohesin (Arguello‐Miranda et al, 2017; Jonak et al, 2017). However, throughout metaphase II‐to‐anaphase II transition Mps1 remained at the inner kinetochore of separating sister chromatids in mouse oocytes, and only there and not elsewhere (Fig 2D). Mps1 kinetochore localization was detected at high levels at the very onset of anaphase II during a very short time window and, as oocytes progressed through anaphase II, Mps1 disappeared quickly, resulting in some kinetochores that had already lost their Mps1 staining and others still harboring Mps1, with a high variability between kinetochores within one oocyte, and oocytes themselves (Fig 2D and E). Altogether, we think it is unlikely that deprotection of centromeric cohesin in oocytes is brought about by degradation of Mps1, because sister chromatids separate before Mps1 disappears completely. Again, this is in agreement with our previous data that kinetochore localization of Mps1 is not required to prevent precocious sister chromatid separation (El Yakoubi et al, 2017).
Is Mps1‐dependent Sgo2 required for cohesin protection in metaphase II?
To await fertilization, oocytes have to maintain a cell cycle arrest—also named cytostatic factor or CSF arrest (Schmidt et al, 2006)—for an extended time in metaphase II. Sgo2 localized by Mps1 kinase to the chromatid junction may have a role in preventing precocious sister chromatid separation during this arrest. To test this hypothesis, we inhibited Mps1 from GVBD onwards and performed chromosome spreads immediately after entry into metaphase II, or upon extended CSF arrest. We observed some precocious separation of sister chromatids (PSCS) in around 40 % of oocytes, such as described before, due to reduction of Sgo2 required for centromeric cohesin protection in meiosis I (El Yakoubi et al, 2017). This PSCS is due to loss of protection in meiosis I already and does not indicate loss of protection in meiosis II. Importantly, the percentage of oocytes with single sister chromatids, and the percentage of single sisters per oocytes did not increase upon prolonged CSF arrest compared to oocytes fixed at entry into metaphase II. This shows that Sgo2 visible at the chromatid junction alone is not necessary to prevent PSCS in meiosis II, even under prolonged arrest conditions (Fig 2F). Because there was no increase in PSCS, we wanted to know whether this correlated with Sgo2 levels remaining stable throughout prolonged CSF arrest in control conditions. Unlike what we expected, Sgo2 levels even slightly increased during extended CSF arrest, both at the centromere and at the chromatid junction (Figs 2G and, EV2E and F). Hence, it remains a possibility that Sgo2 at the chromatid junction, when present, has to be inactivated for deprotection of centromeric cohesin at anaphase II onset. At this stage, our data do not allow us to conclude that there is no protection of centromeric cohesin in meiosis II. Even though Sgo2 at the chromatid junction is not essential to prevent precocious cleavage of centromeric Rec8, it may still serve as a backup system to prevent PSCS during CSF arrest should separase inhibition fail. But our data show that under normal conditions, Mps1‐dependent Sgo2 is not essential to prevent PSCS in meiosis II.
Bipolar tension is dispensable for step‐wise cohesin removal
Sgo2 is found to co‐localize with Rec8 between individualized kinetochores in metaphase II‐arrested oocytes (Chambon et al, 2013b). It remained possible that an increase in bipolar tension at anaphase II onset would be required to remove Sgo2 at the chromatid junction (when present) and bring about deprotection of centromeric Rec8. To clarify this point, we set out to test whether indeed the way tension forces are applied in meiosis I or meiosis II determines if the meiotic cohesin subunit Rec8 is removed from chromosome arms or from the centromere region.
First, to determine whether attachment of chromosomes in meiosis I to both poles of the bipolar spindle is a prerequisite for arm cohesin removal, we treated prometaphase I mouse oocytes with the Eg5 inhibitor STLC to induce monopolar spindles, where bivalents with fused sister kinetochores were attached on monopolar spindles (Vallot et al, 2018). Individual kinetochore fibers forming end‐on attachments were clearly visible by high‐resolution confocal microscopy of cold‐treated spindles (Fig 3A). Missing tension prevents timely anaphase I onset, because Aurora B‐dependent error correction leads to spindle assembly checkpoint (SAC) activation (Vallot et al, 2018); therefore, we added Reversine in metaphase I to override the SAC and allow anaphase I onset in oocytes harboring monopolar spindles. Short Reversine treatment just before anaphase I onset does not interfere with cohesin protection in meiosis I (El Yakoubi et al, 2017). Upon SAC override, bivalents separated into dyads on monopolar spindles, and arm cohesin was removed, as visualized by Rec8 staining (Fig 3B and C). When we examined bivalents that separated into dyads on a monopolar spindle, we observed that sister kinetochores that appeared fused together were clearly separated once arm cohesin was gone (Fig 3C), further confirming that sister kinetochore individualization does not require bipolar attachment. Rec8 was not removed from the centromere region holding sister chromatids together, and no sister chromatid segregation was observed (Fig 3C).
Figure 3. Step‐wise cohesin removal occurs independently of tension forces applied by a bipolar spindle.
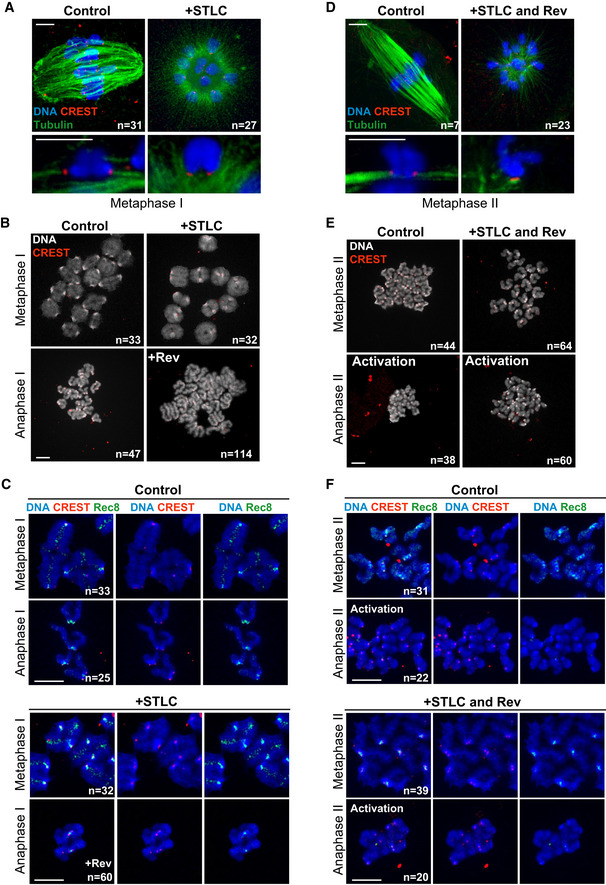
-
A–FOn the left, meiosis I, on the right, meiosis II.
-
A, DOocytes in meiosis I (6 h after GVBD) or meiosis II (left and right panels, respectively) were treated with STLC for 2.5 h and fixed for whole‐mount immunofluorescence. Cold‐stable microtubules were stained with an antibody against tubulin (green), centromeres with CREST (red) and DNA with Hoechst (blue).
-
B, EChromosome spreads at metaphase and anaphase of meiosis I (6 and 9 h after GVBD) or meiosis II (left and right panels, respectively), in the presence or absence of STLC (for 2.5 h) and Reversine (+Rev., for 1 h) as indicated. Spreads were stained with CREST serum (red) and Hoechst (gray). Single chromosomes are present in anaphase I and single sister chromatids in anaphase II.
-
C, FChromosome spreads exactly as described in (B and E), except that spreads were additionally stained with an antibody against Rec8 (green). The CREST signal appears in red and DNA in blue. Rev: Reversine.
Data information: Scale bar indicates 5 µm in (A and D), and 10 µm in (B, C, E and F). In all panels, n indicates the number of oocytes with the shown phenotype and the total number of oocytes analyzed. See also Fig EV3.
To address whether application of bipolar tension on dyads in meiosis II is required for centromeric cohesin removal, we performed the same experiment as above, but in meiosis II. Monopolar spindles were obtained through STLC treatment of metaphase II‐arrested oocytes, SAC response was inhibited through Reversine treatment, and oocytes were activated to undergo the second meiotic division. Microtubule attachments to the monopolar spindle were verified by high‐resolution confocal microscopy of cold‐stable microtubule fibers (Fig 3D). Remarkably, our experiment showed that sister chromatid segregation and hence, centromeric cohesin removal, does not require bipolar attachment of dyads (Fig 3E). Centromeric Rec8 was undetectable upon activation, independently of spindles being mono‐ or bipolar (Fig 3F). Accordingly, sister chromatids came apart on a monopolar spindle, contradicting the hypothesis that tension‐dependent removal of cohesin protection through bipolar attachment is required for centromeric cohesin cleavage, at least in mouse oocytes.
It remained a possibility that unlike degradation, inactivation of Mps1 is required for deprotection in case Sgo2 at the chromatid junction is present. Inactivation of Mps1 by Reversine may have been key for the sister chromatid separation on monopolar spindles in meiosis II observed above. In this case, we would induce deprotection on monopolar spindles in meiosis II by our use of Reversine for SAC override. We repeated the experiment in Mps1ΔN oocytes, which are SAC deficient, hence allowing SAC override without interfering with Mps1 kinase activity. As shown previously, Mps1ΔN oocytes underwent meiosis I in an accelerated manner, mis‐segregated whole chromosomes, and arrested in metaphase II (Hached et al, 2011). Upon activation in the presence or absence of STLC, oocytes entered anaphase II, and importantly, centromeric cohesin was removed and sister chromatids were segregating (Fig EV3). Therefore, neither bipolar spindle tension nor Mps1 kinase inactivation is required for Rec8 removal and sister separation in meiosis II.
Figure EV3. Inactivation of Mps1 kinase activity is not required for sister chromatid segregation on monopolar spindles (related to Fig 3).
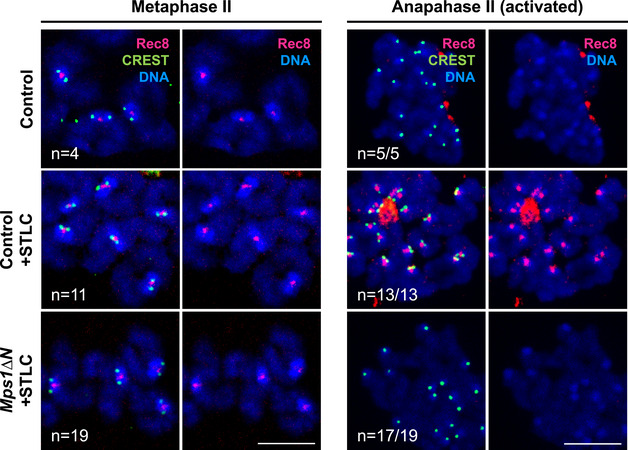
Chromosome spreads obtained from control or Mps1ΔN oocytes in vitro matured oocytes, in metaphase II and in anaphase II, in the presence or absence of prior STLC treatment (for 2.5 h), as indicated. Spreads were stained with Rec8 antibody (pink), CREST (green) and Hoechst (blue).
Data information: In all panels, n indicates the number of oocytes with the shown phenotype and the total number of oocytes analyzed. Scale bar, 10 µm.
Individualization of sister kinetochores depends on separase
Our data indicate that bipolar tension is not essential for allowing sister chromatid separation in oocyte meiosis II. We hypothesized that alternatively, kinetochore individualization in meiosis I determines whether centromeric cohesin can be removed in the following meiosis II division. It was attractive to speculate that separase activity is required for individualizing sister kinetochores in anaphase I and that this is the key event to allow centromeric cohesin removal in meiosis II. To address this hypothesis, we first asked whether oocytes without separase contained fused or individualized kinetochores, using mice harboring an oocyte‐specific invalidation of separase (Sep −/−). Oocytes without separase progress into meiosis II, because loss of separase does not prevent progression through meiosis I into meiosis II, even though chromosome segregation does not take place. Cyclin B1 and securin accumulate as oocytes progress into metaphase I, are degraded, and re‐accumulate in control and Sep −/− oocytes with similar kinetics (Fig EV4A) (Kudo et al, 2006). Furthermore, upon hormonal stimulation of female mice to induce meiotic maturation of oocytes in vivo, control and Sep −/− oocytes move from ovaries into the oviduct and are found as cumulus‐enclosed oocytes awaiting fertilization (Fig 4A). In Sep −/− oocytes harvested from oviducts after in vivo maturation, or matured from GV onwards in vitro, bivalents were still attached in a monopolar manner and sister kinetochores of bivalents remained fused together even though oocytes were in metaphase II (Fig 4B and C). This result indicates that separase is required for sister kinetochore individualization prior to metaphase II in oocytes matured in vivo and in vitro.
Figure EV4. Schemes of cell cycle progression in oocytes devoid of separase, and rescue experiment performed in this study (related to Fig 4 and 5).
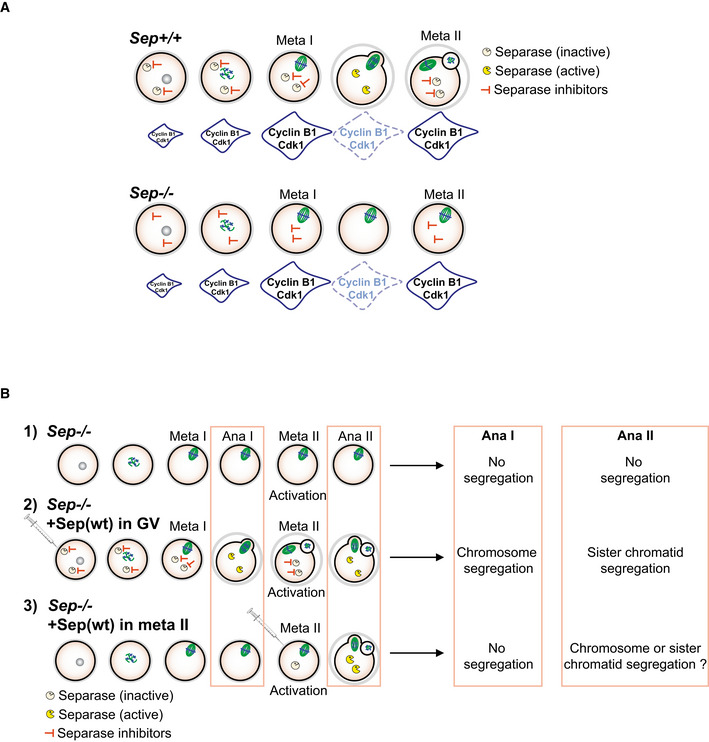
- Scheme summarizing published data on cell cycle progression in oocytes devoid of separase. Cdk1 activity accumulates, disappears, and re‐accumulates on time. The APC/C substrate cyclin B1 is degraded on time in the absence of separase, and re‐accumulates as oocytes progress into meiosis II, even though no polar body extrusion is observed, due to a role of separase in polar body extrusion, which is independent of its cleavage activity. The SAC is not activated, because chromosomes are correctly attached without separase. See text for references.
- Rescue experiments performed in this study: 1) Sep −/− oocytes without rescue. 2) Sep −/− oocytes rescued from GV onwards (Fig 4). 3) Sep −/− oocytes rescued from metaphase II onwards. The question we asked was whether chromosomes or sister chromatids are separated when separase is absent in meiosis I, and present in meiosis II.
Figure 4. Oocytes without separase can be activated to undergo meiosis II and separate sister chromatids when rescued with wild‐type separase from GV onwards.
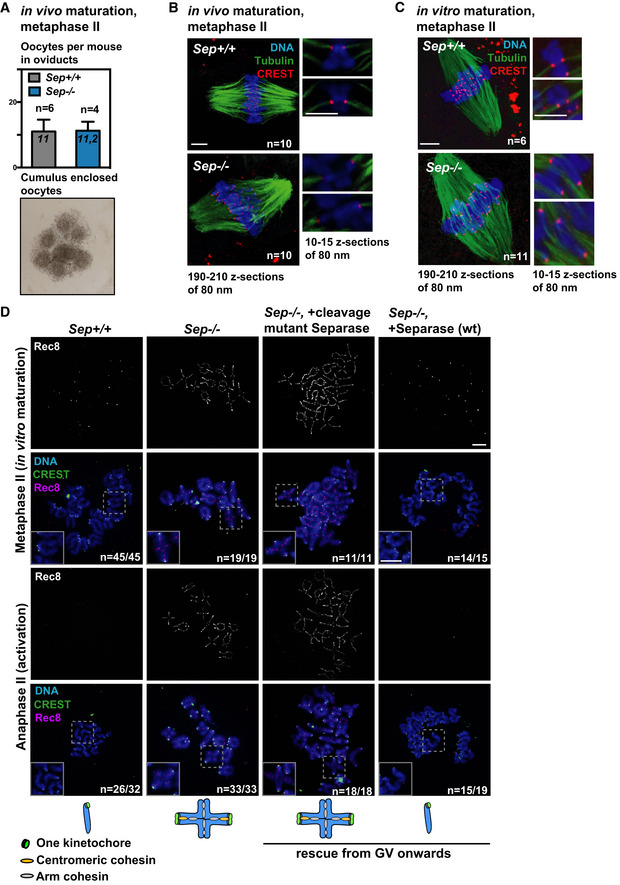
- Metaphase II oocytes from mice carrying a conditional, oocyte‐specific invalidation of separase (Separaseflox / flox Zp3 Cre +, or Sep −/−) and oocytes from litter mates (Separaseflox / flox, or Sep +/+) were obtained after superovulation. In vivo matured control (Sep +/+) and Sep −/− oocytes are found in oviducts at comparable numbers, enclosed by cumulus cells. Example of cumulus‐enclosed oocytes from the oviduct is shown below.
- Whole‐mount immunofluorescence to stain cold‐stable microtubule fibers after in vivo maturation. Microtubules were stained with anti‐tubulin antibody (green), centromeres with CREST serum (red) and DNA with Hoechst (blue). Bivalents are attached in a monopolar fashion. Approximate number of z‐sections used for overlays to visualize the whole spindle (on the left) or individual chromosomes (magnifications on the right) are indicated.
- Same as (B), except that oocytes of the indicated genotypes were obtained after in vitro maturation from GV onwards.
- Metaphase‐to‐anaphase transition of meiosis II in oocytes of the indicated genotype cultured in vitro and injected with the separase constructs as indicated, in GV. The schemes indicate the chromosome figures observed upon activation. Chromosome spreads are stained with CREST serum (green), and Rec8 antibody (pink). DNA was stained with DAPI (blue).
Data information: In (A), n indicates the number of mice and a mean number of oocytes per mouse of each genotype, error bars are + s.e.m. In (B and C) n indicates number of oocytes analyzed. In (D) n indicates the number of oocytes with the shown phenotype (e.g., upon successful activation) and the total number of oocytes analyzed. Scale bars indicate 5 µm in (B and C), and 10 µm in (D). See also Fig EV4A.
Not surprisingly, in the absence of separase activity, oocytes were unable to segregate chromosomes, or sister chromatids upon activation (Figs 4D and EV4B). Meiosis I chromosome segregation, kinetochore individualization, and meiosis II sister chromatid segregation were rescued when separase was re‐expressed from GV onwards, but not when a catalytically inactive separase mutant was used. This shows that separase is essential not only to remove arm cohesin (Kudo et al, 2006), but also to remove centromeric cohesin in mouse oocyte meiosis II (Figs 4D and EV4B). Our data additionally indicate that separase is required for kinetochore individualization in meiosis I.
Bivalents with fused kinetochores separate into dyads, not sister chromatids in meiosis II
We set out to address whether separase activity during the first meiotic division—which would bring about kinetochore individualization—is required for deprotection and separation of sister chromatids in meiosis II. For this, we asked how Sep −/− oocytes segregate bivalents when rescued with exogenously expressed separase only in meiosis II, by injecting CSF‐arrested oocytes with mRNA coding for wild‐type separase. Injected metaphase II‐arrested Sep −/− oocytes were activated to undergo anaphase II and then examined by chromosome spreads to address whether chromosomes or sister chromatids were separated (Fig EV4B). Remarkably, under these conditions, chromosomes were separated and Rec8 was removed from chromosome arms, whereas sister chromatids remained paired with centromeric Rec8 that was not removed by separase (Fig 5A). Hence, if separase is absent in meiosis I but present in meiosis II, it removes arm cohesin instead of centromeric cohesin in meiosis II.
Figure 5. Separase activity during meiosis I is required for centromeric cohesin removal in meiosis II.
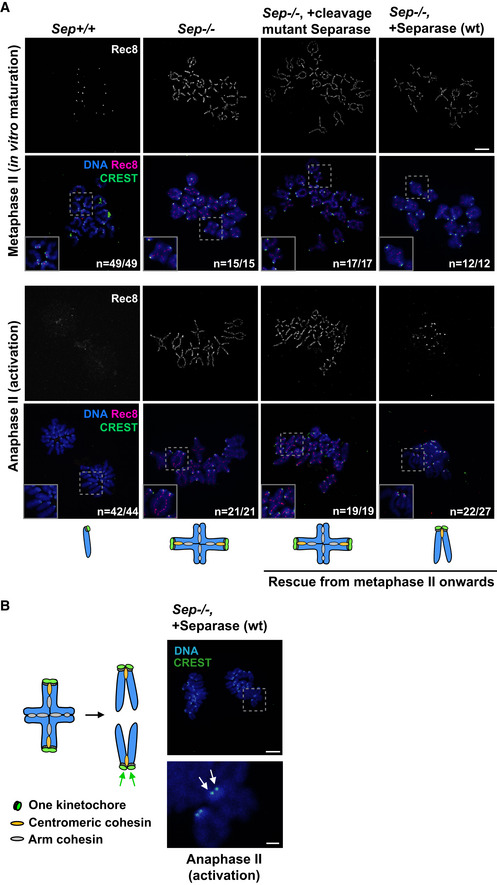
- Sep +/+ or Sep −/− oocytes were matured in vitro until metaphase II. Sep −/− oocytes were injected with wild‐type separase or cleavage mutant separase encoding mRNAs in metaphase II. Oocytes were either fixed in metaphase II or activated and fixed in anaphase II. Spreads were stained with Rec8 antibody (red), CREST serum (green), and DAPI (blue) to label DNA.
- Representative image of kinetochore individualization in Sep −/− oocytes injected with wild‐type separase in metaphase II and fixed after activation, from (A). Shown is staining with CREST serum (green), and DAPI (blue) to label DNA. Arrows indicate separated sister kinetochores. The scheme on the left illustrates the individualization of sister kinetochores (green arrows) at anaphase II.
Data information: n indicates the number of oocytes with the shown phenotype (e.g. in anaphase II, after successful activation) and the total number of oocytes analyzed Scale bars: 10 µm, except bottom panel in (B), where the scale bar is 2 μm. See also Fig EV4B.
Sister kinetochore individualization can also occur in meiosis II
Closer examination of sister kinetochores showed that arm cohesin removal and separation of chromosomes in meiosis II in Sep −/− oocytes rescued from metaphase II onwards also led to sister kinetochore individualization, such as usually observed in meiosis I (Fig 5A and B). Injecting catalytically inactive separase did not lead to kinetochore individualization upon activation (Fig 5A), confirming that in addition to arm cohesin removal, sister kinetochore individualization requires separase activity. Both events can occur in meiosis II, if separase is absent in meiosis I, but our data indicate that separase cannot induce sister kinetochore individualization and remove centromeric cohesin at the same time.
Bivalents with individualized kinetochores separate into sister chromatids in meiosis II
We propose that separase activity at anaphase I is required for kinetochore individualization, and this individualization is a prerequisite for deprotection of centromeric cohesin in meiosis II. In this case, oocytes harboring bivalents in metaphase II with individualized kinetochores such as usually the case for dyads should not segregate chromosomes, but sister chromatids.
Overexpression of mitotic Sgo1 was shown to interfere with arm cohesin removal in mouse oocytes in a previous study, by recruiting PP2A to chromosome arms, hence resulting in the presence of bivalents in meiosis II (Xu et al, 2009). In oocytes, Sgo2 and not Sgo1 is essential for protecting centromeric cohesin (Llano et al, 2008), hence we refrained from using Sgo1 to avoid creating artifacts and asked whether overexpression of meiotic Sgo2 equally allowed us to obtain bivalents in meiosis II. Our data indicated that Sgo2 does not regulate sister kinetochore individualization, as individualization of kinetochores in meiosis I was not due to removal of Sgo2 from the chromatid junction (Fig 1E and F). Consequently, overexpression of Sgo2 was expected to lead to failures in separating chromosomes, yet permit individualization of sister kinetochores, allowing us to obtain bivalents in meiosis II with individualized sister kinetochores. To address this issue, we cultured oocytes overexpressing GFP‐Sgo2 from GV onwards. GFP‐Sgo2 localized to whole chromosomes in prometaphase and metaphase I, as visualized by following the first meiotic division by live imaging (Figs 6A and EV5), similar to what has been observed before (Rattani et al, 2013; Rattani et al, 2017). Indeed GFP‐Sgo2 overexpression prohibited complete cohesin removal from chromosome arms (Fig 6B). As a result, some bivalents that were not able to separate in meiosis I were present in metaphase II in roughly half of the injected oocytes (Fig 6C). Importantly though, passing through meiosis I induced the individualization of sister kinetochores on all chromosomes, including the bivalents that did not separate (Fig 6C). This confirms that indeed, individualization of kinetochores in meiosis I was not affected by Sgo2 overexpression.
Figure 6. Centromeric cohesin is not protected on bivalents with individualized kinetochores in meiosis II.
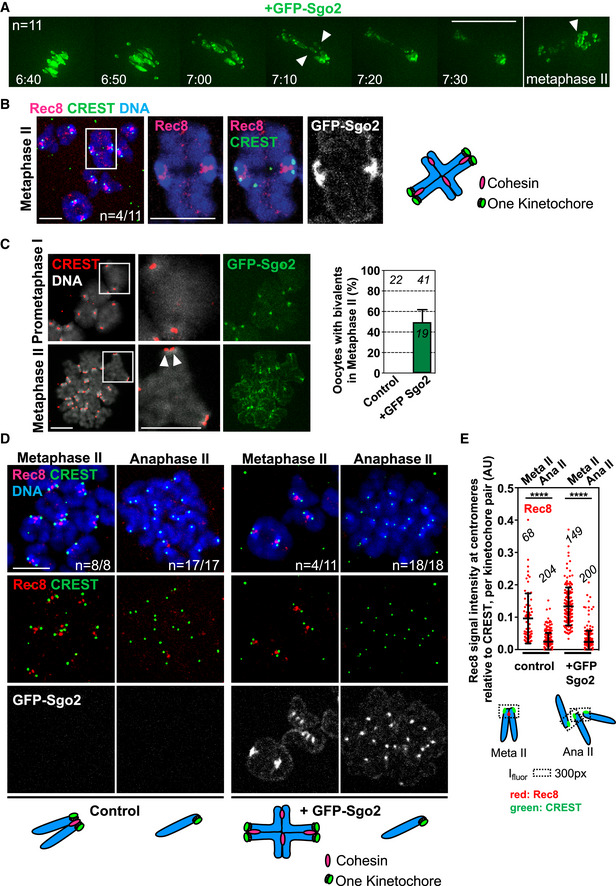
- Live imaging movie of GFP‐Sgo2 expressing oocytes (from GV onwards) undergoing meiosis I. The green channel to visualize GFP‐Sgo2 localized to chromosomes is shown. Timepoints were taken every 10 min, shown is a montage of selected frames such as indicated (hours: time after GVBD) at the metaphase‐to‐anaphase transition of meiosis I and in metaphase II. Each timepoint is comprised of an overlay of 11 z‐sections of 3 μm for GFP. The arrow heads indicate bivalents.
- Representative images of a chromosome spread showing Rec8 localization on a bivalent chromosome in metaphase II of a GFP‐Sgo2 expressing oocyte. Spreads were stained with Rec8 antibody (pink), CREST serum (green), Hoechst for DNA (blue), GFP‐Sgo2 was visualized by GFP fluorescence (gray). The scheme on the right shows a bivalent with individualized kinetochores and some Rec8 staining on arms.
- Metaphase I (4 h after GVBD) and II spreads after GFP‐Sgo2 expression (from GV onwards). Spreads were stained with CREST serum (red), Hoechst for DNA (gray), GFP‐Sgo2 was visualized by GFP fluorescence (green). Arrowheads mark separated sister kinetochores. On the right, graph showing percentage of metaphase II spreads containing bivalent chromosomes.
- Chromosome spreads of oocytes expressing GFP‐Sgo2 (from GV onwards) in metaphase II, and upon activation. Spreads were labeled with Rec8 antibody (shown in pink (top) and red (below), far‐red secondary antibody was used), CREST serum (pseudo‐colored in green), and Hoechst (blue). GFP direct fluorescence was imaged for GFP‐Sgo2 detection. Below the corresponding chromosome figures are represented in the schemes.
- Quantification of Rec8 signal found around and in between CREST signals that were close to each other. Below: scheme illustrating how Rec8 signals (in red) were measured at the indicated meiotic stages. Ifluor stands for a mean fluorescence intensity of the area within the box (dotted lines). Meta: metaphase, ana: anaphase.
Data information: In (A–C) n indicates the number of control non‐injected and GFP‐Sgo2‐expressing oocytes analyzed, in (D), the number of oocytes with the shown phenotype and the total number of oocytes analyzed. On each graph, mean is shown, error bars are + s.e.m. in (C) and ± SD in (E), asterisks indicate significant difference (****P < 0.0001) according to Mann–Whitney U‐test. AU, arbitrary units. The number of analyzed oocytes in (C) and of analyzed kinetochore pairs in (E) is indicated. Scale bars: (A) 50 μm, (B–D) 10 μm. See also Fig EV5 for additional timepoints and channels of the movie in (A).
Figure EV5. GFP‐Sgo2 overexpressing oocyte progressing through meiosis I into metaphase II (related to Fig 6).
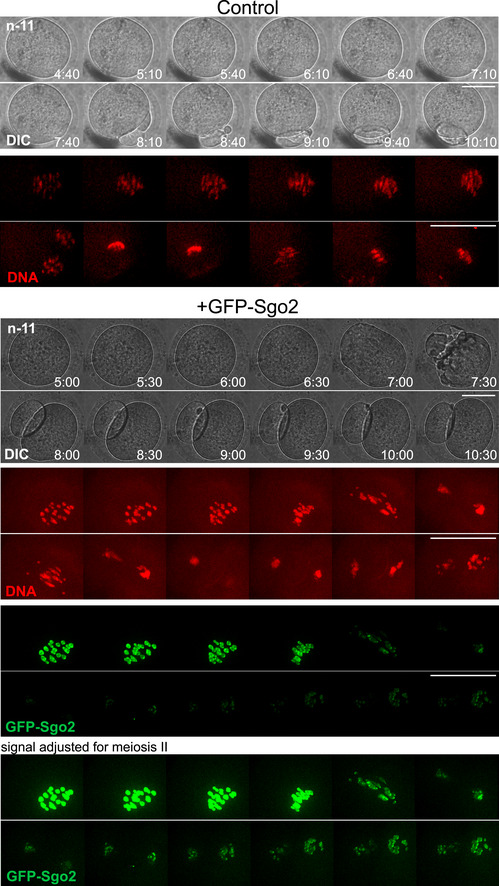
Montage of live imaging movie of control and GFP‐Sgo2 expressing oocytes (from GV onwards) undergoing meiosis I. Chromosome movements were followed by staining DNA with SirDNA (far red, shown in red), GFP‐Sgo2 appears in green. Shown are the DNA, GFP, and DIC channels, each frame comprised of an overlay of 11 z‐sections of 3 μm for GFP and far‐red channels, and 1 z‐section in DIC. Timepoints were taken every 10 min, shown are selected timepoints such as indicated. For the control, the GFP channel has been omitted, because it is devoid of any signal. For GFP‐Sgo2 expressing oocytes, two different settings for brightness are shown, to visualize either meiosis I or meiosis II in an optimal manner, as the signal decreases after anaphase in meiosis I.
Data information: n indicates the number of oocytes analyzed. Scale bars, 50 µm.
Upon activation, dyad separation in meiosis II was not perturbed by GFP‐Sgo2 overexpression, even though GFP‐Sgo2 was able to localize to the chromatid junction in metaphase II (Fig 6D). Thus, if centromeric cohesin was still protected in meiosis II in a Sgo2‐dependent manner, the underlying molecular mechanism must be more complex than mere localization of Sgo2 to the chromatid junction. Crucially though, upon activation, bivalents separated into sister chromatids, because no dyads were observed in any of the anaphase II spreads upon activation. All detectable Rec8, on arms and in the centromere region, was removed (Fig 6D and E). In conclusion, individualization of sister kinetochores in meiosis I leads to centromeric cohesin removal in meiosis II, even on bivalents.
Discussion
Our study was motivated by a quest to understand how the decision to remove centromeric Rec8 only in meiosis II is taken in mammalian oocytes. None of the current models provided a satisfying answer to this question yet. It was proposed that protection of centromeric cohesin is mediated by recruitment of Sgo2/PP2A to the fraction of Rec8 that is not cleaved, and centromeric cohesin is deprotected by physical removal of Sgo2/PP2A in meiosis II. Bipolar tension was suggested to move Sgo2 together with PP2A away from centromeric Rec8 in both male and female meiosis II (Gomez et al, 2007; Lee et al, 2008). This model was very attractive, because it was the only one explaining how centromeric cohesin would be deprotected in meiosis II, but not meiosis I. However, our results demonstrate that sister chromatid segregation can take place on monopolar spindles, hence without bipolar tension, contradicting the tension model (Fig 7A), and in agreement with a recent study (V Mengoli et al, 2021). Sgo2 and PP2A still co‐localize with Rec8 inbetween sister chromatids in mouse oocyte meiosis II (Wassmann, 2013) and, as we show here, anaphase II onset can take place without removing or inactivating Mps1, the kinase required for Sgo2 localization at the chromatid junction in meiosis II. Furthermore, endogenous Sgo2 and exogenously expressed Sgo2 localize to the same region in metaphase II, without preventing sister chromatid segregation in anaphase II. Altogether, our data indicate that either (i) Sgo2 is unable to protect centromeric cohesin in meiosis II, or (ii) Sgo2 has to be inactivated exactly at anaphase II onset. In the first situation, cohesin cleavage in metaphase II would be regulated only through control of separase activity. In this case, the protective function of Sgo2 has to be inactivated prior to entry into meiosis II, but not before inactivation of separase at exit from meiosis I, hence after kinetochore individualization. In the second case, in addition to activation of separase at anaphase II onset, Sgo2 localized to the chromatid junction has to be inactivated through some other, yet to be identified mechanism. Interestingly, in S. cerevisiae it has been shown that Sgo1 still protects centromeric Rec8 in meiosis II, and degradation of Sgo1 is indeed necessary for anaphase II onset (Arguello‐Miranda et al, 2017; Jonak et al, 2017). We show here that in oocytes, overexpression of Sgo2 does not prevent sister chromatid segregation in meiosis II, and endogenous Sgo2 remains localized to the centromere region throughout anaphase II, indicating that Sgo2 when present in meiosis II is inactivated by other means than degradation at anaphase II onset. Since inhibition of Mps1 kinase activity and the resulting loss of Sgo2 from the chromatid junction in meiosis II does not further influence chromatid segregation, we think that the function of Sgo2 which is visible at the chromatid junction and dependent on Mps1 is not essential for meiosis II under normal conditions.
Figure 7. Kinetochore individualization in meiosis I is required for step‐wise cohesin removal.
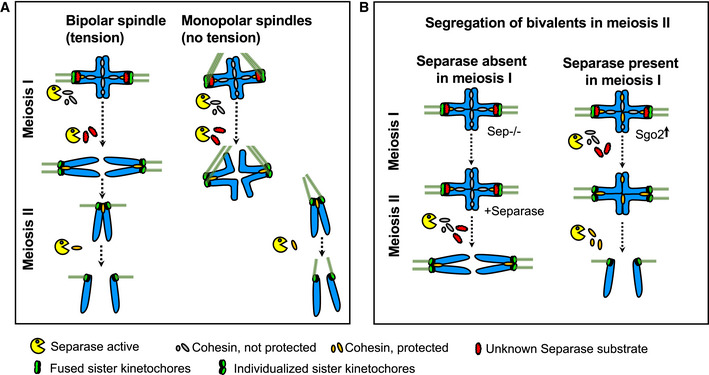
- On monopolar spindles, as on bipolar spindles, chromosomes segregate in meiosis I, and sister chromatids in meiosis II.
- Model of how separase‐dependent kinetochore individualization primes bivalents for deprotection of centromeric cohesin. If separase is absent in meiosis I, arm cohesin and not centromeric cohesin is removed in meiosis II. Under these conditions, sister kinetochores individualize in meiosis II instead of meiosis I, demonstrating that step‐wise cohesin removal (first arm cohesin, then centromeric cohesin) depends on the kinetochore structure. Without kinetochore individualization, centromeric cohesin cannot be cleaved in the subsequent division. See Discussion for details.
Oocytes have to remain in CSF arrest for extended periods of time to await fertilization. Unlike SAC‐induced metaphase arrest, the CSF arrest is dynamic, with fluctuating APC/C activity, to maintain just the right amount of cyclin B‐Cdk1 kinase activity to keep a metaphase state, but at the same time being able to quickly undergo anaphase II onset upon fertilization (Wu & Kornbluth, 2008). In mitosis, separase is tightly inhibited by cyclin B/Cdk1 and securin (Stemmann et al, 2006), and an inhibitor composed of Mad2‐Sgo2 (Hellmuth & Stemmann, 2020). Sgo2‐Mad2‐dependent inhibition of separase requires active SAC signaling, which in CSF‐arrested oocytes should already be turned off, because all kinetochores are correctly attached (Hellmuth & Stemmann, 2020). For this reason, we think that separase inhibition in CSF‐arrested oocytes should not depend on Sgo2‐Mad2. It was suggested that the main inhibitory mechanism impinging on separase in metaphase II is securin (even though securin knock‐out mice are fertile, and securin protein levels are much lower than in meiosis I (Wang et al, 2001; Marangos & Carroll, 2008; Nabti et al, 2008)). Independent of securin or cyclin B inhibiting separase in meiosis II, both are subjected to APC/C‐induced degradation; hence, upon prolonged CSF arrest with some APC/C activity, separase might become active over time. For this reason, keeping separase in check through APC/C inhibition as the only means to prevent precocious centromeric cohesin removal in CSF‐arrested oocytes seems risky. We hypothesize that there is either another unknown mechanism ensuring separase inhibition in CSF‐arrested oocytes which is independent of the APC/C, or that centromeric cohesin protection is still in place in meiosis II prior to anaphase II onset. As long as separase is tightly controlled throughout CSF arrest, protection of centromeric cohesin may not be necessary. But although Mps1‐dependent Sgo2 is not essential in meiosis II, our data do not provide evidence that there is no protection at all any more, as it may only become essential under conditions where separase control is impaired. We can speculate that other inhibitory mechanisms exist, such as additional inhibitors or activators of separase, or posttranslational modifications of Sgo2 or Rec8 itself that are required for cleavage of centromeric Rec8 in meiosis II. Due to the specificities of CSF arrest, additional layers of control that are still unknown may have evolved in oocytes to ensure timely activation of separase and/or accessibility of centromeric Rec8.
Independently of whether centromeric cohesin is still protected or not in meiosis II, we show here that the ability of oocytes to cleave centromeric Rec8 in meiosis II depends on the presence of separase cleavage activity not only in meiosis II, but also in meiosis I (Fig 7B). Based on our observation that sister kinetochores come apart already in anaphase I, when they are still attached to the same pole, we asked whether the decision to remove centromeric cohesin or not is taken already before entry into meiosis II. In other words, the chromosome itself is carrying the information of which “step” of step‐wise cohesin removal (from arms or the centromere region) has to be executed. We found that bivalents segregating in meiosis I are getting "prepared" for sister separation in meiosis II. This preparation corresponds to the visible kinetochore individualization we describe here, taking place in anaphase I. By inhibiting arm cohesin removal through conditional knock‐out of separase, we obtained metaphase II oocytes harboring bivalents that are attached with mono‐oriented sister kinetochores, such as in metaphase I. This is not surprising, because bivalents are held together by chiasmata which allow the establishment of tension‐bearing attachments on co‐oriented sister kinetochores and increase the likelihood of sister kinetochores being attached to the same pole (Herbert et al, 2015). Under these conditions, the SAC in meiosis II is satisfied and oocytes can be activated because, even though bivalents and not dyads are present, they are attached and under tension. Crucially though, bivalents with fused kinetochores separate into dyads whereas bivalents with individualized kinetochores separate into sister chromatids. Furthermore, the bivalents separated into dyads behave like in meiosis I—they individualize kinetochores, resulting in dyads such as usually observed upon exit from meiosis I. Our data are in agreement with a recent study, showing that bivalents transferred from meiosis I into meiosis II oocytes behaved as if they were in meiosis I and segregated into dyads (preprint: Ogushi et al, 2020).
We found that kinetochore individualization depends on cleavage activity of separase. What is the substrate of separase that is holding kinetochores together until arm cohesin has been removed in anaphase I? At this point, we ignore its identity, but one attractive candidate is a separate fraction of Rec8, which is distinct from Rec8 on arms and Rec8 that is protected by Sgo2 and holding sister chromatids together until anaphase II onset. Indeed, a third fraction of Rec8 has been proposed to exist in the centromere region and to confer loss of sister kinetochore co‐orientation and meiosis II cohesin protection upon cleavage by separase after meiosis I in a very recent study (preprint: Ogushi et al, 2020). According to our data, this unknown separase substrate does not seem to be protected by pericentromeric Sgo2, because loss of Sgo2 due to Mps1 inhibition does not lead to precocious sister kinetochore individualization and overexpression of Sgo2 does not prevent kinetochore individualization. How cleavage of this substrate is prohibited until late anaphase I when arm cohesin has been cleaved already is therefore a mystery. Loss of Sgo2 in Sgo2 knock‐out oocytes results in separation of sister chromatids instead of dyads in meiosis I (Llano et al, 2008), even though kinetochores should remain fused according to our hypothesis. Hence, kinetochore fusion cannot substitute for centromeric cohesin or centromeric cohesin protection to hold sisters together and prevent their precocious separation in meiosis I.
In conclusion, the decision to protect or deprotect centromeric cohesin is not due to the fact that oocytes are in meiosis I or meiosis II, nor mono‐ or bipolar attachment to the spindle, but lies within the chromosome itself. Importantly, we do not exclude that inactivation of Sgo2, or co‐localization with I2PP2A/Set (Chambon et al, 2013b) contributes to cleavage of centromeric Rec8 in meiosis II, but we propose that the key event that determines whether centromeric cohesin can be cleaved or not is sister kinetochore individualization in meiosis I. These processes cannot be studied in mitotic cells, because even though Rec8 forms cohesive complexes when co‐expressed with Stag3, these meiotic cohesin complexes are still removed by the mitotic prophase pathway and protected by Sgo1 (Wolf et al, 2018), which in mammals is a different protein and requires Bub1 kinase for localization, unlike Sgo2 (Marston & Wassmann, 2017). Hence, also in the future, elaborate and technically challenging experiments in meiocytes will be required to gain further insights into the underlying molecular processes for cohesin protection and deprotection in meiosis.
Materials and Methods
Animals
Mice were maintained under temperature, humidity, and light‐controlled conditions under the authorization C75‐05‐13 at UMR7622 in a conventional mouse facility, with food and water access ad libitum. The project was submitted to ethical review according to the French law for animal experimentation (authorization B‐75‐1308). Adult CD‐1 mice were purchased (Janvier, France) and C57BL/6 mice of the indicated genotypes were bred in our animal facility. Mice were not involved in any procedures except for genotyping hormone injection for superovulation (see below) prior to being sacrificed by cervical dislocation between 8 and 16 weeks of age, to dissect ovaries and harvest GV stage oocytes.
Mouse oocyte culture
Oocytes were harvested in M2 medium (Merck Millipore, MR‐015P) supplied with 100 µg/ml dibutyryl cyclic AMP (dbcAMP; Sigma‐Aldrich, D0260) to keep them arrested in GV stage until release through several wash steps into M2 medium without dbcAmp. Only oocytes undergoing GVBD up to 90 min after release were used. For microinjections, oocytes were kept 2–3 h arrested before release. For longer incubations (more than 3 h) oocytes were put into M16 medium (Merck Millipore) containing dbcAmp, in CO2. All oocyte culture was done in medium drops covered with mineral oil (Sigma‐Aldrich, M8410). To obtain oocytes in anaphase II, in vitro cultured metaphase II oocytes were artificially activated. Metaphase II oocytes were placed into homemade M16 medium without CaCl2 for 30 min to 1 h prior to being placed into the activation medium (M16 medium without CaCl2 supplied with 100 mM Strontium chloride, Sigma‐Aldrich 204463), for 1 h (except for protein detection during early anaphase II in Fig 2D incubation time was reduced to 25 min), in a CO2 incubator. To obtain in vivo matured oocytes in metaphase II, adult female mice were hormonally stimulated with a first injection of 3.3 UI of pregnant mare serum gonadotropin (PMSG; Abbexa LTD, abx260389), followed by a second injection 48 h later with 5 UI of chorionic gonadotropin (HcG, Abbexa LTD, abx260092). 12–16 h after the second injection, oocytes containing cumulus cells were collected from oviducts and put into M2 medium. To collect oocytes, the medium was supplied with hyaluronidase at 0.1 mg/ml (Sigma‐Aldrich, H4272‐30MG) for 5–10 min and oocytes were harvested by mouth pipetting.
Drug treatment
All inhibitors were dissolved in DMSO (Sigma‐Aldrich, D2650) and added to M2 or M16 medium (Merck Millipore, MR‐10P). STLC (Sigma‐Aldrich, 164739) was used at 1.5 and 5 µM for meiosis I and meiosis II experiments, respectively, and Reversine (Cayman Chemical, 10004412) was used at 0.5 µM and additionally added to the oil used to cover the culture droplets.
In vitro transcription and microinjections
Microinjection pipettes were self‐made, using a magnetic puller (Narishige; PN‐31). Oocytes were manipulated on an inverted Nikon Eclipse Ti microscope, with a holding pipette (Eppendorf, 5178108.000), and injections were done using a FemtoJet Microinjector pump (Eppendorf) with continuous flow. mRNAs for injection into GV‐ or CSF‐arrested metaphase II oocytes were transcribed using the mMessage mMachine T3 Kit (Invitrogen, AM1348) and purified using the RNeasy Mini Kit (Qiagen, 74104). Plasmids to express separase have been described (Kudo et al, 2006). GFP‐Sgo2 was transcribed from the plasmid obtained by sub‐cloning mouse Sgo2 (gift from K. Nasmyth) into pRN3‐EGFP‐C1 vector (Hached et al, 2011).
Chromosome spreads and immunofluorescence
Oocytes were fixed at the indicated times after GVBD. To obtain metaphase II oocytes, oocytes were fixed 16–20 h after GVBD, or after in vivo maturation, where indicated. Prior to fixation, the Zona pellucida of oocytes was removed in successive baths in acidic Tyrode's solution (homemade) without mineral oil, and left to recover. For chromosome spreads, oocytes were fixed in 0.65–1% paraformaldehyde (Sigma‐Aldrich, 441244), 0.15% Triton‐X100 (Sigma‐Aldrich, T8787), and 3 mM DTT (Sigma‐Aldrich, D9779) (Chambon et al, 2013a). For whole‐mount staining of stable spindle microtubules oocytes were incubated 2–6 min in a cold treatment solution (80 mM PIPES [Euromedex, 1124], 1 mM MgCl2 [Euromedex, 2189‐C]) on top of an ice‐water bath. They were immediately fixed for 30 min in BRB80 buffer containing 0.3% Triton‐X100 and 1.9% formaldehyde (Sigma‐Aldrich, F1635). After several washes, oocytes were incubated over night at 4°C in a PBS‐BSA 3% solution supplied with 0.1% Triton‐X100 (Vallot et al, 2018).
The following primary antibodies were used at the indicated concentrations: human CREST serum auto‐immune antibody (Immunovision, HCT‐100, at 1:50), mouse monoclonal anti‐PP2A C subunit clone 1D6 Alexa Fluor 488 conjugate (Sigma‐Aldrich, 05‐421‐AF488, 1:50), rabbit polyclonal anti‐Sgo2 antibody (gift from José Luis Barbero, 1:50), rabbit polyclonal CenpA antibody (Cell Signaling, #2048S, 1:50), rabbit polyclonal anti Histone H3K9me3 Chip‐grade antibody (Abcam, ab 8898, 1:200), rabbit polyclonal anti‐Rec8 (gift from Scott Keeney, 1:50), mouse monoclonal anti‐α‐tubulin (DM1A) coupled to FITC (Sigma‐Aldrich, F2168, 1:100) and rabbit polyclonal anti‐Mps1 (gift from Hongtao Yu, 1:50).
Secondary antibodies were used at the following concentrations: donkey anti‐human CY3 (709‐166‐149, Jackson Immuno Research, 1:200), donkey anti‐human Alexa Fluor 488 (709‐546‐149, Jackson Immuno Research, 1:200), donkey anti‐mouse CY3 (715‐166‐151, Jackson Immuno Research, 1:200), donkey anti‐rabbit CY3 (715‐166‐152, Jackson Immuno Research, 1:200), donkey anti‐rabbit Alexa Fluor 488 (711‐546‐152, Jackson Immuno Research, 1:200), donkey anti‐mouse Alexa Fluor 488 (715‐546‐150, Jackson Immuno Research, 1:200), donkey anti‐human Alexa Fluor 647 (709‐606‐149, Jackson Immuno Research, 1:200), donkey anti‐rabbit Alexa Fluor 647 (711‐606‐152, Jackson Immuno Research, 1:200) and donkey anti‐mouse Alexa Fluor 647 (715‐606‐150, Jackson Immuno Research, 1:200).
To stain chromosomes, Hoechst 33342 (Invitrogen, H21492) at 50 µg/ml was used during secondary antibody incubation with AF1 Citifluor mounting medium (Biovalley, AF1‐100), or alternatively, IF Hardset + DAPI (VECTASHIELD) (Eurobio H‐1200) was used to mount the slides. Staining for chromosomes with propidium iodide (Sigma, P4864, at 1:200) was done 10 min before mounting the slides.
Image acquisition and image treatment
To image chromosome spreads, an inverted Zeiss Axiovert 200M microscope with a 100X/1.4 NA oil objective coupled to an EMCD camera was used. 6 z‐sections with 0.4 µm interval were taken. Live imaging was performed on the same microscope using a Plan APO (63×/1.4 NA) oil objective (Zeiss). Prior to acquisition, oocytes were preincubated in M2 media containing 1 µM SirDNA (far‐red DNA labeling probe, Spirochrome, SC007), for 1 h. Oocytes in chambers were prepared for imaging as described previously (Nikalayevich et al, 2018). Time lapse was set up for 10 h with 10 min intervals. 11 Z‐stacks (3 µm optical section spacing) were acquired in 491 nm channel for GFP and 640 nm channel for SirDNA and 1 z‐section for the DIC image. To image whole‐mount oocytes by immunofluorescence, we used an inverted Leica laser scanning confocal microscope TCS SP5 II with a 63× oil immersion objective (HCX Plan APO CS, NA 1.4). Scan speed was 400 Hz and Z‐section interval was 0.08 µm (Vallot et al, 2018). Pictures of cumulus were taken through a Zeiss Stereomicroscope Stemi2000 ocular and Asus ZenFone5Z camera (Sony IMX363 12MP). Image acquisitions were done with Metamorph or Leica Software. Images of whole‐mount oocytes or spreads were processed using Fiji or ImageJ software (NIH). No manipulations were performed other than brightness and contrast adjustments, that were applied with the same settings to images being compared. For artificial 3D‐rendering in Fig EV2 Arivis Vision 4‐D software was used (isosurface setting).
Quantifications and statistical analysis
Quantifications were done using ImageJ. The mean fluorescence intensities were measured at centromeres on sum‐projected images (by drawing a box around centromeres, as depicted on the scheme next to each dot plot). The mean fluorescence intensities (Ifluor bg) were corrected to background (bg) and normalized to CREST signals as follows: Ifluor (normalized) = [Ifluor − Ifluor bg]/[Ifluor CREST − Ifluor CREST(bg)]. Data plots and statistical analysis were obtained with PRISM6 software. Quantification results were compared using nonparametric Mann–Whitney U‐test built into PRISM6. All experiments were performed at least in two experimentally independent repeats. Shown are results from all replicates performed.
Author contributions
Most experiments in Figs 1, 2, 6, and EV2, EV3, and EV5 were performed by YG, experiments in Fig 3 by LK, experiments in Fig 5 by SAT, and experiments in Fig 4 and EV1A by SAT and DC. WEY and EB performed the experiment in Figs 2B, EV1B, EV2C, and D. DC provided expert technical help throughout the project. Statistical analysis was done by LK, YG, and SAT, figures were prepared by YG, LK, SAT, and KW. Overall supervision, funding acquisition, and project administration were done by KW. The manuscript was written by KW with substantial input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We are grateful to Wolfgang Zachariae for discussion and to Marie‐Hélène Verlhac and M‐Emilie Terret for advice on oocyte culture; we thank Safia El Jailani, Camille Gauthier, and Alison Kem‐Seng for preliminary experiments during their undergraduate internships, Sophie Gournet for help with oocyte schemes, and members of the MOM group for advice and discussion. We furthermore thank Scott Keeney for Rec8 antibody, José Barberos for Sgo2 antibody, Jan van Deursen for the Bub1 kinase‐dead mouse strain, Kim Nasmyth for the separase conditional knock‐out mouse strain and mouse Sgo2 plasmid, and Hongtao Yu for Mps1 antibody. LK was the recipient of a PhD fellowship by FRM (Fondation de la Recherche Medicale, ECO 20170637505), WEY was supported by a postdoctoral fellowship by the Fondation ARC pour la Recherche sur le Cancer. The KW Lab obtained funding for this project by the ANR (ANR‐16‐CE92‐0007‐01, ANR‐19‐CE13‐0015), FRM (Equipe FRM DEQ 20160334921), through "Emergence" funding by Paris Sorbonne Université (OoCyclins) and core funding by the CNRS and Sorbonne Université.
The EMBO Journal (2021) 40: e106797.
See also V Mengoli et al (April 2021)
Data availability
This study includes no data deposited in external repositories.
References
- Arguello‐Miranda O, Zagoriy I, Mengoli V, Rojas J, Jonak K, Oz T, Graf P, Zachariae W (2017) Casein kinase 1 coordinates cohesin cleavage, gametogenesis, and exit from M phase in meiosis II. Dev Cell 40: 37–52 [DOI] [PubMed] [Google Scholar]
- Chambon JP, Hached K, Wassmann K (2013a) Chromosome spreads with centromere staining in mouse oocytes. Methods Mol Biol 957: 203–212 [DOI] [PubMed] [Google Scholar]
- Chambon JP, Touati AS, Berneau S, Hebras C, Groeme R, Cladière D, Dumollard R, McDougall A, Wassmann K (2013b) The PP2A inhibitor I2PP2A is essential for sister chromatid segregation in meiosis II. Curr Biol 23: 485–490 [DOI] [PubMed] [Google Scholar]
- Chang HY, Jennings PC, Stewart J, Verrills NM, Jones KT (2011) Essential role of protein phosphatase 2A in metaphase II arrest and activation of mouse eggs shown by okadaic acid, dominant negative protein phosphatase 2A, and FTY720. J Biol Chem 286: 14705–14712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang T, Duncan FE, Schindler K, Schultz RM, Lampson MA (2010) Evidence that weakened centromere cohesion is a leading cause of age‐related aneuploidy in oocytes. Curr Biol 20: 1522–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clift D, Marston AL (2011) The role of shugoshin in meiotic chromosome segregation. Cytogenet Genome Res 133: 234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duro E, Marston AL (2015) From equator to pole: splitting chromosomes in mitosis and meiosis. Genes Dev 29: 109–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yakoubi W, Buffin E, Cladiere D, Gryaznova Y, Berenguer I, Touati SA, Gomez R, Suja JA, van Deursen JM, Wassmann K (2017) Mps1 kinase‐dependent Sgo2 centromere localisation mediates cohesin protection in mouse oocyte meiosis I. Nat Commun 8: 694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez R, Valdeolmillos A, Parra MT, Viera A, Carreiro C, Roncal F, Rufas JS, Barbero JL, Suja JA (2007) Mammalian SGO2 appears at the inner centromere domain and redistributes depending on tension across centromeres during meiosis II and mitosis. EMBO Rep 8: 173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hached K, Xie SZ, Buffin E, Cladiere D, Rachez C, Sacras M, Sorger PK, Wassmann K (2011) Mps1 at kinetochores is essential for female mouse meiosis I. Development 138: 2261–2271 [DOI] [PubMed] [Google Scholar]
- Hellmuth S, Gomez HL, Pendas AM, Stemmann O (2020) Securin‐independent regulation of Separase by checkpoint‐induced shugoshin‐MAD2. Nature 580: 536–541 [DOI] [PubMed] [Google Scholar]
- Hellmuth S, Stemmann O (2020) Separase‐triggered apoptosis enforces minimal length of mitosis. Nature 580: 542–547 [DOI] [PubMed] [Google Scholar]
- Herbert M, Kalleas D, Cooney D, Lamb M, Lister L (2015) Meiosis and maternal aging: insights from aneuploid oocytes and trisomy births. Cold Spring Harb Perspect Biol 7: a017970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro T, Tanaka K, Sakuno T, Watanabe Y (2010) Shugoshin‐PP2A counteracts casein‐kinase‐1‐dependent cleavage of Rec8 by Separase. Nat Cell Biol 12: 500–506 [DOI] [PubMed] [Google Scholar]
- Jonak K, Zagoriy I, Oz T, Graf P, Rojas J, Mengoli V, Zachariae W (2017) APC/C‐Cdc20 mediates deprotection of centromeric cohesin at meiosis II in yeast. Cell Cycle 16: 1145–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katis VL, Lipp JJ, Imre R, Bogdanova A, Okaz E, Habermann B, Mechtler K, Nasmyth K, Zachariae W (2010) Rec8 phosphorylation by casein kinase 1 and Cdc7‐Dbf4 kinase regulates cohesin cleavage by Separase during meiosis. Dev Cell 18: 397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating L, Touati SA, Wassmann K (2020) A PP2A‐B56‐centered view on metaphase‐to‐anaphase transition in mouse oocyte meiosis I. Cells 9: 390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ishiguro K, Nambu A, Akiyoshi B, Yokobayashi S, Kagami A, Ishiguro T, Pendas AM, Takeda N, Sakakibara Y et al (2015) Meikin is a conserved regulator of meiosis‐I‐specific kinetochore function. Nature 517: 466–471 [DOI] [PubMed] [Google Scholar]
- Kitajima TS, Ohsugi M, Ellenberg J (2011) Complete kinetochore tracking reveals error‐prone homologous chromosome biorientation in mammalian oocytes. Cell 146: 568–581 [DOI] [PubMed] [Google Scholar]
- Kudo NR, Wassmann K, Anger M, Schuh M, Wirth KG, Xu H, Helmhart W, Kudo H, McKay M, Maro B et al (2006) Resolution of chiasmata in oocytes requires Separase‐mediated proteolysis. Cell 126: 135–146 [DOI] [PubMed] [Google Scholar]
- Lee J, Okada K, Ogushi S, Miyano T, Miyake M, Yamashita M (2006) Loss of Rec8 from chromosome arm and centromere region is required for homologous chromosome separation and sister chromatid separation, respectively, in mammalian meiosis. Cell Cycle 5: 1448–1455 [DOI] [PubMed] [Google Scholar]
- Lee J, Kitajima TS, Tanno Y, Yoshida K, Morita T, Miyano T, Miyake M, Watanabe Y (2008) Unified mode of centromeric protection by shugoshin in mammalian oocytes and somatic cells. Nat Cell Biol 10: 42–52 [DOI] [PubMed] [Google Scholar]
- Lister LM, Kouznetsova A, Hyslop LA, Kalleas D, Pace SL, Barel JC, Nathan A, Floros V, Adelfalk C, Watanabe Y et al (2010) Age‐related meiotic segregation errors in Mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr Biol 20: 1511–1521 [DOI] [PubMed] [Google Scholar]
- Llano E, Gomez R, Gutierrez‐Caballero C, Herran Y, Sanchez‐Martin M, Vazquez‐Quinones L, Hernandez T, de Alava E, Cuadrado A, Barbero JL et al (2008) Shugoshin‐2 is essential for the completion of meiosis but not for mitotic cell division in mice. Genes Dev 22: 2400–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailhes JB, Hilliard C, Fuseler JW, London SN (2003) Okadaic acid, an inhibitor of protein phosphatase 1 and 2A, induces premature separation of sister chromatids during meiosis I and aneuploidy in mouse oocytes in vitro . Chromosome Res 11: 619–631 [DOI] [PubMed] [Google Scholar]
- Marangos P, Carroll J (2008) Securin regulates entry into M‐phase by modulating the stability of cyclin B. Nat Cell Biol 10: 445–451 [DOI] [PubMed] [Google Scholar]
- Marston AL, Wassmann K (2017) Multiple duties for spindle assembly checkpoint kinases in meiosis. Front Cell Dev Biol 5: 109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengoli V, Jonak K, Lyzak O, Lamb M, Lister LM, Lodge C, Rojas J, Zagoriy I, Herbert M, Zachariae W (2021) Deprotection of centromeric cohesin at meiosis II requires APC/C activity but not kinetochore tension. EMBO J 40: e106812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabti I, Reis A, Levasseur M, Stemmann O, Jones KT (2008) Securin and not CDK1/cyclin B1 regulates sister chromatid disjunction during meiosis II in mouse eggs. Dev Biol 321: 379–386 [DOI] [PubMed] [Google Scholar]
- Nikalayevich E, Bouftas N, Wassmann K (2018) Detection of separase activity using a cleavage sensor in live mouse oocytes. Methods Mol Biol 1818: 99–112 [DOI] [PubMed] [Google Scholar]
- Ogushi S, Rattani A, Godwin J, Metson J, Schermelleh L, Nasmyth K (2020) Loss of sister kinetochore co‐orientation and peri‐centromeric cohesin protection after meiosis i depends on cleavage of centromeric REC8. bioRxiv 10.1101/2020.02.06.935171 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M, Siomos MF, Nasmyth K (2003) Un menage a quatre: the molecular biology of chromosome segregation in meiosis. Cell 112: 423–440 [DOI] [PubMed] [Google Scholar]
- Rattani A, Wolna M, Ploquin M, Helmhart W, Morrone S, Mayer B, Godwin J, Xu W, Stemmann O, Pendas A et al (2013) Sgol2 provides a regulatory platform that coordinates essential cell cycle processes during meiosis I in oocytes. eLife 2: e01133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattani A, Ballesteros Mejia R, Roberts K, Roig MB, Godwin J, Hopkins M, Eguren M, Sanchez‐Pulido L, Okaz E, Ogushi S et al (2017) APC/C(Cdh1) enables removal of shugoshin‐2 from the arms of bivalent chromosomes by moderating cyclin‐dependent kinase activity. Curr Biol 27: 1462–1476.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke RM, Jeganathan KB, Malureanu L, Harrison AM, van Deursen JM (2012) Bub1 kinase activity drives error correction and mitotic checkpoint control but not tumor suppression. J Cell Biol 199: 931–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers E, Bishop JD, Waddle JA, Schumacher JM, Lin R (2002) The aurora kinase AIR‐2 functions in the release of chromosome cohesion in Caenorhabditis elegans meiosis. J Cell Biol 157: 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpf C, Cipak L, Dudas A, Benko Z, Pozgajova M, Riedel CG, Ammerer G, Mechtler K, Gregan J (2010) Casein kinase 1 is required for efficient removal of Rec8 during meiosis I. Cell Cycle 9: 2657–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Rauh NR, Nigg EA, Mayer TU (2006) Cytostatic factor: an activity that puts the cell cycle on hold. J Cell Sci 119: 1213–1218 [DOI] [PubMed] [Google Scholar]
- Stemmann O, Gorr IH, Boos D (2006) Anaphase topsy‐turvy: Cdk1 a securin, Separase a CKI. Cell Cycle 5: 11–13 [DOI] [PubMed] [Google Scholar]
- Vallot A, Leontiou I, Cladiere D, El Yakoubi W, Bolte S, Buffin E, Wassmann K (2018) Tension‐induced error correction and not kinetochore attachment status activates the SAC in an Aurora‐B/C‐dependent manner in oocytes. Curr Biol 28: 130–139.e3 [DOI] [PubMed] [Google Scholar]
- Wang Z, Yu R, Melmed S (2001) Mice lacking pituitary tumor transforming gene show testicular and splenic hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell cycle progression, and premature centromere division. Mol Endocrinol 15: 1870–1879 [DOI] [PubMed] [Google Scholar]
- Wassmann K (2013) Sister chromatid segregation in meiosis II: deprotection through phosphorylation. Cell Cycle 12: 1352–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf PG, Ramos AC, Kenzel J, Neumann B, Stemmann O (2018) Studying meiotic cohesin in somatic cells reveals that Rec8‐cohesin requires Stag3 and is regulated by Wapl and Sororin. J Cell Sci 131: jcs212100 [DOI] [PubMed] [Google Scholar]
- Wu JQ, Kornbluth S (2008) Across the meiotic divide ‐ CSF activity in the post‐Emi2/XErp1 era. J Cell Sci 121: 3509–3514 [DOI] [PubMed] [Google Scholar]
- Xu Z, Cetin B, Anger M, Cho US, Helmhart W, Nasmyth K, Xu W (2009) Structure and function of the PP2A‐shugoshin interaction. Mol Cell 35: 426–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinska AP, Holubcova Z, Blayney M, Elder K, Schuh M (2015) Sister kinetochore splitting and precocious disintegration of bivalents could explain the maternal age effect. eLife 4: e11389 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.