

Abstract
Interleukin (IL)‐33 cytokine plays a critical role in allergic diseases and cancer. IL‐33 also has a nuclear localization signal. However, the nuclear function of IL‐33 and its impact on cancer is unknown. Here, we demonstrate that nuclear IL‐33‐mediated activation of SMAD signaling pathway in epithelial cells is essential for cancer development in chronic inflammation. Using RNA and ChIP sequencing, we found that nuclear IL‐33 repressed the expression of an inhibitory SMAD, Smad6, by interacting with its transcription factor, RUNX2. IL‐33 was highly expressed in the skin and pancreatic epithelial cells in chronic inflammation, leading to a markedly repressed Smad6 expression as well as dramatically upregulated p‐SMAD2/3 and p‐SMAD1/5 in the epithelial cells. Blocking TGF‐β/SMAD signaling attenuated the IL‐33‐induced cell proliferation in vitro and inhibited IL‐33‐dependent epidermal hyperplasia and skin cancer development in vivo. IL‐33 and SMAD signaling were upregulated in human skin cancer, pancreatitis, and pancreatitis‐associated pancreatic cancer. Collectively, our findings reveal that nuclear IL‐33/SMAD signaling is a cell‐autonomous tumor‐promoting axis in chronic inflammation, which can be targeted by small‐molecule inhibitors for cancer treatment and prevention.
Keywords: chronic inflammation, Nuclear Interleukin‐33, pancreatic cancer, skin cancer, SMAD signaling
Subject Categories: Cancer, Immunology, Signal Transduction
Tumor‐promoting activities of interleukin‐33 are independent of its cytokine function, but instead involve direct regulation of RUNX2‐dependent Smad6 expression in the cell nucleus.
Introduction
Chronic inflammation and its associated tumor‐promoting environment are the hallmarks of many cancer types (Grivennikov et al, 2010; Todoric et al, 2016). Cancer‐associated chronic inflammation contains immune cells and factors including regulatory T cells, myeloid‐derived suppressor cells, interleukin (IL)‐10, and transforming growth factor‐beta (TGF‐β) that exclude antitumor cytotoxic immunity (Grivennikov et al, 2010; Todoric et al, 2016). Thus, cancers that contain chronic inflammatory microenvironment are resistant to conventional chemotherapies and novel immunotherapies including immune checkpoint blockade resulting in poor prognosis (Brahmer et al, 2012; Johnson et al, 2017). To overcome this challenge, new approaches to prevent and treat cancer in the context of chronic inflammation are urgently needed.
IL‐33 is an epithelium‐derived cytokine that is upregulated early on in cancer‐prone chronic inflammation (Ameri et al, 2019). As a member of the IL‐1 family of cytokines, IL‐33 is implicated in the pathogenesis of allergic diseases by inducing a type 2 immune response (Saenz et al, 2008; Sy & Siracusa, 2016; Wang et al, 2018). In addition, IL‐33 has been associated with pro‐ and antitumor functions in cancer (Ameri et al, 2019; Moral et al, 2020). Upon cellular stress, injury, or necrosis, IL‐33 is rapidly released to “alarm” the immune system (Molofsky et al, 2015). Secreted IL‐33 binds to its cognate receptor, interleukin‐1 receptor‐like 1 (IL1RL1), or suppressor of tumorigenesis 2 (ST2 (also known as IL‐1R4)), generating a potent pro‐inflammatory stimulus particularly in the context of chronic inflammation (Kumar et al, 1995; Oshikawa et al, 2002; Moussion et al, 2008). Notably, IL‐33 protein also has a nuclear localization signal and a homeodomain (helix–turn–helix‐like motif) that can bind to heterochromatin in the nucleus, similar to nuclear proteins (Carriere et al, 2007; Ali et al, 2011). At baseline, IL‐33 resides in the nucleus of the cells in which it is expressed. Therefore, IL‐33 may have a dual function in epithelial cancers, acting as a cytokine when released from the cell and as a nuclear factor within the cell with transcriptional regulatory properties.
TGF‐β, bone morphogenetic protein (BMP), and SMAD signaling pathway are involved in many cellular processes, including tumorigenesis in chronic inflammation (Massague et al, 2000; Shi & Massague, 2003; Grivennikov et al, 2010; Massague, 2012). TGF‐β/BMP ligands bind to their receptors on the cell surface and activate SMAD2/3 or SMAD1/5 proteins through phosphorylation, followed by their translocation into the nucleolus by Co‐SMAD (SMAD4; Massague et al, 2005). SMAD6/7 are inhibitory SMADs (I‐SMAD) that either block SMAD4 binding to other SMAD proteins or inactivate TGF‐β/BMP receptor serine/threonine kinases (Imamura et al, 1997; Massague et al, 2005). SMAD6 can block TGF‐β/BMP‐mediated tumorigenesis and is associated with an increased survival rate among patients with squamous cell carcinoma (SCC) (Mangone et al, 2010). TGF‐β/BMP signaling is highly upregulated in pancreatic ductal adenocarcinoma (PDAC; Bardeesy et al, 2006), which commonly arises in the context of chronic pancreatitis and is characterized by rapid progression and profound resistance to immunotherapy (Melzer et al, 2017; Shen et al, 2017). Thus, blocking TGF‐β/BMP/SMAD signaling may provide an effective strategy to suppress carcinogenesis in chronic inflammation.
Herein, we demonstrate that IL‐33 induces a tumor‐promoting environment in chronic inflammation of skin and pancreas independent of its cytokine function. IL‐33 was highly expressed and localized in the nuclei of skin and pancreatic epithelial cells in chronic inflammation. Nuclear IL‐33 repressed Smad6 gene expression by blocking the transcription activity of RUNX2, which led to highly phosphorylated SMAD2/3 (p‐SMAD2/3) and p‐SMAD1/5. TGF‐β/SMAD signaling pathway activation was required for IL‐33‐induced cell proliferation and tumor development. Furthermore, we demonstrated that upregulated IL‐33 and SMAD signaling were associated with skin cancer, pancreatitis, and pancreatitis‐associated PDAC in humans. We conclude that nuclear IL‐33/SMAD signaling creates a cell‐autonomous axis essential for tumor promotion in chronic inflammatory conditions of the skin and pancreas.
Results
IL‐33 promotes tumor development in chronic skin inflammation independent of ST2
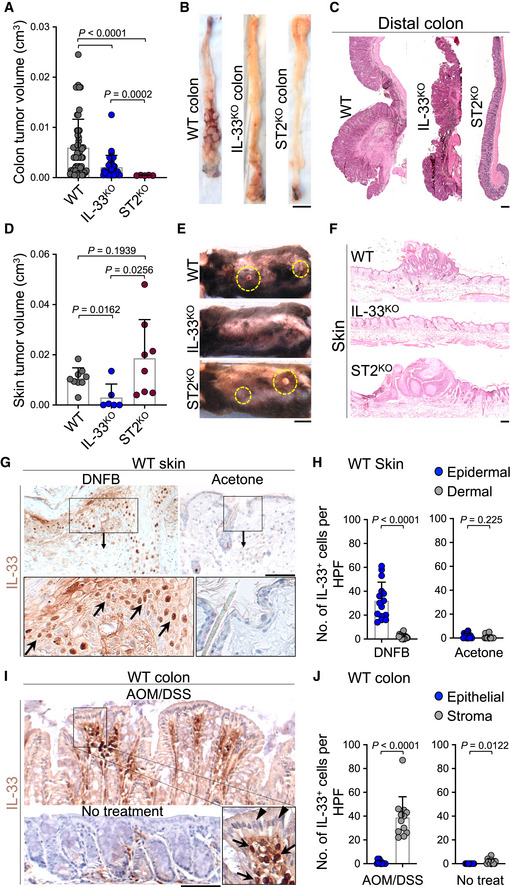
To elucidate the impact of IL‐33 on cancer development in chronic inflammation, we subjected Il33 knockout (IL‐33tm1b/tm1b or IL‐33KO), Il1rl1 knockout (ST2KO), and wild‐type (WT) mice to an established model of colitis‐induced colorectal cancer. Mice received five treatment cycles of intraperitoneal injection with azoxymethane (AOM) followed by a colitis‐causing agent, dextran sodium sulfate (DSS), added to the drinking water over 5 days (Appendix Fig S1A). IL‐33KO and ST2KO mice developed fewer and smaller colorectal tumors compared with WT mice (Fig 1A and B). Moreover, crypt architecture was better preserved in both IL‐33KO and ST2KO compared with the WT colon (Fig 1C). Colitis‐induced shortening of the colon length was significantly attenuated in ST2KO compared with WT mice (Appendix Fig S1B). Next, we examined the impact of IL‐33 on tumor promotion in the context of chronic skin inflammation (Ameri et al, 2019). IL‐33KO, ST2KO, and WT mice were treated with 7,12‐dimethylbenz(a)anthracene (DMBA, a carcinogen) once followed by 2,4‐Dinitro‐1‐fluorobenzene (DNFB, a contact allergen) in acetone on the back skin three times a week for 30 weeks (Appendix Fig S1C; Ameri et al, 2019). Although IL‐33KO mice were protected from DMBA‐DNFB‐induced skin carcinogenesis (Ameri et al, 2019), ST2KO mice developed large skin tumors similar to WT mice (Fig 1D–F). In addition, there was no difference between ST2KO and WT mice in skin tumor onset and the number of tumors that developed over time, while IL‐33KO showed significantly delayed tumor onset and fewer tumors compared with ST2KO and WT mice over time (Appendix Fig S1D and E). To determine the reason for the discordant skin cancer yet concordant colorectal cancer outcomes between IL‐33KO and ST2KO mice, we examined the cellular source of IL‐33 in the inflamed skin versus colon tissue. Topical DNFB treatment led to significantly elevated IL‐33 levels localized in the nuclei of keratinocytes, which are cancer forming cells in the skin (Fig 1G and H, and Appendix Fig S1F). In stark contrast, IL‐33 was mostly overexpressed by stromal cells, but not colon epithelial cells, in AOM/DSS‐induced colitis (Fig 1I and J, and Appendix Fig S1F). These findings suggest that IL‐33 acts as a nuclear protein in skin cancer development independent of its receptor, ST2, while it acts as a cytokine to promote colorectal cancer in an ST2‐dependent manner.
Figure 1. IL‐33 promotes skin cancer development in an ST2‐independent manner.
-
A–CColorectal tumor outcomes in WT (n = 7), IL‐33KO (n = 4), and ST2KO (n = 3) mice at the completion of the AOM/DSS carcinogenesis protocol. (A) Tumor volumes at the endpoint (each dot represents a tumor, mice with no tumor are marked as zero), (B) representative images of the exposed colonic lumen (scale bar: 1 cm), and (C) representative images of H&E‐stained distal colon in each group (scale bar: 100 μm).
-
D–FSkin tumor outcomes in WT (n = 7), IL‐33KO (n = 6), and ST2KO (n = 7) mice at the completion of the DMBA/DNFB carcinogenesis protocol. (D) Tumor volumes at the endpoint (each dot represents a tumor, mice with no tumor are marked as zero), (E) representative images of the back skin (yellow circles highlight the skin tumors, scale bar: 1 cm), and (F) representative images of H&E‐stained back skin of the mice in each group (scale bar: 100 μm).
-
GRepresentative images of IL‐33 immunostaining on WT skin treated with DNFB or acetone control. Arrows in the inset highlight IL‐33+ nuclei in epidermal keratinocytes (scale bar: 100 μm).
-
HQuantification of nuclear IL‐33+ epidermal and dermal cells in the DNFB‐ versus acetone‐treated WT skin. Dots represent cell counts from three randomly selected high power field (HPF) images per sample (n = 5 per group).
-
IRepresentative images of IL‐33 immunostaining on WT colon at the completion of the AOM/DSS carcinogen protocol compared with no treatment control. Arrows in the inset point to IL‐33+ stromal cells and arrowheads point to lack of IL‐33 nuclear stain in colonic epithelial cells (scale bar: 100 μm).
-
JQuantification of nuclear IL‐33+ epithelial and stromal cells of colon treated with AOM/DSS versus no treatment. Dots represent cell counts from three randomly selected HPF images per sample (n = 4 per group).
Data information: Graphs show mean + SD, unpaired t‐test.
Nuclear IL‐33 suppresses Smad6 expression via interaction with RUNX2
To investigate the nuclear function of IL‐33 in chronic inflammation, we performed RNA sequencing on the epidermal keratinocytes isolated from the back skin of IL‐33KO and WT mice treated with DNFB for 22 days (Appendix Fig S2A and B) (Ameri et al, 2019). 203 genes showed differential expressions in IL‐33KO compared with WT epidermal keratinocytes, which belonged to several developmental pathways (Fig 2A, Appendix Fig S2C and D, and Appendix Table S1). Interestingly, IL‐33KO showed negative regulation of the TGF‐β/BMP signaling pathway and restricted SMAD proteins phosphorylation by Gene Set Enrichment Analysis (GSEA, Appendix Fig S2E–G). To further examine the nuclear function of IL‐33, we generated IL‐33 full length and IL‐33 cytokine domain only constructs (Fig 2B and Appendix Fig S3A). IL‐33 full length localized into the nucleus while IL‐33 cytokine domain localized in the cytoplasm of a mouse keratinocyte cell line (Pam212, Appendix Fig S3B). Chromatin immunoprecipitation (ChIP) sequencing identified 2455 genes with differential peaks in IL‐33 full length compared with IL‐33 cytokine domain expressing Pam212 cells (Fig 2B). By cross comparing the RNA‐Seq and ChIP‐Seq data, we narrowed down the nuclear IL‐33 candidate targets to 7 genes, all of which showed increased expression upon IL‐33 loss in the skin keratinocytes (Fig 2B and C, Appendix Fig S3C). Among these genes, we focused on Smad6 because the DNFB‐treated IL‐33KO epidermis showed downregulation of the SMAD signaling pathway compared with WT by GSEA. IL‐33 full length, but not IL‐33 cytokine domain, bound to Smad6 regulatory region (Appendix Fig S3C). To verify that Smad6 was a target of IL‐33, we performed qPCR on the skin of WT mice treated with DNFB versus acetone (carrier control). Il33 was highly increased and Smad6 was markedly decreased in DNFB‐treated skin compared with acetone‐treated controls (Fig 2D). Importantly, the IL‐33 full length, but not cytokine domain, suppressed Smad6 expression in Pam212 cells compared with HA‐empty vector control (Fig 2E and Appendix Fig S3A). Neither IL‐33 full length nor cytokine domain impacted the expression of a control gene, Pdcd4, in Pam212 cells (Fig 2E). To determine the mechanism by which IL‐33 repressed Smad6 expression, we examined whether nuclear IL‐33 interacted with the Smad6 transcription factor, RUNX2 (Wang et al, 2007). Immunoprecipitation of endogenous nuclear IL‐33, which was highly expressed in a breast cancer cell line (PyMT), revealed a strong binding between IL‐33 and endogenous RUNX2 (Fig 2F). Overexpression of IL‐33 full length or cytokine domain in Pam212 cells revealed that IL‐33 full length, but not cytokine domain, bound to RUNX2 (Fig 2G). Furthermore, IL‐33 full length, compared with IL‐33 cytokine domain, decreased the binding of RUNX2 to the Smad6 promoter region (Fig 2H). Consistent with these findings, RUNX2 target genes were highly expressed in IL‐33KO compared with WT epidermis treated with DNFB (Appendix Fig S3D). These results indicate that nuclear IL‐33 regulates Smad6 expression via its interaction with RUNX2.
Figure 2. Nuclear IL‐33 blocks Smad6 expression via interaction with RUNX2 transcription factor.

- Heatmap of differentially expressed genes between IL‐33KO (n = 3) and WT (n = 3) epidermis after topical treatment with DNFB. Representative genes in differentially affected biological pathways are listed on the heatmap (the full list of differentially expressed genes is shown in Appendix Table S1).
- Schematic of IL‐33 full length and IL‐33 cytokine domain constructs (top). Venn diagram of differential genes shared between RNA‐Seq and ChIP‐Seq results (bottom). For ChIP‐Seq, Pam212 cells transfected with IL‐33 full length versus IL‐33 cytokine domain were compared. Any gene with a peak in HA‐empty vector control was excluded from the final analysis.
- RNA‐Seq result of the seven differential genes shared between RNA‐Seq and ChIP‐Seq results. Significance analysis comparing the two groups was performed using DESeq2 R package.
- Il33 and Smad6 expression levels in DNFB‐treated skin relative to acetone‐treated controls (n = 9 in DNFB group, n = 7 in acetone group for Il33, n = 10 in DNFB group, and n = 9 in acetone group for Smad6).
- Smad6 and Pdcd4 (negative control gene) expression levels upon IL‐33 full length versus cytokine domain expression compared with HA‐empty vector expression in Pam212 cells (n = 4 in each group).
- Immunoblot for interaction between endogenous IL‐33 and RUNX2. Lysate from PyMT and Pam212 cell lines were subjected to immunoprecipitation with an anti‐IL‐33 antibody followed by immunoblot analysis. Immunoprecipitation with an anti‐IgG antibody on an equal amount of PyMT lysate is shown as a negative control. Data represent two independent experiments with similar results.
- Immunoblot for interaction between nuclear IL‐33 and RUNX2. After Pam212 cells were transfected with IL‐33 full length or cytokine domain for 24 h, cell lysates were subjected to immunoprecipitation with anti‐IL‐33 antibody followed by immunoblot analysis.
- ChIP‐qPCR assay for Smad6 in the presence of IL‐33 full length or cytokine domain using an anti‐RUNX2 antibody. After Pam212 cells were transfected with IL‐33 full length or cytokine domain for 24 h, cell lysates were subjected to chromatin immunoprecipitation with anti‐RUNX2 antibody and eluted RUNX2‐bound chromatin was used for qPCR with primers against Smad6 promoter region. Anti‐IgG ChIP‐qPCR results are shown as negative controls (n = 4 per group).
Data information: GAPDH is used as the control housekeeping protein in (F) and (G). Graphs show mean + SD, NS: not significant, unpaired t‐test.
Source data are available online for this figure.
Nuclear IL‐33 induces epithelial cell proliferation via TGF‐β/SMAD signaling pathway
As SMAD6 is an inhibitory SMAD protein, we examined whether IL‐33 can suppress the TGF‐β/SMAD signaling pathway. IL‐33 induction in DNFB‐treated WT skin (Figs 1G, H and 2D) was accompanied by a reduction in SMAD6 protein and a marked increase in phosphorylated SMAD2/3 (p‐SMAD2/3) and p‐SMAD1/5 compared with acetone‐treated WT skin (Fig 3A, Appendix Fig S4A and B). Furthermore, SMAD6 levels were restored and p‐SMAD2/3 and p‐SMAD1/5 were greatly reduced in DNFB‐treated IL‐33KO compared with WT skin (Appendix Fig S4C and D). In contrast, no IL‐33‐mediated regulation of SMAD proteins was observed in the AOM/DSS‐treated colon of IL‐33KO compared with WT mice (Appendix Fig S4E and F). These findings demonstrate that nuclear IL‐33 represses Smad6 expression and activates SMAD signaling pathway when expressed by the epithelial cells in inflamed tissue.
Figure 3. Nuclear IL‐33 promotes cell proliferation by upregulating TGF‐β/SMAD signaling pathway.
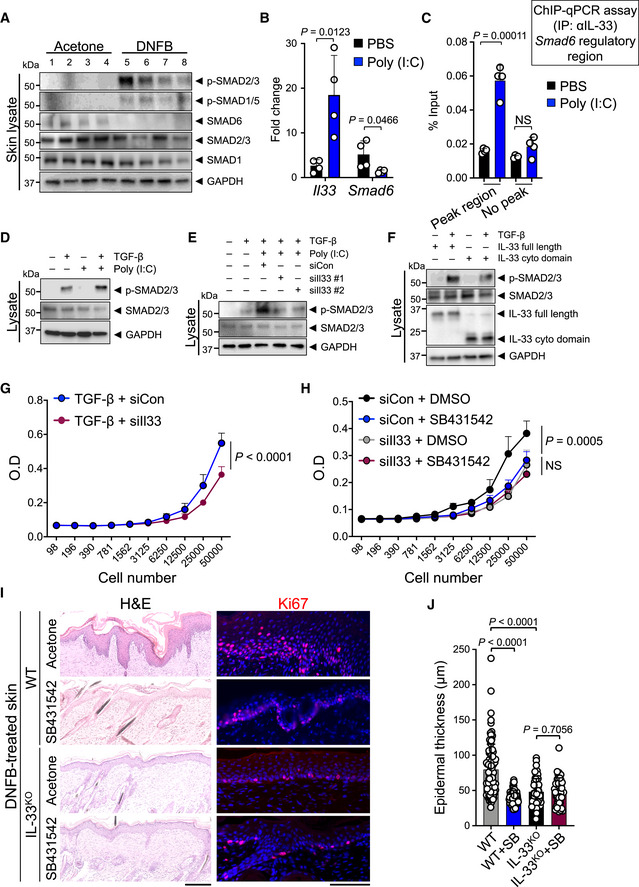
- Immunoblot of p‐SMAD2/3, p‐SMAD1/5, SMAD6, SMAD2/3, and SMAD1 proteins in DNFB‐ versus acetone‐treated WT skin (n = 4 in each group).
- Quantitative PCR (qPCR) of Il33 and Smad6 mRNA in Pam212 cells treated with poly (I:C) or PBS (carrier control, n = 4 in each group).
- ChIP‐qPCR assay for Smad6 in the presence of poly (I:C) or PBS using an anti‐IL‐33 antibody. After Pam212 cells were transfected with poly (I:C) for 24 h, cell lysates were subjected to chromatin immunoprecipitation with anti‐IL‐33 antibody and eluted IL‐33‐bound chromatin was used for qPCR with Smad6 regulatory peak region (63,968k) primers and Smad6 no peak region (63,955k) primers (n = 4 in each group).
- Immunoblot of p‐SMAD2/3 and SMAD2/3 proteins in response to TGF‐β and poly (I:C) treatment. Pam212 cells were treated with poly (I:C) or PBS followed by incubation with and without TGF‐β (5nM). Data represent three independent experiments with similar results.
- Immunoblot of p‐SMAD2/3 and SMAD2/3 proteins in response to TGF‐β + poly (I:C) after knocking down of IL‐33. Pam212 cells were transfected with siIl33 knockdown construct or siRNA control (siCon) for 30 h followed by incubation with poly (I:C) and TGF‐β. Data represent three independent experiments with similar results.
- Immunoblot of p‐SMAD2/3 and SMAD2/3 proteins upon the expression of IL‐33 full length or cytokine domain. Pam212 cells were transfected with IL‐33 full length or cytokine domain for 24 h followed by incubation with and without TGF‐β. Data represent three independent experiments with similar results.
- Impact of IL‐33 knockdown on cell proliferation in response to TGF‐β. Pam212 cells were treated with siIl33 or siCon followed by TGF‐β treatment (n = 7 in each group).
- Impact of IL‐33 knockdown and SB431542 treatment on cell proliferation. Pam212 cells were treated with siIl33 or siCon in combination with SB431542 or DMSO (carrier control, n = 7 in each group).
- Representative images of H&E‐ and Ki67‐stained back skin of IL‐33KO and WT mice treated with 22‐day DNFB protocol in combination with SB431542 or acetone (carrier control). Note the epidermal thickness, keratinocytes proliferation, and dermal inflammation in each group (scale bars: 100 μm).
- The epidermal thickness of DNFB‐treated IL‐33KO and WT skin after treatment with SB431542 or acetone. Each dot represents the average of three measurements in an HPF image. 10 random HPF images per skin sample are included (n = 10 for WT with acetone, n = 8 for WT with SB431542, n = 6 for IL‐33KO with acetone, n = 5 for IL‐33KO with SB431542 per group).
Data information: GAPDH is used as the control housekeeping protein in (A, D‐F). Graphs show mean + SD, NS: not significant, unpaired t‐test.
Source data are available online for this figure.
Next, we examined whether IL‐33 can directly regulate TGF‐β/SMAD signaling within the epithelial cells using polyinosinic‐polycytidylic acid (poly (I:C)), a TLR3 agonist that can induce IL‐33 and elicit an inflammatory response in epithelial cells (Polumuri et al, 2012; Natarajan et al, 2016). Poly (I:C) treatment led to Il33 induction and concomitant Smad6 repression in Pam212 cells (Fig 3B). Poly (I:C) treatment led to increased IL‐33 protein levels in the nuclei of Pam212 cells (Appendix Fig S5A). Poly (I:C)‐induced endogenous IL‐33 bound specifically to Smad6 gene regulatory region corresponding to a peak in our ChIP‐Seq results (Fig 3C and Appendix Fig S5B). We tested the impact of endogenous IL‐33 on RUNX2 binding to Smad6 promoter using siRNA against IL‐33 (siIl33) to knock down IL‐33 levels in Pam212 cells. RUNX2 binding to Smad6 promoter was increased after IL‐33 knockdown (Appendix Fig S5C). In contrast, IL‐33 induction by poly (I:C) led to a markedly reduced RUNX2 binding to Smad6 promoter, which was partially rescued by IL‐33 knockdown in poly (I:C)‐treated cells (Appendix Fig S5C). Accordingly, poly (I:C) highly augmented the effect of TGF‐β treatment on Pam212 cells as shown by increased p‐SMAD2/3 levels even though endogenous SMAD2/3 levels remained unchanged (Fig 3D and Appendix Fig S5D). Importantly, poly (I:C)‐induced p‐SMAD2/3 upregulation was reversed when IL‐33 was knocked down using siIl33 (Fig 3E and Appendix Fig S5E). The overexpression of IL‐33 full length, but not cytokine domain, in TGF‐β‐treated Pam212 cells increased p‐SMAD2/3 levels (Fig 3F and Appendix Fig S5F). These data demonstrate that nuclear IL‐33 promotes TGF‐β/SMAD signaling within the epithelial cells.
TGF‐β/SMAD signaling pathway can promote cell proliferation (Zhang et al, 2017a). Therefore, we examined whether IL‐33 regulates the TGF‐β/SMAD signaling effect on epithelial cell proliferation. Knocking down the endogenous IL‐33 levels in TGF‐β‐treated Pam212 cells resulted in a significant reduction in cell proliferation compared with TGF‐β plus control siRNA‐treated group (Fig 3G and Appendix Fig S6A). Endogenous TGF‐β expression in Pam212 cells was not affected by knocking down IL‐33 (Appendix Fig S6B). Treatment of Pam212 cells with SB431542, an inhibitor of TGF‐β receptor (Hjelmeland et al, 2004; James et al, 2010), significantly reduced Ki67+ proliferating cells (Appendix Fig S6C and D). SB431542 treatment reduced Pam212 cell proliferation to the same degree as knocking down IL‐33 (Fig 3H). Importantly, knocking down IL‐33 in Pam212 cells that were treated with SB431542 did not further reduce cell proliferation (Fig 3H). These results indicate that IL‐33 regulates cell proliferation via TGF‐β/SMAD signaling pathway. To investigate the interplay of IL‐33 and TGF‐β/SMAD signaling in promoting cell proliferation in vivo, WT and IL‐33KO mice were treated with DNFB plus either SB431542 (test) or acetone (control) for 22 days. DNFB‐induced epidermal hyperplasia and keratinocyte proliferation in WT mice was significantly attenuated in IL‐33KO mice (Fig 3I and J) (Ameri et al, 2019). However, no change in epidermal keratinocyte apoptosis was observed (Appendix Fig S6E). SB431542 treatment markedly decreased the epidermal hyperplasia and keratinocyte proliferation in DNFB‐treated WT mice (Fig 3I and J). However, SB431542 treatment had no impact on the epidermal hyperplasia or keratinocyte proliferation in DNFB‐treated IL‐33KO mice (Fig 3I and J). These findings demonstrate that nuclear IL‐33 promotes epithelial cell proliferation via TGF‐β/SMAD signaling pathway.
Nuclear IL‐33/SMAD signaling pathway promotes skin carcinogenesis
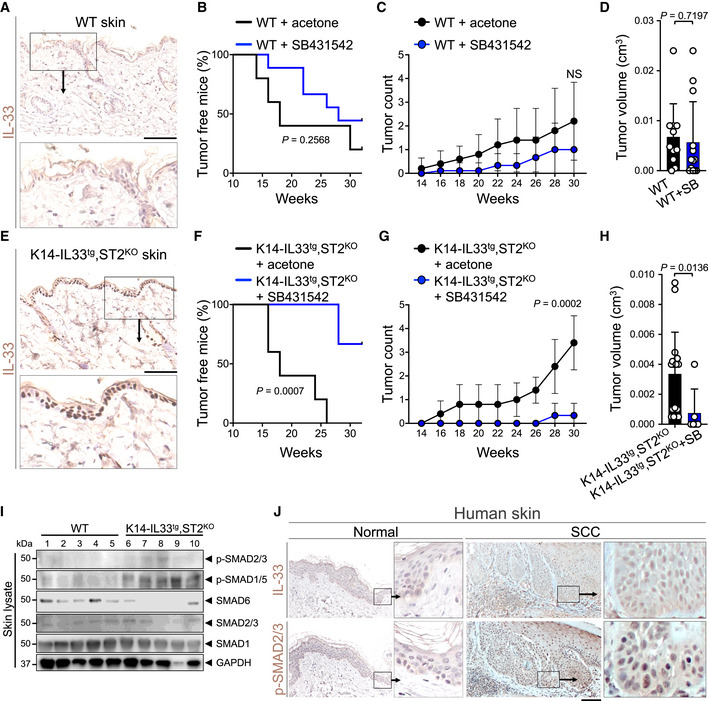
To examine the specific impact of nuclear IL‐33/SMAD signaling on skin cancer, we used the standard skin carcinogenesis protocol in which mice were treated with one dose of DMBA followed by topical application of 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA) three times a week for 30 weeks (Appendix Fig S7A). TPA treatment caused a significantly less IL‐33 induction in the skin compared with DNFB treatment (Appendix Fig S7B). Considering the low levels of nuclear IL‐33 in WT skin keratinocytes (Fig 4A), blocking the TGF‐β/SMAD signaling pathway by SB431542 over the 30‐week DMBA/TPA treatment had no significant impact on skin tumor onset, tumor numbers over time, or endpoint tumor volumes in WT mice (Fig 4B–D and Appendix Fig S7C). To study the impact of TGF‐β/SMAD signaling on skin cancer development in the presence of increased nuclear IL‐33 while excluding IL‐33 cytokine function, we generated K14‐IL33tg,ST2KO mice that overexpressed IL‐33 specifically in their skin keratinocytes but lacked IL‐33 cytokine receptor (ST2) on all their cells (Fig 4E). K14‐IL33tg,ST2KO mice treated with SB431542 showed markedly increased tumor latency and developed significantly fewer skin tumors compared with acetone‐treated mice in response to DMBA/TPA treatment (Fig 4F and G). Moreover, skin tumors that developed in K14‐IL33tg,ST2KO mice treated with DMBA/TPA and SB431542 were smaller than those developed in DMBA/TPA‐treated K14‐IL33tg,ST2KO controls (Fig 4H and Appendix Fig S7C). SB431542 treatment reduced the number of p‐SMAD2/3+ and Ki67+ epidermal keratinocytes without impacting apoptosis in DMBA/TPA‐treated K14‐IL33tg,ST2KO skin compared with the acetone‐treated controls (Appendix Fig S7D and E). Consistent with the specific requirement for TGF‐β/SMAD signaling in K14‐IL33tg,ST2KO skin tumor promotion, DMBA/TPA‐treated K14‐IL33tg,ST2KO skin had higher expression of both p‐SMAD2/3 and p‐SMAD1/5 and lower expression of SMAD6 compared with WT mice (Fig 4I and Appendix Fig 8A). These results demonstrate that IL‐33 induction promotes skin cancer development via the TGF‐β/SMAD signaling pathway. To determine whether IL‐33/SMAD signaling was also active in human skin cancer, we examined IL‐33 and p‐SMAD2/3 protein levels in human skin cancer versus normal tissues. Consistent with our findings in mice, IL‐33 and p‐SMAD2/3 were significantly upregulated in the nuclei of human cutaneous squamous cell carcinoma (SCC) cells compared with normal skin keratinocytes (Fig 4J and Appendix Fig S8B and C).
Figure 4. Nuclear IL‐33/SMAD signaling promotes skin cancer development.
-
ARepresentative images of IL‐33 immunostaining on WT skin. Inset highlights the IL‐33‐negative nuclei in the epidermis (scale bar: 100 μm).
-
B–DSkin tumor outcomes in WT mice treated with DMBA/TPA carcinogenesis protocol in combination with SB431542 (test, n = 9) or acetone (carrier control, n = 5). (B) Tumor latency (log‐rank test), (C) skin tumor counts per mouse over time, and (D) skin tumor volume at the completion of the DMBA/TPA carcinogenesis protocol.
-
ERepresentative images of IL‐33 immunostaining on K14‐IL33tg,ST2KO skin. Inset highlights the IL‐33‐positive nuclei in the epidermis (scale bar: 100 μm).
-
F–HSkin tumor outcomes in K14‐IL33tg,ST2KO mice treated with DMBA/TPA carcinogenesis protocol in combination with SB431542 (test, n = 6) or acetone (control, n = 5). (F) Tumor latency (log‐rank test), (G) skin tumor counts per mouse over time, and (H) skin tumor volume at the completion of the DMBA/TPA carcinogenesis protocol.
-
ISMAD signaling proteins levels in WT and K14‐IL33tg,ST2KO skin treated with acetone (control) at the completion of the DMBA/TPA carcinogenesis protocol. Tissue lysates prepared from the whole back skin of the animals were subjected to immunoblot with p‐SMAD2/3, p‐SMAD1/5, SMAD6, SMAD2/3, and SMAD1 antibodies. GAPDH is used as the control housekeeping protein (n = 5 in each group).
-
JRepresentative images of IL‐33 and p‐SMAD2/3 immunostaining on the adjacent sections of human squamous cell carcinoma (SCC) and normal skin (scale bar: 100 μm).
Data information: Graphs show mean ± SD, NS: not significant, unpaired t‐test.
Source data are available online for this figure.
Nuclear IL‐33 promotes pancreatic cancer development in chronic pancreatitis
To extend our findings to other epithelial cancers affected by chronic inflammation, we investigated the role of nuclear IL‐33 in pancreatic cancer. Chronic pancreatitis is a common risk factor for developing pancreatic cancer (Bansal & Sonnenberg, 1995; Guerra et al, 2007). To examine the expression and localization of IL‐33 in cancer‐prone chronic pancreatitis, we treated WT mice with intraperitoneal injections of caerulein in phosphate‐buffered saline (PBS) or PBS alone hourly for 6 h per day, three days per week for 4 weeks (Appendix Fig S9A) (Lin et al, 2014). Caerulein treatment led to the development of chronic pancreatitis and a significant upregulation of IL‐33 in the WT pancreas (Appendix Fig S9B and C). IL‐33 and p‐SMAD2/3 were highly increased in the nuclei of pancreatic acinar cells in caerulein compared with PBS‐treated WT mice (Fig 5A). Consistent with the nuclear function of IL‐33 in epithelial cells, Il33 upregulation associated with a marked reduction in Smad6 expression in caerulein compared with the PBS‐treated pancreas (Fig 5B). Furthermore, p‐SMAD2/3 and p‐SMAD1/5 were increased and SMAD6 was decreased in caerulein compared with PBS‐treated pancreas (Fig 5C and Appendix Fig S9D). These results suggest that nuclear IL‐33 via the induction of TGF‐β/SMAD signaling may promote the transformation of pancreatic acinar cells to PDAC in the context of chronic pancreatitis.
Figure 5. Nuclear IL‐33 promotes pancreatic cancer development.
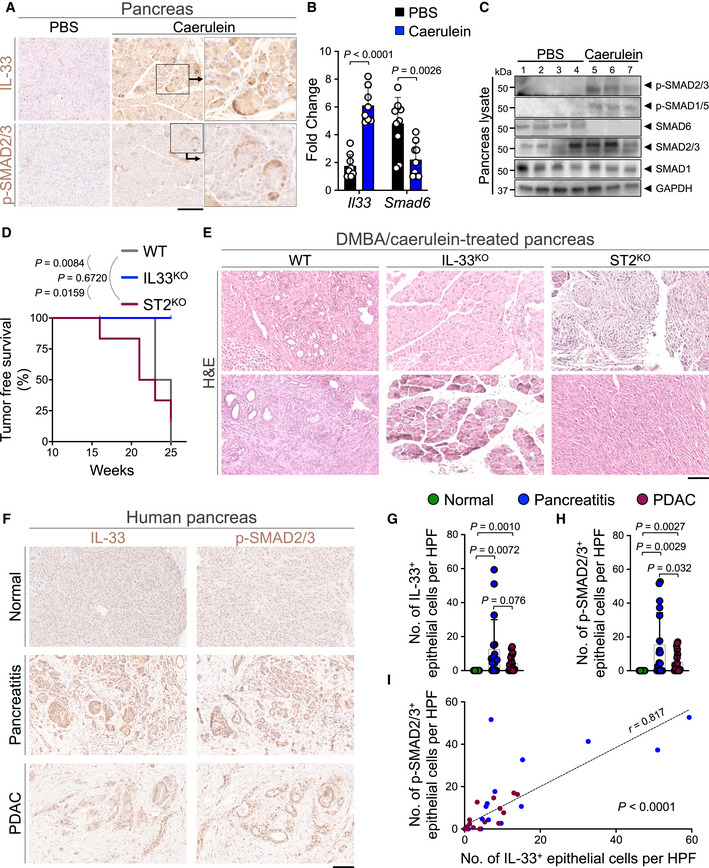
-
ARepresentative images of IL‐33 and p‐SMAD2/3 immunostaining on WT pancreas treated with caerulein or PBS (carrier control) to induce chronic pancreatitis (scale bar: 100 μm).
-
BIl33 and Smad6 mRNA levels in caerulein‐treated WT pancreas compared with PBS‐treated controls at the completion of chronic pancreatitis protocol (n = 8 in caerulein and n = 9 in PBS group for Il33, n = 9 in caerulein and n = 10 in PBS group for Smad6, unpaired t‐test).
-
CImmunoblot for p‐SMAD2/3, p‐SMAD1/5, SMAD6, SMAD2/3, and SMAD1 proteins in lysates from caerulein (n = 3)‐ and PBS (n = 4)‐treated WT pancreas. GAPDH is used as the control housekeeping protein.
-
DPancreatic tumor‐free survival of WT (n = 4), IL‐33KO (n = 5), and ST2KO (n = 6) mice at the completion of the DMBA/caerulein carcinogenesis protocol (log‐rank test).
-
ERepresentative H&E images of two pancreatic tissues from WT, IL‐33KO, and ST2KO groups treated with DMBA/caerulein (scale bar: 100 μm).
-
FRepresentative images of IL‐33 and p‐SMAD2/3 immunostaining on adjacent sections of matched normal pancreas, chronic pancreatitis, and PDAC collected from pancreatic cancer patients (scale bar: 100 μm).
-
G, HQuantification of nuclear (G) IL‐33+ and (H) p‐SMAD2/3+ epithelial cells per HPF in the matched human pancreatic tissues. Each dot represents the average cell counts across three randomly selected HPF images per sample (n = 18 patients, paired t‐test).
-
ICorrelation between IL‐33+ and p‐SMAD2/3+ epithelial cell counts across normal pancreas, chronic pancreatitis, and PDAC samples (n = 54 matched samples from 18 patients, t‐test for the Pearson correlation coefficient).
Data information: Graphs show mean + SD.
Source data are available online for this figure.
To examine the impact of nuclear IL‐33 on pancreatic cancer development in chronic pancreatitis, IL‐33KO, ST2KO, and WT mice received an intrapancreatic injection of DMBA followed by intraperitoneal injections of caerulein once a day, three‐time a week for 25 weeks (Appendix Fig S9E; Guerra et al, 2007; Scarlett et al, 2011). DMBA/caerulein‐treated WT mice developed PDAC while IL‐33KO mice only showed mild pancreatitis with no pancreatic cancer detectable at the completion of the 25‐week DMBA/caerulein treatment (Fig 5D and E, and Appendix Fig S9F). DMBA/caerulein treatment caused severe pancreatitis in ST2KO mice, which led to their early demise compared with IL‐33KO animals (Fig 5D). Post‐mortem analysis revealed severe fibrotic tumors in the pancreas of ST2KO mice (Fig 5E and Appendix Fig S9F). To determine whether IL‐33/SMAD signaling is active in pancreatitis‐associated PDAC in humans, we examined IL‐33 and p‐SMAD2/3 expression in the nuclei of the epithelial cells across 18 matched samples of the normal pancreas, pancreatitis, and pancreatitis‐associated PDAC. IL‐33 and p‐SMAD2/3 were highly expressed in the epithelial cells in pancreatitis and pancreatitis‐associated PDAC samples (Fig 5F–H). Moreover, the number of IL‐33+ epithelial cells was positively correlated with the number of p‐SMAD2/3+ epithelial cells across the samples (Fig 5I). Collectively, these outcomes demonstrate that the nuclear IL‐33/SMAD signaling axis promotes cancer development in the context of chronic inflammation in the skin and pancreas.
Discussion
Our findings reveal that IL‐33 has a tumor‐promoting role as a nuclear protein, which is independent of its cytokine function. Nuclear IL‐33 binds transcription factor RUNX2 thereby preventing RUNX2 from binding to Smad6 promoter. By suppressing Smad6 expression, nuclear IL‐33 elevates p‐SMAD2/3 and p‐SMAD1/5 levels in the skin and pancreatic epithelial cells during chronic inflammation. The IL‐33‐dependent activation of the TGF‐β/SMAD signaling pathway accelerates epithelial cell proliferation and contributes to cancer development in chronic inflammation. Therefore, blocking the nuclear IL‐33/SMAD signaling axis with small molecule inhibitors is a novel strategy for cancer treatment and prevention, which can impact a large array of malignancies with increased IL‐33 and SMAD signaling within the cancer cells.
Previous studies have shown that IL‐33 can act as a transcriptional repressor by inhibiting NF‐κB protein and thereby dampening its transcription activity in vitro and function as histone methyltransferase in endothelial cells (Carriere et al, 2007; Ali et al, 2011). In addition, the cell‐intrinsic role of IL‐33 in regulatory T cells is found to be essential for their suppressive function in the tumor microenvironment (Hatzioannou et al, 2020). However, IL‐33 is mostly known as an epithelium‐derived alarmin cytokine in allergic diseases and cancer (He et al, 2017; Chan et al, 2019). We demonstrate that the N‐terminal domain of IL‐33, which interacts with RUNX2, plays an essential role in nuclear IL‐33’s regulation of the SMAD signaling pathway. Thus, our discovery of IL‐33 nuclear function in epithelial cells of skin and pancreas provides novel insights into the pathogenesis of cancer and inflammation in these organs. Ultimately, our work reveals that nuclear IL‐33 functions to potentiate TGF‐β/SMAD signaling in epithelial cells by repressing Smad6 expression via interaction with RUNX2. The novel cell‐autonomous effect of IL‐33 in cancer cells suggests an innovative therapeutic path to blocking cancer development in chronic inflammation by targeting the IL‐33/SMAD signaling axis with small molecules instead of antibodies against IL‐33 cytokine.
Our work provides a much‐needed insight into the pathogenesis of pancreatic cancer. It is well established that pancreatic cancer arises in the context of chronic inflammation and contains a severely immunosuppressive environment nonresponsive to immunotherapeutics (Torphy et al, 2018). TGF‐β/SMAD signaling is found to be upregulated in PDAC (Perera & Bardeesy, 2015). This signaling pathway can induce cell proliferation, angiogenesis, and epithelial–mesenchymal transition (Jakowlew, 2006; Colak & Ten Dijke, 2017). In addition, the inhibition of SMAD signaling reduces PD‐L1 and PD‐L2 expression, which indicates the importance of this signaling pathway in creating an immunosuppressive microenvironment (Martinez et al, 2014). However, it is still unclear how this pathway is regulated in pancreatic cancer. Our research provides a mechanism for this upregulation by demonstrating that nuclear IL‐33, which is induced in the inflamed epithelia, promotes TGF‐β/SMAD signaling within the cancer cells. The majority of IL‐33 and p‐SMAD2/3 proteins are localized in the nuclei of the pancreatic acinar cells in cancer‐prone chronic pancreatitis of mice and humans. Pancreatic acinar cells are considered the prime origin of PDAC (Guerra et al, 2007). Therefore, our findings suggest that the IL‐33/SMAD signaling axis is involved in the transformation of acinar cells into cancer cells that form in the context of chronic pancreatitis.
Previous studies have provided conflicting views of IL‐33’s role in cancer development. The IL‐33/ST2 axis has been shown to promote intestinal tumorigenesis and metastasis of colorectal cancer, suggesting that IL‐33 plays a tumor‐promoting role in the colon (Maywald et al, 2015; He et al, 2017; Zhang et al, 2017b; Ameri et al, 2019). However, these findings have been countered by evidence that IL‐33 inhibits colorectal tumor growth, suggesting a tumor‐suppressing role instead (Malik et al, 2016; Chen et al, 2018). These studies have focused on the cytokine function of IL‐33. Based on our results, we propose a need to distinguish the cellular source of IL‐33 (epithelial versus stromal) and its cell‐autonomous versus cytokine function across malignancies in which IL‐33 has been implicated in order to reconcile the divergent conclusions. Interestingly, a recent report shows that IL‐33/TGF‐β cytokine loop between tumor‐initiating cells and macrophages promotes skin cancer progression (Taniguchi et al, 2020). Our findings suggest that nuclear IL‐33 can further amplify TGF‐β signaling within the tumor‐initiating cells. Ultimately, understanding the distinct contributions of nuclear versus secreted IL‐33 is key to developing proper therapeutic strategies directed at IL‐33 in cancer and inflammatory diseases.
Materials and Methods
Human samples
De‐identified slides of formalin‐fixed paraffin‐embedded human tissue sections were obtained from the Department of Pathology at Massachusetts General Hospital.
Animal studies
All mice were housed under pathogen‐free conditions in an animal facility at Massachusetts General Hospital in accordance with animal care regulations. WT C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). K14‐IL33tg mice were purchased from the Transgenic Inc. (Kobe, Japan). IL‐33KO mice were a gift from Dr. Marco Colonna, and ST2KO mice were a gift from Dr. Peter Nigrovic. All mouse strains are on the C57BL/6 background. Age‐ and gender‐matched mice were used in all experiments.
Skin chronic inflammation
Four‐ to six‐week‐old male and female mice were shaved on their abdomen and sensitized to 50 μl 0.5% 1‐Fluoro‐2,4‐dinitrobenzene (DNFB, Millipore Sigma, St. Louis, MO, catalog no. 42085) dissolved in acetone plus olive oil at a 3:1 ratio (refer to as acetone). Two days after first sensitization, mice were re‐sensitized to 50 μl 0.25% DNFB on their abdomen. After 5 days, mice were challenged with 100 μl 0.25% DNFB on their back skin, which was repeated three times per week for 22 days. The skin rash was monitored over the duration of the study.
Skin carcinogenesis in chronic inflammation
For DMBA/DNFB carcinogenesis protocol (Ameri et al, 2019), four‐ to six‐week‐old male and female mice were shaved on their abdomen and sensitized to 50 μl 0.5% DNFB. Two days later, mice were re‐sensitized to 50 μl 0.25% DNFB on the abdomen. Five days later, mice were treated with 100 μg 7,12‐dimethylbenz(a)anthracene (DMBA, Millipore Sigma, catalog no. D3254) in 200 μl acetone on their back skin. After a week, mice were challenged with 100 μl 0.25% DNFB on their back skin, which was continued three times per week for 30 weeks at the dosage that maintained mild erythematous dermatitis in WT mice. Skin rash and tumor development were monitored and recorded weekly over the duration of the study. For DMBA/TPA carcinogenesis protocol, mice were treated with 6 μg 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA, Millipore Sigma, catalog no. P1585) in 200 μl acetone instead of DNFB in DMBA/DNFB protocol without abdominal sensitization.
Skin treatment with SB431542
To inhibit the TGF‐β/SMAD signaling pathway (Mordasky Markell et al, 2010), mice were treated with 10 μM SB431542 (Millipore Sigma, catalog no. 301836‐41‐9) in 200 μl acetone or 200 μL acetone alone to their back skin twice a week. SB431542/acetone treatments were given in between TPA or DNFB doses.
Colitis‐associated colorectal carcinogenesis
Mice were weighed and injected intraperitoneally with 10 mg/kg Azoxymethane (AOM, Millipore Sigma, catalog no. A5486) in 100 μL PBS. One week later, they received a 2% dextran sodium sulfate (DSS, Millipore Sigma, catalog no. 42867) solution in their drinking water for 5 days. After DSS administration, drinking water was changed to regular water for two weeks. This AOM/DSS protocol was repeated for five cycles. Mice were harvested 20 weeks after initial AOM injection.
Chronic pancreatitis
Mice were weighed and injected with 50 μg/kg caerulein (BACHEM, Torrance, CA, catalog no. 4030451) in 100 μl of PBS intraperitoneally every hour for 6 h, three days per week for 4 weeks and mice were harvested.
Pancreatic cancer in chronic pancreatitis
Mice were anesthetized with isoflurane, and the body and tail of the pancreas were exposed through a right lateral subcostal incision in the abdominal wall and exteriorization of the spleen. 1 mg DMBA in 50 μL PBS with 0.001% of the Tween‐20 solution was injected into the tail of the pancreas using a 25‐G needle (Scarlett et al, 2011). The spleen and pancreas were returned into the abdominal cavity, and the peritoneum/muscle layers and skin were sutured and covered with CURAD ointment and petrolatum gauze for a week. One week later, mice were injected with 50 μg/kg caerulein in 100 μl of PBS once a day, three days a week, until harvesting. Mice were monitored for any sign of distress, weight loss, or palpable abdominal mass. Mice were harvested 25 weeks after DMBA treatment.
Cell lines, plasmids, and transfection
Pam212 cells (Mouse keratinocyte cell line) were grown at 37°C in DMEM (Thermo Fisher Scientific, Waltham, MA, catalog no. 11995065) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, catalog no. 26140079), 1X penicillin–streptomycin–glutamine (Thermo Fisher Scientific, catalog no. 10378016), 1X MEM non‐essential amino acid solution (Thermo Fisher Scientific, catalog no. 11140050), 1X HEPES (Thermo Fisher Scientific, catalog no. 15630080), and 0.1% 2‐Mercaptoethanol (Thermo Fisher Scientific, catalog no. 21985023). PyMT cell lines (derived from a primary breast tumor of MMTV‐PyMTtg mouse on a C57BL/6 background) were grown at 37°C in RPMI Medium 1640 (Thermo Fisher Scientific, catalog no. 11875093) supplemented with 10% fetal bovine serum, 1X penicillin–streptomycin–glutamine and 0.1% 2‐Mercaptoethanol. cDNAs for mouse IL‐33 full length and IL‐33 cytokine domain were subcloned in pcDNA3.1‐HA vector. Transfections of plasmids or Poly (I:C) (Invivogen, San Diego, CA, catalog no. tlrl‐pic) in Pam212 cells were performed using Lipofectamine 2000 (Thermo Fisher Scientific, catalog no. 11668019) according to the manufacturer’s instructions.
Western blot
Cell lysates were prepared in LIPA buffer (Thermo Fisher Scientific, catalog no. 89900) consisting of 1X protease inhibitor cocktail, EDTA‐free (Thermo Fisher Scientific, catalog no. A32955). Mice tissues were meshed and lysed by 0.1% Tween‐20 (Millipore Sigma, catalog no. P1379) in PBS. Mice tissues were frozen by liquid nitrogen and thaw by incubation at 37°C for further lysis. Cell lysates were sonicated for 20 s, and then, they were centrifugated at 17,000 g. After checking the protein concentration of each sample, the same amount of proteins was loaded into Mini‐PROTEIN TGXTM Gels (BIO‐RAD, Hercules, CA, catalog no. 456‐1083 or 456‐1086) with 1X Tris/Glycine/SDS buffer (BIO‐RAD, catalog no. 1610732). A few minutes later (according to protein size), they were transferred to Immobilon–P membrane (Millipore Sigma, catalog no. IPVH00010) with Transfer buffer (Boston Bioproducts, Ashland, MA, catalog no. BP‐190). Then, they were incubated 3% bovine serum albumin (Thermo Fisher Scientific, catalog no. BP1600) or 5% Skim milk (BD biosciences, San Jose, CA, catalog no. 232100) in 1X Tris‐Buffered Saline (Boston Bioproducts, catalog no. BM301X) containing 0.1% Tween, called TBS‐T for 30 min. After the membrane was washed with TBS‐T, they were subjected to immunoblot with proper antibodies overnight at 4°C. The following day, they were incubated with proper secondary antibodies and were developed with Pierce ECL Western blotting substrate kit (Thermo Fisher Scientific, catalog no. 32106). First and secondary antibodies are found in Appendix Table S2.
Western blot quantification
Western blot bands were quantified with cSeries Capture software (Azure biosystem, Dublin, CA, catalog no. Azure 600). Each band quantity was calculated by measuring band intensity minus the background. Total proteins were normalized based on GAPDH levels. p‐SMAD2/3 and p‐SMAD1/5 were normalized based on endogenous SMAD2/3 and SMAD1 levels, respectively. Pictures of full Western blot gels are provided as a source data file.
Immunoprecipitation
Cell lysates were prepared in buffer A consisting of 50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.5% NP‐40 and 1X protease inhibitor cocktail. After confirming the concentration of total proteins, the cell lysates were incubated with IL‐33 (Nessy‐1) overnight at 4°C and incubated with 30 μl of 50% slurry of protein A‐conjugated Sepharose (Cell Signaling Technology, Danvers, MA, catalog no. 9863S) for the next 2 h. The samples were precipitated, washed three times with buffer A containing 300 mM NaCl. Samples were added to 25 μl SDS sampling buffer (Boston Bioproducts, catalog no. BP‐111R) and boiled at 95°C for 10 min. Samples were subjected to SDS–PAGE followed by immunoblot analysis.
RNA‐Seq
IL‐33KO and WT mice were treated with DNFB‐induced skin inflammation protocol (Appendix Fig S2A) and epidermis was isolated from the back skin. RNA was extracted using an RNeasy micro kit (Qiagen, Hilden, Germany, catalog no. 74004) and quantified using Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA). Libraries were prepared by Novogene (Sacramento, CA) using the NEBNext Ultra RNA Library Prep kit for Illumina (New England Biolabs, Ipswich, MA, catalog no. E7770). Sequencing was performed by Novogene using the Illumina NovaSeq6000 System. Reads were aligned to the mouse reference genome (mm10) using STAR. Differential expression analysis was performed by Novogene using the DESeq2R package. Each group includes three mice. Original data are available in the NCBI Gene Expression Omnibus (GEO) with accession number GSE149579 (RNA‐Seq).
ChIP‐qPCR assay
Pam212 cells that had been transfected with plasmids were fixed in 1% formaldehyde (Millipore Sigma, catalog no. F8775) for 10 min and were washed by cold PBS. Cells were lysed with buffer contained with 2.5% of glycerol, 50 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, 0.5% NP‐40, and 0.25% Triton X‐100. After centrifugation at 400 g, cell lysates were resuspended in buffer A contained with 1 mM Tris–HCl (pH 7.9), 20 mM NaCl, and 0.5 mM EDTA and incubated at room temperature for 10 min. Following the second centrifugation at 400 g, cells were sonicated in sonication buffer containing 10 mM HEPES, 1 mM EDTA, and 0.5% SDS for 30 min to achieve chromatin fragmentation. Following centrifugation at 16,200 g, proteins were immunoprecipitated with anti‐RUNX2 or anti‐IL‐33 antibody at 4°C overnight. The next day, the samples were incubated with 30 μl 50% slurry of ChIP‐Grade Protein G agarose beads (Cell Signaling Technology, catalog no. 9007S) for 2 h. Samples were precipitated, washed three times with wash buffer containing 20 mM Tris–HCl (pH 7.9), 10 mM NaCl, 2 mM EDTA, 0.1% SDS, and Triton X‐100. After the last wash step, samples were eluted with 100 μl of TE buffer contained with 100 mM Tris–HCl (pH 8), 1 mM EDTA, and 1% SDS three times. De‐crosslinking was performed by overnight incubation with 15 μl of 3 M NaCl at 65°C. Precipitated DNA was purified by ChIP DNA Clean & Concentrator (ZYMO research, Irvine, CA, catalog no. D5205) and subjected to quantitative PCR.
ChIP‐Seq
Mouse Pam212 cell line was transfected with HA‐IL‐33 full length or HA‐IL‐33 cytokine domain or HA‐empty vector. Transfections of these vectors were performed using Lipofectamine 2000 according to the manufacturer’s instructions. After 24 h, each cell lysate underwent ChIP assay using anti‐HA antibody and eluted DNA was purified by ChIP DNA Clean & Concentrator to obtain 50 ng DNA for each sample. Sequencing was performed by Novogene using the Illumina NovaSeq6000 System. For the raw data which passed the quality control, the adapter sequences and low‐quality bases were trimmed off to get clean data. The MACS2 software was used to predict the fragment sizes of IP experiments. The fragment sizes were used in the peak calling. MACS2 software (threshold q value = 0.05) was used to perform peak calling and find the peak‐related genes (Zhang et al, 2008). Original data are available in the NCBI Gene Expression Omnibus (GEO) with accession number GSE149579 (ChIP‐Seq).
Histology, immunohistochemistry, and immunofluorescence
Tissue samples were collected and fixed in 4% paraformaldehyde (Millipore Sigma, catalog no. P6148) overnight at 4°C. Next, tissues were dehydrated in ethanol, processed, and embedded in paraffin. 5 μm sections paraffin‐embedded tissues were placed on slides, deparaffinized, and stained with H&E. For immunohistochemistry, antigen retrieval was performed in 500 μl of antigen unmasking solution (VECTOR Laboratories, Burlingame, CA, catalog no. H3300) diluted in 50 ml water using Cuisinart pressure cooker for 20 min at high pressure. Slides were washed three times for 3 min each in 1X TBS with 0.025% Triton X‐100. Sections were blocked with 1% bovine serum albumin (Thermo Fisher Scientific, catalog no. BP1600) and 5% goat serum (Millipore Sigma, catalog no. G9023) for 1 h. Slides were stained overnight at 4°C with a primary antibody diluted in TBS containing 1% BSA (Appendix Table S2). The following day, slides were washed as above and incubated in 100 μl biotinylated secondary antibody (VECTOR Laboratories, catalog no. PK‐6200) for 30 min. After washing, slides were incubated with 100 μl mixture of reagent A and B from VECTASTAIN Elite ABC universal kit Peroxidase (VECTOR Laboratories, catalog no. PK‐6200) for 30 min. After washing, slides were incubated with 100 μl ImmPACT DAB chromogen stating (VECTOR Laboratories, catalog no. SK‐4105) for a few minutes. Finally, slides were dehydrated in ethanol and xylene and mounted with a coverslip using 3 drops of mounting media. For immunofluorescence staining, rehydrated tissue sections were permeated with 1XPBS supplemented with 0.2% Triton X‐100 for 5 min. Antigen retrieval was performed similar to immunohistochemistry. Slides were washed three times for 3 min each in 1XPBS with 0.1% Tween‐20. Sections were blocked with 1% bovine serum albumin and 5% goat serum for 1 h. The slides were stained overnight at 4°C with primary antibodies (Appendix Table S2). The following day, slides were washed as above and incubated for 2 h at room temperature with secondary antibodies conjugated to fluorochromes (Appendix Table S2). Next, slides were incubated with 1:2,000 DAPI (Thermo Fisher Scientific, catalog no. D3571) for 5 min at room temperature, then washed as above. Slides were mounted with Prolong Gold Antifade Reagent (Thermo Fisher Scientific, catalog no. P36930). The number of positive cells was counted in randomly selected high power field (HPF, 200× magnification) images in a blinded manner by a trained investigator. Clinical samples were reviewed by a pathologist.
Quantitative PCR
Mouse dorsal skin samples were homogenized and lysed in RLT lysis solution (QIAGEN, catalog no. 79216)/0.1% MeOH using Mini‐BeadBeater‐8 (BioSpec Products, Inc., Bartlesville, OK). Total RNA was extracted using an RNeasy micro kit and quantified using NanoDrop ND‐1100 (NanoDrop Technologies, Wilmington, DE). cDNA samples were synthesized from 1 μg of total RNA using Invitrogen SuperScripts III Reverse Transcriptase (Thermo Fisher Scientific, catalog no. 18080085). Expression levels of all cDNA samples were determined with LightCycler 480 system (Roche Molecular Systems Inc., Pleasanton, CA) using iTaq Universal SYBR green supermix (Bio‐Rad Laboratories, Hercules, CA, catalog no. 1725121) or TaqMan Universal Master Mix II (Thermo Fisher Scientific, catalog no. 44‐400‐40). Primer sequences for SYBR green and TaqMan assays are listed in Appendix Table S2. Quantitative real‐time PCR for SYBR green analyses were performed in a final reaction volume of 25 μl consisting of 5 μl of cDNA of the respective sample and 12.5 μl of SYBR green master mix mixed with the corresponding primers (2 μM) for each gene. The TaqMan analysis was performed in a final reaction of 10 μl with 4.5 μl cDNA and 5.5 μl TaqMan master mix and corresponding primers (20 μM). All assays were conducted in triplicate and normalized to Gapdh expression.
PCR
DNA was isolated from mouse tissue using KAPA Express Extract buffer and KAPA Express extract Enzyme from Mouse genotyping kit (Kapa Biosystems Inc, Wilmington, MA, catalog no. KK7302). After tissue lysis, PCR was performed with a 2X KAPA2G Fast genotyping mix from the same kit using primers listed in Appendix Table S2.
Proliferation assay
Pam212 cells were transfected with siIl33 (QIAGEN, catalog no. SI00870443) or siCon (QIAGEN, catalog no. 1022076) using Lipofectamine RNAiMax Transfection reagent (Thermo Fisher Scientific, catalog no. 13778075) according to the manufacturer’s instructions. The following day, cells were divided into 96‐well plates with a serial dilution of cells. The next day, the cells were treated with 5 nM of TGF‐β (Cell Signaling Technology, catalog no. 5231LF) or 10 μM of SB431542 for 3 h. After incubation, the proliferation assay was performed by CellTiter 96 Non‐radioactive Cell proliferation assay (Promega, Madison, WI, catalog no. G4001) according to the manufacturer’s instructions. O.D was recorded as the measure of cell proliferation in each well. Absorbance (O.D) reading is directly proportional to the cell proliferation in each well.
Nuclear extraction
Pam212 cell line was transfected with poly (I:C) using Lipofectamine 2000 according to the manufacturer’s instructions. After 8 h, cells were lysed using Nuclear Extraction Kit (Novus Biologicals, Littleton, CO, catalog no. NBP2‐29447). Briefly, cells were washed with 1XPBS supplemented with PMSF (Novus biologicals, catalog no. KC‐407). Cells were centrifuged for 5 min at 100 g. Cell pellets were resuspended in 1× cold hypotonic buffer (Novus biologicals, catalog no. KC‐401) and incubated on ice for 15 min. After adding 10 μl of the 10% detergent solution (Novus biologicals, catalog no. KC‐403), cell lysates were centrifuged 10 min at 17,000 g. The supernatants were kept as a cytoplasmic fraction, and the cell pellets were resuspended in 100 μl nuclear lysis buffer (Novus biologicals, catalog no. KC‐402). Lysates from the pellets were sonicated for a few seconds and incubated for 10 min at 4°C. Then, lysates were centrifuged at 17,000 g for 10 min. The supernatants were kept as the nuclear fraction.
Statistical analysis
Unpaired t‐test was used as the test of significance for tumor volume, counts, epidermal thickness, count of nuclear protein staining, RNA and protein expression levels, and other quantitative measurements. The log‐rank test was used as the test of significance for time to tumor onset outcomes. A paired t‐test was used for the comparison of IL‐33+ and pSMAD2/3+ cell counts, and t‐test for the Pearson correlation coefficient was used to determine significance in the correlation between IL‐33+ and pSMAD2/3+ counts across matched human pancreatic samples. P‐value < 0.05 is considered significant. Bar graphs show mean ± SD.
Study approval
Massachusetts General Hospital IRBs approved the analysis of de‐identified clinical samples. Massachusetts General Hospital IACUC approved animal studies.
Author contributions
Conception and design of the experiment: JHP, AHA, and SD. Experiments and data analysis: JHP, AHA, KED, and DNC. Data interpretation and manuscript writing: JHP and SD. Clinical samples and human tissue sample analysis: MK and MM‐K.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgments
We thank Dr. Marco Colonna for IL‐33KO mice and Dr. Peter Nigrovic for ST2KO mice. We thank Kenneth Ngo for his technical assistance. IL‐33KO mice were generated with support from Mucosal Immunology Studies Team (MIST) (U01; RFA‐AI‐15‐023). JHP was supported by the National Research Foundation of Korea (NRF‐2019R1A6A3A03031609). AHA was supported by the Howard Hughes Medical Institute. SD holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. JHP, AHA, KED, DNC, and SD were supported by grants from the Burroughs Wellcome Fund, Arthur, Sandra, and Sarah Irving Fund for Gastrointestinal Immuno‐Oncology, Sidney Kimmel Foundation, and NIH (K08AR068619 and R01AR076013).
The EMBO Journal (2021) 40: e106151.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. RNA‐Seq and ChIP‐Seq data have been deposited to the NCBI Gene Expression Omnibus (GEO, accession number GSE149579; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE149579).
References
- Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, Martin MU (2011) The dual function cytokine IL‐33 interacts with the transcription factor NF‐kappaB to dampen NF‐kappaB‐stimulated gene transcription. J Immunol 187: 1609–1616 [DOI] [PubMed] [Google Scholar]
- Ameri AH, Moradi Tuchayi S, Zaalberg A, Park JH, Ngo KH, Li T, Lopez E, Colonna M, Lee RT, Mino‐Kenudson M et al (2019) IL‐33/regulatory T cell axis triggers the development of a tumor‐promoting immune environment in chronic inflammation. Proc Natl Acad Sci USA 116: 2646–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal P, Sonnenberg A (1995) Pancreatitis is a risk factor for pancreatic cancer. Gastroenterology 109: 247–251 [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D et al (2006) Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 20: 3130–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al (2012) Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. The New England journal of medicine 366: 2455–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP (2007) IL‐33, the IL‐1‐like cytokine ligand for ST2 receptor, is a chromatin‐associated nuclear factor in vivo. Proc Natl Acad Sci USA 104: 282–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan BCL, Lam CWK, Tam LS, Wong CK (2019) IL33: roles in allergic inflammation and therapeutic perspectives. Front Immunol 10: 364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Lu K, Timko NJ, Weir DM, Zhu Z, Qin C, Mann JD, Bai Q, Xiao H, Nicholl MB et al (2018) IL‐33 notably inhibits the growth of colon cancer cells. Oncol Lett 16: 769–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak S, Ten Dijke P (2017) Targeting TGF‐beta Signaling in Cancer. Trends Cancer 3: 56–71 [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140: 883–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez‐Gallego L, Dubus P, Sandgren EP, Barbacid M (2007) Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K‐Ras oncogenes in adult mice. Cancer Cell 11: 291–302 [DOI] [PubMed] [Google Scholar]
- Hatzioannou A, Banos A, Sakelaropoulos T, Fedonidis C, Vidali MS, Kohne M, Handler K, Boon L, Henriques A, Koliaraki V et al (2020) An intrinsic role of IL‐33 in Treg cell‐mediated tumor immunoevasion. Nat Immunol 21: 75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Chen L, Souto FO, Canasto‐Chibuque C, Bongers G, Deshpande M, Harpaz N, Ko HM, Kelley K, Furtado GC et al (2017) Epithelial‐derived IL‐33 promotes intestinal tumorigenesis in Apc (Min/+) mice. Sci Rep 7: 5520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmeland MD, Hjelmeland AB, Sathornsumetee S, Reese ED, Herbstreith MH, Laping NJ, Friedman HS, Bigner DD, Wang XF, Rich JN (2004) SB‐431542, a small molecule transforming growth factor‐beta‐receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol Cancer Ther 3: 737–745 [PubMed] [Google Scholar]
- Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K (1997) Smad6 inhibits signalling by the TGF‐beta superfamily. Nature 389: 622–626 [DOI] [PubMed] [Google Scholar]
- Jakowlew SB (2006) Transforming growth factor‐beta in cancer and metastasis. Cancer Metastasis Rev 25: 435–457 [DOI] [PubMed] [Google Scholar]
- James D, Nam HS, Seandel M, Nolan D, Janovitz T, Tomishima M, Studer L, Lee G, Lyden D, Benezra R et al (2010) Expansion and maintenance of human embryonic stem cell‐derived endothelial cells by TGFbeta inhibition is Id1 dependent. Nat Biotechnol 28: 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA 3rd, Yarchoan M, Lee V, Laheru DA, Jaffee EM (2017) Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin Cancer Res 23: 1656–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Minnich MD, Young PR (1995) ST2/T1 protein functionally binds to two secreted proteins from Balb/c 3T3 and human umbilical vein endothelial cells but does not bind interleukin 1. J Biol Chem 270: 27905–27913 [DOI] [PubMed] [Google Scholar]
- Lin WR, Yen TH, Lim SN, Perng MD, Lin CY, Su MY, Yeh CT, Chiu CT (2014) Granulocyte colony‐stimulating factor reduces fibrosis in a mouse model of chronic pancreatitis. PLoS One 9: e116229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik A, Sharma D, Zhu Q, Karki R, Guy CS, Vogel P, Kanneganti TD (2016) IL‐33 regulates the IgA‐microbiota axis to restrain IL‐1alpha‐dependent colitis and tumorigenesis. J Clin Investig 126: 4469–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangone FR, Walder F, Maistro S, Pasini FS, Lehn CN, Carvalho MB, Brentani MM, Snitcovsky I, Federico MH (2010) Smad2 and Smad6 as predictors of overall survival in oral squamous cell carcinoma patients. Mol Cancer 9: 106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez VG, Hidalgo L, Valencia J, Hernandez‐Lopez C, Entrena A, del Amo BG, Zapata AG, Vicente A, Sacedon R, Varas A (2014) Autocrine activation of canonical BMP signaling regulates PD‐L1 and PD‐L2 expression in human dendritic cells. Eur J Immunol 44: 1031–1038 [DOI] [PubMed] [Google Scholar]
- Massague J (2012) TGFbeta signalling in context. Nat Rev Mol Cell Biol 13: 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS (2000) TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103: 295–309 [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19: 2783–2810 [DOI] [PubMed] [Google Scholar]
- Maywald RL, Doerner SK, Pastorelli L, De Salvo C, Benton SM, Dawson EP, Lanza DG, Berger NA, Markowitz SD, Lenz HJ et al (2015) IL‐33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci USA 112: E2487–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer C, Hass R, von der Ohe J, Lehnert H, Ungefroren H (2017) The role of TGF‐beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun Signal 15: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AB, Savage AK, Locksley RM (2015) Interleukin‐33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 42: 1005–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, Ramnarain A, Gasmi B, Gururajan M, Redmond D et al (2020) ILC2s amplify PD‐1 blockade by activating tissue‐specific cancer immunity. Nature 579: 130–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordasky Markell L, Perez‐Lorenzo R, Masiuk KE, Kennett MJ, Glick AB (2010) Use of a TGFbeta type I receptor inhibitor in mouse skin carcinogenesis reveals a dual role for TGFbeta signaling in tumor promotion and progression. Carcinogenesis 31: 2127–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussion C, Ortega N, Girard JP (2008) The IL‐1‐like cytokine IL‐33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel 'alarmin'? PLoS One 3: e3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan C, Yao SY, Sriram S (2016) TLR3 Agonist Poly‐IC Induces IL‐33 and Promotes Myelin Repair. PLoS One 11: e0152163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshikawa K, Yanagisawa K, Tominaga S, Sugiyama Y (2002) Expression and function of the ST2 gene in a murine model of allergic airway inflammation. Clin Exp Allergy 32: 1520–1526 [DOI] [PubMed] [Google Scholar]
- Perera RM, Bardeesy N (2015) Pancreatic cancer metabolism: breaking it down to build it back up. Cancer Discov 5: 1247–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polumuri SK, Jayakar GG, Shirey KA, Roberts ZJ, Perkins DJ, Pitha PM, Vogel SN (2012) Transcriptional regulation of murine IL‐33 by TLR and non‐TLR agonists. J Immunol 189: 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz SA, Taylor BC, Artis D (2008) Welcome to the neighborhood: epithelial cell‐derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev 226: 172–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlett CJ, Colvin EK, Pinese M, Chang DK, Morey AL, Musgrove EA, Pajic M, Apte M, Henshall SM, Sutherland RL et al (2011) Recruitment and activation of pancreatic stellate cells from the bone marrow in pancreatic cancer: a model of tumor‐host interaction. PLoS One 6: e26088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Tao GQ, Zhang Y, Cai B, Sun J, Tian ZQ (2017) TGF‐beta in pancreatic cancer initiation and progression: two sides of the same coin. Cell Biosci 7: 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J (2003) Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 113: 685–700 [DOI] [PubMed] [Google Scholar]
- Sy CB, Siracusa MC (2016) The therapeutic potential of targeting cytokine alarmins to treat allergic airway inflammation. Front Physiol 7: 214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi S, Elhance A, Van Duzer A, Kumar S, Leitenberger JJ, Oshimori N (2020) Tumor‐initiating cells establish an IL‐33‐TGF‐beta niche signaling loop to promote cancer progression. Science 369: eaay1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todoric J, Antonucci L, Karin M (2016) Targeting inflammation in cancer prevention and therapy. Cancer Prev Res (Phila) 9: 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torphy RJ, Zhu Y, Schulick RD (2018) Immunotherapy for pancreatic cancer: Barriers and breakthroughs. Ann Gastroenterol Surg 2: 274–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Wei X, Zhu T, Zhang M, Shen R, Xing L, O'Keefe RJ, Chen D (2007) Bone morphogenetic protein 2 activates Smad6 gene transcription through bone‐specific transcription factor Runx2. J Biol Chem 282: 10742–10748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Li Y, Lv Z, Chen Y, Li Y, Huang K, Corrigan CJ, Ying S (2018) Bronchial allergen challenge of patients with atopic asthma triggers an alarmin (IL‐33, TSLP, and IL‐25) response in the airways epithelium and submucosa. J Immunol 201: 2221–2231 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Alexander PB, Wang XF (2017a) TGF‐beta family signaling in the control of cell proliferation and survival. Cold Spring Harb Perspect Biol 9: a022145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Davis C, Shah S, Hughes D, Ryan JC, Altomare D, Pena MM (2017b) IL‐33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinog 56: 272–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. RNA‐Seq and ChIP‐Seq data have been deposited to the NCBI Gene Expression Omnibus (GEO, accession number GSE149579; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE149579).