

Abstract
The pathogenesis of the SARS‐CoV‐2 virus initiates through recognition of the angiotensin‐converting enzyme 2 (ACE2) receptor of the host cells by the receptor‐binding domain (RBD) located at the spikes of the virus. Here, using molecular dynamics simulations, we have demonstrated the allosteric crosstalk within the RBD in the apo‐ and the ACE2 receptor‐bound states, revealing the contribution of the dynamics‐based correlated motions and the electrostatic energy perturbations to this crosstalk. While allostery, based on correlated motions, dominates inherent distal communication in the apo‐RBD, the electrostatic energy perturbations determine favorable pairwise crosstalk within the RBD residues upon binding to ACE2. Interestingly, the allosteric path is composed of residues which are evolutionarily conserved within closely related coronaviruses, pointing toward the biological relevance of the communication and its potential as a target for drug development.
Keywords: electrostatics‐mediated crosstalk, intrinsic dynamic allostery, MD simulation, receptor‐binding domain, SARS‐CoV‐2

Abbreviations
ACE2, angiotensin‐converting enzyme 2
DCCM, Dynamics Cross‐Correlation Mapping
LA, linoleic acid
PC, principal components
RBD, receptor‐binding domain
RBM, receptor‐binding motif
RMSF, root mean square fluctuations
S, spike protein
SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Currently, the world is encountering a battle with a novel coronavirus, the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), a close relative of the previously known SARS‐Cov. Following its emergence, it has created a pandemic situation worldwide within no time owing to its alarming high rate of transmission [1]. SARS‐CoV‐2 is a single‐stranded RNA virus with four major constituent proteins—spike protein (S), membrane protein (M), envelope protein (E), and nucleocapsid protein (N) [2]. As the name implies, the infection majorly induces pneumonia accompanied by life‐threatening acute respiratory distress [3, 4].
Cell entry in particular is an important step of cross‐species transmission. The spike protein of SARS‐CoV‐2 plays the key role of attachment to the angiotensin‐converting enzyme 2 (ACE2) receptor of the host cells and mediates viral entry. The recently published crystal structure of the SARS‐CoV‐2 spike protein receptor‐binding domain (RBD) in complex with the ACE2 receptor [5] revealed that the RBD's receptor‐binding motif (RBM; residues C480‐C488) interacts with the long N‐terminal helix of ACE2 (PDB code: 6M0J). The transmembrane S that forms homotrimers sticking out from the viral capsid mediates SARS‐CoV‐2 entry into host cells. S consists of two active subunits essential for host cell ACE2 binding (S1 subunit) and viral envelope as well as cellular membrane fusion (S2 subunit). The S1 subunit includes the RBD and helps in stabilization of the prefusion state [6]. The association with the host‐cell receptor is regulated by the RBD of the spike protein. Following receptor recognition, proteolytic cleavage between S1 and S2 subunits of the spike protein occurs by a host protease, which subsequently releases the spike fusion peptide and, thereby, promotes viral entry [7]. Furthermore, cryo‐EM structures of the SARS‐CoV‐2 spike protein display significant mobility and dynamics in the S1 subunit, particularly around the RBD revealing two distinct conformational states, a ‘down’ conformation, segregated from ACE2 binding, and an ‘up’ conformation, responsive to the ACE2 receptor [6]. Clearly, successful infection of viruses is reliant on the effective complex formation between the receptor and the viral spike protein, which makes it an obvious drug target. However, in addition to the contact residues, studies have indicated potential role of noncontact residues and allostery in the spike protein–receptor interaction [8, 9].
To better understand the etiology of the infection, we have focused on the RBD of the spike protein and its binding to the ACE2 receptor [8, 10], information on which may provide important clues for designing drugs to combat the virus. Our study aims to understand the hidden allosteric crosstalks in the RBD and their possible role in the formation of the spike protein complex with the ACE2 receptor. Here, we report a novel allosteric site in the RBD and the allosteric crosstalk between the ACE2 binding residues of the RBD and the distal residues correspond to an allosteric site of the RBD using molecular dynamics (MD) simulation followed by extensive analysis of both dynamics and energy terms at the atomic level. MD simulation has long been implicated as a suitable procedure to explore the allosteric paths of information exchange between amino acid residues of a protein by elucidating intermediate protein conformations that often occur in pico‐ or nano‐second time scale which are often difficult to capture with other techniques. It has already been reported that the interface dynamics of ACE2‐SARS‐CoV‐2/SARS‐CoV spike protein interactions show truly unique mechanisms [11]. The conformational dynamics and changes in energetics within the RBD before and after binding to the ACE2 receptor revealed the hidden communication that shapes the crosstalk pathway leading to recognition and subsequent internalization of the virus.
Insight gained from our results on the underlying mechanism of viral infection offers the idea that modification or inhibition of the allosteric communication would hinder host receptor recognition, and therefore, it may be considered as a possible intervention pathway for infection prevention.
Materials and methods
Molecular dynamics simulation studied (MD)
The ACE2‐bound (PDB ID: 6M0J) and free RBD of SARS‐CoV‐2 crystal structure (extracted from 6M0J) (Fig. S1) were equilibrated for 1ns with capped N and C termini by N‐methyl amide and acetyl groups for both. Topologies for the 2‐acetamido‐2‐deoxy‐beta‐D‐glucopyranose (NAG) residues were build using module of AmberTools 18 (AMBER 2018, University of California, San Francisco) and were converted to the GROMACS format using the ACPYPE software [12]. MD simulations were performed in with both the free and bound spike protein with the (GROMACS 5.1.2 software, University of Groningen, Royal Institute of Technology, Uppsala University) [13] using the Amber99SB‐Ildn force field [14] and the TIP3P water model [15] and centering the protein in the box, and placed it at least 1.0 nm from the defined cubic box edge. The titrable residue protonation states were defined using multi‐conformation continuum electrostatics method [16] and also based on PROPKA [17] web server (http://nbcr‐222.ucsd.edu/pdb2pqr_2.0.0) with respect to pH 7.2. All Arg, Asp, Glu, and Lys residues were charged, and His residues were neutral. The systems were neutralized by adding 150 mm NaCl by substituting appropriate number of solvent molecules to mimic experimental salt condition [5] using the parameters provided by Amber99SB‐Ildn force field. Before simulation, the initial coordinates were relaxed by 1000 steps steepest‐descent energy minimization followed by two‐step equilibration, namely NVT equilibration followed by NPT equilibration for 10 ns each. Temperature was controlled through velocity rescaling [18] at 310 K with a time constant of 0.1 ps, and pressure was controlled using Parrinello–Rahman barostat [19] at 1 bar. The particle mesh Ewald algorithm [20] was applied to calculate long‐range electrostatic interactions. The cutoff for short‐range electrostatics and van der Waals interactions was 1.2 nm. MD simulations of 1 μs each for both the free and the ACE2‐bound RBD were performed along with steps were recorded at every 2 psand analyzed results. The RMSD, root mean square fluctuations (RMSF) and principal components (PC) were calculated using the tools provided by GROMACS. Convergence of the simulation for both the apo‐RBD and ACE2‐bound RBD was checked from respective RMSD comparison (Fig. S2). Here, two individual sets of 1 µs MD simulations for each system (either apo‐RBD or ACE2‐bound RBD) were performed (Table S1). Allosteric drug pocket was predicted using VLS3D [21] and PockDrug [22] web servers.
Dynamic Cross‐correlation Mapping (DCCM)
Region of RBD with coupled dynamics was identified by the network analysis of correlated motions. Cross‐correlation maps were used to identify the regions that move in or out of phase during the simulations [23]. The elements of the matrix (Cij) were obtained from their position vector (r) as shown in Eq. 1:
| (1) |
where i and j correspond to any two residues; r i and rj are position vectors of i and j; and the angle brackets denote an ensemble average. Inter‐atomic cross‐correlation fluctuations between any two pairs of residues of RBD (all atoms were considered) in its apo‐ and ACE2‐bound state were calculated by using this expression and were represented graphically by the Dynamics Cross‐Correlation Mapping (DCCM) for the RBD residues only. The value of Cij can vary from −1 (strongly anti‐correlated motion) to +1 (strongly correlated motion).
Path analysis from correlated motions
Hierarchical clustering was implemented for each correlation network to produce cumulative nodal clusters, or communities those are strongly intra‐connected yet slightly inter‐connected, using a related betweenness clustering algorithm to that developed by Girvan and Newman [24]. Nevertheless, as is standard for unweighted networks, rather than selecting the partitions with the highest modularity ranking, we selected the partition nearest to the highest modularity score that consisted of the smallest number of overall communities. This excluded the typical circumstance where several small communities with modularity values of similarly high scores were created. The density of connections per node which was assessed by the node centralities were calculated as described by equation:
| (2) |
where xi represents the centrality of the node i, Aij is the ijth entry of the adjacent matrix A, λ is actually a constant, and G represents all nodes. Aij ≠ 0 if node i and j are linked, and it will be equal to , where wij is the edge weight. For every i (i∈G) is equivalent to defining the Eigen values and eigenvectors of matrix A. Considering a pair of nodes optimal (shortest) and suboptimal (near however farther than optimized one) linking node paths were established employing the previously mentioned algorithm [25].
Order parameter (S 2)
Backbone N‐H vectors were selected to calculate S 2 over the period of trajectory, which represent dynamics of protein, with a value 1 indicating complete rigidity and a value toward 0 represents enhanced dynamics.
| (3) |
where μ1, μ2, and μ3 are the x, y, and z components of the relevant bond vector scaled to unit magnitude, μ, respectively [26]. Angular brackets indicate averaging over the snapshots.
Energy perturbation
The perturbation in residue‐wise nonbonded interaction energy between free RBD and its bound state with ACE2 was described as:
| (4) |
The nonbonded interactions () include both electrostatic () and van der Waals () interactions and were modeled using a Coulomb and Lennard‐Jones potential function, respectively. For ith residue, the can be further broken down in terms of contributions from RBD, ACE2, and water in the following manner:
| (5) |
| (6) |
where , , and , denote the change in the average nonbonded interaction energy over the whole trajectories between the ACE2‐bound and unbound state of RBD due to the interactions between the ith residue and RBD/ACE2/water, respectively. A large cutoff of 2 nm was applied for computing interaction energy with water molecules. For nonbonded interaction, no cutoff was used. Every 10 time steps, we stopped overall translation and rotation during the MD simulations to prevent all kinetic energy to be associated with center of mass motions and overall rotations. All energy calculations were performed over 1 μs of MD simulations.
Path analysis from electrostatic energy contributions
The perturbation in pairwise electrostatic interactions :
| (7) |
The contributions due to LJ and electrostatic (Coulomb) nonbonded interactions to nonbonded energy were calculated separately, but the LJ terms were found to be numerically much smaller than the respective electrostatic ones, so we have focused on the electrostatic interactions. The interaction energy between two residues i and j is the sum of the nonbonded interaction energies already defined in a force field where:
| (8) |
| (9) |
Energy networks were built by considering the amino acid residues as nodes. A weighted edge was made between any pair of residues i and j by considering the interaction energy as the weight. Energy‐Hubs are defined as nodes that have a higher degree or connectivity in the network.
Betweenness‐centrality [27] was computed using the following equation:
| (10) |
where BC(v) is betweenness‐centrality of residue ν.n is the number of residues within network, σij(v) is the number of shortest paths between residue i and j that pass through residue ν and σij is the total number of shortest paths from i to j. Dijkstra's algorithm [28] was applied to calculate the shortest path between residues i and j.
Results
Conformational dynamics of the free and the ACE2‐bound RBD
To probe deeper into the dynamics of the free and the ACE2‐bound RBD of the SARS‐CoV‐2 spike protein, the Cα atoms based RMSF and the order parameter (S2) considering the N‐H vectors [26] of the RBD were calculated from the respective trajectories. The overall RMSF value of the RBD in the ACE2‐bound state manifested a lower value relative to that of the free RBD, implicating higher rigidity of the RBD after binding to the ACE2 compared to its apo‐form. This suggests the ACE2‐directed stabilization of the RBD in the ACE2‐bound state by the selection of particular conformation from the fast time scale (ps‐ns) regime.
In this regard, the residues exhibiting remarkably distinguishable RMSF values between the two states (i.e., the free and the ACE2‐bound RBD) are clearly those regions that display highly dynamic motifs in the free RBD (an RMSF cutoff of > 0.25 nm was set to define regions of high flexibility): the binding loop (containing residues 474–486), and another distal or allosteric loop (consisting residues 358–376). It may be noted that the underlying reason behind using the term allosteric loop was that the identified region is distal from the ACE2 binding site (i.e., it is situated toward the S2 domain of the spike protein) [6, 29].
Significantly, when the dynamics of the apo‐RBD was compared to the bound RBD, the fluctuation of the allosteric loop decreased considerably along with the ACE2 binding loop (Fig. 1A–C). This diminution in the fluctuation could be expected for the binding loop due to its interaction with the ACE2 but the simultaneous decrement in the dynamics of the distal loop pointed toward the allosteric involvement through correlated motion [23]. Interestingly, a slightly enhanced flexibility was observed in another distal loop (containing residues 384–390) of the RBD upon binding to the ACE2 (Fig. 1).
Fig. 1.
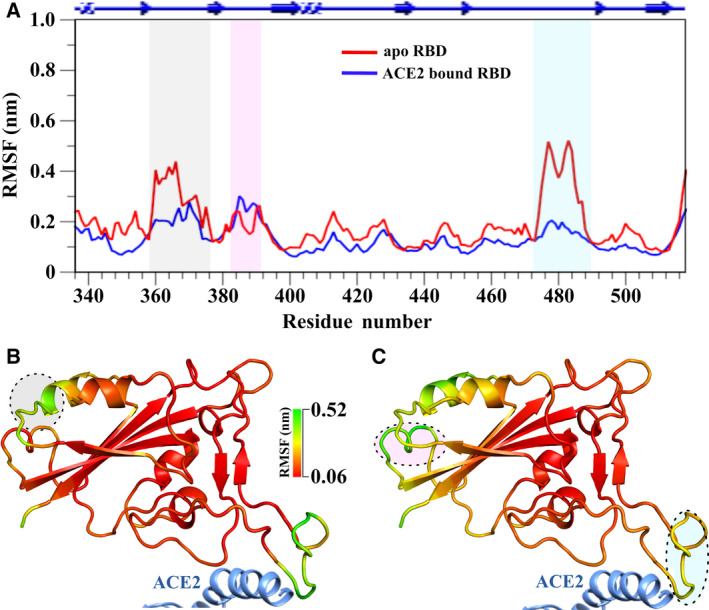
Comparison of Cα atom dynamics. (A) Residue‐wise Cα atom RMSF for both free and ACE2‐bound RBD of SARS‐CoV‐2. Significant changes (above cutoff value of 0.2 nm) were marked with different colored columns. RMSF values were mapped onto the crystal structures (PDB ID: 6M0J) of respective (B) free, and (C) ACE2‐bound RBD. A PyMol spectrum bar‐based color scheme was used to point out differential fluctuating regions of both and is indicated by circles correspond to the same color used for (A).
Analysis of order parameters (S2) based on N‐H vectors singularly complied with the conclusions of the RMSF analysis. Similar allosteric fluctuations in both the above‐mentioned distal loops were also identified along with an overall decrement in the dynamics of the ACE2‐bound RBD and also with the distinguishable alterations in the flexibility of the binding region (between residues 474–486) (Fig. S3). The S2 actually represents flexibility and rigidity of a protein with the values from 0 to 1, where values close to 0 and 1 indicate high flexibility and rigidity respectively. A good agreement between the simulated and experimental order parameters was observed using equation 3 (see Materials and method section) and applying the same methodology [26].
Modulation of allosteric communication within the RBD upon receptor binding
Dynamics Cross‐Correlation Mapping has gained significant attention for the determination of correlated allosteric motions among amino acids. This is actually a residue‐wise correlation matrix‐based approach where the correlated and the anti‐correlated motions are scored between 1 and −1 (strong: |Cij| = 1.0–0.7; moderate: |Cij| = 0.7–0.5; and weak: |Cij| = 0.3–0.5). A number of noticeable differences in correlation were found when DCCM of both the apo‐ and the ACE2‐bound RBD were compared. The free RBD consisted of several strongly correlated and moderately anti‐correlated motions within the amino acids, which were either missing or became weakly correlated in the ACE2‐bound RBD (Fig. 2A,B). Interestingly, the binding loop (residues from 474 to 486) and the allosteric loop (residues from 358 to 376) showed a moderate anti‐correlated motion to each other. In other word, these loops were moving apart from each other in a correlated manner. This kind of anti‐correlated motion almost disappeared in the ACE2‐bound RBD. This could be attributed to the structural flexibility of the apo‐RBD which was lost in the ACE2‐bound RBD owing to its conformational rigidity in the bound state.
Fig. 2.
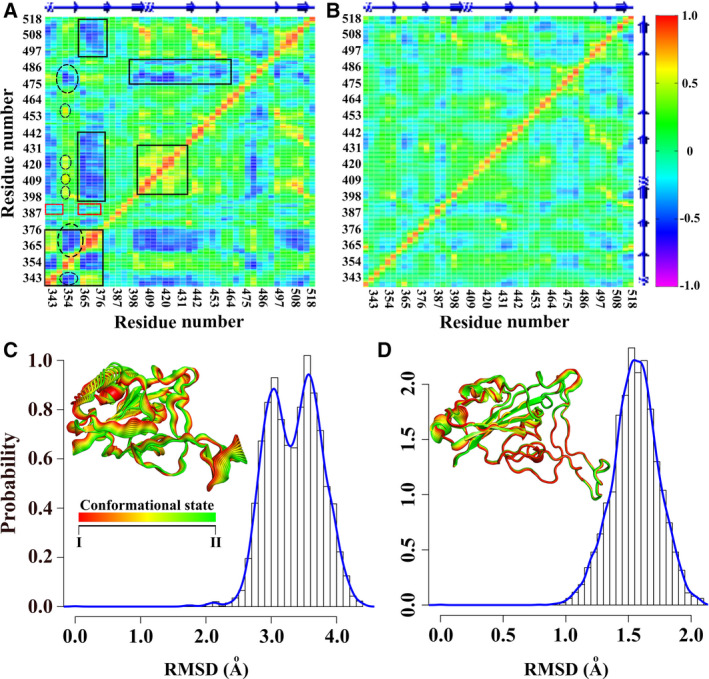
Correlated motions and dynamics. DCCM of (A) apo‐ and (B) ACE2‐bound RBD are shown. Differential residue‐wise correlations in both the form of RBD are indicated by box. Allosteric communication path is marked with broken circle. RMSD probability reflected two populations in (C) apo‐ and one (D) ACE2‐bound RBD. The 3D projections of both (calculated from PCA) are shown in inset. Apo‐RBD dynamics reflects conformational alterations between two states from ps‐ns time scale regime of dynamics.
For further verification, the RMSD distribution and the PC of both the free and the ACE2‐bound RBD were calculated throughout the whole trajectories. The RMSD distribution comparative study between the two forms revealed strikingly divergent populations (Fig. 2C,D). Two distinct populations were observed for the apo‐RBD while the ACE2‐bound RBD noticeable displayed a single RMSD population and that were in agreement with the PC analysis results. The PC analysis of the free RBD identified two major distinct groups (PC1[39.09%]‐PC2[12.19%]) correspond to dynamic alteration between conformational state I and state II which is distinctly different in the case of the ACE2‐bound RBD (PC1[17.26%]‐PC2[15.58%]) (Figs 2C,D and S4A,B).
Both the RMSD distribution and the PC analysis results clearly indicated the flexible conformational alterations of the apo‐RBD between two conformational states which were not apparent for the ACE2‐bound RBD, simultaneously reestablishing the dynamic rigidity of the RBD upon the ACE2 binding.
Correlated motion‐based allosteric communication in the free RBD
Subsequently, we analyzed the path of the free RBD (the ACE2‐bound RBD was excluded from calculation as it showed negligible cross‐correlated motions in the DCCM) through which allosteric crosstalk propagates from the binding loop (the selected residue is P479) to the identified allosteric loop (the selected residue is D364). It is to be noted that both the residues were selected based on their highest betweenness‐centrality values (Fig. S4C) within the highly fluctuating above mentioned regions. Interestingly, the identified crosstalk was found to propagate between the residues P479 and D364 through (a) the hydrogen‐bonded residues Y423 and D398 in one path (Fig. S5A,B) and (b) the residues A419 to I410 in another path (Fig. S5C) remaining all other constituent residues (like V362, A363, F338, G339, E340, I410, A419, K458, G476, and T478) of the predicted path same (see Materials and method for details). Note that, the constituent residues of the path are evolutionarily conserved with closely related coronaviruses [30]. Based on crystal structure (PDB ID: 6M0J), this hydrogen‐bonded communication seems to act like a bridge between the binding and the allosteric regions of the apo‐RBD. Hence, we postulate that inherent allosteric crosstalk in the apo‐RBD is a significant factor in ACE2 recognition.
Hidden electrostatic perturbation revealed favorable allosteric crosstalk in the ACE2‐bound RBD
The role of hidden contributions (e.g., electrostatic energy perturbation) in molecular recognitions is another important contributor that remains yet to be investigated in the case of the ACE2‐bound RBD. Hence, the residue‐wise nonbonded energy contributions in terms of the electrostatic () and the van der Waals () interactions of the RBD toward the ACE2 binding were calculated. Our results successfully captured the binding residues of the RBD with significantly high electrostatic energies, particularly, K417. Similarly, binding residues along with some proximal to the binding site showed the VDW contributions as expected, for example L455, F456, N487, Y489, Q493, T500, N501, and Y505 (Fig. 3A–D) [5, 31]. It was reported that due to the mutation at the residues L455, F456, Q493, and N501 their binding affinity for the ACE2 was abolished [32]. The residue‐wise electrostatic contributions (up to ~ 165 kcal·mol−1) are much higher compared to the van der Waals (up to ~ 12 kcal·mol−1). Hence, undoubtedly in the recognition of the ACE2 by the RBD, the electrostatic energy is the predominant factor in term of the nonbonded energies.
Fig. 3.
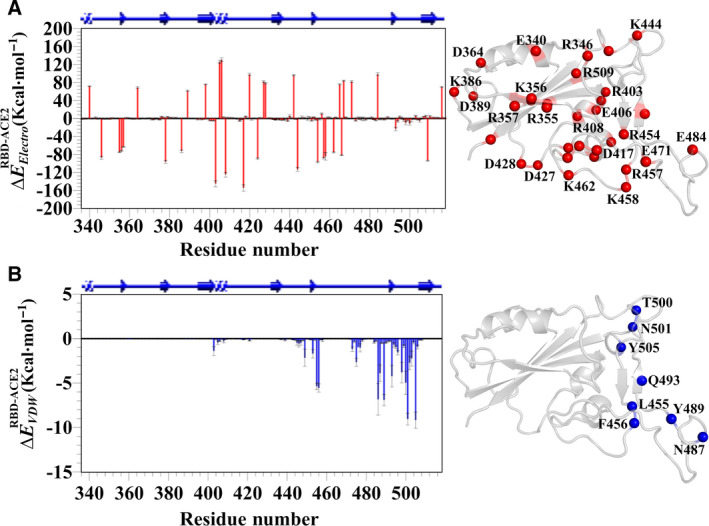
Residue‐wise nonbonded energy contributions. Residue‐wise (A) electrostatic and (C) van der Waals contributions of RBD toward ACE2 binding are presented. Contributions of (B) electrostatic and (D) VDW are mapped on crystal structures and represented in red and blue spheres, respectively.
While scrutinizing the residue‐wise electrostatic contributions of the RBD toward the ACE2, it was found that some distal residues like D364 and K386 exhibited significant electrostatic contributions (Fig. 3B). Notably, we have already predicted the allosteric involvement of the residue D364 in the free RBD, so we further dissected the residue pairwise electrostatic interactions within the RBD in both the free and the ACE2‐bound state to build a connectivity network between the ACE2 binding residue K417 and the allosteric site residue D364 (see Materials and method for details). As a result, the allosteric crosstalk in the RBD propagated from the residue K417 to the residue D364 via the residues R408 to R355 to K356 (Fig. 4A). Notably, except the binding residue K417, all other constituent partners show evolutionary conservation within closely related coronaviruses [30].
Fig. 4.
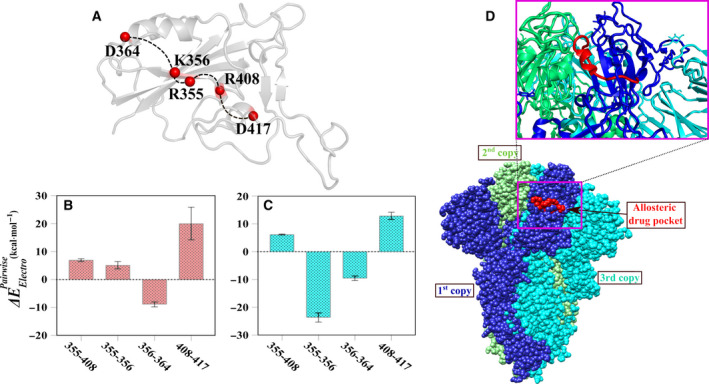
Electrostatic defined allosteric communication path and pairwise energy perturbations. (A) Represented allosteric crosstalk based on electrostatic energy. Pairwise electrostatic perturbation of (B) free and (C) ACE2‐bound RBD are shown. Residues within the path (calculated from electrostatic energy contributions) are selected for pairwise electrostatic energy calculations and are represented in red spheres. Path is indicated by broken black lines. (D) The allosteric drug pocket is shown in red sphere and marked. Trimers are represented by different colors in sphere model and marked the respective copies (PDB ID: 6VXX). A close‐up view of the pocket is given in the upper panel.
Remarkably, the unfavorable pairwise electrostatic interactions of R355‐K356 pair in the apo‐RBD ( = 5.17 ± 1.31 kcal·mol−1) became favorable in the ACE2‐bound state ( = −23.65 ± 1.64 kcal·mol−1). Interestingly, average pairwise contribution for K417‐R408 pair of the RBD decreased upon complex formation with the ACE2 (for the free RBD = 20.07 ± 5.4 kcal·mol−1 and for the ACE2‐bound RBD = 13±1.3 kcal·mol−1) (Fig. 4B,C). Conclusively, the results indicated the hidden role of the residue pairwise energy perturbations in the recognition of the ACE2 by the RBD.
Therefore, we have initially identified an allosteric site in the free RBD from the dynamic correlated motion analysis which can be a site‐specific target for allosteric drugs or antibodies. Formation of the stable complex as reported earlier [5] can also be verified from the noticeably high residue‐wise electrostatic contributions compared to the van der Waals energy. Note that, we predicted the same allosteric site in the RBD based on electrostatic energies too and the residue pairwise electrostatic energies of the RBD became favorable in some pairs after binding to the ACE2 receptor. It may be noted here that the allosteric druggable pocket predicted by available web servers includes the residues identified as allosteric site in this study (Fig. S6A). The predicted allosteric site is well accessible in closed state conformation of the RBD. Due to conformational exchange between up and down states, the allosteric site remains unhindered from any other motif or domain of the spike protein trimer (Fig. 4D). On the contrary to the dynamic cross‐correlation, the existence of favorable communication (between the binding site to the allosteric regions precisely, toward the S2 domain) based on pairwise electrostatic energies in the ACE2‐bound RBD can be extrapolated to the hidden transmission of the signal for the next queue process, that is, the cleavage between S1 and S2 domains of the spike protein after binding to the ACE2 [6, 29].
Discussion
Allosteric regulation in proteins is one of the most important areas in the field of allosteric drug development, and it was proposed that allosteric communications were achieved either by correlated motions of the amino acids or by fundamental nonbonded energy contributions [23, 33]. Here, we found that, following binding to the ACE2, dynamics of the RBD decreases resulting in stabilization of the ACE2‐bound RBD by the selection of a particular conformational state from the fast time scale (ps‐ns) regime. This observation also suggests the decrement in the overall RBD backbone entropy factor (as S2 is directly related to conformational entropy[34]) upon binding to the ACE2 receptor and formation of a stable complex. We have shown that the crosstalk mediated by the cross‐correlated motions between the allosteric and the binding loops dominated in the free RBD representing inherent allostery present in the apo‐form. The allosteric paths derived from both dynamic cross‐correlations and electrostatic energies can be claimed relevant for biological functions considering evolutionary conservation of the residues involved within closely related coronaviruses. Moreover, this allosteric site is not glycosylated as reported by Watanabe et al. [35] using mass spectrometry of recombinant glycosylated spike trimers. Conclusively, this allosteric site could be a very good drug target.
Recent high‐resolution structures of the transmembrane spike protein of SARS‐CoV‐2 show [6, 36] intertwined arrangement of three monomers. Each monomer consists of sub‐domains S1 and S2. RBD that interacts with the ACE2 receptor of host belongs to the S1 subunit, while the S2 subunit anchors on the viral membrane (Fig. S7). It is conceivable that the trimer of spike protein needs to function in a concerted way. However, the architecture of an individual monomer of spike protein is extended in shape placing S2 domain away from the S1 (comprising RBD). Interestingly, the criss‐cross arrangement of monomers in trimeric spike places the allosteric site of RBD of one monomer in close proximity of the S2 domain of another copy (Fig. S7). The allosteric communication likely transmits from RBD of one monomer to S2 of another copy within the trimer (Video S1). Based on our results, it is tempting to propose that inter‐monomer communication plays a key role in orchestrating conformational dynamics of spike trimer in a concerted manner.
Taken together, our study suggests that the allostery in the RBD plays the most important role during ACE2 recognition followed by host cell insertion. Based on our results, we propose that the allosteric region of the ACE2‐binding site of the RBD reported here can be a potential allosteric drug target in addition to the binding interface between the viral spike protein and the host ACE2 receptor [37]. In this context, a cryo‐EM study [38] published during the preparation of this paper needs to be mentioned. This study reported that linoleic acid (LA) binding to a distal site of the RBM locks the SARS‐CoV‐2 spike trimer in a closed conformation, which is incompatible with ACE2‐receptor binding. Remarkably, the LA binding pocket exactly matches with the amino acid stretch (residues 358–376) identified as an allosteric druggable site in our study (Fig. S6B). Thus, this structural study provides experimental verification of our result. At the same time, our study offers a fundamental basis for direct crosstalk between the RBM and the allosteric site which could not be established from the structure.
In the current SARS‐CoV‐2 outbreak, designing drugs (e.g., peptide, small molecules) or antibody against SARS‐CoV‐2 is a pressing need. The residue‐wise dynamics as well as energy information provided in this study would be beneficial for that purpose.
Author contributions
SB and JS conceived the project. SB designed and performed the computational work. SB, RB, and JS analyzed the data and wrote the paper.
Supporting information
Fig. S1. Structural representation of RBD.
Fig. S2. Convergence of the MD simulation.
Fig. S3. Order Parameter comparison.
Fig. S4. PCA and Betweenness‐centrality of RBD.
Fig. S5. Allosteric communication path derived from correlated residues and H‐bind occurrence probability of apo‐RBD.
Fig. S6. Allosteric drug pocket.
Fig. S7. RBD of S1 and S2 domain of Spike protein.
Table S1. Details of the MD simulations. For both apo‐ and ACE2‐bound RBD two sets of 1000 ns long MD simulations were performed.
Video S1. Conformational alteration of allosteric loop along with binding loop derived from final simulated structures (i.e.1 µs state) of both apo‐ and ACE2‐bound RBD.
Acknowledgements
This work was supported by SERB, DST (India) sponsored project, and CSIR‐Indian Institute of Chemical Biology, Kolkata, India. We acknowledge the Central Instrument Facility of CSIR‐Indian Institute of Chemical Biology of CSIR‐Indian Institute of Chemical Biology, and CSIR‐4Pi for super computer facility for supporting our computational work. SB and RB acknowledge UGC and CSIR, India, respectively, for awarding senior research fellowship.
Edited by Michael Bubb
Contributor Information
Sayan Bhattacharjee, Email: xsayany@gmail.com.
Jayati Sengupta, Email: jayati@iicb.res.in.
References
- 1. Thompson R (2020) Pandemic potential of 2019‐nCoV. Lancet Infect Dis 20, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, Meng J, Zhu Z, Zhang Z, Wang J et al. (2020) Genome composition and divergence of the novel coronavirus (2019‐nCoV) originating in China. Cell Host Microbe 27, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gibson PG, Qin L and Puah SH (2020) COVID‐19 acute respiratory distress syndrome (ARDS): clinical features and differences from typical pre‐COVID‐19 ARDS. Med J Aust 213, 54–56.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Torres Acosta MA and Singer BD (2020) Pathogenesis of COVID‐19‐induced ARDS: implications for an ageing population. Eur Respir J 56, 2002049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L et al. (2020) Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature 581, 215–220. [DOI] [PubMed] [Google Scholar]
- 6. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT and Veesler D (2020) Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 181, 281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jaimes JA, Millet JK and Whittaker GR (2020) Proteolytic cleavage of the SARS‐CoV‐2 spike protein and the role of the novel S1/S2 Site. iScience 23, 101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Letko M, Marzi A and Munster V (2020) Functional assessment of cell entry and receptor usage for SARS‐CoV‐2 and other lineage B betacoronaviruses. Nat Microbiol 5, 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Di Paola L and Giuliani A (2020) Mapping active allosteric loci SARS‐CoV Spike Proteins by means of Protein Contact Networks .
- 10. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ali A and Vijayan R (2020) Dynamics of the ACE2‐SARS‐CoV‐2/SARS‐CoV spike protein interface reveal unique mechanisms. Sci Rep 10, 14214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sousa da Silva AW and Vranken WF (2012) ACPYPE ‐ AnteChamber PYthon Parser interfacE. BMC Res Notes 5, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE and Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- 14. Lindorff‐Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO and Shaw DE (2010) Improved side‐chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Price DJ and Brooks CL 3rd (2004) A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys 121, 10096–10103. [DOI] [PubMed] [Google Scholar]
- 16. Georgescu RE, Alexov EG and Gunner MR (2002) Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins. Biophys J 83, 1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Olsson MH, Sondergaard CR, Rostkowski M and Jensen JH (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput 7, 525–537. [DOI] [PubMed] [Google Scholar]
- 18. Bussi G, Donadio D and Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126, 014101. [DOI] [PubMed] [Google Scholar]
- 19. Parrinello M and Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52, 7182–7190. [Google Scholar]
- 20. Essmann U, Perera L, Berkowitz ML, Darden T, Lee H and Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103, 8577–8593. [Google Scholar]
- 21. Villoutreix BO, Renault N, Lagorce D, Sperandio O, Montes M and Miteva MA (2007) Free resources to assist structure‐based virtual ligand screening experiments. Curr Protein Pept Sci 8, 381–411. [DOI] [PubMed] [Google Scholar]
- 22. Hussein HA, Borrel A, Geneix C, Petitjean M, Regad L and Camproux AC (2015) PockDrug‐Server: a new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res 43, W436–W442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghosh A and Vishveshwara S (2007) A study of communication pathways in methionyl‐ tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc Natl Acad Sci USA 104, 15711–15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Girvan M and Newman ME (2002) Community structure in social and biological networks. Proc Natl Acad Sci USA 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yen JY (1971) Finding the K shortest loopless paths in a network. Manage Sci 17, 712–716. [Google Scholar]
- 26. Trbovic N, Cho JH, Abel R, Friesner RA, Rance M and Palmer AG 3rd (2009) Protein side‐chain dynamics and residual conformational entropy. J Am Chem Soc 131, 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brandes U (2001) A faster algorithm for betweenness centrality. J Math Sociol 25, 163–177. [Google Scholar]
- 28. Dijkstra EW (1959) A note on two problems in connexion with graphs. Numer Math 1, 269–271. [Google Scholar]
- 29. Simmons G, Zmora P, Gierer S, Heurich A and Pohlmann S (2013) Proteolytic activation of the SARS‐coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res 100, 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Y, Guo Y, Pan Y and Zhao ZJ (2020) Structure analysis of the receptor binding of 2019‐nCoV. Biochem Biophys Res Commun 525, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yan R, Zhang Y, Li Y, Xia L, Guo Y and Zhou Q (2020) Structural basis for the recognition of SARS‐CoV‐2 by full‐length human ACE2. Science 367, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yi C, Sun X, Ye J, Ding L, Liu M, Yang Z, Lu X, Zhang Y, Ma L, Gu W et al. (2020) Key residues of the receptor binding motif in the spike protein of SARS‐CoV‐2 that interact with ACE2 and neutralizing antibodies. Cell Mol Immunol 17, 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vijayabaskar MS and Vishveshwara S (2010) Interaction energy based protein structure networks. Biophys J 99, 3704–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharp KA, O'Brien E, Kasinath V and Wand AJ (2015) On the relationship between NMR‐derived amide order parameters and protein backbone entropy changes. Proteins 83, 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Watanabe Y, Allen JD, Wrapp D, McLellan JS and Crispin M (2020) Site‐specific glycan analysis of the SARS‐CoV‐2 spike. Science 369, 330–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS and McLellan JS (2020) Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Han Y and Kral P (2020) Computational design of ACE2‐based peptide inhibitors of SARS‐CoV‐2. ACS Nano 14, 5143–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Toelzer C, Gupta K, Yadav SKN, Borucu U, Davidson AD, Kavanagh Williamson M, Shoemark DK, Garzoni F, Staufer O, Milligan R et al. (2020) Free fatty acid binding pocket in the locked structure of SARS‐CoV‐2 spike protein. Science 370, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Structural representation of RBD.
Fig. S2. Convergence of the MD simulation.
Fig. S3. Order Parameter comparison.
Fig. S4. PCA and Betweenness‐centrality of RBD.
Fig. S5. Allosteric communication path derived from correlated residues and H‐bind occurrence probability of apo‐RBD.
Fig. S6. Allosteric drug pocket.
Fig. S7. RBD of S1 and S2 domain of Spike protein.
Table S1. Details of the MD simulations. For both apo‐ and ACE2‐bound RBD two sets of 1000 ns long MD simulations were performed.
Video S1. Conformational alteration of allosteric loop along with binding loop derived from final simulated structures (i.e.1 µs state) of both apo‐ and ACE2‐bound RBD.