

Abstract
Protein termini are determinants of protein stability. Proteins bearing degradation signals, or degrons, at their amino‐ or carboxyl‐termini are eliminated by the N‐ or C‐degron pathways, respectively. We aimed to elucidate the function of C‐degron pathways and to unveil how normal proteomes are exempt from C‐degron pathway‐mediated destruction. Our data reveal that C‐degron pathways remove mislocalized cellular proteins and cleavage products of deubiquitinating enzymes. Furthermore, the C‐degron and N‐degron pathways cooperate in protein removal. Proteome analysis revealed a shortfall in normal proteins targeted by C‐degron pathways, but not of defective proteins, suggesting proteolysis‐based immunity as a constraint for protein evolution/selection. Our work highlights the importance of protein termini for protein quality surveillance, and the relationship between the functional proteome and protein degradation pathways.
Keywords: C‐degron pathway, CRL2 ubiquitin ligase, N‐degron pathway, protein spatial quality control, protein termini
Subject Categories: Post-translational Modifications, Proteolysis & Proteomics
Proteome‐wide analyses suggest functions of C‐terminal degradation signals in protein quality surveillance as well as interplay with N‐degron‐dependent mechanisms.

Introduction
Faithful interpretation of the genome into the proteome is essential for life, yet proteome fidelity is constantly challenged by genetic mutations, mistranscription and mistranslation, faulty segregation, erroneous folding, and post‐translational oxidative damage (Harper & Bennett, 2016; Hipp et al, 2019). For instance, the decoding of diverse sorting signals to route proteins to different cellular compartments involves highly complex processes, which entails a risk of mistargeting (Wang & Li, 2014; Vitali et al, 2018). The failure rates of protein segregation are as high as ~ 10% for several membrane proteins (Kim et al, 2002; Rane et al, 2010). Accumulation of mislocalized or defective proteins is cytotoxic and has been implicated in various neurodegenerative disorders (Hetz & Mollereau, 2014; Dubnikov et al, 2017). To defend the proteome, robust proteolysis‐mediated quality control systems have evolved to specifically remove abnormal proteins from cells (Chen et al, 2011; Pohl & Dikic, 2019).
Nevertheless, the aforementioned proteolytic mechanisms also represent a threat to the normal proteome. Coexistence of these proteolytic systems and the normal proteome imposes an evolutionary pressure on both. An effective balance must be achieved between the proteolytic systems having to effectively and efficiently eliminate only unwanted proteins but leave necessary proteins intact with the need for the functional proteome to evolve evasive mechanisms to prevent proteolytic destruction. A deeper understanding of the former process, i.e., the mechanism by which proteolytic systems recognize and clear aberrant proteins, is quickly emerging (Petroski & Deshaies, 2005; Williamson et al, 2013; Zheng & Shabek, 2017). However, our comprehension of the “compatibility” between protein surveillance systems and the functional proteome, and how functional proteins prevent proteolysis, remains limited.
Protein termini are determinants of protein degradation (Varshavsky, 2019). Proteins containing N‐terminal degradation signals (N‐degrons) are degraded by N‐degron pathways (Sriram et al, 2011; Varshavsky, 2011). N‐degrons are created by proteolytic cleavage or enzymatic N‐terminal‐modification of proteins (Hwang et al, 2010; Piatkov et al, 2014). We previously discovered a CRL2 ubiquitin ligase‐mediated C‐degron pathway, DesCEND (Destruction via C‐End Degron), that selectively eliminates proteins with deviant C‐termini, i.e., C‐degrons (Lin et al, 2015; Lin et al, 2018). In line with its function in protein quality surveillance, this C‐degron pathway eliminates truncated selenoproteins generated by translation miscoding and the N‐terminal fragment of USP1 from autocleavage. C‐degrons are portable short motifs and comprise a few conserved residues, with the remainder being rather flexible. Glycine is frequently the last residue of C‐degrons. An extreme C‐terminal position and structural accessibility are absolutely essential for C‐degrons to trigger degradation. CRL2 exploits interchangeable BC‐box proteins to target distinct C‐degrons, i.e., KLHDC2, KLHDC3, and APPBP2 recognize the diGly, R__G and _R_G_ C‐degrons, respectively (Koren et al, 2018; Lin et al, 2018; Rusnac et al, 2018). A detailed characterization of C‐degrons and additional functions of C‐degron pathways has yet to be achieved.
In this study, we developed a context‐independent random peptide platform to characterize the features of C‐degrons. We further demonstrate novel functions of C‐degron pathways in protein spatial quality control and clearing the products of deubiquitinating enzymes. We characterize the first example of a single protein simultaneously harboring both an N‐degron and a C‐degron, and we showcase two modes of collaborative action between N‐degron and C‐degron pathways in protein elimination. Lastly, we reveal that coronaviruses have adapted to resist proteolysis attack by C‐degron pathways and provide evidence to support that avoiding C‐degron‐mediated degradation shapes eukaryotic protein evolution/selection.
Results
C‐degron surrounding sequences affect the degree of degradation
The conventional approach to characterizing degrons is via identification of consensus residues from alignments of multiple degrons coupled with mutagenesis analysis. Nevertheless, it remains challenging to uncover complex or excluded features when the number of known degron sequences is limited. Furthermore, conclusions drawn from specific mutagenesis analyses can be template‐ and amino acid‐dependent, yet it is laborious to perform mutagenesis on multiple templates and to change each targeted residue to various different amino acids.
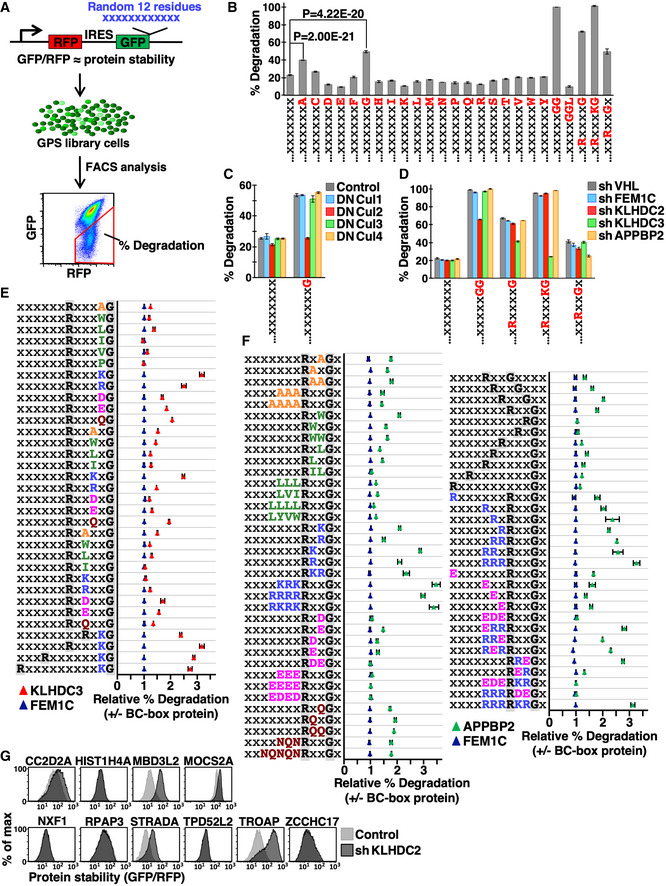
To evaluate unbiasedly the role of the extreme C‐termini of protein sequences for their stability, we adapted a “context‐independent” Global Protein Stability (GPS) random peptide platform. GPS is a fluorescence‐based reporter for measuring protein stability with single‐cell resolution (Yen et al, 2008). We fused a 12‐residue random peptide library at the C‐terminus of the fluorescence tag GFP, with another fluorescent tag (RFP) serving as a control to normalize protein synthesis (Fig 1A). We introduced our GPS random peptide library into the genome of human HEK293T cells via lentiviral transduction at a low multiplicity of infection (MOI) so that each GPS reporter cell only carried one unique GFP fusion peptide. The GFP/RFP ratio, as measured by fluorescence‐activated cell sorting (FACS), represented a proxy for the stability of GFP fusion proteins. Cells carrying specific peptides that promote GFP degradation display decreased GFP/RFP ratios, and the percentage of library cells with a reduced GFP/RFP ratio (% degradation, Fig 1A) reflects the percentage of random peptides in the library that can function as C‐degrons. Instead of examining loss‐of‐activity by mutating the targeted feature on a particular template sequence, this method compares activities with or without “adding‐in” a targeted feature under randomized sequence backgrounds, thereby enabling the effect of this targeted feature to be assessed across a heterogeneous pool. To avoid data biases due to uneven or insufficient variant representation, we constructed multiple independent peptide libraries for each examined C‐terminal residue or motif, and then analyzed % degradation for each library by multiple 105‐sized random samplings (Fig 1B–F). Since the sample space of all 12‐residue peptides (20X, where X is the number of random amino acids and ranges between 5 and 12) far exceeds the size of random sampling (105), the small standard deviations in Fig 1 further support that the features uncovered by our assay are truly independent of the identity of their neighboring residues.
Figure 1. Sequences nearby C‐degrons affect the potency of degradation.
-
AA schematic depiction of our GPS random peptide assay. The GPS reporter relies on the expression of two fluorescent proteins from a single promoter, enabled by an internal ribosome entry site (IRES). A 12‐residue peptide library was fused to the C‐terminus of GFP, whereas RFP serves as an internal control. The GFP/RFP ratio represents protein stability. The GPS peptide library was delivered into cells as a single copy so that each reporter cell expressed a unique GFP‐peptide fusion construct. The “% degradation”, i.e., the % of library cells displaying a decreased GFP/RFP ratio, reflects the % of peptides within the library that promote GFP degradation.
-
BThe % degradation of random peptide libraries terminating in the indicated amino acids or C‐terminal motifs labeled at bottom. X represents any amino acid residue. To illustrate the reliability of this approach, libraries terminating in each specific amino acid or motif were independently constructed three times and each independent library was analyzed by FACS in triplicate (three random samplings with 105 cells each time). Data are presented as mean ± standard deviation from the nine replicates described above. Since the sample space of all 12‐residue peptides far exceeds the size of random sampling, the probability of retrieving repeated peptides in random samples is extremely low. Thus, those random samples are expected to carry distinct peptides and should be considered as biological replicates. P‐values were calculated by unpaired t‐test.
-
CGPS reporter cells expressing peptide libraries ending with a random (X) or Gly (G) residue were treated with dominant‐negative (DN) cullins and analyzed. Data are presented as mean ± standard deviation from nine replicates.
-
DGPS reporter cells carrying indicated libraries were treated with shRNAs against various BC‐box proteins and analyzed. VHL and FEM1C serve as unrelated BC‐box protein controls. Data are presented as mean ± standard deviation from nine replicates.
-
E, FThe relative % degradation of indicated libraries in cells with or without assigned BC‐box proteins. A value of 1 indicates no difference. BC‐box proteins were depleted by shRNA‐mediated knockdown and FEM1C served as an irrelevant BC‐box protein control. Amino acids with different biophysical properties are marked with different colors, i.e., small aliphatic, hydrophobic, basic, acidic and amide residues are orange, green, blue, magenta, and crimson, respectively. Libraries terminating in each specific motif were independently constructed twice and each independent library was analyzed in triplicate. Data are presented as mean ± standard deviation from six replicates.
-
GGPS reporter cells expressing indicated diGly‐ending full‐length human proteins were treated with or without an shRNA against KLHDC2 and analyzed by FACS.
Approximately 23% of peptides ending in random amino acids (i.e., X12) promoted GFP degradation (Fig 1B), highlighting the potency of protein C‐termini in controlling protein stability. To assess the role of the last C‐terminal residue for protein half‐lives, we constructed twenty GFP‐random peptide libraries, each terminating with a distinct amino acid. Consistent with our observations that C‐degrons frequently end with Gly and occasionally Ala (Fig EV1A; Lin et al, 2018), we found that random peptides ending in Gly or Ala were more likely to stimulate degradation, respectively (Fig 1B). Moreover, Gly‐dependent degradation was mediated by the CRL2 ubiquitin ligase, consistent with CRL2 playing a dominant role in C‐degron pathways (Fig 1C; Lin et al, 2015). The KLHDC2, KLHDC3, and APPBP2 substrates we identified contain a conserved C‐terminal diGly, R__G or _R_G_ motif, respectively (Fig EV1A). Next, we examined if the diGly, R__G and _R_G_ motifs are sufficient to predict corresponding BC‐box protein‐mediated degradation. As expected, random peptides terminating in these motifs significantly stimulated degradation (Fig 1B right), and the respective degradation was inhibited only by knocking down their corresponding BC‐box proteins (Fig 1D). More than 95% of diGly‐ending random peptides stimulated degradation, whereas capping diGly with Leu blocked degradation (Fig 1B), suggesting that exposure of the terminal diGly motif alone is typically adequate for KLHDC2 degron recognition. We were unable to completely abolish KLHDC2 function by shRNA‐mediated knockdown (Fig 1D) because KLHDC2 binds diGly degrons with exceptionally high affinities (~ nM) (Rusnac et al, 2018). The upstream sequences of diGly peptides that promoted degradation (as recovered by single‐cell sorting) are rather diverse. These flanking sequences do not define but rather tune the potency of degradation (Table EV1).
Figure EV1. The extreme C‐termini of proteins affect protein stability.

- The last 10 residues of KLHDC2, KLHDC3, and APPBP2 substrates identified from a CRL2 substrate screen (Lin et al, 2018). The critical diGly, R__G and _R_G motifs demonstrated by mutagenesis are indicated. The minimal lengths of some degrons were mapped and are underlined.
- GPS reporter cells expressing human proteins with a C‐terminal R__KG motif were treated with or without an shRNA against KLHDC3 and analyzed.
- PCA on the amino acid frequency of the last 50 residues of normal human proteins. The terminal (−1) and the final 10 residues (−2 to −10) are labeled red and blue, respectively, with the remainder marked yellow.
- The relative frequency of Gly and diGly in the final 50 residues of functional and aberrant human proteins. The human protein sequences analyzed were downloaded from the Uniprot database in (A) and (B).
- The relative frequency of diGly at the C‐termini of indicated proteomes.
- Comparisons of protein stability among wild‐type (original) and mutant proteins (indicated at right). The original extreme C‐terminal residue of each protein is indicated in parentheses.
The R__G and _R_G_ motifs alone are insufficient to define KLHDC3‐ and APPBP2‐targeted degrons, respectively (Fig 1B and D). We applied an add‐in approach to determine their missing features. We appointed specified positions with specific amino acids, with the rest of the positions remaining indiscriminate, and then compared % degradation in cells with or without assigned BC‐box proteins (Fig 1E and F). For KLHDC3 degrons, the residue directly upstream of Gly−1 (i.e., the −2 position) is crucial. It has to be either charged or polar, with positively charged amino acids being particularly appropriate (Fig 1E). All random peptides terminating with the R___KG motif promoted degradation (Fig 1B) and the R__KG motif‐stimulated degradation is KLHDC3‐mediated (Fig 1D). The spacing between Arg and Gly only mildly affected degradation (Fig 1E, bottom four rows). In contrast, Arg‐Gly spacing for APPBP2 degrons was rigorous, with two intervening residues being favored (Fig 1F, right panel, rows 4–11 from top). The number of residues downstream of Gly can range from one to three (Fig 1F, right panel, rows 2–4 from top). The electrical property near the Arg and Gly residues significantly influenced APPBP2‐dependent degradation, with a positive charge being advantageous but a negative charge being detrimental (Fig 1F, left panel, rows 6–21 from bottom). Consistently, Asp and Glu were absent from previously identified APPBP2 degrons (Fig EV1A). Taken together, KLHDC3 and APPBP2 degrons comprise a conserved R__G motif and the remaining residues are “comparatively” changeable. Thus, rather than having a strictly conserved sequence of residues, these adjacent variable residues can tune degradation. It would be difficult to uncover this variability by conventional sequence comparison of infrequently encountered substrate or by taking a mutagenesis approach that is template‐dependent, but it is readily apparent from GPS random peptide library assays.
Stochastic peptides terminated with diGly or R___KG effectively triggered KLHDC2‐ or KLHDC3‐mediated GFP degradation, respectively (Table EV1, Fig 1B and D). We wondered if functional human proteins bearing these C‐terminal motifs are KLHDC2 or KLHDC3 substrates. Surprisingly, only half of the diGly‐ending proteins we examined (5/10) are KLHDC2 substrates (Fig 1G). All three of the cytosolic R__KG‐ending proteins that we tested were not targeted by KLHDC3 (Fig EV1B). Our data suggest that for functional proteins, harboring C‐degrons does not guarantee degradation. Thus, mechanisms must exist to prevent such functional proteins from being eliminated by the C‐degron pathway, such as by structural masking of terminal diGly, as in ubiquitin and SUMO2 (Lin et al, 2018), or other scenarios that we discuss further below.
Gly/C‐degron shortfall is limited to functional eukaryotic proteomes
It has been shown that C‐degrons are depleted in functional eukaryotic proteomes (Koren et al, 2018), but whether this phenomenon is universal or specific to a subset of proteins remains unclear. To investigate the effect of the C‐degron pathway on the protein C‐terminome, we performed principle component analysis (PCA) on the amino acid frequency of the final 50 residues of all functional human proteins. We found that amino acid usage of the last five residues deviated from the pattern for the other residues, with the terminal residue (−1) exhibiting the most dramatic difference (Figs 2A and EV1C). Gly is the least preferred amino acid, and Ala is the third least preferred (Table EV2). Intriguingly, this Gly deficit is limited to the last residue of normal proteins conserved across eukaryotic proteomes, but does not occur in abnormal proteins produced from translational frameshift or readthrough errors, NMD (nonsense‐mediated mRNA decay) transcripts, or small peptides encoded by upstream open reading frames (uORFs) (Figs 2B top, C, and EV1D left). The pattern for the diGly motif (the KLHDC2 degron) is similar, but is more exaggerated (Figs 2B bottom, and EV1D right, E). We did not perform the same analysis for the R__G and _R_G_ motifs because the KLHDC3 and APPBP2 degrons are more complex and their ability to stimulate degradation of functional proteins is low (Figs 1E–G and EV1B).
Figure 2. Gly‐ and diGly‐ending proteins are scarce in functional proteomes.
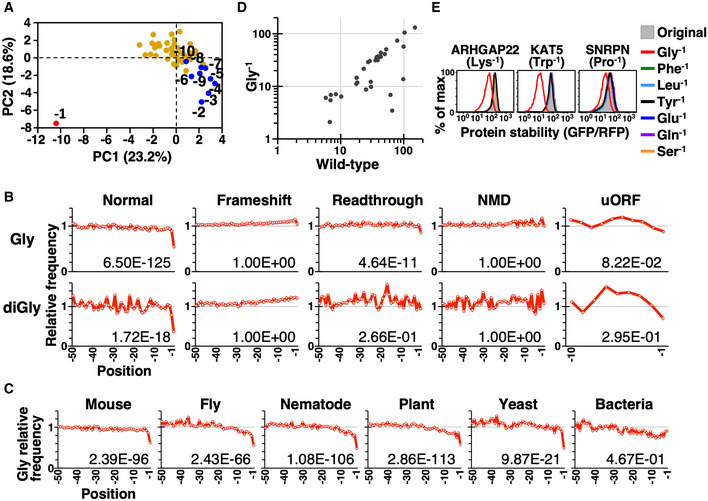
- PCA on the amino acid frequency of the last 50 residues of functional human proteins. Human protein coding sequences were downloaded from NCBI GeneBank. The terminal (−1) and the final 10 residues (−2 to −10) are labeled red and blue, respectively, with the remainder marked yellow.
- The relative frequency of Gly (top) and diGly (bottom) in the final 50 residues of functional and aberrant human proteins. We only analyzed the last 10 residues of peptides translated from uORFs due to their inherently smaller size. The number on each graph indicates the left‐tailed FDR (false discovery rate)‐adjusted P‐value of the extreme terminal (−1) residue. Analyses using human sequences downloaded from Uniprot database are shown in Fig EV1C and D.
- The relative frequency of Gly at the C‐termini of indicated proteomes, i.e., mouse (Mus musculus), fly (Drosophila melanogaster), nematode (Caenorhabditis elegans), plant (Arabidopsis thaliana), yeast (Saccharomyces cerevisiae), and bacteria (Escherichia coli).
- Stability (mean GFP/RFP) comparisons between the wild‐type protein and its mutant in which the terminal residue has been changed to Gly. Detailed information on tested proteins is presented in Table EV3.
- Comparisons of protein stability among wild‐type protein and the mutants indicated on the right.
The shortfall of terminal Gly or diGly in functional proteomes may be linked to avoidance of C‐degron‐mediated protein elimination. To test this hypothesis directly, we selected a panel of cytosolic human proteins and mutated their non‐Gly terminal residues into Gly and then examined their protein half‐lives. Mutation to Gly frequently reduced the stability of those proteins (18/36 proteins showed ≥ 15% reduction in stability), indicative of a direct causal relationship between having a Gly terminus and protein instability (Fig 2D, Table EV3). This relationship is Gly‐specific (Figs 2E and EV1F). These data collectively suggest that C‐degron pathway avoidance may be a driving force for proteome evolution. Thus, in order to prevent the cellular defense mechanism against erroneous proteins targeting functional proteins, strategies have evolved for the proteome to evade C‐degron pathways.
C‐degron pathway eliminates mislocalized cellular proteins
To identify the substrates of C‐degron pathways, we analyzed the C‐termini of human proteins localized in different cellular compartments (Fig 3A). Gly−1 is scare among cytosolic and nuclear proteins. Intriguingly, we noted a Gly−1 shortage only for membrane proteins having C‐termini facing the cytosol (inward), but not for secreted proteins or membrane proteins having their C‐termini facing the extracellular space or ER lumen (outward), suggesting that spatial quarantine protects proteins with C‐degrons. We employed model substrates to test if the distribution of a protein in cells dictates its accessibility to C‐degron pathways. Firefly luciferase (FLuc) and Gaussia luciferase (GLuc) are naturally cytosolic and secreted proteins, respectively (Fig EV2A). We compared their activities with or without fusion with a diGly degron. Although the difference in luciferase activity for FLuc with or without the diGly degron is > 480‐fold (mean ± standard deviation; 1.18E + 04 ± 1.38E + 03 versus 5.69E + 06 ± (< 0.01)), it is only two‐fold for GLuc (1.49E + 07 ± 1.73E + 05 versus 3.00E + 07 ± (< 0.01)), supporting the notion of localization‐restricted degradation. Next, we selected a set of multi‐pass transmembrane proteins that are not native KLHDC2 substrates to test the impact of substrate C‐terminal topology for C‐degron pathway‐mediated degradation (Fig EV2B and C). N‐terminal GFP tagging does not affect membrane anchoring of these transmembrane proteins (Fig EV2D). Consistently, we found that C‐terminal diGly degron‐tagging only triggered the destruction of membrane proteins having inward‐facing but not outward‐facing C‐termini (Fig 3B and C), and this degradation required the activity of p97/VCP ATPase (Fig 3D; Meyer et al, 2012).
Figure 3. The subcellular distribution of a protein dictates its accessibility to the C‐degron pathway.
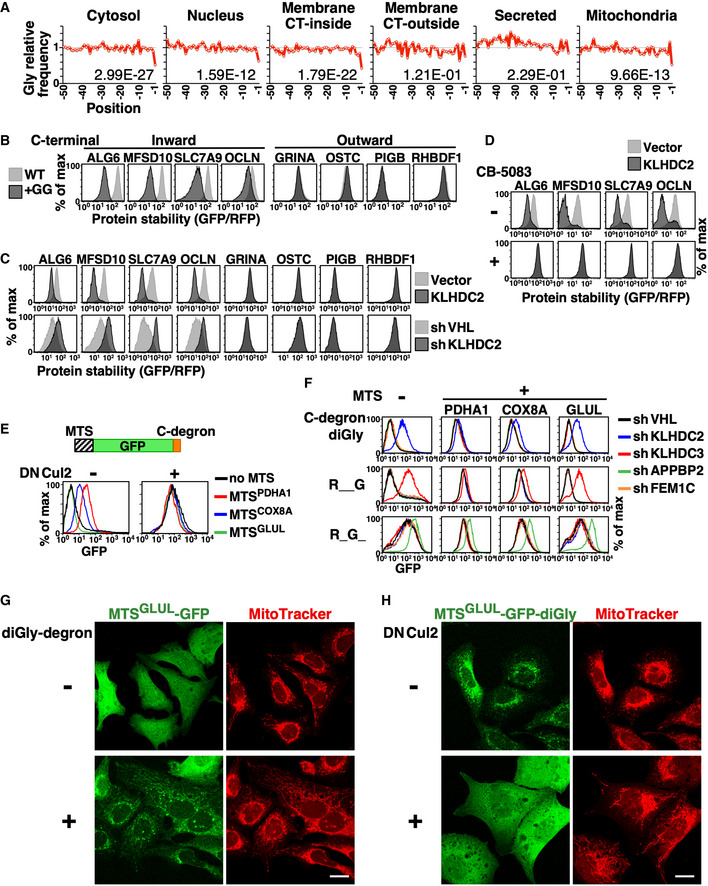
- The relative frequency of Gly in the final 50 residues of functional human proteins with different subcellular localizations. The number on each graph denotes the left‐tailed P‐value of the −1 residue. CT‐inside and CT‐outside represent membrane proteins with their C‐termini facing the cytosol or extracellular space, respectively.
- Comparison of stabilities between wild‐type and diGly degron‐tagged membrane proteins. The C‐terminal topology of tested proteins is indicated at top.
- GPS reporter cells expressing indicated membrane proteins with a C‐terminal diGly degron were treated with or without KLHDC2 overexpression or knockdown and then analyzed.
- Stability analysis of C‐terminal diGly degron‐tagged membrane proteins with or without KLHDC2 overexpression and CB‐5083 treatment (to inhibit p97/VCP).
- Abundance analysis of GFP‐diGly degron fusion proteins with or without N‐terminal tagging with the MTSs of PDHA1, COX8A, or GLUL, and with (+) or without (−) blockage of C‐degron pathways by dominant‐negative Cul2 (DNCul2).
- Abundance comparison of MTS‐GFP‐degron fusion proteins in cells treated with shRNAs against various BC‐box proteins. The MTS and C‐degrons used are labeled at top and left, respectively. The MTS and C‐degron sequences are provided in the Materials and Methods.
- Confocal images of U2OS cells expressing MTSGLUL‐GFP proteins with or without a C‐terminal diGly degron. MitoTracker staining shows mitochondria. Scale bar = 20 µm. DAPI staining and merged images are shown in Appendix Fig S1.
- Confocal images of U2OS cells expressing MTSGLUL‐GFP‐diGly degron fusion proteins co‐stained with MitoTracker, and with or without DNCul2 treatment. Scale bar = 20 µm.
Figure EV2. Model substrates used in Fig 3 .
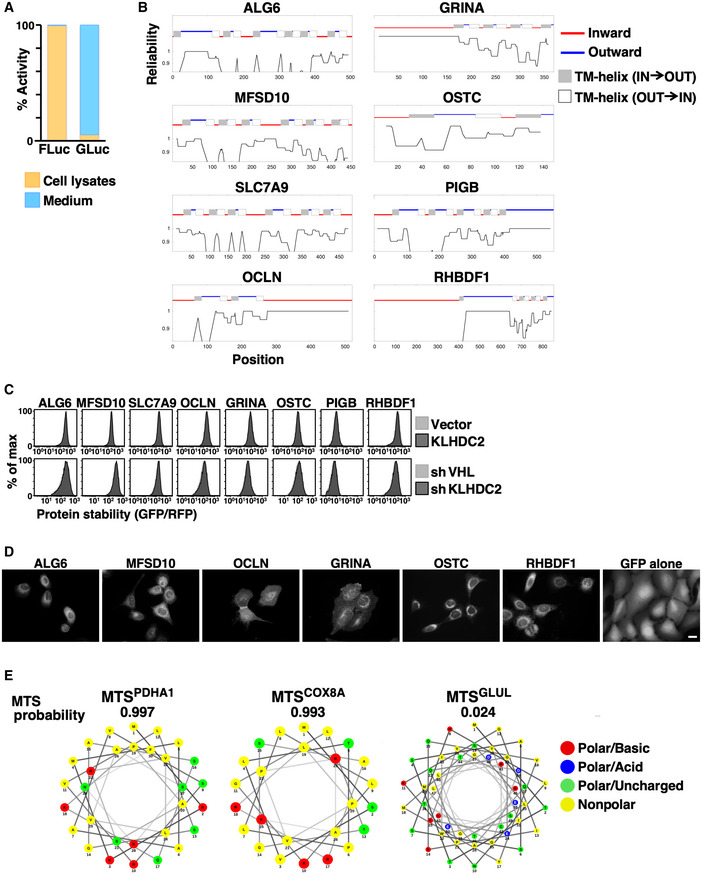
- The luciferase activity of cytosolic (cell lysate) and secreted (medium) FLuc and GLuc.
- The topology of indicated membrane proteins predicted by the TOPCONS web server (http://topcons.cbr.su.se/) (Tsirigos et al, 2015).
- Stability analysis of indicated membrane proteins with or without KLHDC2 overexpression or knockdown. The C‐terminal topology of proteins is indicated at top.
- Live‐cell images of U2OS cells stably expressing GFP or N‐terminal GFP‐tagged membrane proteins. Scale bar = 20 µm.
- The MTS of PDHA1, COX8A, and GLUL was scored and analyzed using MitoFates (http://mitf.cbrc.jp/MitoFates/cgi‐bin/top.cgi) (Fukasawa et al, 2015) and NetWheels (http://lbqp.unb.br/NetWheels/) (preprint: Mól et al, 2018). The MTS is typically localized at protein N‐termini and is composed of an amphipathic helix with alternating hydrophobic and positively charged amino acids.
Rather surprisingly, we noted a Gly−1 scarcity for mitochondrial proteins (Fig 3A). Unlike secreted and membrane proteins that are transported by co‐translational targeting, mitochondrial proteins are synthesized in cytosol and then post‐translationally delivered to mitochondria. Therefore, we investigated if the C‐degron pathway clears mitochondrial proteins that erroneously remain in cytosol. Most proteins that are destined for mitochondria have a mitochondrial targeting sequence (MTS) at their N‐terminus (Pfanner & Geissler, 2001). We created model mitochondrial substrates by fusing the MTSs from PDHA1, COX8A, and GLUL proteins to the N‐terminus of a GFP‐degron fusion reporter (Fig 3E). The efficiency of these MTSs for mitochondrial targeting differ, with that of GLUL (MTSGLUL) being the weakest (Fig EV2E, Appendix Fig S1; Matthews et al, 2010). We then compared the abundance of these mitochondrial GFP‐degron fusion proteins in the presence or absence of functional C‐degron pathways (Fig 3E and F). Supporting the notion of localization‐selective degradation, the degree of difference was inversely correlated with the efficiency of the MTS tag, i.e., it was positively correlated with the amount of misplaced cytosolic protein. MTSGLUL‐GFP was localized everywhere in cells, whereas when we added a diGly degron it was only observed in mitochondria (Fig 3G, Appendix Fig S1). Blocking the C‐degron pathway rescued the cytosolic distribution of the MTSGLUL‐GFP‐diGly fusion proteins (Fig 3H).
To examine the function of the C‐degron pathway in protein spatial quality control, we inferred a panel of physiological secreted and mitochondrial substrates of C‐degron pathways based on their C‐terminal sequences and subcellular localizations (Fig 4A). Abolition of the predestined localization of those proteins, either by N‐terminal capping (Fig 4B–F top) or targeted peptide deletion (Fig 4F bottom, G), specifically stimulated C‐degron pathway‐dependent protein destruction. Changing the C‐termini of these proteins by either deletion or masking completely inhibited C‐degron pathway‐mediated degradation (Fig 4C). Although APPBP2 knockdown only modestly stabilized BMP5 and BMP7, this effect was not observed for their C‐degron mutants (∆G) (Fig 4C). In addition, BMP5 and BMP7, but not their C‐degron mutants, were significantly destabilized when the abundance of APPBP2 was elevated (Fig 4D). Moreover, signal peptide‐deleted mutant BMP5 and BMP7 (∆SP) accumulated in the cytosol when APPBP2 was inhibited (Fig 4G). Intracellular protein sorting is known to be error‐prone (Levine et al, 2004; Vitali et al, 2018). Thus, our findings suggest that C‐degron pathways potentially eradicate misplaced cellular proteins caused by failed protein targeting.
Figure 4. C‐degron pathways clear mislocalized cellular proteins.
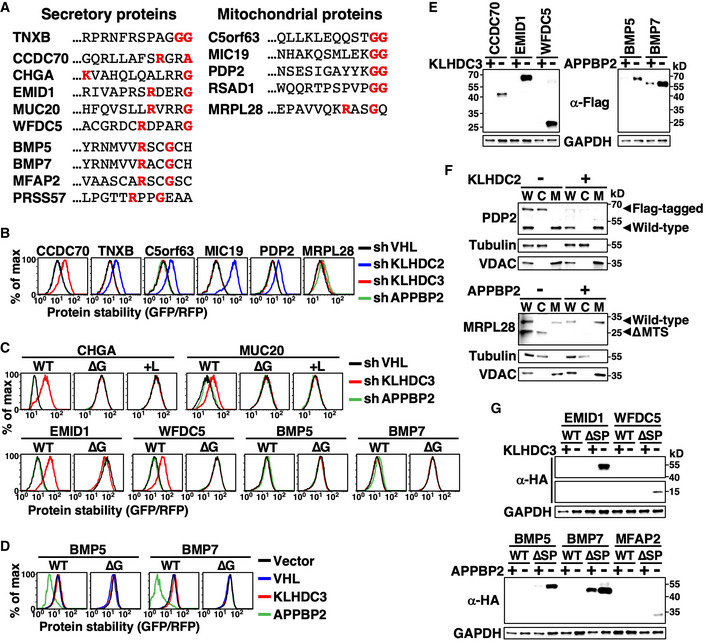
-
AThe extreme C‐terminal sequence of indicated secretory and mitochondrial proteins. The diGly, R__G, and R_G_ motifs are labeled red.
-
BStability analysis of indicated proteins in cells treated with shRNAs against various BC‐box proteins. Since GFP was tagged at the protein N‐terminus and signal peptide and MTSs need to be exposed to be functional, all tested proteins were mislocalized.
-
C, DProtein stability analyses of wild‐type (WT) and mutant proteins lacking their extreme terminal Gly (∆G) or having it masked by adding Leu (+L) in cells with or without BC‐box protein knockdown (C) or overexpression (D).
-
EWestern blot analysis of Flag‐tagged proteins in cells with or without KLHDC3 or APPBP2 expression. The N‐terminal Flag abolished protein targeting. GAPDH serves as a loading control.
-
FFractionation and Western blot analysis of wild‐type and Flag‐tagged PDP2 (top), and wild‐type and MTS‐deleted (∆MTS) MRPL28 (bottom). Flag‐PDP2 and ∆MTS MRPL28 were mislocalized. W, C, and M denote whole cell lysates, cytosol fraction and mitochondrial fraction, respectively. Tubulin and VDAC serve as fractionation quality controls.
-
GProtein abundance analysis of HA‐tagged wild‐type or mutant secretory proteins with their signal peptide deleted (∆SP) in cells with or without KLHDC3 or APPBP2 expression. The HA epitope was inserted immediately after the signal peptide (SP), which had no impact on protein secretion.
Source data are available online for this figure.
Dual‐end inspections safeguard the mitochondrial localization of MIC19
We searched for physiological targets of KLHDC2 by GST pull‐down and mass spectrometry‐based proteomics (Table 1, Table EV4). Consistent with the idea that C‐degron pathways clear mislocalized proteins, we isolated various full‐length diGly‐ending proteins localized at mitochondria or ER membrane, including MIC19.
Table 1.
KLHDC2‐binding proteins identified by GST pull‐down and mass spectrometry.
| Gene a | DiGly end | Protein name | Subcellular location b |
|---|---|---|---|
| AGPAT1 | Yes | 1‐acyl‐sn‐glycerol‐3‐phosphate acyltransferase alpha | Endoplasmic reticulum |
| CC2D2A | Yes | Coiled‐coil and C2 domain‐containing protein 2A | Cytoskeleton; cytosol |
| CHCHD3 | Yes | MICOS complex subunit MIC19 | Mitochondrion |
| DERL2 | Yes | Derlin‐2 | Endoplasmic reticulum membrane; endosome |
| DNAJB14 | Yes | DnaJ homolog subfamily B member 14 | Endoplasmic reticulum membrane; nucleus membrane |
| DNAJC18 | Yes | DnaJ homolog subfamily C member 18 | Membrane |
| FAU | No | Ubiquitin‐like protein FUBI | – |
| H4C1 | Yes | Histone H4 | Nucleus; chromosome |
| MOCS2A | Yes | Molybdopterin synthase sulfur carrier subunit small subunit | Cytoplasm, cytosol |
| NMD3 | Yes | 60S ribosomal export protein NMD3 | Nucleus; cytosol |
| PDGFC | Yes | Platelet‐derived growth factor C | Cytoplasm; secreted; nucleus; cytoplasmic granule; cell membrane |
| PDP2 | Yes | Pyruvate dehydrogenase phosphatase catalytic subunit 2 | Mitochondrion matrix |
| PTOV1 | Yes | Prostate tumor‐overexpressed gene 1 protein | Cell membrane; cytoplasm; nucleus |
| RSAD1 | Yes | Radical S‐adenosyl methionine domain‐containing protein 1 | Mitochondrion |
| SDE2 | No | Replication stress response regulator SDE2 | Nucleus |
| SEC61G | Yes | Protein transport protein Sec61 subunit gamma | Endoplasmic reticulum membrane |
| SELENOK | No | Selenoprotein K | Endoplasmic reticulum membrane; cell membrane |
| SELENOS | No | Selenoprotein S | Endoplasmic reticulum membrane; cytoplasm |
| SLC25A39 | Yes | Solute carrier family 25 member 39 | Mitochondrion inner membrane |
| STRADA | Yes | STE20‐related kinase adapter protein alpha | Cytosol; nucleus |
| USP1 | No | Ubiquitin carboxyl‐terminal hydrolase 1 | Nucleus |
Listed in alphabetical order.
Based on Uniprot GO (cellular component) or Human Protein Atlas annotations.
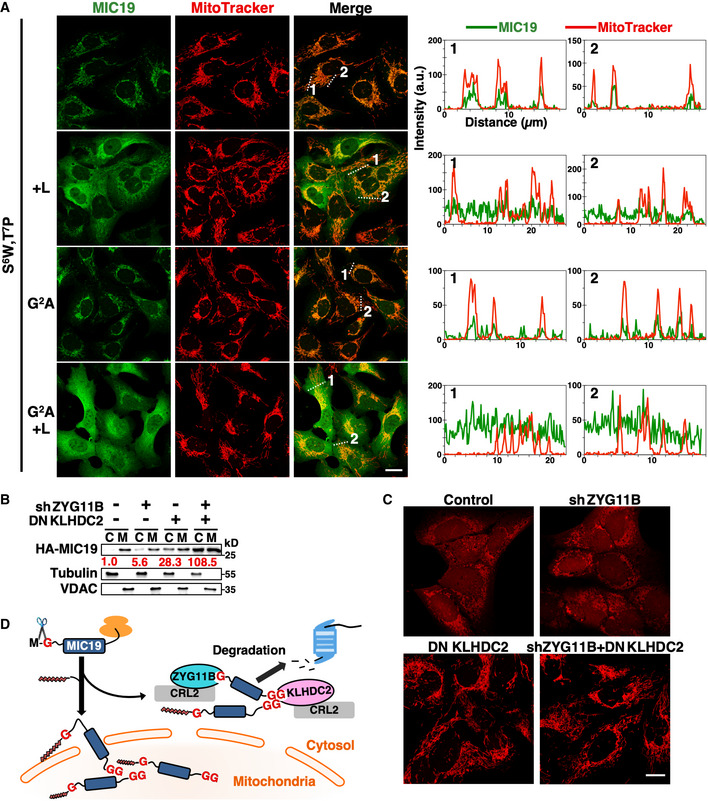
MIC19 is a mitochondrial inner membrane protein crucial for crista integrity and mitochondrial function (Darshi et al, 2011). Downregulation of MIC19 causes fragmented mitochondria, restricted oxygen consumption and glycolysis, and reduced growth rate. MIC19 orthologs are present throughout metazoans, and their C‐terminal diGly tails are highly conserved (Appendix Fig S2). Unlike canonical mitochondrial proteins that rely on MTSs, Gly2 myristoylation‐mediated membrane‐targeting and the CHCH domain contribute to the mitochondrial localization of MIC19 (Fig 5A) (Darshi et al, 2012; Ueda et al, 2019). Consistently, the localization of GFP N‐terminally tagged with the 24‐residue N‐terminal myristoylation motif of MIC19 (MIC19NT) resembled that of an ER membrane protein (Figs 5B and EV3A). We examined the role of the diGly/C‐degron pathway in localization of endogenous MIC19. MIC19 only resides in mitochondria in normal cells. Nevertheless, blocking the C‐degron pathway, either by inhibiting Cul2 or KLHDC2, resulted in trace amounts of MIC19 appearing in the cytosol (Figs 5C and EV3B), suggesting that mitochondrial transport of a proportion of endogenous MIC19 failed. Under normal conditions, that misplaced MIC19 is likely cleared by the C‐degron pathway. Facilitating MIC19 mistargeting by treatment with the myristoylation inhibitor IMP‐1088 further magnified the abundance of cytosolic MIC19 (Figs 5C and EV3B).
Figure 5. MIC19 is regulated by both N‐degron and C‐degron pathways.
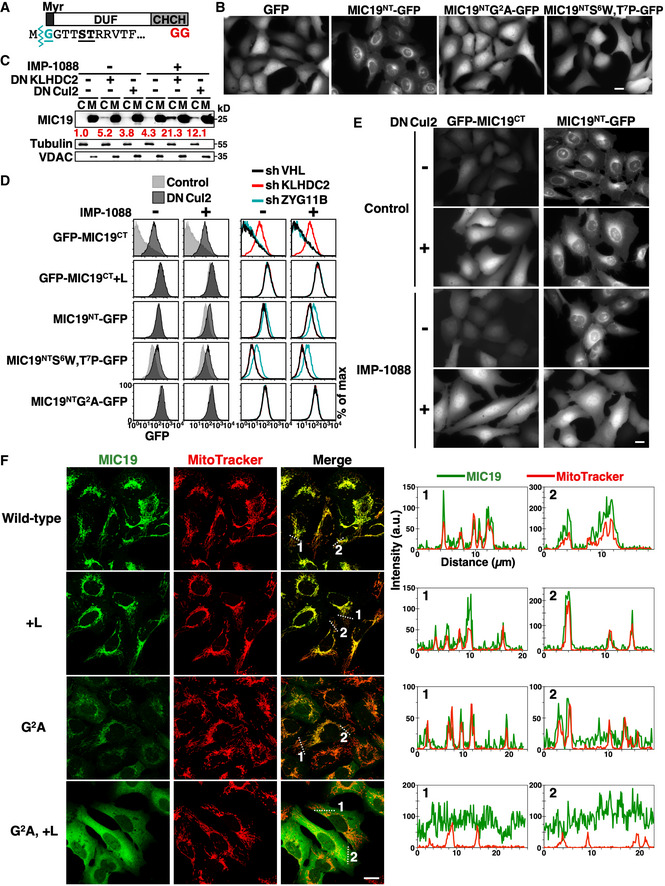
- Schematic diagram of the MIC19 protein. MIC19 contains an N‐terminal myristoylation motif followed by a DUF37 domain (domain of unknown function) and a CHCH (coiled‐coil helix‐coiled‐coil helix) domain and is terminated by a diGly motif. Myristic acid was co‐translationally attached to the N‐terminal Gly2 residue by N‐myristoyltransferases (NMTs) after removal of the initiating Met by methionine aminopeptidases. The N‐terminal sequence of MIC19 is indicated at bottom with the myristoylation site, Gly2, and residues crucial for myristoylation labeled turquoise or underlined, respectively.
- Live‐cell images of U2OS cells stably expressing GFP or GFP N‐terminally tagged with the wild‐type or mutant N‐terminal motif of MIC19 (a.a. 1–24). Scale bar = 20 µm.
- Fractionation and Western blot analysis of endogenous MIC19 in U2OS cells with or without DNKLHDC2, DNCul2, or IMP‐1088 treatments. IMP‐1088 inhibits NMTs. C and M denote cytosol and mitochondrial fractions, respectively. Tubulin and VDAC serve as fractionation quality controls. The relative abundance of cytosolic MIC19/tubulin was normalized to that of lane 1 and is indicated under each lane in red. Another replicate of this experiment is shown in Fig EV3B.
- Abundance comparison of GFP N‐ or C‐terminal tagged with the wild‐type or mutant N‐terminal (a.a. 1–24) or C‐terminal (a.a. 218–227) motif of MIC19 in cells with indicated treatments. Data from sh#1 is shown here.
- Live‐cell images of U2OS cells expressing indicated GFP fusion proteins with or without DNCul2 and IMP‐1088 treatments. The complete set of images containing degron mutant controls is shown in Appendix Fig S3. Scale bar = 20 µm.
- Representative confocal immunofluorescence images of HA‐tagged MIC19 variants with MitoTracker staining in U2OS cells. The fluorescence intensities along the dashed lines are shown as line profile graphs on the right. Scale bar = 20 µm.
Source data are available online for this figure.
Figure EV3. MIC19 bears both N‐ and C‐degrons.
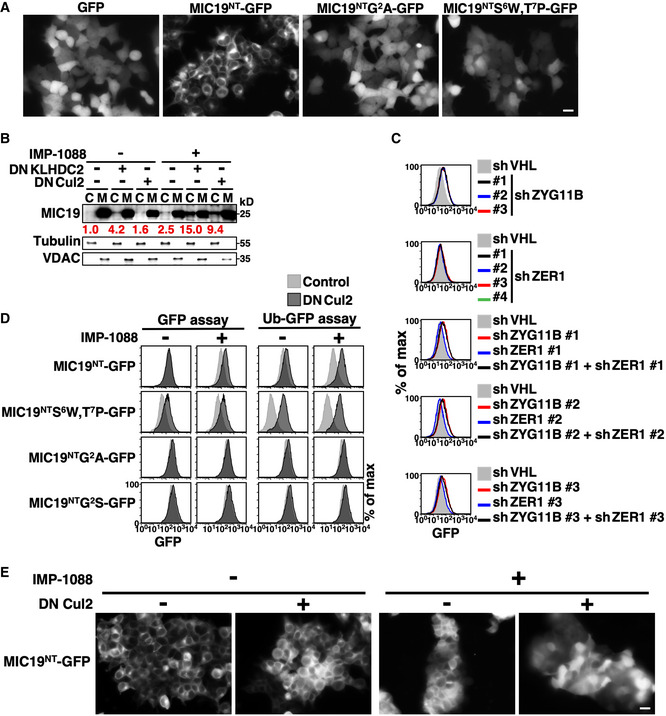
- Live‐cell images of 293T cells stably expressing GFP or GFP N‐terminally tagged with the wild‐type or mutant N‐terminal motif of MIC19 (aa. 1–24). Scale bar = 20 µm.
- Fractionation and Western blot analysis of endogenous MIC19 in U2OS cells with or without DNKLHDC2 or DNCul2 treatments. C and M denote cytosol and mitochondrial fractions, respectively. Tubulin and VDAC serve as fractionation quality controls. The relative abundance of cytosolic MIC19/tubulin was normalized to that of lane 1 and is indicated under each lane in red.
- Abundance analysis of MIC19NT‐GFP in cells treated with multiple shRNAs against ZYG11B, ZER1 or both. ZYG11B and ZER1 are BC‐box proteins involved in Gly/N‐degron recognition (Timms et al, 2019).
- Abundance analysis of MIC19NT‐GFP fusion proteins with (right) or without (left) a N‐terminal ubiquitin. The ubiquitin fusion technique is commonly used to characterize N‐degrons. Proteolytic cleavage of the ubiquitin moiety by Dubs resulted in exposure of MIC19NT.
- Live‐cell images of MIC19NT‐GFP in 293T cells with or without DNCul2 and IMP‐1088 treatments. IMP‐1088 treatment led to aggregations of 293T cells. Scale bar = 20 µm.
Source data are available online for this figure.
Intriguingly, besides having a conspicuous C‐terminal diGly, the N‐terminal myristoylation motif of MIC19 resembles a recently described CRL2‐mediated Gly/N‐degron (Timms et al, 2019; Fig 5A), raising the possibility that mitochondrial localization of MIC19 comes under “dual” surveillance by both the N‐degron and C‐degron pathways. Tagging either the N‐ or C‐terminal motif of MIC19 with GFP promoted CRL2‐mediated degradation, confirming that both “ends” of MIC19 are autonomous CRL2‐dependent degrons (Fig 5D left). However, these two degrons differ in two ways. Firstly, although the C‐degron is insensitive to IMP‐1088 treatment, the N‐degron is stimulated by IMP‐1088, suggesting that myristoylation of Gly2 abolishes N‐degron function. Consistently, we found that a myristoylation‐incapable mutant (MIC19NTS6W,T7P‐GFP) underwent degradation in the absence of the myristoylation inhibitor (Fig 5B and D). Secondly, the two degrons are recognized by distinct substrate receptors/BC‐box proteins, i.e., ZYG11B and KLHDC2 for the N‐degron and C‐degron, respectively (Figs 5D right, and EV3C). Consistent with the pivotal role of Gly in both degrons, mutating Gly2 or capping diGly prevented CRL2‐mediated degradation (Fig 5D). We noticed that N‐degron activity was enhanced when MIC19NT‐GFP was expressed using the ubiquitin fusion technique (Bachmair et al, 1986) (Fig EV3D). Importantly, localization of MIC19NT‐GFP depends on CRL2 activity. When we inhibited myristoylation, MIC19NT‐GFP localized at the ER membrane in wild‐type cells but became globally distributed upon blocking CRL2 function, suggesting that the CRL2‐mediated N‐degron pathway eliminated the misplaced cytosolic fraction of MIC19NT24‐GFP (Figs 5E and EV3E, Appendix Fig S3).
Next, we examined the function of the C‐degron in full‐length MIC19. We placed an HA epitope immediately after the myristoylation motif in order to analyze exogenously introduced MIC19 variants. The HA insertion did not affect mitochondrial delivery of MIC19 (Fig 5F, top row). We used a MIC19 antibody to detect exogenously expressed HA‐MIC19 because the signal from HA antibodies was too weak. We confirmed that levels of endogenous MIC19 were too low to be detected under this staining condition (Appendix Fig S4). Consistent with the idea that the rate of failed mitochondrial targeting is intrinsically low (Fig 5C), solely masking the C‐degron (+L) did not notably affect MIC19 localization (Fig 5F second row). Moreover, the limited amounts of cytosolic MIC19 may be removed by the N‐degron pathway. When we blocked both MIC19 myristoylation and N‐degron activity by means of Gly2 mutation, we observed contrasting distributions for MIC19 with or without a functional C‐degron. In the G2A or G2S background, MIC19 with a C‐degron was primarily localized in mitochondria, whereas MIC19 lacking a C‐degron was globally distributed (Figs 5F and EV4A). We obtained the same outcome by means of a biochemical fractionation assay (Fig EV4B left), suggesting that a C‐degron pathway mediates spatial quality control of MIC19. Mitochondrial enrichment of MIC19 was more apparent by image analysis because the volume of the cytosol is much greater than that of mitochondria (> 10‐fold) (Posakony et al, 1977), and imaging signals reflect protein “concentrations”, whereas fractionation assays measure “total protein abundance”.
Figure EV4. Mitochondrial localization of MIC19 is doubly assured by N‐degron and C‐degron pathways.
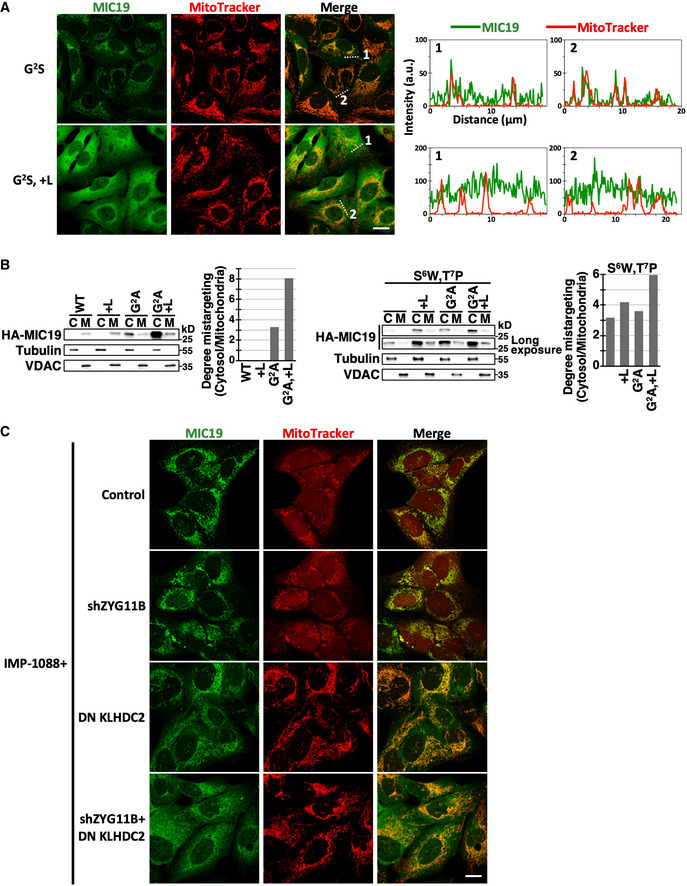
- Confocal immunofluorescence images of indicated HA‐tagged MIC19 variants and MitoTracker staining in U2OS cells. The fluorescence intensities along the dashed lines are shown as line profile graphs on the right. Scale bar = 20 µm.
- Fractionation analysis of HA‐tagged MIC19 variants in U2OS cells. The relative abundance of cytosolic to mitochondrial HA‐MIC19 is quantified and shown on right.
- Confocal immunofluorescence images of wild‐type HA‐MIC19 and MitoTracker staining in U2OS cells treated with IMP‐1088 and with or without inhibition of ZYG11B or KLHDC2. Scale bar = 20 µm.
We applied two approaches to simultaneously appraise the N‐degron and C‐degron of MIC19. Firstly, we exploited the S6W,T7P MIC19 mutation that suppresses myristoylation without compromising the activity of its N‐degron (Fig 5B and D). In the S6W,T7P background, MIC19 with a functional N‐degron and C‐degron remained predominantly concentrated in mitochondria (Fig 6A). Defective N‐ or C‐degrons alone only modestly diminished MIC19 localization, though the effect was more profound for C‐degron disruption. Importantly, we observed appreciably more misplaced MIC19 when both degrons were disrupted, as evidenced by both imaging analysis (Fig 6A) and fractionation assays (Fig EV4B right). Secondly, we impaired the N‐degron or C‐degron pathways in trans by inhibiting ZYG11B or KLHDC2, respectively, and we reached the same conclusion. Significantly more cytosolic MIC19 appeared when both BC‐box proteins were suppressed in IMP‐1088‐treated cells (Figs 6B and EV4C). Importantly, IMP‐1088 treatment caused fragmented mitochondria due to insufficient mitochondrial MIC19 and this mitochondrial abnormality was recovered by inhibiting N‐degron and C‐degron pathways‐mediated proteolysis (Figs 6C and EV4C). Overall, these data indicate that the two end degron pathways act cooperatively to safeguard proper localization of MIC19 (Fig 6D).
Figure 6. Mitochondrial localization of MIC19 is ensured by dual‐end inspections.
- Representative confocal immunofluorescence images of HA‐tagged MIC19 variants with MitoTracker staining in U2OS cells. All MIC19 variants examined carried the S6W,T7P mutation. The fluorescence intensities along the dashed lines are shown as line profile graphs on the right. Scale bar = 20 µm.
- Fractionation analysis of wild‐type HA‐MIC19 in U2OS cells treated with IMP‐1088 and with or without inhibition of ZYG11B or KLHDC2. The normalized relative abundance of cytosolic HA‐MIC19/tubulin is indicated under each lane in red.
- MitoTracker staining of IMP‐1088‐treated U2OS cells with or without inhibition of ZYG11B or KLHDC2. The complete set of images containing MIC19 localizations is shown in Fig EV4C. Scale bar = 20 µm.
- Proposed model. CRL2‐operated “double‐end” inspections safeguard the mitochondrial localization of MIC19.
Source data are available online for this figure.
The diGly/C‐degronKLHDC2 pathway eliminates cleavage products of deubiquitinating enzymes
Apart from diGly‐ending full‐length proteins, we also identified KLHDC2‐binding proteins that do not contain a connate diGly C‐terminal (Table 1, Table EV4). We previously found that the N‐terminal domain (NTD) of USP1 generated by autocleavage is removed by KLHDC2 (Lin et al, 2018). Comparably to USP1, the FAU, SDE2, and TUG proteins contain an ubiquitin‐like (UBL) domain with the characteristic C‐terminal diGly motif and all these proteins are processed by deubiquitinating enzymes (Dubs) (Olvera & Wool, 1993; Habtemichael et al, 2018; Thakran et al, 2018). Dubs‐mediated cleavage results in their UBL‐harboring NTD fragments ending with a diGly tail (Fig 7A; Olvera & Wool, 1993; Jo et al, 2016; Habtemichael et al, 2018). Therefore, we tested the hypothesis that the diGly/C‐degronKLHDC2 pathway eliminates Dubs cleavage products.
Figure 7. The diGly/C‐degron pathway eliminates cleavage products of deubiquitinating enzymes.
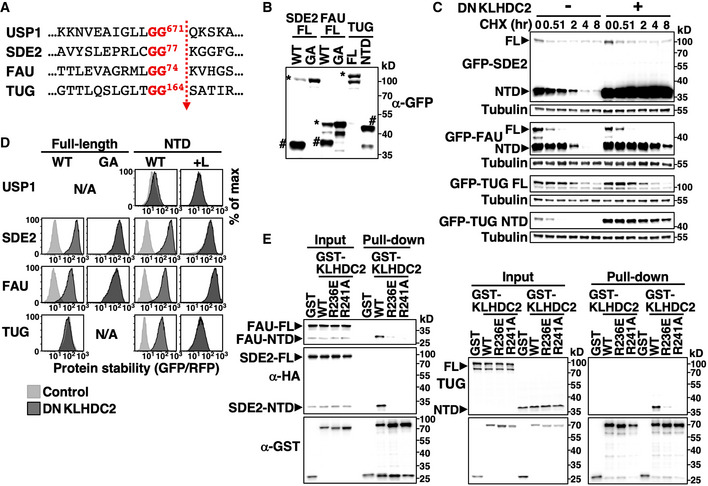
- The cleavage sequence of indicated proteins.
- Western blot analysis of HEK293T cells expressing GFP‐tagged wild‐type (WT) or an uncleavable mutant (diGly to GA) full‐length (FL) version or the N‐terminal domain (NTD) of indicated proteins. Full‐length and NTD proteins are marked by * or # on the blot, respectively.
- Cycloheximide (CHX)‐chase analysis of GFP‐tagged FL and NTD versions of indicated proteins.
- GPS analysis of wild‐type and mutant forms of FL or the NTD of indicated proteins with or without dominant‐negative KLHDC2 (DNKLHDC2) treatment. A C‐terminal Leu was added (+L) to NTDs to cap the diGly degron.
- GST pull‐down assay using cells expressing GST or GST‐tagged wild‐type or mutant KLHDC2 and HA‐tagged SDE2, FAU or TUG.
Source data are available online for this figure.
We detected both full‐length and NTDs of SDE2 and FAU when HEK293T cells expressed wild‐type but not a diGly mutant (GA) version of full‐length proteins, suggesting that SDE2 and FAU were indeed cleaved at the diGly motif by endogenous Dubs (Fig 7B). TUG‐NTD was not observed because TUG is processed by a muscle‐specific isoform of USP25 (isoform M) (Habtemichael et al, 2018). In support of our hypothesis, NTDs but not the full‐length forms of all three proteins are KLHDC2 substrates (Figs 7C and EV5A). Mutating the internal diGly of full‐length proteins or masking the terminal diGly of NTDs completely abrogated KLHDC2‐dependent degradation (Figs 7D and EV5B). Consistently, we observed that wild‐type KLHDC2, but not its mutants defective in diGly‐peptide binding (R236E; R241A) (Rusnac et al, 2018), physically associated with the NTDs of SDE2, FAU, and TUG but not the respective full‐length proteins (Fig 7E). Collectively, these data suggest that the C‐degronKLHDC2 pathway can eliminate diGly‐ending cleavage products of Dubs or other proteases.
Figure EV5. The KLHDC2/C‐degron pathway eliminates Dub products.
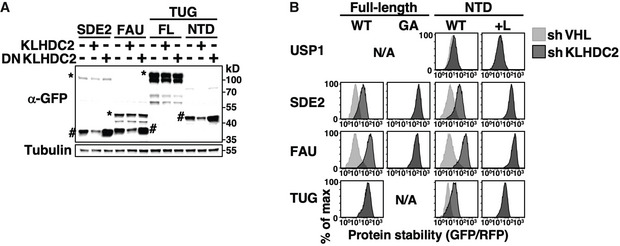
- Western blot analysis of HEK293T cells expressing GFP‐tagged full‐length SDE2, FAU or TUG constructs with or without overexpressing wild‐type or dominant‐negative KLHDC2 as indicated. Full‐length (FL) and the NTDs of proteins are indicated by * or # on the blots, respectively. Tubulin serves as a loading control.
- Stability analysis of wild‐type and mutant forms of FL or NTDs of indicated proteins with or without KLHDC2 knockdown.
Coronaviruses resist proteolytic attack from host C‐degron pathways by editing the surrounding sequence of diGly/C‐degrons
Coronavirus non‐structural proteins (NSPs) are synthesized as long polypeptide chains that are subsequently proteolytically processed into individual NSP proteins by the viral proteases PLpros/NSP3 and 3CLpro. PLpros cleaves at the NSP1‐2, 2‐3 and 3‐4 junctions following a diGly motif (Fig 8A) (Harcourt et al, 2004; Báez‐Santos et al, 2015). We wondered if PLpros‐processed NSP proteins are targeted by KLHDC2. Intriguingly, unlike human proteins cleaved by Dubs, diGly‐terminating NSP1 and NSP2 from SARS‐CoV (severe acute respiratory syndrome coronavirus) escaped the diGly/C‐degron pathway (Fig 8B). We characterized an inactivated NSP1 mutant (R124A, K125A) because wild‐type NSP1 expression is lethal to cells (Lokugamage et al, 2012; Tanaka et al, 2012).
Figure 8. Coronaviral diGly‐ending polypeptides resist C‐degron pathway‐mediated proteolysis.
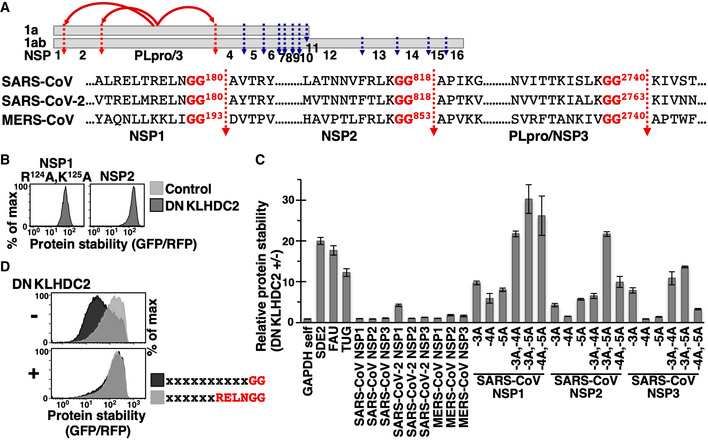
- Schematic depiction of the proteolytic process of coronaviral replicase protein 1a and 1ab by proteases PLpro/NSP3 (labeled red) and 3CLpro (labeled blue). PLpro/NSP3‐mediated cleavage generates NSP1, NSP2, and PLpro/NSP3 proteins ending in the diGly motif. The cleavage sequences of indicated viruses are shown.
- Stability analysis of FL SARS‐CoV NSP1 and NSP2 proteins with or without DNKLHDC2 treatment. We were unable to analyze NSP3 due to its large size (214 kDa).
- Relative stability analysis of GAPDH C‐terminally tagged with the last 12 residues of indicated proteins with or without DNKLHDC2 treatment by GPS assays. The −3, −4, or −5 residue of SARS‐CoV NSP proteins was mutated to Ala (A) as indicated. The experiment was done in triplicate using three independently prepared viruses to express DNKLHDC2. Data are presented as the mean value of GFP/RFP +/− standard deviation from triplicate experiments.
- Global protein stability profiling of GPS reporter cells expressing random peptide libraries ending with a diGly motif or the last six residues of SARS‐CoV NSP1 protein with or without DNKLHDC2 treatment.
To explain their immunity to the C‐degron pathway, we tested if the C‐terminal tails of NSP proteins include autonomous KLHDC2 degrons. In contrast to the 12‐residue C‐terminal motif of SDE2, FAU, or TUG, the C‐termini of coronavirus NSP proteins did not or only mildly promoted KLHDC2‐mediated degradation (Fig 8C), suggesting that the sequences surrounding coronavirus terminal diGly inactivate the KLHDC2 degrons. Consistent with that notion, the stability of GFP‐random peptide libraries ending with the final six residues of SARS‐CoV NSP1 was much greater than for those ending with only diGly, and that difference in stability was completely abolished when KLHDC2 function was blocked (Fig 8D). We found that KLHDC2 degron activity against SARS‐CoV NSPs could be drastically improved by changing as little as a single residue at the −3, −4, or −5 positions to Ala (Fig 8C). Collectively, our data demonstrate differential regulation of cellular and viral diGly‐terminating polypeptides by the diGly/C‐degron pathway and suggest that coronaviruses have adapted to resist proteolytic attack by host cells.
Discussion
Herein, we report physiological functions of C‐degron pathways and demonstrate the importance of protein “ends” in proteolysis‐mediated protein quality inspection. Furthermore, we establish links between the functional proteome and C‐degron pathways.
One selective pressure sculpting proteome evolution is the requirement that proteins have sufficient stability to perform their biological functions. Our results indicate that functional proteomes have evolved multiple tactics to cope with the threat of C‐degron pathway‐mediated proteolysis. Our proteome sequence analyses reveal striking Gly/C‐degron scarcity in functional eukaryotic proteomes but not in defective proteins produced from translation errors. Only ~ 50 full‐length proteins terminating with diGly occur in the human proteome. It is unclear why terminal Gly is depleted in the yeast proteome given that a CRL2 homolog does not exist in yeast. In line with our expectations, the shortfall in terminal Gly is restricted to intracellular proteins accessible to C‐degron pathways. Importantly, mutating the extreme C‐terminal residue of human proteins to Gly mostly triggered degradation, supporting a direct causal relationship between Gly ends and protein instability. In addition to excluding Gly/C‐degrons, proteins may possess particular folding properties to shield latent C‐degrons, such as those we have reported previously for ubiquitin and SUMO2 (Lin et al, 2018). Viral proteins are typically subjected to host protein degradation machineries. Interestingly, our data suggest that the sequences near to the diGly motifs of coronaviral diGly‐ending proteins prevent KLHDC2 attack. These results shed light on the protein quality control systems that support functional proteomes.
Inspired by the significant annotation of secreted and mitochondrial proteins harboring C‐degrons and identification of KLHDC2‐binding proteins localized at mitochondria or ER membrane, we have revealed the function of C‐degron pathways in protein spatial quality control. The targeting signal for protein transport into a given organelle is typically specified by general biophysical properties rather than particular amino acid sequences. For instance, the signal peptide comprises a stretch of primarily hydrophobic helix‐compatible residues, and the MTS forms an amphiphilic α‐helix with an alternating pattern of hydrophobic and positively charged amino acids (von Heijne, 1990). Due to their miscellaneous nature, the efficacy of targeting signals in different proteins varies substantially, with overall failure rates of intracellular protein segregation potentially being as high as a few percent (Kim et al, 2002). We have identified a panel of secreted and mitochondrial proteins that are eliminated by C‐degron pathways upon mislocalization. The C‐termini of aberrant proteins are expected to be mainly stochastic. Why C‐degron pathways target particular C‐terminal motifs and what the diGly, R__G and R_G_ motifs denote remain unclear. We have demonstrated that the “diGly‐targeting” C‐degronKLHDC2 pathway eliminates “diGly‐ending” products of Dubs. Classically, ubiquitin ligases and Dubs are antagonistic in editing protein ubiquitination. We have defined a counter‐intuitive “cooperative” relationship between CRL2KLHDC2 ubiquitin ligase and Dubs in protein removal, i.e., substrates are cleaved by Dubs to expose the diGly C‐degron that triggers KLHDC2‐mediated degradation. The C‐degronKLHDC2 pathway may also eliminate diGly‐terminated products of other proteases besides Dubs.
Our results reveal two types of cooperation between N‐degron and C‐degron pathways in protein elimination (Fig 9A). Firstly, for USP1, SDE2, and TUG, proteolytic cleavage concurrently creates a C‐degron on the N‐terminal fragment and a N‐degron on the C‐terminal fragment (Bogan et al, 2012; Piatkov et al, 2012; Lin et al, 2018; Rageul et al, 2019). The two end degron pathways work conjointly to clear both fragments. Secondly, MIC19 simultaneously possesses both an N‐degron and a C‐degron. These two degrons are normally inactive due to the protein’s localization, but they can be targeted when MIC19 is mislocalized. Coincidentally, both degrons are attacked by CRL2 ubiquitin ligases and Gly is the protagonist in both cases. The N‐degron is inhibited by Gly myristoylation.
Figure 9. Protein termini regulate protein stability.

- Two modes of collaboration between N‐ and C‐degron pathways in protein removal.
- Competition between Gly2 myristoylation and Gly/N‐degron pathway‐mediated degradation.
We noticed that the activity of the MIC19 N‐degron is relatively weak and wondered why MIC19 demands dual‐end‐mediated surveillance. N‐myristoylatransferase enzymes (NMTs) and CRL2ZYG11B compete for the Gly2 residue; the former contributes to MIC19 delivery into mitochondria, and the latter clears cytosolic unmyristoylated MIC19. To optimize mitochondrial entry of MIC19, the biosynthesis pathway, i.e., Gly myristoylation, should take precedence over the quality control pathway, i.e., N‐degron‐mediated proteolysis. The weaker N‐degron extends the time window for myristoylation and successful mitochondrial sorting, but entails potential liberation of MIC19 that has failed to myristoylate. Moreover, successfully myristoylated MIC19 may still not enter mitochondria, so the C‐degron pathway acts as a reinforcement to capture such aberrant MIC19. The competitive dominance of myristoylation can be explained by rapid Gly2 access of ribosome‐associated NMTs (Fig 9B left) (Deichaite et al, 1988; Giglione et al, 2015). Consistent with that notion, we observed enhanced N‐degron‐mediated degradation when we expressed MIC19NT‐GFP using the ubiquitin fusion technique that exposes the Gly residue away from the exit tunnel of nascent peptides on the ribosome (Fig 9B right). Analogous spatial segregation‐mediated prioritization has been proposed to illustrate the conflict for signal peptide binding between SRP (signal recognition particle) and the Bag6 cochaperone (Hessa et al, 2011). Another benefit of dual inspections may be provided by the dissimilar intracellular localizations of KLHDC2 and ZYG11B. Based on the Human Protein Atlas database, KLHDC2 mainly localizes to the nucleoplasm and nuclear membrane, whereas ZYG11B is predominately distributed at Golgi apparatus and intermediate filaments.
Protein termini are ideal for incorporating regulatory elements due to their particular characteristics. They are easy to access, less likely to perturb global protein folding, and mark the start and end of protein synthesis. Our study expands the known functionalities of protein termini to conferring protein fate, and uncovers the impacts of C‐degron pathways on both functional and defective proteomes.
Materials and Methods
Cell culture
HEK293T (ATCC® CRL‐3216) and U2OS (ATCC® HTB‐96) cells were cultured in Dulbecco’s Modified Eagle’s Medium and McCoy’s 5A medium (Gibco), respectively, supplemented with 10% fetal bovine serum (Hyclone), 100 μg/ml of streptomycin, and 100 U/ml of penicillin (Gibco) at 37°C in a 6% CO2 atmosphere. Experiments were performed in HEK293T cells unless otherwise indicated.
To block proteasome function, cells were treated with 1 μM bortezomib (BioVision) for 6 h. To block CRL2 function, cells were either treated with 1 μM MLN4924 (Active Biochem) for 6 h or infected with viruses carrying DNCul2 (multiplicity of infection MOI ~ 10) for 40 h. To inhibit KLHDC2, cells were infected with viruses expressing a dominant‐negative form of KLHDC2 (DNKLHDC2, a.a. 1–362) or shRNAs against KLHDC2 and analyzed 40 h or 88 h thereafter, respectively. DNKLHDC2 is the N‐terminal part of KLHDC2 that only associates with substrates but not Elongin B/C or Cul2. Consequently, DNKLHDC2‐bound proteins cannot be ubiquitinated and then degraded. To study the function of BC‐box proteins, cells were infected with viruses expressing BC‐box proteins or shRNAs against BC‐box proteins (MOI ~ 10) and analyzed 20 h or 88 h thereafter, respectively. To inhibit p97/VCP, cells were treated with 2.5 μM of CB‐5083 (APExBIO) for 6 h. The NMT inhibitor IMP‐1088 (Cayman Chemical) was used at a final concentration of 0.5 μM or 5 μM in HEK293T and U2OS cells, respectively, for 24 h. All exogenous proteins and shRNAs were expressed via lentivirus‐mediated transduction. Constructs and DNAs used in this study are listed in Table EV5.
Transfection, infection, and lentivirus production
Transfection of HEK293T cells was executed using TransIT‐293 transfection reagent (Mirus Bio) according to the manufacturer’s instructions. Virus infection was conducted in medium containing 8 μg/ml polybrene (Sigma‐Aldrich). To prepare lentiviruses, HEK293T cells at approximately 80% confluency were transfected with pRev, pTat, pHIV gag/pol, pVSVG, and the lentiviral construct of interest. Medium was freshly replaced and viruses were harvested 24 and 48 h post‐transfection, respectively.
Generation of GPS reporter cell lines and GPS assay
To generate GPS reporter constructs, the gene of interest was cloned into the pLenti‐GPS vector by Gateway recombination (Invitrogen). To generate GPS reporter cell lines, cells were infected with lentiviruses carrying GPS reporter constructs at a low MOI of ∼ 0.2 and then selected by puromycin (1 μg/ml, Clontech Laboratories). GPS reporter cells were analyzed using a BD LSR Fortessa system (BD Biosciences) operated by BD FACSDiva™ software. The 488‐nm and 561‐nm lasers were used to stimulate GFP and RFP, respectively. The flow rate was 5 × 103~1 × 104 cells/s. No background correction or compensation was applied. Cells were gated by doublet discrimination and as RFP‐positive in order to only measure the fluorescent signals of individual GPS reporter cells. We typically analyzed 20,000 or 100,000 individual cells for reporter cells carrying a single GPS construct or GPS peptide libraries, respectively. FlowJo software was used for FACS analysis.
Context‐independent GPS random peptide platform
Synthesized oligonucleotides (Life Technologies) with 5′ and 3′ linkers encoding 12‐residue random peptides were amplified by PCR and cloned into pDONR by Gibson assembly, followed by transfer into the pLenti‐GPS reporter via Gateway recombination to generate GPS random peptide libraries. NNK degenerate codons (N = A/T/G/C; K = G/T) instead of NNN were used to encode random residues in order to reduce amino acid bias and to decrease the incidence of stop codons from 3/64 to 1/32. To avoid uneven representation of libraries due to biases in synthesis of oligonucleotides, two or three independent libraries were constructed for each examined C‐terminal residue or motif generated from separately synthesized oligonucleotides. The complexity of each random peptide library is approximately 1010.
GPS random peptide library cells were generated by infecting HEK293T cells with lentiviruses carrying GPS random peptide libraries at a low MOI of ~ 0.1 to ensure that each cell only expresses one unique GFP‐peptide fusion. The percentage of GPS library cells with reduced GFP/RFP ratios, as determined by FACS (and represented as “% degradation”), was used as an index to reflect the ability of peptide mixtures to promote protein degradation. It is unfeasible to analyze cells carrying all ~ 1010 random peptides for each library. To ensure the reliability of our results, we performed FACS analyses in triplicate and recorded 100,000 cells each time. Some peptides encoded by random oligonucleotides have fewer than 12 residues due to random insertions of internal TAG stop codons. Thus, the % degradation revealed by FACS (Dobs) represents both full‐length and undesired truncated peptides. We approximated Dobs as a linear combination of Dreal and DA, i.e., the real % degradation of 12‐residue “full‐length” peptides terminated with specified C‐terminal residues and the observed % degradation from GPS library cells carrying the A‐residue random peptide library, respectively
where “A” denotes the number of random residues coded by NNK codons within the 12 residues.
Cycloheximide‐chase assay
Cells were treated with 100 μg/ml cycloheximide (Calbiochem), followed by sample collection at multiple time‐points. Samples were collected by directly lysing cells in 2× Laemmli sample buffer followed by sonication and boiling for 5 min. Protein abundance was analyzed by immunoblotting.
GST pull‐down assay, cell fractionation, and immunoblotting
To detect physical interactions between putative substrates and BC‐box proteins, HEK293T cells stably expressing HA‐tagged substrates were transfected with plasmids carrying GST‐tagged BC‐box proteins for 42 h followed by treatment with 1 μM bortezomib and 1 μM MLN4924 for 6 h to block protein degradation. Cells were then lysed on ice using IP‐lysis buffer (20 mM Tris pH 7.6, 150 mM NaCl and 0.5% IGEPAL CA‐630) supplemented with protease inhibitor cocktails (Roche). Cell extracts were incubated with Glutathione‐Sepharose 4B (GE Healthcare Life Sciences) for 16 h at 4°C. The corresponding Sepharose was washed three times in IP‐lysis buffer, boiled in 2× Laemmli sample buffer, and subjected to immunoblotting.
Cell fractionation was performed by using a cell fractionation kit (Abcam, ab109719) according to the manufacturer’s instructions. In brief, cells were resuspended in buffer A and permeabilized with detergent I. The cell suspension was then spun at 10,000 g for 2 min. The resulting supernatant was collected as the cytosol fraction. The cytosol‐depleted pellet was resuspended in buffer A and solubilized with detergent II. Following centrifugation at 10,000 g for 2 min, the supernatant was collected as the mitochondria‐enriched fraction. Protein abundance in each fraction was analyzed by immunoblotting. VDAC1 and α‐tubulin served as organelle‐specific markers for mitochondria and cytosol, respectively.
For immunoblotting, primary antibodies were purchased from the following suppliers: Flag (Sigma‐Aldrich, M2), GFP (Takara Bio Clontech, JL‐8, 632381), GST (GE Healthcare, 27457701), HA (Abcam, ab130275), α‐tubulin (Thermo Scientific™, MS‐581‐P), GAPDH (GeneTex, 100118), PDP2 (GeneTex, 44905), MRPL28 (GeneTex, 115439), VDAC1 (Abcam, ab14734), and MIC19 (Abcam, ab224565).
Cell staining
To detect ectopically‐expressed MIC19, a HA tag was inserted directly after the myristoylation motif (a.a. 1–14) of MIC19. We have confirmed that the HA tag does not affect mitochondrial targeting of MIC19. U2OS cells stably expressing HA‐MIC19 were grown on coverslips coated with poly‐l‐lysine (Sigma‐Aldrich). Mitochondrial staining was performed by incubating cells with growth medium containing 25 nM of MitoTracker probe (Thermo Fisher) for 45 min at 37°C. After rinses with PBS, cells were fixed in 4% paraformaldehyde for 15 min at room temperature, followed by three washes in 0.2% Triton X‐100/PBS. The coverslips were blocked with blocking buffer (2% BSA in 0.2% Triton X‐100/PBS) for 1 h and then incubated overnight with a MIC19 primary antibody (Abcam, ab224565) (1:100 dilution in blocking buffer) at 4°C. Following three washes in 0.2% Triton X‐100/PBS, cells were incubated with Alexa Fluor‐conjugated secondary antibody (Thermo Fisher, A27034) (1:100 dilution in blocking buffer) for 1 h at room temperature in the dark. The coverslips were mounted on glass slides using Fluoroshield DAPI mounting medium (Sigma‐Aldrich). Images were captured using a scanning laser confocal microscope (ZEISS LSM710). For quantification analysis, all images were acquired using the same device parameters.
Luciferase assay
Luciferase activities were measured in triplicate using a Pierce™ Firefly Luciferase Glow Assay kit (Thermo Fisher) and BioLux® Gaussia Luciferase Assay kit (NEB) according to the manufacturers’ instructions. To measure the activity of secreted luciferases, the conditioned media of HEK293T cells stably expressing FLuc or GLuc were collected. To measure cytosolic luciferase activities, HEK293T cells expressing FLuc or GLu were lysed to prepare cell lysates. Chemiluminescence signals were detected using a SpectraMax Paradigm Multi‐Mode Microplate Reader (Molecular Devices).
Affinity purification‐mass spectrometry
For mass spectrometry (MS)‐based identification of KLHDC2 substrates, HEK293T cells stably expressing GST‐tagged DNKLHDC2 (a.a. 1–362) were treated with 1 μM bortezomib and 1 μM MLN4924 for 6 h to block protein degradation before cell harvesting. Cells were lysed on ice in IP‐lysis buffer (20 mM Tris pH 7.6, 150 mM NaCl and 0.5% IGEPAL CA‐630) supplemented with protease inhibitors (Roche). Cell lysates were spun at 16,000 g and 4°C for 15 min to remove cell debris. Cell compartmentalization was disrupted under this lysis condition. The clarified lysate was then incubated with Glutathione‐Sepharose 4B (GE Healthcare Life Science) for 16 h at 4°C. The corresponding Sepharose was washed five times with washing buffer (20 mM Tris pH 7.6, 300 mM NaCl, 1% IGEPAL CA‐6300, and 0.5% sodium deoxycholate). KLHDC2‐binding proteins were eluted with 2% SDS, reduced with DTT, and alkylated with iodoacetamide, followed by trypsin digestion for 16 h at 37°C. The tryptic peptides were cleaned up using C18 zip‐tips and subjected to MS analysis.
MS data were acquired on an Orbitrap Fusion mass spectrometer (Thermo Scientific) equipped with an EASY‐nLC 1200 system (Thermo Scientific) and a nanoelectrospray ion source (New Objective, Inc.). Samples dissolved in 0.1% formic acid were injected onto a self‐packed precolumn (150 µm I.D. × 30 mm, 5 µm, 200 Å) at a flow rate of 10 μl/min. Chromatographic separation was performed on a self‐packed reverse‐phase C18 nano‐column (75 μm I.D. × 200 mm, 3 μm, 100 Å) using 0.1% formic acid in water as mobile phase A and 0.1% formic acid in 80% acetonitrile as mobile phase B, operated at a flow rate of 300 nl/min. The LC gradient was applied from 2% buffer B at 2 min to 40% buffer B at 40 min. Electrospray voltage was maintained at 1.8 kV and the capillary temperature was set at 275°C. Full MS survey scans were executed in the mass range of m/z 320 to 1,600 (AGC target at 5 × 105) with lock mass, resolution of 120,000 (at m/z 200), and a maximum injection time of 50 ms. The MS/MS were run in top speed mode with 3 s cycles, and the dynamic exclusion duration was set to 60 s with a 10 ppm tolerance around the selected precursor and its isotopes. The precursor ion isolation was performed with a mass selecting quadrupole, and the isolation window was set to m/z 2.0. Monoisotopic precursor ion selection was enabled and 1+ charge‐state ions were rejected for MS/MS. The MS/MS analyses were carried out with the collision induced dissociation (CID) mode and with a normalization collision energy (NCE) of 30%. The maximum injection time for spectral acquisition was 150 ms and the automatic gain control (AGC) target values for MS/MS scans were set at 5 × 104.
For data analysis, all MS/MS spectra were converted to mgf format from an experiment RAW file using msConvert (version 3.0.18165, ProteoWizard), and then analyzed using Mascot for MS/MS ion search. The search parameters included an error tolerance for precursor ions and the MS/MS fragment ions in spectra of 10 ppm and 0.6 Da, respectively. The enzyme cutting site was customized at the C‐terminal of lysine, arginine, and glycine, with missed cleavages numbering two. The variable post‐translational modifications in the search parameters were assigned to include oxidation of methionine and carbamidomethylation of cysteine. Peptides were identified with the false discovery rate (FDR) < 1%.
Targeting sequences for shRNAs
APPBP2: GCCTTCAGTTGTGTACTCT
FEM1C: GTAACAGTTGTTTCATAA
KLHDC2: CTTGGTGTCTGGGTATATA
KLHDC3: TGGAAAAAGATTGAACCGA
VHL: TGGCTCAACTTCGACGGCG
ZER1: ACAAAGTAAGTGGTGACAA (#1); AACTGCGAGATGTTCCTCA (#2); TCCAGCTACTACAAGTTCA (#3); GGGATCGAGGTTTCCTACA (#4)
ZYG11B: AGCAAGAATCCTTCAAGGT (#1); CTGTTGACCTGATTTCTGA (#2);
AGACCGACTCAAGTCTCTA (#3)
C‐degrons used for targeting model substrate degradation
KLHDC2 diGly degron: LRGPSPPPMAGG
KLHDC3 R___G degron: ASKERARPCQRG
APPBP2 R_G_ degron: EMALSPPRSWGQ
Mitochondria targeting sequences used for targeting GFP to mitochondria
MTSPDHA1: MRKMLAAVSRVLSGASQKPASRVLVASRNF
MTSCOX8A: MSVLTPLLLRGLTGSARRLPVPRAK
MTSGLUL: MTTSASSHLNKGIKQVYMSLPQGEKVQAMYIWIDGTGEGLRCKTRTLDSE
Bioinformatics analysis of anomalous amino acid distribution at protein C‐termini
Collection and processing of protein sequences
Human protein coding sequences were downloaded from NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/, download date: 18 Feb. 2017) and (https://www.uniprot.org/, download date: 05 Aug. 2020). The protein coding sequences of Mus musculus (GRCm38.p5), Drosophila melanogaster (BDGP6), Caenorhabditis elegans (WBcel235), Arabidopsis thaliana (Araport11_genes.201606), Saccharomyces cerevisiae (R64‐1‐1), and Escherichia coli (MG1655) were obtained from the Ensembl BioMart data mining tool (https://www.ensembl.org/biomart/martview/), the TAIR Arabidopsis Resource (https://www.arabidopsis.org/) or NCBI GenBank. Redundant sequences and sequences shorter than 50 amino acids were removed. The total numbers of protein sequences analyzed in human (NCBI), human (Uniprot), mouse, fly, nematode, plant, yeast, and E. coli are 36,644, 20,221, 41,504, 21,763, 27,644, 40,366, 6,484, and 4,028, respectively.
Protein sequences produced by NMD were collected from BioMart with the following criteria: gene biotype = protein coding and transcript type = nonsense‐mediated decay (GRCh38.p12, release 92). Protein sequences from uORFs were decoded from the TISdb database (http://tisdb.human.cornell.edu/) (Wan & Qian, 2014). Since protein products translated from uORFs are relatively small, we kept unique sequences that are equal to or longer than 10 amino acids. The total numbers of NMD and uORF proteins/peptides analyzed are 11,225, and 3,901, respectively.
To prepare abnormal protein sequences, human transcript sequences were downloaded from Ensembl (download date: 3 Jan. 2017) and non‐protein transcripts were removed. We considered two types of abnormal protein sequences derived from the normal mRNA transcripts. Protein translation may undergo frameshifts when the reading frame is shifted at the +1 or −1 position, so the downstream amino acid sequences are completely altered. In addition, protein translation may also undergo readthrough error when ribosomes skip the first stop codon they encounter and instead continue peptide elongation until they reach the next stop codon. The procedures for generating these two types of aberrant protein sequences are described in detail below:
Frameshifted protein sequences
We retained 36,284 mRNA transcripts satisfying the following criteria as templates for generating frameshifted protein sequences: (1) they possessed RefSeq IDs according to the NCBI database; (2) they encoded proteins, as the RefSeq IDs were converted to Ensemble protein IDs according to the file “gene2ensemble” from NCBI, ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2ensembl; (3) the encoded proteins had ≥ 50 amino acids. For each template transcript, we generated 1,000 frameshifted protein sequences by (1) randomly selecting the positions of the initially disrupted reading frames; (2) randomly selecting the phases of frameshifts (+1 or −1); and (3) discarding sequences shorter than 50 amino acids. One representative was randomly drawn from among the 1,000 frameshifted protein sequences for each transcript. This sampling scheme is preferable to exhaustively generating the frameshifted sequences starting from all positions.
Readthrough protein sequences
We selected 39,216 transcripts as templates according to the following criteria: (1) they encode proteins; (2) they contain 3′UTR regions; (3) they have stop codons in their 3′UTR regions; (4) the translated proteins are longer than 50 amino acids; and (5) the translated amino acid sequences are unique. The native stop codons of these transcript templates were skipped, resulting in generation of longer peptide sequences terminating at the first downstream stop codon in the 3′UTR region.
Protein sequences of human transmembrane proteins with different C‐terminal tail topologies were downloaded from the Human Transmembrane Proteome database (http://htp.enzim.hu/, download date: 7 Mar. 2018). The numbers of sequences corresponding to “C‐terminal inside of membrane” and “C‐terminal outside of membrane” are 4,052, and 1,360, respectively. Sequences of human secreted proteins were collected from VerSeDa (the vertebrate secretome database) (http://genomics.cicbiogune.es/VerSeDa/index.php, download date: 29 Jan. 2018) (Cortazar et al, 2017), with the following criteria: “only” extracellular and SignalP ≥ 0.5 in order exclude proteins with multiple subcellular localizations or that are secreted via unconventional pathways. Cytosolic, nuclear, and mitochondrial proteins were collected based on the Human Protein Atlas (https://www.proteinatlas.org/) and MitoProteome (http://www.mitoproteome.org/). Cytosolic proteins do not include those located at endoplasmic reticulum, Golgi, endosomes, lysosomes, peroxisomes, mitochondria, vesicles, or lipid droplets. Proteins with multiple localizations or shorter than 50 amino acids were excluded. The total numbers of sequences from cytosolic, nuclear, mitochondrial, and secreted proteins analyzed are 7,197, 5,983, 3,363, and 2,218, respectively.
Statistical assessment of the prominence and significance of amino acid bias at the C‐termini of proteins
We extracted the last 50 amino acid sequences of proteins and counted the occurrence frequency of each amino acid residue at each position. Two statistical assessments were performed to quantify the disparity of the amino acid frequencies at the terminal position relative to those of all other positions. First, the occurrence frequencies of amino acid residues at each position were treated as a 20‐dimensional vector, and principal component analysis (PCA) was applied to project those vectors onto a two‐dimensional space. Second, the P‐value whereby each amino acid residue was under‐ or over‐represented at the C‐terminal compared to a background distribution derived from the protein data over multiple positions was computed. Here, the background (null) model is the multinomial distribution F = (F 1, …, F 20), where each Fi represents the relative occurrence frequency of amino acid residue i according to the background distribution. Suppose among n proteins that amino acid residue i appears at the terminal position in ni proteins. Then, ni follows a binomial distribution and the probability of under‐representation is the left‐tail probability according to the cumulative distribution function (CDF) of ni:
where denotes the number of proteins where amino acid residue i appears at the terminal position from the empirical data. The probability of over‐representation is the right‐tail probability of the same distribution. can be approximated by the CDF function of a Poisson distribution:
where λi = nFi.
We computed the P‐values of under‐ and over‐representations for each amino acid at the C‐terminal for the following classes of proteins or peptide sequences: (1) functional full‐length proteins; (2) functional proteins with specific localizations; (3) frameshifted protein sequences; (4) stop codon readthrough protein sequences; (5) NMD protein sequences; and (6) peptide products from uORFs. The P‐values of background amino acid frequencies of classes (1)–(5) were derived from all positions of all functional full‐length proteins. The P‐values of background amino acid frequencies of class (6) were derived from all positions of the uORF peptides alone due to the differential amino acid usages between functional proteins and uORF peptides. FDR (false discovery rate)‐adjusted P‐values were calculated for multi‐hypotheses test correction. For the P‐values pertaining to the amino acid frequencies of the last residues of proteins (e.g., Table EV2), there were 20 hypotheses corresponding to all amino acids. For the P‐values pertaining to the amino acid frequencies of the last 50 residues of proteins (e.g., Fig 2), there were 50 × 20 = 1,000 hypotheses corresponding to all combinations of amino acids and positions.
Author contributions
C‐WY characterized C‐degrons, human C‐degron‐harboring proteins and MIC19. W‐CH carried out GST pull‐down analyses to search for KLHDC2 substrates and characterized cellular and viral diGly‐ending polypeptides. P‐HH performed mass spectrometry analyses; K‐HY, PW‐CH, and Y‐NC performed bioinformatic analyses; L‐CW and S‐CC performed mutagenesis analyses and GPS random peptide assays to characterize C‐degrons; H‐CL developed GPS random peptide assays and characterized C‐degron‐bearing model substrates; and C‐HY supervised bioinformatic and statistical analyses. H‐CSY designed experiments, analyzed data, supervised the project, and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank S. Baker and S. Makino for SARS‐CoV nsp constructs, S.P. Li for confocal image analysis, M.D. Tsai for mass spectrometry analysis, the Data Science Statistical Cooperation Center of Academia Sinica (AS‐IA‐108‐L02) for statistical support, and H. Cheng, K.L. Hsu, C.L. Lin, S.C. Lin, S.T. Lin, J. O’Brien, and H.N. Wu for suggestions and assistance. Image analyses were performed at the Imaging Core of the Institute of Molecular Biology at Academia Sinica. Mass spectrometric analyses were supported by the Taiwan Protein Project and performed at the Mass Spectrometry Core Facility at the Genomics Research Center, Academia Sinica. This work was supported by MOST grants 107‐2321‐B‐001‐021 and 108‐2321‐B‐001‐006 from the National Science Council of Taiwan, and an Investigator Award from Academia Sinica.
The EMBO Journal (2021) 40: e105846.
Data availability
This study includes no data deposited in external repositories.
References
- Bachmair A, Finley D, Varshavsky A (1986) In vivo half‐life of a protein is a function of its amino‐terminal residue. Science 234: 179–186 [DOI] [PubMed] [Google Scholar]
- Báez‐Santos YM, St. John SE, Mesecar AD (2015) The SARS‐coronavirus papain‐like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115: 21–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan JS, Rubin BR, Yu C, Loffler MG, Orme CM, Belman JP, McNally LJ, Hao M, Cresswell JA (2012) Endoproteolytic cleavage of TUG protein regulates GLUT4 glucose transporter translocation. J Biol Chem 287: 23932–23947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Retzlaff M, Roos T, Frydman J (2011) Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol 3: a004374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazar AR, Oguiza JA, Aransay AM, Lavin JL (2017) VerSeDa: vertebrate secretome database. Database (Oxford) 2017: baw171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darshi M, Mendiola VL, Mackey MR, Murphy AN, Koller A, Perkins GA, Ellisman MH, Taylor SS (2011) ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J Biol Chem 286: 2918–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darshi M, Trinh KN, Murphy AN, Taylor SS (2012) Targeting and import mechanism of coiled‐coil helix coiled‐coil helix domain‐containing protein 3 (ChChd3) into the mitochondrial intermembrane space. J Biol Chem 287: 39480–39491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deichaite I, Casson LP, Ling HP, Resh MD (1988) In vitro synthesis of pp60v‐src: myristylation in a cell‐free system. Mol Cell Biol 8: 4295–4301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnikov T, Ben‐Gedalya T, Cohen E (2017) Protein quality control in health and disease. Cold Spring Harb Perspect Biol 9: a023523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa Y, Tsuji J, Fu SC, Tomii K, Horton P, Imai K (2015) MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics 14: 1113–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglione C, Fieulaine S, Meinnel T (2015) N‐terminal protein modifications: bringing back into play the ribosome. Biochimie 114: 134–146 [DOI] [PubMed] [Google Scholar]
- Habtemichael EN, Li DT, Alcazar‐Roman A, Westergaard XO, Li M, Petersen MC, Li H, DeVries SG, Li E, Julca‐Zevallos O et al (2018) Usp25m protease regulates ubiquitin‐like processing of TUG proteins to control GLUT4 glucose transporter translocation in adipocytes. J Biol Chem 293: 10466–10486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt BH, Jukneliene D, Kanjanahaluethai A, Bechill J, Severson KM, Smith CM, Rota PA, Baker SC (2004) Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain‐like protease activity. J Virol 78: 13600–13612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Bennett EJ (2016) Proteome complexity and the forces that drive proteome imbalance. Nature 537: 328–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G (1990) The signal peptide. J Membr Biol 115: 195–201 [DOI] [PubMed] [Google Scholar]
- Hessa T, Sharma A, Mariappan M, Eshleman HD, Gutierrez E, Hegde RS (2011) Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature 475: 394–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15: 233–249 [DOI] [PubMed] [Google Scholar]
- Hipp MS, Kasturi P, Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20: 421–435 [DOI] [PubMed] [Google Scholar]
- Hwang CS, Shemorry A, Varshavsky A (2010) N‐terminal acetylation of cellular proteins creates specific degradation signals. Science 327: 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo U, Cai W, Wang J, Kwon Y, D'Andrea AD, Kim H (2016) PCNA‐Dependent cleavage and degradation of SDE2 regulates response to replication stress. PLoS Genet 12: e1006465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Mitra D, Salerno JR, Hegde RS (2002) Signal sequences control gating of the protein translocation channel in a substrate‐specific manner. Dev Cell 2: 207–217 [DOI] [PubMed] [Google Scholar]
- Koren I, Timms RT, Kula T, Xu Q, Li MZ, Elledge SJ (2018) The Eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C‐terminal degrons. Cell 173: 1622–1635.e1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine CG, Mitra D, Sharma A, Smith CL, Hegde RS (2004) The efficiency of protein compartmentalization into the secretory pathway. Mol Biol Cell 16: 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Ho SC, Chen YY, Khoo KH, Hsu PH, Yen HC (2015) SELENOPROTEINS. CRL2 aids elimination of truncated selenoproteins produced by failed UGA/Sec decoding. Science 349: 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Yeh CW, Chen YF, Lee TT, Hsieh PY, Rusnac DV, Lin SY, Elledge SJ, Zheng N, Yen HS (2018) C‐terminal end‐directed protein elimination by CRL2 ubiquitin ligases. Mol Cell 70: 602–613.e603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokugamage KG, Narayanan K, Huang C, Makino S (2012) Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J Virol 86: 13598–13608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews GD, Gur N, Koopman WJH, Pines O, Vardimon L (2010) Weak mitochondrial targeting sequence determines tissue‐specific subcellular localization of glutamine synthetase in liver and brain cells. J Cell Sci 123: 351–359 [DOI] [PubMed] [Google Scholar]
- Meyer H, Bug M, Bremer S (2012) Emerging functions of the VCP/p97 AAA‐ATPase in the ubiquitin system. Nat Cell Biol 14: 117–123 [DOI] [PubMed] [Google Scholar]
- Mól AR, Castro MS, Fontes W (2018) NetWheels: a web application to create high quality peptide helical wheel and net projections. bioRxiv 10.1101/416347 [PREPRINT] [DOI] [Google Scholar]
- Olvera J, Wool IG (1993) The carboxyl extension of a ubiquitin‐like protein is rat ribosomal protein S30. J Biol Chem 268: 17967–17974 [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ (2005) Function and regulation of cullin–RING ubiquitin ligases. Nat Rev Mol Cell Biol 6: 9–20 [DOI] [PubMed] [Google Scholar]
- Pfanner N, Geissler A (2001) Versatility of the mitochondrial protein import machinery. Nat Rev Mol Cell Biol 2: 339–349 [DOI] [PubMed] [Google Scholar]
- Piatkov KI, Colnaghi L, Bekes M, Varshavsky A, Huang TT (2012) The auto‐generated fragment of the Usp1 deubiquitylase is a physiological substrate of the N‐end rule pathway. Mol Cell 48: 926–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkov KI, Oh JH, Liu Y, Varshavsky A (2014) Calpain‐generated natural protein fragments as short‐lived substrates of the N‐end rule pathway. Proc Natl Acad Sci USA 111: E817–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl C, Dikic I (2019) Cellular quality control by the ubiquitin‐proteasome system and autophagy. Science 366: 818–822 [DOI] [PubMed] [Google Scholar]
- Posakony JW, England JM, Attardi G (1977) Mitochondrial growth and division during the cell cycle in HeLa cells. J Cell Biol 74: 468–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rageul J, Park JJ, Jo U, Weinheimer AS, Vu TTM, Kim H (2019) Conditional degradation of SDE2 by the Arg/N‐End rule pathway regulates stress response at replication forks. Nucleic Acids Res 47: 3996–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane NS, Chakrabarti O, Feigenbaum L, Hegde RS (2010) Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J Cell Biol 188: 515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnac D‐V, Lin H‐C, Canzani D, Tien KX, Hinds TR, Tsue AF, Bush MF, Yen H‐CS, Zheng N (2018) Recognition of the diglycine C‐end degron by CRL2KLHDC2 ubiquitin ligase. Mol Cell 72: 813–822.e814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram SM, Kim BY, Kwon YT (2011) The N‐end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Biol 12: 735–747 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kamitani W, DeDiego ML, Enjuanes L, Matsuura Y (2012) Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J Virol 86: 11128–11137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakran P, Pandit PA, Datta S, Kolathur KK, Pleiss JA, Mishra SK (2018) Sde2 is an intron‐specific pre‐mRNA splicing regulator activated by ubiquitin‐like processing. EMBO J 37: 89–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms RT, Zhang Z, Rhee DY, Harper JW, Koren I, Elledge SJ (2019) A glycine‐specific N‐degron pathway mediates the quality control of protein N‐myristoylation. Science 365: eaaw4912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirigos KD, Peters C, Shu N, Kall L, Elofsson A (2015) The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res 43: W401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda E, Tamura Y, Sakaue H, Kawano S, Kakuta C, Matsumoto S, Endo T (2019) Myristoyl group‐aided protein import into the mitochondrial intermembrane space. Sci Rep 9: 1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A (2011) The N‐end rule pathway and regulation by proteolysis. Protein Sci 20: 1298–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A (2019) N‐degron and C‐degron pathways of protein degradation. Proc Natl Acad Sci USA 116: 358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali DG, Sinzel M, Bulthuis EP, Kolb A, Zabel S, Mehlhorn DG, Figueiredo Costa B, Farkas Á, Clancy A, Schuldiner M et al (2018) The GET pathway can increase the risk of mitochondrial outer membrane proteins to be mistargeted to the ER. J Cell Sci 131: jcs211110 [DOI] [PubMed] [Google Scholar]
- Wan J, Qian SB (2014) TISdb: a database for alternative translation initiation in mammalian cells. Nucleic Acids Res 42: D845–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li S (2014) Protein mislocalization: mechanisms, functions and clinical applications in cancer. Biochim Biophys Acta 1846: 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Werner A, Rape M (2013) The Colossus of ubiquitylation: decrypting a cellular code. Mol Cell 49: 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ (2008) Global protein stability profiling in mammalian cells. Science 322: 918–923 [DOI] [PubMed] [Google Scholar]
- Zheng N, Shabek N (2017) Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem 86: 129–157 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
This study includes no data deposited in external repositories.