

Abstract
The emergence of a variety of coronaviruses (CoVs) in the last decades has posed huge threats to human health. Especially, the ongoing pandemic of coronavirus disease 2019 (COVID‐19) caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has led to more than 70 million infections and over 1.6 million of deaths worldwide in the past few months. None of the efficacious antiviral agents against human CoVs have been approved yet. 3C‐like protease (3CLpro) is an attractive target for antiviral intervention due to its essential role in processing polyproteins translated from viral RNA, and its conserved structural feature and substrate specificity among CoVs in spite of the sequence variation. This review focuses on all available crystal structures of 12 CoV 3CLpros and their inhibitors, and intends to provide a comprehensive understanding of this protease from multiple aspects including its structural features, substrate specificity, inhibitor binding modes, and more importantly, to recapitulate the similarity and diversity among different CoV 3CLpros and the structure–activity relationship of various types of inhibitors. Such an attempt could gain a deep insight into the inhibition mechanisms and drive future structure‐based drug discovery targeting 3CLpros.
Keywords: 3C‐like protease, binding modes, coronavirus, inhibitors, structure and function
1. INTRODUCTION
The recognition of coronaviruses (CoVs) could be initiated from 1947 as the first prototype murine CoV (strain, JHM), has been discovered. 1 The human CoV 229E (HCoV‐229E) was identified as the first HCoV in 1966 and HCoV‐OC43 recovered next after that.2, 3 It is noteworthy that while HCoV‐229E and HCoV‐OC43 infecting healthy volunteers to cause a common cold, an impression was first created on HCoVs as relatively harmless respiratory pathogens.2, 4, 5 Such a mild impression was, however, heavily exacerbated by the sudden invasion of severe acute respiratory syndrome (SARS)‐CoV in 2002–2003, which swept 29 countries with a fatality rate of 7% as reported by the World Health Organization (WHO). 6 Additionally, the HCoV‐NL63 and HCoV‐HKU1 were identified from a child with bronchiolitis in the Netherlands 7 and an adult with chronic pulmonary disease in Hong Kong in 2004–2005, respectively. 8 The Middle East respiratory syndrome coronavirus (MERS‐CoV, formerly known as HCoV‐EMC) became epidemic in Middle Eastern countries in 2012 and a total of 2266 laboratory‐confirmed cases of MERS with a fatality rate of 35.3% were reported up to December, 2018. 9 Very recently, a new SARS‐CoV‐2 has become globally pandemic, 10 with 70,476,836 worldwide confirmed cases including 1,599,922 deaths as reported by the WHO at December 15, 2020. 11 These data not only reminds us the huge hazard of HCoVs but also alerts the possible emergence of another new HCoV types in future. Unfortunately, none of the efficacious antiviral agents for the treatment of the highly pathogenic HCoVs have been approved yet. Thus, the development of effective anti‐HCoV chemotherapy becomes extremely urgent, which, as we consider, should be benefited by a systematic and deep exploration covering of all known occurred HCoVs. Such an effort could also provide useful information for the intervention of the present COVID‐19 as well as possible HCoV infections outbreak in the future.
According to the phylogenetic clustering provided by the International Committee for Taxonomy of Viruses (ICTV), the new classification of CoVs (Class Pisoniviricetes → order Nidovirales → suborder Cornidovirineae → family Coronaviridae → subfamily Orthocoronavirinae) defined a total of 45 known species and replaced the traditional groups (1, 2, and 3) by four major genera of Alphacoronavirus (αCoV), Betacoronavirus (βCoV), Gammacoronavirus (γCoV), and Deltacoronavirus (δCoV).12, 13, 14 Among the known seven HCoVs, HCoV‐229E and HCoV‐NL63 belong to αCoVs while the remaining five including the newly reported SARS‐CoV‐2 are βCoVs. 15 These HCoVs are all believed to be transmitted from animals, of which bats are the major source. 16 Besides human beings, a variety of hosts such as feline, porcine, bat, mouse, and avian can also be infected with CoVs, given the names of Feline Infectious Peritonitis Virus (FIPV), Porcine Epidemic Diarrhea Virus (PEDV), Porcine Transmissible Gastroenteritis Virus (TGEV), BatCoV‐HKU4 (BtCoV‐HKU4), Mouse Hepatitis Virus A59 (MHV), and Avian Infectious Bronchitis Virus (IBV), respectively.
Featured with an overall spherical structure with coronal protrusions surrounding the surface (Figure 1A), CoVs represent a family of positive‐sense, single‐stranded, enveloped RNA viruses with the largest viral RNA genomes known to date (26–32 kb).13, 17 Generally speaking, the genome contains a variable number (6–11) of open reading frames (ORFs) with a 5ʹ cap structure and a 3ʹ poly (A) tail, playing an important role in the translation of replicase polyproteins.18, 19 The one‐third of genomic RNA from 3ʹ terminus encodes four canonical structural proteins: a spike glycoprotein (S), an envelope small membrane protein (E), a membrane protein (M), and a nucleoprotein (N) for virion assembly (Figure 1A). An additional hemagglutinin‐esterase (HE) also exists in a minority of CoVs (e.g., HCoV‐OC43, HCoV‐HKU1, and MHV).20, 21, 22 Locating at the two‐thirds of genome from the 5ʹ terminus, two overlapping ORFs (ORF1a and ORF1b) encode the replicase polyproteins, pp1a and pp1ab.15, 16 These two polyproteins are cleaved by a papain‐like cysteine protease (PLpro) and a 3C‐like protease (3CLpro) encoded in ORF1a to generate 16 mature nonstructural proteins (nsps) for genome transcription and replication (Figure 1B). 23
Figure 1.
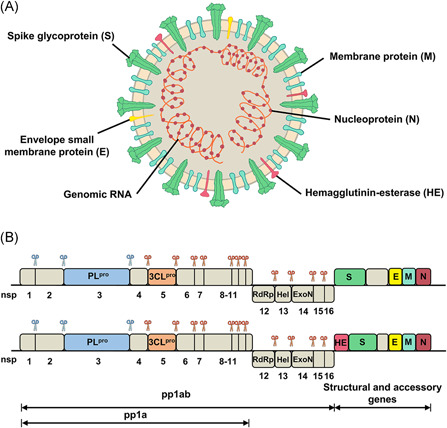
Structure and genome organization of coronaviruses. In panel A, structural proteins of coronaviruses are marked. In panel B, cleavage positions of PLpro and 3CLpro are indicated by scissors colored blue and orange, respectively [Color figure can be viewed at wileyonlinelibrary.com]
3CLpro cleaves 11 out of the 14 conserved sites. 24 The indispensable role in the life cycle of CoVs and none of the known human proteases possessing a similar cleavage specificity make 3CLpro a highly prospective target for anti‐CoV drug development. Accordingly, some reviews have been published to attract the researchers' attention to the 3CLpro target.13, 15, 25, 26, 27, 28, 29, 30, 31, 32 However, a specific and timely review that sheds light on the similarity and diversity of 3CLpros among different CoVs from the aspects of the structure–function relationship, the ligand binding modes, and the structure‐based inhibitor design is still highly necessary. In this regard, we would like to provide a comprehensive review of the published information of 3CLpro in a total of 12 CoVs (6 human and 6 nonhuman CoVs) since crystal structures of these 12 CoV 3CLpros have been determined. The 3D structure of HCoV‐OC43 3CLpro has not determined and thus was not included in this review. Such information could facilitate the design and discovery of more drug‐like 3CLpro inhibitors with a potential of broad‐spectrum activities against CoVs.
2. THE STRUCTURE, CONFORMATIONAL CHANGE, AND THEIR CORRELATION TO THE BIOLOGICAL FUNCTION OF 3CLpro
Since the outbreak of SARS in 2003, motivation has been driven to visualize the 3D structure of 3CLpro from various CoVs. In Protein Data Bank (PDB), a total of 315 crystal structures have been deposited for 6 human CoV 3CLpros (SARS‐CoV, HCoV‐HKU1, HCoV‐229E, HCoV‐NL63, MERS‐CoV, and the latest SARS‐CoV‐2) and 6 nonhuman CoV 3CLpros (IBV, TGEV, BtCoV‐HKU4, FIPV, PEDV, and MHV).24, 33, 34, 35, 36, 37, 38, 39, 40, 41 Structural biology studies on SARS‐CoV and SARS‐CoV‐2 3CLpros contribute the majority of the structures (110 and 167, respectively, Figure 2). Among the 167 structures of SARS‐CoV‐2 3CLpro, 111 structures were resulted from a large XChem crystallographic fragment screening performed in the Diamond Light Source (UK). 43 These fragments and the related complex structures are, however, not involved in the detailed discussion hereinafter because of the lack of definite inhibitory activity against SARS‐CoV‐2 3CLpro. In the pairwise alignment among these 12 CoV 3CLpros by ESPript, 42 the averaged sequence identity is 51%, with the highest value (96%) for SARS‐CoV versus SARS‐CoV‐2 and the lowest one (39%) for SARS‐CoV versus IBV. Despite the extensive sequence variation, overall structures of these 3CLpros share significant similarity and the minor deviations exist in domain III. 44
Figure 2.
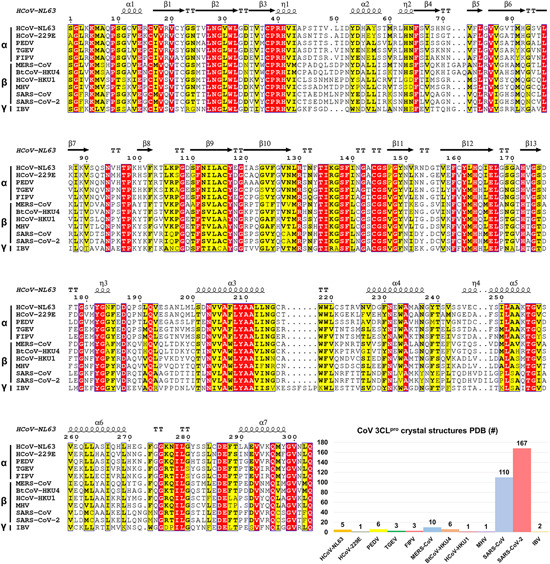
Sequence alignment of 12 CoV 3CLpros by ESPript 42 and the number of crystal structures of 3CLpro in PDB [Color figure can be viewed at wileyonlinelibrary.com]
2.1. The quaternary structure of mature 3CLpro
In general, the 3CLpro protomer consists of an N‐finger and a chymotrypsin‐like two‐domains fold (domains I and II) at the N‐terminus, a globular five‐helices domain III at the C‐terminus, and a long loop connecting domains II and III. 33 A shallow‐surfaced cleft between domains I and II, contributed by 7 and 6 antiparallel β sheets from two domains, constructs the native substrate‐binding site including the catalytic Cys‐His dyad (Figure 3A). The monomeric form of 3CLpro mutants (e.g., the G11A, S139A, and R298A mutants of SARS‐CoV 3CLpro) captured by X‐ray protein crystallography45, 46, 47 only represents an inactive state while dimerization is required to execute the proteolytic activity of wide‐type 3CLpro.
Figure 3.
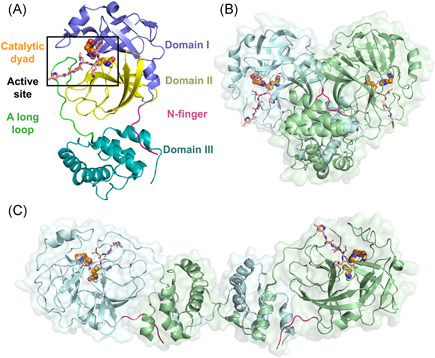
3D structures of 3CLpro. (A) Protomer, (B) homo‐dimer (PDB ID: 2AMQ), and (C) domain‐swapped dimer of homo‐octamer (PDB ID: 3IWM). The bound inhibitors are indicated by sticks and the N‐finger is colored magenta. Two catalytic residues are colored orange and were shown as spheres [Color figure can be viewed at wileyonlinelibrary.com]
The homo‐dimer is the most common form for the crystal structures of catalytically active 3CLpros, and it is stabilized by the contacts between two domain III and modulated by two N‐fingers (residues 1–7) squeezed in between the two protomers (Figure 3B).17, 32, 33, 36 The partial or complete deletion of N‐finger residues (e.g., the deletion of residues 1–5 in TGEV 3CLpro, 48 residues 1–4 49 or 1–750, 51 in SARS‐CoV 3CLpro, or residues 1–8 in PEDV 3CL 40 ) almost completely depleted the proteases' activity. While the crucial role of N‐finger residues in maintaining the enzyme activity was revealed by these studies, the underlying mechanism is, however, under debate. Chen et al. 50 suggested that the N‐finger deletion was indispensable for proteolytic activity but not for the dimerization, whereas the others stated that the N‐finger deletion significantly destroyed the dimerization and the truncations resulted in the monomeric form of the protease.40, 49, 52 The more recent NMR experiment by Zhong et al. 53 observed that the SARS‐CoV 3CLpro without the N‐finger formed a new type of inactive dimer which is different from the native active dimer, implying the crucial role of the N‐finger in the enzymatically active dimer formation and explaining the abovementioned controversy. Additionally, the first N‐terminal residue, Ser1, was highlighted as it formed interactions with Phe140 and Glu166 from the neighboring promoter to stabilize the S1 subsite of the ligand‐binding pocket in the dimeric SARS‐CoV 3CLpro. 54
In addition to the homo‐dimer, a homo‐octamer of SARS‐CoV 3CLpro which was formed by the self‐assembling of four domain‐swapped dimers (Figure 3C), was also observed, and speculated to be a novel super‐active state because it has much higher proteolytic activity than the dimer does at low protein concentrations. 55 Such a domain‐swapped configuration, which is rather constructed by the packing of the last four helices (residues 227–306) from domain III, is certainly different from the commonly seen dimer.
The equilibrium between monomeric and dimeric 3CLpro has been extensively investigated by utilizing various methodologies including analytical ultracentrifuge,40, 49, 56, 57 gel filtration size exclusive chromatography,40, 49, 50, 53, 57, 58, 59 small‐angle X‐ray scattering, chemical cross‐linking and enzyme kinetics, 60 dynamic light scattering, 17 and so forth. For instance, Anand et al. 17 found the coexistence of monomers (~65%) and dimers (~35%) in the diluted solutions of HCoV‐229E and TGEV 3CLpros. The dissociation constant (K d ) of SARS‐CoV 3CLpro dimer has been reported in the nanomolar range.56, 57, 61 By removing the N‐terminal His‐tag of SARS‐CoV 3CLpro expressed in Escherichia coli, Kuo et al. 62 revealed that the K d of the untagged dimer was 15 nM. The measured K d of MERS‐CoV 3CLpro dimer was ~52 μM. 63 As a result, the activity of dimer was dependent on not only the protein concentration but also the binding affinity between two protomers, which is in line with the observation that the dimer was constantly present only if a high concentration was reached. 55 The activity of active octamer, on the other hand, was proposed to be active once formed, which may provide the explanation for the higher activity of the octamer than that of the dimer. 55 Interestingly, the substrate‐induced dimerization has been also found. For example, the binding of peptidic substrate enables to induce the dimerization of SARS‐CoV 3CLpro,64, 65, 66, 67 and is particularly required for the formation of more weakly associated MERS‐CoV 3CLpro dimer. 68 It should be noted that the detailed in vivo information of 3CLpro in the viral polyproteins and its maturation process is still elusive.27, 56, 67 Nevertheless, the observation of super‐novel octamer and substrate‐binding induced dimerization suggested that the functional mechanism of 3CLpro could be much more complicated than what has been supposed.
2.2. The substrate‐binding site
The substrate‐binding site, also called the active site, of 3CLpro displays intrinsic structural dynamics upon the binding of substrates or inhibitors. In comparison to the large cavity maintained in the inactive monomer, the structure of the active site in the 3CLpro dimer is converted to a catalytically competent conformation containing an intact oxyanion hole and a substrate‐binding pocket (Figure 4A,B). The long substrate‐binding pocket is composed of two deeply buried subsites (S1 and S2), a shallow hydrophobic subsite (S4), and several extended solvent‐accessible subsites (S3, S5, and S1ʹ). At the S1ʹ subsite, the cysteine of the Cys‐His dyad acts as a nucleophile while the histidine serves as a base and proton acceptor. 39 Meanwhile, at the S1 subsite, the oxyanion hole is formed by the main chain nitrogen of Gly143, Ser144, and Cys145 and is an essential and conserved functional component of 3CLpro.
Figure 4.
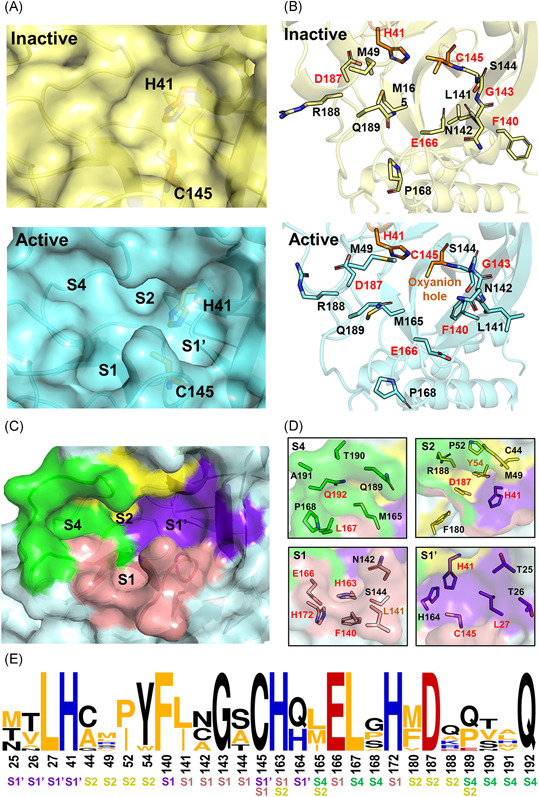
Characterization of the substrate‐binding site of 3CLpro. (A) Expanded view of the substrate‐binding site of SARS‐CoV 3CLpro in its inactive and active forms (PDB ID: 1UJ1 and 2AMQ). (B) Detailed configurations of all residues involved in the binding site. Conserved residues are highlighted in red. (C,D) Residue configurations of each subsite of SARS‐CoV‐2 3CLpro (PDB ID: 6LU7). (E) The sequence logo69, 70 representation of the conservation of residues forming the binding site. The color coding is defined according to the chemical properties of amino acids (polar or neutral: black, basic: blue, acidic: red, and hydrophobic: orange) and the subsite legend is given under each amino acid [Color figure can be viewed at wileyonlinelibrary.com]
In the SARS‐CoV‐2 3CLpro, the S1 subsite is mainly composed of the side chains of Phe140/Leu141/Asn142/Gly143/Ser144/His163/Glu166/His172, while the hydrophobic S2 subsite is constituted by the side chains of His41/Cys44/Met49/Pro52/Tyr54 and the alkyl portion of the side chains of Asp187/Gln189 (Figure 4C,D). In addition, the side chains of Met165/Pro168/Gln192 and the main chains of Gln189/Thr190/Ala191 contribute to the subsite of S4, and the side chains of Thr25/Leu27/His41/Cys145/H164 belong to the extensive S1ʹ subsite. These residues are well conserved in 12 CoV 3CLpros, in particular in the S1 subsite (Figure 4E).
Based on the structural biology studies on SARS‐CoV 3CLpro, two switch models, including the pH‐triggered activity switch and the gate‐regulated switch, have been proposed for the transition from an inactive state to an active one of 3CLpro. The molecular dynamics simulations by Chen et al. suggested that the two protomers in the SARS‐CoV 3CLpro dimer were asymmetric and only one protomer was active at a time, namely half‐site activity. 71 The crystal structure analysis by Yang et al. 33 confirmed the half‐site activity of SARS‐CoV 3CLpro at pH 6 and found that the increase in pH (up to 7.3–8.5) could prompt both protomers to adopt the catalytically competent conformation. Similarly, Tan et al. observed that the maximal enzymatic activity of SARS‐CoV 3CLpro was reached at pH 7 but it was reduced to ~50% at pH 6. 72 Recently, the asymmetric conformation for the two protomers has been also observed in SARS‐CoV‐2 3CLpro. 73
On the other hand, different conformations of S1 and S2 subsites of the SARS‐CoV 3CLpro were observed upon the binding of peptidyl inhibitors. 37 For instance, the side chain of Asn142 flipped over and acted as a lid above the P1 position of the substrate in accompany with the main‐chain movement of residues 141–143, working as a half of a gate to cover the S1 subsite (PDB ID: 2AMQ, Figure 4B). 37 Met49, on the opposite side, protruded from the S2 subsite and Gln189 moved upwards to interact with the P2 portion of the substrate or inhibitor to form the other half of the gate. This is the so‐called gate‐regulated switch induced by the substrate/inhibitor binding.
3. SUBSTRATE SPECIFICITY AND REACTION WITH 3CLpro
3.1. The catalytic mechanism for the cleavage of polyproteins by 3CLpro
Inferred from the structure and autolytic cleavage (auto‐processing) analysis of the SARS‐CoV 3CLpro, 32 a universe nucleophilic‐type reaction mechanism has been proposed for the cleavage of polyproteins by 3CLpro. 74 As shown in Figure 5, the Cys‐thiol of the catalytic dyad is first deprotonated under the assistance of the nearby histidine (step I). The anionic sulfur next attacks the carbonyl carbon of the scissile amide bond (II). Subsequently, a peptide product is released with an amine terminus while the histidine restores the deprotonated form (III). Finally, the generated thioester undergoes hydrolysis to release the carboxylic acid (IV) and the catalytic dyad is recovered to the initial state ready for the next proteolytic cycle. Recently, Wang et al. 44 used an integrated computational and X‐ray crystallography approach to investigate the catalysis mechanism of SARS‐CoV and MERS‐CoV 3CLpros, providing detailed insight into the catalytic reaction process. In addition, it has been suggested that the zwitterionic Cys145‐His41 dyad instead of its neutral form triggered the reaction in step I for both SARS‐CoV75, 76 and SARS‐CoV‐2 3CLpros.63, 77
Figure 5.
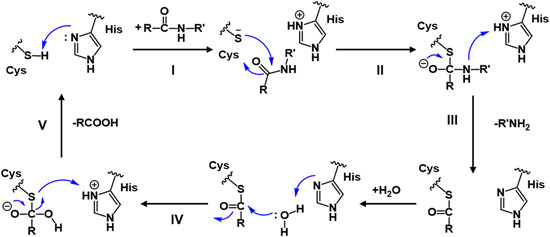
Schematic view of the catalytic mechanism underlying natural substrate hydrolysis by 3CLpro [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Substrate selectivity: Similarity and diversity
As 3CLpro cleaves 11 sites including two auto‐cleavage sites on the polyproteins, ample opportunities are provided to explore its substrate specificity based on the sequence of the polyproteins. Following the Schechter–Berger nomenclature, residues of the peptide substrate binding to the subsites of 3CLpro, termed Sn, …, S1, S1ʹ, …, Snʹ, are accordingly defined as Pn, …, P1, P1ʹ, …, Pnʹ. 78 It has been revealed that all CoV 3CLpros confer extremely high specificity for the P1 residue of glutamine in the substrates.32, 79 A comprehensive study on the residues close to the cleavage site enables us to design or discover more potent inhibitors, which fit perfectly into the substrate‐binding pocket.
The sequence logos generated by Weblogo69, 70 on 12 CoV polyproteins collected from Uniprot indicated a general degree of specificity of each residue on the substrate that follows an order of P1 > P2 > P1ʹ > P4 > P3 (Figure 6). In addition, these Pn positions feature their own preferences. For instance, while Gln is often specific for the P1 position, in HCoV‐NL63 and HCoV‐HKU1 3CLpro, His appears at P1 in one of 11 cleavage sites (Figure 6). The P2 position is mainly occupied by various hydrophobic residues such as Leu, Val, Met, Ile, and Phe, among which Leu is the most frequent one. The P1ʹ position, on the other hand, is favored by small‐sized residues like Ser and Ala, while either small polar (Thr and Ser) or hydrophobic (Val and Ala) residues are located at the P4 position. For the P3 position, the specificity is relatively low. Thr, Val, and even positive‐charged residues are acceptable for this position.
Figure 6.
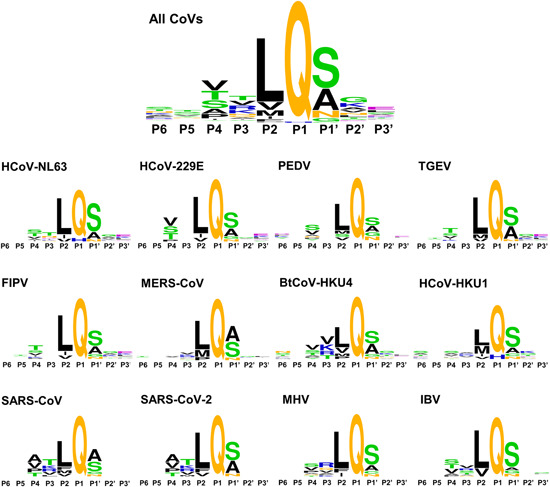
Sequence logos indicating the amino acid specificity in the cleavage site of substrates for 12 CoV 3CLpros [Color figure can be viewed at wileyonlinelibrary.com]
The residue specificity of individual Pn position varies in the substrates for different CoV 3CLpros. As shown in Figure 6, the residue preference at the P3 and P4 positions of MERS‐CoV and HCoV‐HKU1 3CLpros is extremely low. Moreover, the substrates of HCoV‐229E, PEDV, TGEV, and FIPV 3CLpros are even deficient of residue specificity at the P3 position. The sequence logos of SARS‐CoV and SARS‐CoV‐2 3CLpros substrates share significant similarity but a subtle difference exists at the P1ʹ position. In summary, it is revealed that 12 CoV 3CLpros require a high sequence specificity at the P1, P2, and P1ʹ positions of the substrates while the preferences for the remaining Pn's residues are less demanding. It is thus suggested that the development of potent and broad‐spectrum inhibitors of 3CLpros could focus largely on the optimization for improving the binding of the inhibitors to S1, S2, and S1ʹ subsites.
4. CLASSIFICATION OF 3CLpro INHIBITORS
4.1. The binding kinetics of 3CLpro inhibitors
As shown in Figure 7, 3CLpro inhibitors bind to the protease with noncovalent, irreversible covalent, or reversible covalent manners. For noncovalent inhibitors, the equilibrium constant (K i ) is defined as the ratio of unbinding and binding rate constants (k− 1/k 1). Mechanistically, the covalent inhibitors interact with the protease in two steps: the inhibitors first approach to the protease to form a noncovalent complex such that the warhead is in close proximity to the catalytic residue (K i = k −1/k 1, the same as that of the noncovalent inhibitor), and then form a covalent complex by reacting with the catalytic cysteine. As the covalent binding is formed reversibly, the overall dissociation constant K i * is represented by taking into account of the first‐step K i and the second‐step kinetics k 2/k −2 as summarized in Figure 7D. 80 For irreversible covalent inhibitors, k 2 is referred as the inactivation rate constant (k inact ) and k −2 is near or equal to zero. Overall, the recommended measurement for covalent binders is k on = k 2/K i (k inact/K i for irreversible covalent inhibitors). 80 But due to the time‐consuming measurement of K i and k inact , the IC50 or EC50 value is often used for analysis and discussion.81, 82
Figure 7.
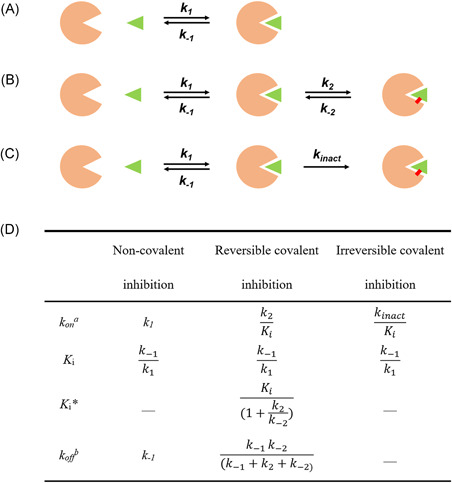
A schematic representation of mode of action of 3CLpro inhibitors: (A) noncovalent, (B) irreversible covalent, and (C) reversible covalent inhibitors. The green triangle, orange sector, and red bold line represent the inhibitor, 3CLpro, and covalent bond, respectively. (D) Kinetics parameters for inhibitors binding with 3CLpro. (a k on : the ligand binding rate constant, b k off : the ligand disassociate rate constant) [Color figure can be viewed at wileyonlinelibrary.com]
4.2. Design of 3CLpro inhibitors by mimicking substrates
Known 3CLpro inhibitors can be classified into two types: peptidyl inhibitors and small molecule‐based inhibitors. Both types can bind to 3CLpro through noncovalent and covalent linking. While the latter are generally discovered through high‐throughput screening of compounds and natural products, the former are designed by mimicking the peptide substrates. According to the substrate specificity and catalytic mechanism, the substrate residues attached with various chemical warheads are generally utilized to design reversible and irreversible covalent inhibitors to compete with the native substrates. Such inhibitors are also called peptidomimetic inhibitors with a covalent mode of action.
With this strategy, inhibitors should be highly similar to substrates to maintain the key interactions of the substrates with the protease. Special attention should be paid to the modification at the P1 position of inhibitors due to the almost absolute specificity of CoV 3CLpro for glutamine. Additionally, the reactivity as well as selectivity of the chemical warhead with the catalytic cysteine of 3CLpro needs to be optimized so as to obtain the more drug‐like covalent inhibitors. To date, the reported chemical warheads (irreversible and reversible) of 3CLpro inhibitors include chloromethyl ketones (CMKs), epoxy ketones, Michael acceptors, aldehydes, ketones with leaving groups, aldehyde bisulfite adducts, nitriles, and alpha‐ketoamides (Figure 8).
Figure 8.
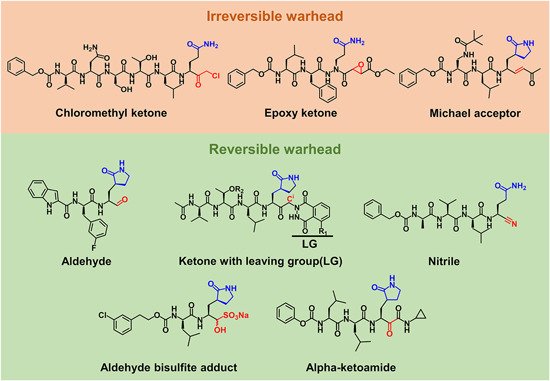
Structures of representative 3CLpro peptidomimetic inhibitors with the highlights on the P1 position (blue) and warheads (red) [Color figure can be viewed at wileyonlinelibrary.com]
5. DETAILED BINDING MODES OF 3CLpro INHIBITORS
In this section, the discovery or development process of inhibitors together with their binding modes toward 3CLpro was elaborated through the analysis of crystal structures as well as the structure–activity relationship. It proceeds following the inhibitor classification into peptidyl and small molecule‐based ones.
5.1. Peptides
5.1.1. Peptidyl chloromethyl ketone
A substrate‐analog CMK inhibitor 1 (Cbz‐VNSTLQ‐CMK, Figure 9A), whose sequence was derived from P6 to P1 residues of the N‐terminal auto‐cleavage site of TGEV 3CLpro, was co‐crystallized with the TGEV 3CLpro (PDB ID: 1P9U) 17 and SARS‐CoV 3CLpro (PDB ID: 1UK4) 33 in 2003. In these complex structures, the methylene of the reactive group of CMK formed a covalent bond with the Sγ atom of catalytic Cys144 and Cys145 in TGEV and SARS‐CoV 3CLpros, respectively, which for the first time provided insights into the inhibitor binding mode of 3CLpro.
Figure 9.
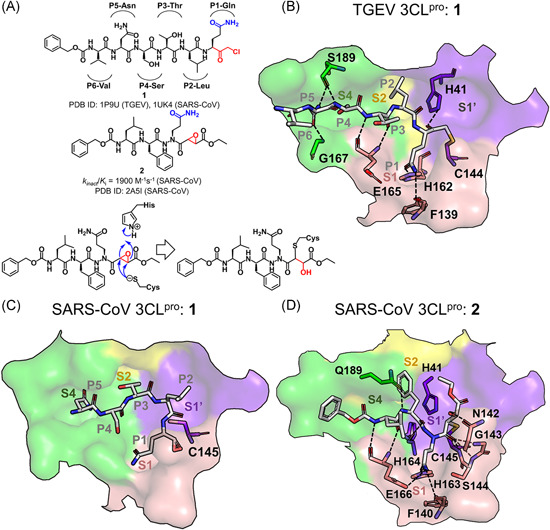
Chloromethyl ketone (CMK) and aza‐peptide epoxide (APE) inhibitors and their complex structures with 3CLpro. (A) Chemical structures of CMK and APE, and the proposed molecular mechanism of 3CLpro inhibited by APE. Under each chemical structure are shown the reported activity data and the available PDB ID of the crystal structure(s) with 3CLpro(s). The CoV type is given in the bracket. (B–D) Detailed configuration of the inhibitors interacting with the S1ʹ‐S4 subsites of 3CLpro in the crystal structures. The subsites are colored purple (S1ʹ), pink (S1), yellow (S2), and green (S4) here and in the figures hereinafter. The 3CLpro residues involved in the interactions are shown in sticks and colored according to their belonging subsites. Hydrogen bonds are shown with dashed lines [Color figure can be viewed at wileyonlinelibrary.com]
Residues, P1‐Gln, P2‐Leu, and P4‐Ser, of inhibitor 1 fitted well into the corresponding subsites of the TGEV 3CLpro, in accord with their crucial roles in the ligand binding with the protease, whereas the side chain of P3‐Thr oriented out to the bulk solvent and P6‐Val had no direct interaction with the protease residues (Figure 9B). The high specificity of the P1 position was dominantly shown via the hydrogen bonding of P1‐Glu side chain with His162 imidazole and Phe139 main chain, and the moderate specificity of P2 and P4 (Figure 6) was associated with the side‐chain hydrophobicity of the former and a small side chain of the latter. Meanwhile, an antiparallel β‐sheet‐like configuration was formed between the P3‐Thr to P5‐Asn of the inhibitor and the protease residues of 165–167, stabilized by the inhibitor‐protease main‐chain/main‐chain hydrogen bonds of P3‐Thr‐Glu165 and a side‐chain/main‐chain hydrogen bond of P5‐Asn‐Gly167. Both P5‐Asn and P4‐Ser also contributed to the hydrogen bonding interactions with the main chain of Ser189. In contrast to the well‐established hydrogen bonds in the TGEV 3CLpro, the P2‐Leu side chain of inhibitor 1 failed to match into the S2 subsite of the SARS‐CoV 3CLpro because of the position shift of P3‐Thr and the dislocation of subsequent residues, which, indeed, suggested the less restricted requirement for the SARS‐CoV 3CLpro than the TGEV 3CLpro to recognize the P2 residue (Figure 9C).
5.1.2. Aza‐peptide epoxide
Given the high reactivity but low specificity of CMK inhibitors that inhibited both serine and cysteine peptidases, an aza‐peptide epoxide (APE) inhibitor 2 (Cbz‐Leu‐Phe‐AGln‐(S,S)EP‐COOEt) displaying excellent specificity toward clan CD cysteine peptidase was identified as a 3CLpro inhibitor (k inact /K i = 1900 M−1 s−1). 83 The crystal structure of the SARS‐CoV 3CLpro in complex with 2 (PDB ID: 2A5I) revealed the covalent linking between the epoxide C3 atom of the inhibitor and the Sγ atom of the catalytic Cys145. The proposed reaction mechanism of 2 with 3CLpro is provided in Figure 9A.83, 84, 85
The binding mode of inhibitor 2 with the SARS‐CoV 3CLpro (Figure 9D) behaved more substrate‐like compared to that of 1 (Figure 9B). Similar to the interactions of 1 with the TGEV 3CLpro, the P1‐Gln side chain of 2 interacted with His163, Phe140 (equivalent to His162 and Phe139 in the TGEV 3CLpro), and Glu166 of SARS‐CoV 3CLpro through forming hydrogen bonds. Moreover, the P3‐Leu of 2 also formed main‐chain/main‐chain hydrogen bonds with Glu166. On the other hand, unlike 1 in the TGEV 3CLpro, the P2‐Phe main chain established a hydrogen bond with the side chain of Gln189, the carbonyl oxygen atom in the scissile peptide bond of P1‐P1ʹ in 2 contacted directly with the amide hydrogen atoms of Gly143 and Cys145 in the oxyanion hole, and meanwhile, the opening of the epoxide ring contributed a hydroxyl group to form a hydrogen bond with Asn142.
5.1.3. Peptides with a Michael acceptor
An interacting pattern similar to that 1 with the TGEV 3CLpro has been found in human rhinovirus (HRV) serotype 2 3Cpro in complex with rupintrivir (compound 3; AG7088; PDB ID: 1CQQ) 86 which has entered clinical trials for the treatment of a common cold. Since CoV 3CLpro and picornavirus 3Cpro possess a similar substrate specificity in P1–P1ʹ and P4, compound 3 was thought to inhibit CoV 3CLpros too. 17 Unfortunately, it failed to inhibit CoV 3CLpros owe to its too long P2‐p‐fluorobenzyl to fit the S2 subsite. 87 Its derivatives were then designed by shortening the side chain of P2‐residue while keeping trans‐α,β‐unsaturated ethyl ester as a Michael acceptor (Figure 10A).
Figure 10.
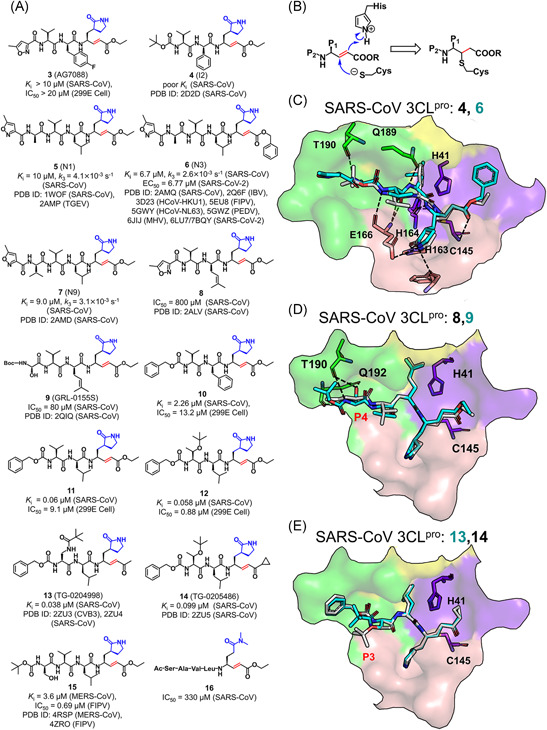
(A,B) Chemical structures of inhibitors containing Michael acceptors and the molecular mechanism of 3CLpro inhibited by these inhibitors. (C–E) Binding modes of the representative inhibitors with a Michael acceptor with 3CLpro. The superposed inhibitors are distinguished by different colors [Color figure can be viewed at wileyonlinelibrary.com]
With the proposed covalent reaction mechanism shown in Figure 10B,32, 88 the crystal structures of the SARS‐CoV 3CLpro in complex with the derivatives of 4–7 (also called I2, N1, N3, and N9; PDB ID: 2D2D, 1WOF, 2AMQ, and 2AMD, respectively) and of the TGEV 3CLpro in complex with 5 (PDB ID: 2AMP) all showed the covalent linking between the Cβ atom of the Michael acceptors and the Sγ atom of the catalytic cysteine (Figure 10C). The inhibitor 4 used a phenyl group to replace the fluorobenzyl at the P2 position, which allowed it to dock into the SARS‐CoV 3CLpro (PDB ID: 2D2D) but still yielded a poor K i , owe to the insufficient interaction of the P2 portion with the S2 subsite and the failure of the planar P4 butyloxycarbonyl engaging with the S4 subsite. In the next structural modification, a more flexible leucine was applied to replace the rather rigid phenyl group at the P2 position and various hydrophobic groups were substituted at the P4 position to improve the inhibitory activity. 37 Intriguingly, the derivative 6 has also been verified as a potential broad‐spectrum inhibitor 37 against multiple CoV 3CLpros such as IBV, 34 FIPV, 39 PEDV, 88 NL63, 35 MHV, 41 and SARS‐CoV‐2 89 3CLpros.
The comparison of compounds 8 and 9 demonstrated the influence of the side‐chain substitution at the P4 position on the inhibitory activity. The substituent changed from a methyl isoxazole in 8 (IC50 = 800 μM) to a Boc‐Ser in 9 (IC50 = 80 μM) led to a significant enhancement in the inhibitory activity against the SARS‐CoV 3CLpro. 90 The resulting crystal structures (PDB ID: 2ALV and 2QIQ) revealed that the Boc‐Ser in 9 formed three hydrogen bonds with Thr190 and Gln192 whereas the oxazole in 8 only had one hydrogen bond with Gln192 (Figure 10D).
For compounds 10–12, 91 the replacement of the methyl isoxazole by a benzoxy at the P4 and the fluorobenzyl by a benzyl at the P2 were shown to have >4‐fold increase in the inhibitory activity (3 → 10), which could be further improved by using leucine group at the P2 position (11). Although the P3 residue is poorly conserved and known to orient toward the solvent environment, a bulkier group such as a lipophilic tert‐butyl group in 12 and 14 or an N‐methylpivalamide group in 13 gained more than tenfold in potency. 92 The crystal structures of the SARS‐CoV 3CLpro bound with 13 and 14 (PDB ID: 2ZU4 and 2ZU5) indicated that both bulkier groups at the P3 position formed hydrophobic interactions with the benzene ring at the P4 position while their backbones fitted well into the S3 and S4 subsites, respectively (Figure 10E). In addition, the modification at the P1ʹ position using α,β‐unsaturated ketone (13) and α,β‐unsaturated ketone cyclopropane (14), introduced a slight influence on the inhibitory activity (K i = 0.058 μM for 12, 0.038 μM for 13 and 0.099 μM for 14).
Besides the SARS‐CoV 3CLpro, the complex structures of compound 15 with FIPV and MERS‐CoV 3CLpros (PDB ID: 4ZRO and 4RSP) were also determined, demonstrating the binding mode of 15 similar to that of 3 with the SARS‐CoV 3CLpro.68, 93 Additionally, the S2 subsite in these two CoV 3CLpros is suitable to accept the side chain of the P2‐Leu residue rather than a larger alkyl or phenylalanine group, providing an explanation for the less inhibitory activity of 8 against the FIPV (52.23%) and MERS‐CoV 3CLpro (21%) compared to those of 15 against the two proteases (both 99%) at a concentration of 50 μM.68, 93
5.1.4. Peptides with an aldehyde
Although the ethyl ester is widely used as a Michael acceptor in covalent inhibitors (Figure 10), it could be easily hydrolyzed by carboxylesterase in plasma. A replacement of the vinyl ethyl ester group with an aldehyde group has been found to yield a stable profile of the compound in mouse, rat, and human plasma. 91 Accordingly, the replacement of the P2‐Leu and α,β‐ethyl ester group in 14 with more lipophilic cyclohexyl alanine and aldehyde yielded compound 17 with improved antiviral activity in cells (Figure 11A). Subsequently, Akaji et al. 94 confirmed that with the same peptide sequence, the inhibitor 18 with a warhead of aldehyde displayed better activity against SARS‐CoV R188I 3CLpro compared to 16 which contains the vinyl ethyl ester group. Compounds 19 and 20 were resulted by the optimization at the P1, P2, and P4 positions of a series of peptidyl aldehydes including 18. 94 In the SAR studies, the cyclization of the side chain of P1‐Glu has been revealed to efficiently improve the potency of inhibitors. The use of an imidazole ring improved the inhibitory activity by >6‐fold as compared to the placement of N,N‐dimethylacetamide at the P1 position (18 → 19). In addition, compound 24 with P1‐(S)‐γ‐lactam showed relatively superior inhibitory activity against the SARS‐CoV 3CLpro than 23 with P1‐(S)‐δ‐lactam does. 44 At the P2 position, a bulkier cyclohexyl group introduced in 20 led to a great improvement in inhibitory activity (>80‐fold) as compared to 19 possessing the P2‐Leu residue. In contrast, the deletion of the P5 portion to reduce molecular weight and the introduction of a more hindered secondary alcohol at the P4 position made the tetrapeptide (compound 21, IC50 = 98 nM) keep the inhibitory activity similar to that of the pentapeptide (compound 20). Thus, it is again demonstrated that modifications at the P1 and P2 positions remarkably change the inhibitory activity of the compounds.
Figure 11.
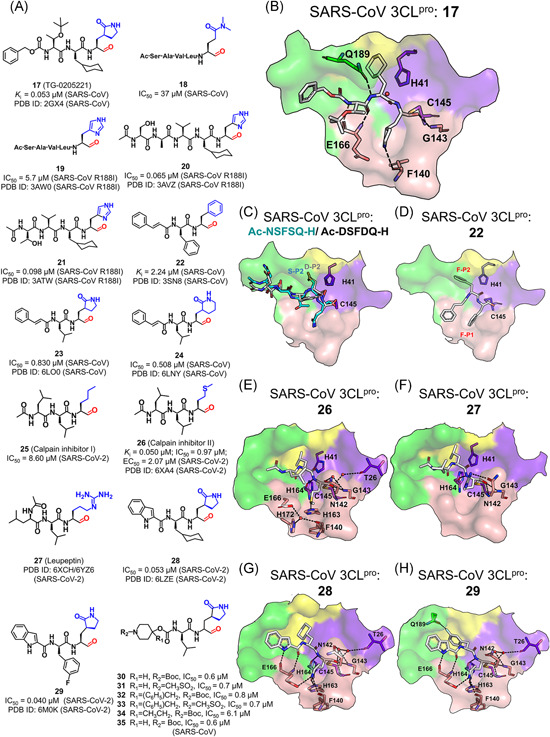
Peptides with an aldehyde: (A) Chemical structures and (B–F) ligand binding modes with various 3CLpros [Color figure can be viewed at wileyonlinelibrary.com]
While the crystal structure of the SARS‐CoV 3CLpro in complex with 17 (PDB ID: 2GX4) exhibited a covalent bond formed between the inhibitor and the protease, the structures of SARS‐CoV R188I 3CLpro bound with 19, 20, and 21 (3AW0, 3AVZ and 3ATW) all showed that the carbonyl carbon of the aldehyde group was in close proximity to the Cys145 Sγ (~2.30 Å) and the electron density could be fitted by an sp2 carbonyl carbon, suggesting no evident covalent bond formed between them. The kinetics experiment also verified that aldehyde‐containing inhibitors compete with the substrate and feature reversible covalent binding. 94 Except the warhead region, the binding modes of these inhibitors are similar. In addition, the cyclohexyl at the P2 position almost occupies the whole S2 subsite, making the main chains of 17, 20, and 21 tightly attach to the active site cleft, which partly explained that the inhibitory activity of these compounds is superior to that of 19 (Figure 11B).
An interesting observation is that the leucine‐preferred S2 subsite could accommodate serine and aspartic acid. For example, several pentapeptides with a warhead of aldehyde bind to the SARS‐CoV 3CLpro with K i values in the micromolar range (Ac‐ESTLQ‐H, Ac‐NSFSQ‐H, Ac‐DSFDQ‐H, and Ac‐NSTSQ‐H; PDB ID: 3SNE, 3SNC, 3SNB, and 3SNA, respectively; Figure 11C). In contrast, the P2‐Leu substitution with serine or aspartic acid in native substrates significantly decreased the cleavage efficiency. 95 Moreover, the relatively hydrophilic but quite restricted P1 position could be placed with phenylalanine in the case of Cm‐FF‐H which contains an aldehyde and is an inhibitor of the SARS‐CoV 3CLpro (22, K i = 2.24 μM, PDB ID: 3SN8, Figure 11D). 95 Therefore, the strict specificity of SARS‐CoV 3CLpro on the P1 and P2 positions might be overruled for the class of the aldehyde‐containing inhibitors.
The overruling of the P1‐Gln specificity is also seen in the newly released crystal structure of the SARS‐CoV‐2 3CLpro in complex with calpain inhibitor II (26; 6XA4), which revealed that a methionine side chain occupied the S1 subsite. 96 At the S1 subsite, the sulfur atom of P1‐Met of 26 (IC50 = 0.97 μM) formed a weak hydrogen bond with His163, and such a hydrogen bond is also found with other peptidyl inhibitors (Figure 11E). When the sulfur atom was replaced with a carbon atom, the inhibitory activity of the resulting compound, calpain inhibitor I (25), was almost tenfold weaker (IC50 = 8.60 μM), validating the significant impact of this key hydrogen bond on the potency. 96 Besides, the crystal structure of SARS‐CoV‐2 3CLpro in complex with leupeptin (27), an aldehyde‐containing protease inhibitor, revealed that the P1‐Arg side chain of leupeptin formed a hydrogen bond with the side chain of Asn142 without entering the S1 subsite (PDB ID: 6YZ6; Figure 11F). This thus implies the tolerance of other residues except glutamine at the P1 position of the 3CLpro inhibitors.
Recently, two aldehyde‐containing compounds, 28 and 29, showed excellent inhibitory activity against the SARS‐CoV‐2 3CLpro. 97 In the two compounds, an (S)‐γ‐lactam ring which yielded the best activity in previous studies 91 was introduced at the P1 position, a cyclohexyl or 3‐fluorophenyl group was placed at the P2 position and an indole group was at the P3 position to form additional hydrogen bonds with the S4 subsite (Figure 11G,H). The resulting crystal structures of the SARS‐CoV‐2 3CLpro in complex with compounds (PDB ID: 6LZE and 6M0K) showed the binding mode of the two compounds similar to other peptidyl inhibitors. The subtle difference between two complex structures is that an additional hydrogen bond was formed between the 3‐fluorophenyl group of 29 to Gln189 while it is absent in the complex of 28.
5.1.5. Peptides with a phthalhydrazido group
Keto‐glutamine peptidyl inhibitors (37–44) containing a phthalhydrazido group at the P1ʹ‐position represent another class of reversible covalent inhibitors against the SARS‐CoV 3CLpro (Figure 12A). 98 Initiated by a reversible covalent inhibitor (36) of the Hepatitis A virus 3Cpro, peptides attaching to the keto‐glutamine warhead yielded enhanced inhibitory activities against the SARS‐CoV 3CLpro with IC50 values ranging from 0.65 to 70 μM. Compared to compounds 37–40, 41–44 featuring an imidazole ring at the P1 position further improved the potency by more than tenfold, which again verified the significant impact of the P1‐imidazole ring on the potency.
Figure 12.
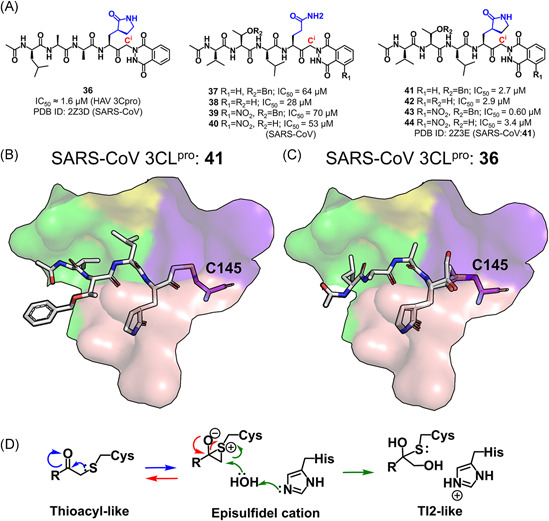
(A) Chemical structures of peptides containing a phthalhydrazido group. (B) A thioacyl‐like covalent bond formed in the SARS‐CoV 3CLpro:41 complex. (C) An episulfidel cation and a Tl2‐like configuration seen in the SARS‐CoV 3CLpro:36 complex. (D) A proposed mechanism for the reaction of the episulfidel cation and the generation of the tetrahedral intermediate in the SARS‐CoV 3CLpro:36 complex [Color figure can be viewed at wileyonlinelibrary.com]
Crystal structures of the SARS‐CoV 3CLpro in complex with 36 and 41 (PDB ID: 2Z3D and 2Z3E) were determined to explore the binding mode of keto‐glutamine peptides with the protease. 99 In the complexes, the Sγ atom of Cys145, instead of the phthalhydrazide group, made a covalent bond to the Ci atom of the inhibitors. Intriguingly, besides the commonly seen thioacyl‐like covalent bond observed in the SARS‐CoV 3CLpro:41 complex structure (Figure 12B), an episulfide cation (thiiranium ring) 100 as well as a second tetrahedral intermediate (TI2)‐like configuration were found in the SARS‐CoV 3CLpro:36 structure (Figure 12C), probably representing the intermediate states in the hydrolysis process of substrates (Figure 12D). 99
5.1.6. Peptides with a nitrile group
A series of nitrile‐contained peptidomimetic inhibitors (45–48, Figure 13A) have been reported to possess broad‐spectrum inhibition against 3CLpros. 101 Structures of the SARS‐CoV 3CLpro co‐crystallized with these inhibitors (PDB ID: 3VB7, 3VB4, 3VB5, and 3VB6) confirmed the covalent bond of the nitrile warhead with the Cys145 Sγ. Toward the SARS‐CoV 3CLpro, compound 47 containing a protective group of carboxybenzyl (Cbz) was tenfold more potent than 45 or 46 with a 5‐methylisoxazole‐3‐carboxyl (Mic) or tert‐buty‐loxycarbonyl (Boc) group, demonstrating the higher potency of the Cbz group at the P4 position. This could be explained by the hydrophobic interactions formed through the aromatic ring of Cbz with Pro168 and P3‐Val (Figure 13B). In addition, 46 also inhibited 3CLpro from other CoVs including HCoV‐229E, NL63, OC43, HKU1, and IBV, with the IC50 value ranging from 1.3 to 3.7 μM. The size increase of the compound to a hexapeptide Cbz‐TSAVLQ‐CN (48) resulted in a decreased inhibitory activity (IC50 = 39 ± 1 μM).
Figure 13.
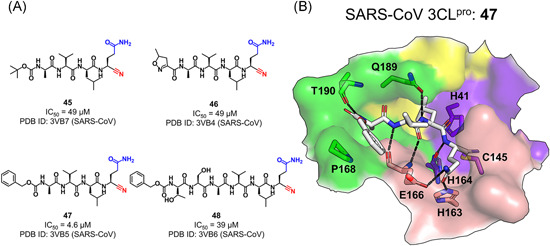
Peptides with a nitrile group: (A) chemical structures and (B) the binding mode of the representative compound (47) with the SARS‐CoV 3CLpro[Color figure can be viewed at wileyonlinelibrary.com]
5.1.7. Peptides with an aldehyde bisulfite adduct
GC376 (49), an inhibitor of the FIPV 3CLpro, is the first inhibitor of coronavirus protease with the successful demonstration of clinical efficacy102, 103 and it also has an excellent inhibitory effect on the SARS‐CoV‐2 3CLpro,104, 105 indicating its potential utility as a broad‐spectrum antiviral agent for the treatment of human CoV infections. A series of aldehyde bisulfite adducts were designed on the basis of GC376 and they were identified to inhibit the MERS‐CoV 3CLpro (Figure 14A). 24 An examination of the crystal structure of the MERS‐CoV 3CLpro in complex with GC376 (PDB ID: 5WKJ) suggested that the aldehyde bisulfite adduct first converted into a peptidyl aldehyde, and then reacted with Cys148 to form a tetrahedral hemi‐thioacetal (Figure 14B), which was also seen in complex structures of the TGEV, 106 PEDV, 107 and SARS‐CoV‐2 3CLpro bound with GC376. 96 A replacement of the P3‐benzyl with m‐Cl phenethyl substituent (GC813, 50) extended the “cap” of the peptide and oriented the phenyl ring toward the S4 subsite, which slightly improved the inhibitory activity against the MERS‐CoV 3CLpro (PDB ID: 5WKK, Figure 14B). Attachment of a piperidine ring to the peptidyl chain generated dipeptidyl inhibitors (51–56) that achieved comparable in vitro potency as the aldehydes (30–35 in Figure 11A) toward the MERS‐CoV 3CLpro. Among these compounds, the substitution of the R1 group which interacted with the S3 subsite had a greater influence on the inhibitory activity than the substitution of the R2 group did. The latter occupied the P4 position and was supposed to interact with the S4 subsite. Crystal structures of the MERS‐CoV 3CLpro bound with two effective inhibitors (53 and 55; PDB ID: 5WKL and 5WKM, respectively) demonstrated that the piperidine ring with the R2 group was missed. This together with the identification of the R1 group at the S3 subsite suggested that the R2 group oriented to the S4 subsite but had weak interactions with the subsite (Figure 14C).
Figure 14.
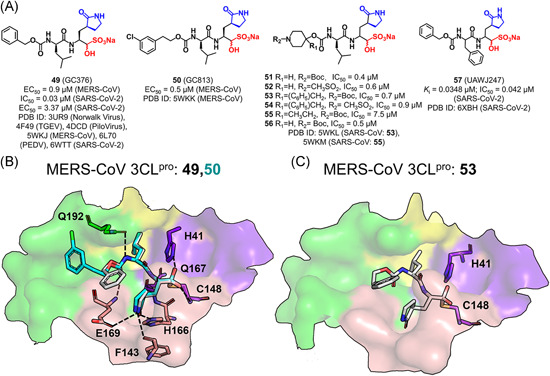
Peptides with an aldehyde bisulfite adduct: (A) chemical structures and (B, C) the ligand binding modes with 3CLpro[Color figure can be viewed at wileyonlinelibrary.com]
5.1.8. Peptides with an alpha‐ketoamide
Recently, α‐ketoamides as novel broad‐spectrum inhibitors of CoV 3CLpros have been recognized (Figure 15A). 96 While crystal structures of the SARS‐CoV (PDB ID: 5N19) and HCoV‐NL63 (PDB ID: 6FV2) 3CLpro bound with 58 identified the covalent bond between the α‐keto‐carbon and Cys145, it inhibited the former protease with an IC50 value of 1.95 μM but had no inhibitory effect on the latter (IC50 > 50 μM). 108 The underlying mechanism is that the much smaller and less plastic S2 subsite of the HCoV‐NL63 3CLpro, caused by the rigid Pro189 instead of Gln189 in the SARS‐CoV 3CLpro (Figure 15B,C), could not fully accommodate the P2‐benzyl group. Accordingly, the substitution of the P2‐benzyl group (58) by an isobutyl one (59) significantly improved the inhibitory activity against the HCoV‐NL63 3CLpro (IC50 = 1.08 μM vs. > 50 μM) due to the fact that the structurally small and flexible P2‐isobutyl group fitted well into the shallow S2 subsite (PDB ID: 6FV1).
Figure 15.
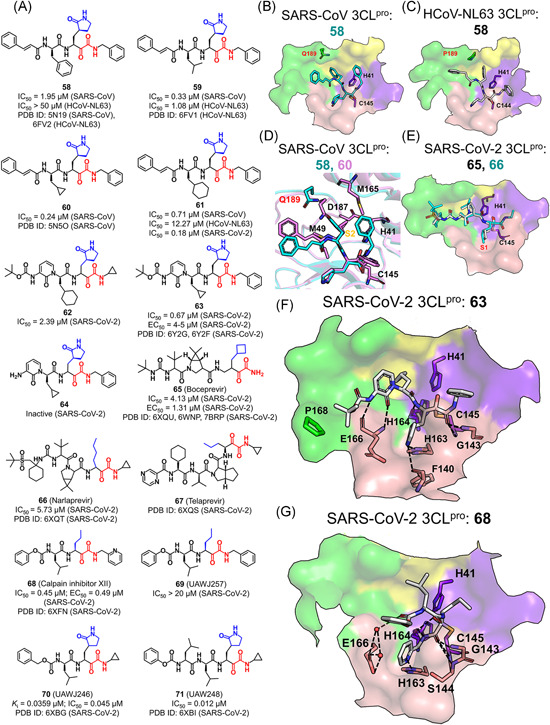
Peptides with an alpha‐ketoamide: (A) chemical structures and (B–G) the ligand binding modes with various 3CLpros [Color figure can be viewed at wileyonlinelibrary.com]
The P2‐cyclopropylmethyl substitution yielded the best inhibitory activity against the SARS‐CoV 3CLpro (60, IC50 = 0.24 μM). Although such a modification is small, the S2 subsite of the SARS‐CoV 3CLpro tightly engaged in the P2 moiety owe to the conformational variability of Gln189. As a result, the cyclopropylmethyl group deeply entered the S2 subsite to form hydrophobic interactions with Met49, Met165, and the Cβ of Asp187 (PDB ID: 5N5O, Figure 15D). In addition, a replacement of the phenyl group by a cyclohexyl one (61) resulted in a significant improvement of the inhibitory activity toward both SARS‐CoV (IC50 = 0.7 μM) and HCoV‐NL63 3CLpros (IC50 = 12.3 μM). Moreover, 61 was found to be a potential broad‐spectrum inhibitor of 3CLpros, as it exhibited excellent antiviral activity against not only HCoV‐229E (EC50 = 1.8 μM) and MERS‐CoV (EC50 = 0.4 nM) in Huh7 cells, but also MERS‐CoV (EC50 = 5 μM) and SARS‐CoV (EC50 = 1.8–2.1 μM) in Vero E6 cells without a toxicity issue. 108
Recently, the lead compound 61 was under a modification so as to inhibit the SARS‐CoV‐2 3CLpro. 73 Substitution of the P3–P2 amide bond and the hydrophobic cinnamoyl moiety with a pyridine ring and a less hydrophobic Boc group, respectively, the resulting compound 62 increased the plasma half‐life by ~3‐fold in mice (from 0.3 to 1.0 h) and in vitro kinetic plasma solubility by a factor of ~19 (from 6 to 112 μM), and reduced the binding to mouse plasma protein from 99% to 97%. 73 However, such a modification decreased the inhibitory activity against the SARS‐CoV‐2 3CLpro (IC50 = 2.39 μM). Subsequently, researches were focused on the improvement of the antiviral activity of the compound toward β‐CoVs. A replacement of the P2‐cyclohexyl group with a smaller cyclopropyl (63) recovered the inhibitory activity against the SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV 3CLpros with IC50 values of 0.67, 0.90, and 0.58 μM, respectively. The antiviral activity against the SARS‐CoV‐2 in human Calu3 cells was measured and the resulting EC50 was 4–5 μM. As compared to 63, the lack of hydrophobic and bulky Boc group in compound 64 resulted in the inactivity against SARS‐CoV‐2, implying the importance of the hydrophobic moiety at the P4 position.
A crystal structure of the SARS‐CoV‐2 3CLpro in complex with 63 (PDB ID: 6Y2F) revealed the same thiohemiketal form for the covalent bond between the α‐keto warhead and Cys145 as that in the structure of 58 or 60 bound SARS‐CoV‐2 3CLpro (Figure 15D, F). Unlike the protective groups in 58 or 60, the P3‐Boc group in 63 stayed close to Pro168 rather than the S4 subsite. It is noteworthy that the α‐ketoamide warhead could form two hydrogen bonds with the catalytic Cys‐His dyad, while aldehydes and Michael acceptors only form a single hydrogen bond with the catalytic dyad. 73
The substrate plasticity (at P1 and P2 positions) existing in the aldehyde inhibitors was also seen in the inhibitors with the alpha‐ketoamide warhead. Nonclassical alpha‐ketoamide peptidyl inhibitors including two FDA‐approved serine protease inhibitors, boceprevir (65) and narlaprevir (66), for the treatment of hepatitis C virus infections, and one calpain inhibitor XII (68) 96 had shown inhibitory activities against the SARS‐CoV‐2 3CLpro. 73 The resulting crystal structures (PDB ID: 6XQU, 6XQT, and 6XQS) revealed that the hydrophobic side chains at the P1 position of 65–67 projected well into the S1 subsite (Figure 15E). Differentiated from 65–67 which adopted a nonextended pose, the P1ʹ‐pyridine of 68 entered the S1 subsite with the P1‐norvaline accommodated in the S1ʹ subsite, the P2‐valine pointing to the solvent, and the P3‐carboxybenzyl group folding back close to the S1 subsite (Figure 15G, PDB ID: 6XFN). 96 The 3CLpro‐68 hydrogen bonds were formed between the P1ʹ‐pyridine nitrogen atom and His163, the P1ʹ main chain and Gly143/Ser144/Cys145, and the water molecule connected P3‐carboxybenzyl oxygen with Glu166 (Figure 15G). A replacement of the P1ʹ‐pyridine group in 68 (IC50 = 0.45 μM) with a phenyl one in 69 (IC50 > 20 μM) led to undetectable inhibition toward the SARS‐CoV‐2 3CLpro, suggesting the importance of the hydrogen bond to His163.
The enzymatic assay and antiviral experiment by Ma et al. 104 found that GC376 (49) was a potent inhibitor of the SARS‐CoV‐2 3CLpro (IC50 = 0.033 μM, EC50 = 3.37 μM). 104 Either the benzyl substitution at the P2 position (57, IC50 = 0.042 μM) or the use of the cyclopropyl capped alpha‐ketoamide warhead instead of the aldehyde bisulfite adduct (70, IC50 = 0.045 μM) had a slight change in the inhibitory activity, and the addition of a P3‐substitution on 70 improved the potency (71, IC50 = 0.012 μM). 96 Combining the data of these four compounds together with 25, 26, 68, and 69, the authors suggested that the inhibitory effect was dominantly determined by the substitution at the P1 position and the reactive warhead, followed by the P2 substitution whereas the modification at the P1ʹ, P3, and P4 position provided the limited contribution. 96
5.2. Small molecules
The determined 3CLpro structures in complex with small molecule‐based inhibitors are significantly fewer than those of the proteases bound with peptidyl inhibitors. In contrast to the general similarity among the binding modes of the peptidyl inhibitors, small molecule inhibitors displayed various binding modes.
5.2.1. Benzotriazole‐based inhibitors
Two series of small molecules bearing a benzotriazole moiety have been found to inhibit 3CLpro. Benzotriazole esters (72 and 73) represent one class of small molecule‐based covalent inhibitors of the SARS‐CoV 3CLpro, with an IC50 value of <5 and 0.1 μM, respectively (Figure 16A). They were first identified as the intermediate product in the search for derivatives of lopinavir by Wu et al., 109 and displayed no cell toxicity at a concentration of 100 μM. 109 With the proposed reaction mechanism shown in Figure 16B, the two compounds acylated the catalytic Cys and the resulting thioester product was subsequently hydrolyzed. The yielded benzoic acid was found to approach the hydrophobic S2 or S1 subsite (PDB ID: 2V6N and 2VJ1, Figure 16C,D). 110
Figure 16.
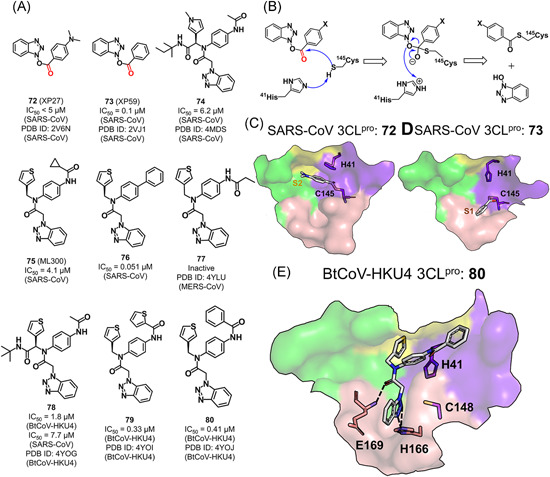
Benzotriazole‐based inhibitors: (A) chemical structures and (B) the proposed mechanism for the benzotriazole ester of inhibitors reacted with the protease. (C–E) The binding modes of the representative benzotriazole‐based inhibitors with various 3CLpros [Color figure can be viewed at wileyonlinelibrary.com]
The second class of the inhibitors (74–80) are noncovalent inhibitors without any warhead resulted from a screening campaign on the NIH Molecular Libraries Probe Production Centers Network. 111 The identification of the diamide compound 74 (IC50 = 6.2 μM) as an inhibitor of 3CLpro and its complex structure with the SARS‐CoV 3CLpro (PDB ID: 4MDS) suggested a unique ligand binding mode in which side chains of Gln189 and Met49 were rearranged to regenerate the S2–S4 subsites. 111 The analog ML300 (75, IC50 = 4.1 μM) and compound 76 (IC50 = 0.051 μM) improved the inhibitory activity by truncating the P3 group and changing the P2–P1ʹ moiety, respectively.
Subsequently, compounds 78–80 were designed to inhibit the BtCoV‐HKU4 3CLpro with a similar binding mode as well as structure–activity relationship (Figure 16E, PDB ID: 4YOG, 4YOI, and 4YOJ). 38 These compounds displayed more potent inhibitory activities toward the BtCoV‐HKU4 3CLpro over the SARS‐CoV 3CLpro, which could be attributed to the difference of the subsites (e.g., S2 and S4) in the two 3CLpros. Compound 77 designed by the same research group, on the other hand, could bind to the MERS‐CoV 3CLpro (PDB ID: 4YLU) but inhibited the 3CLpro only at high concentrations. 68 Given the observation from the analytical ultracentrifugation studies that the MERS‐CoV 3CLpro weakly formed the dimer configuration with the requirement of ligand induction, 68 the MERS‐CoV 3CLpro was suggested to have different properties from other 3CLpros of β‐CoVs.
5.2.2. Pyridyl‐based inhibitors
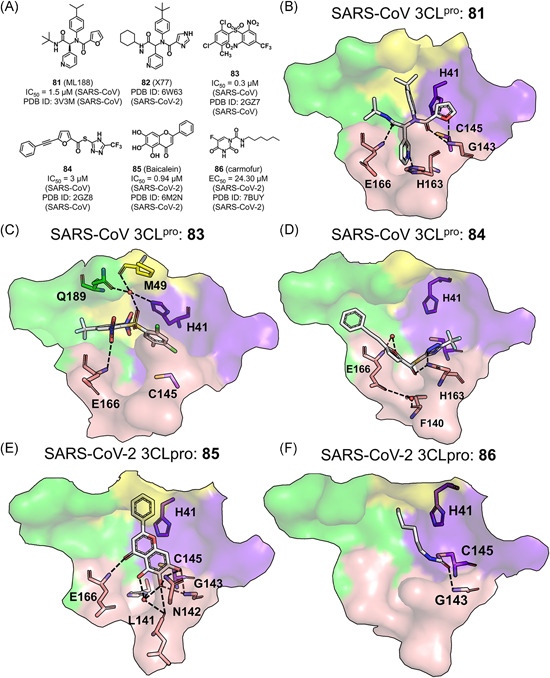
A noncovalent pyridyl‐based compound, ML188 (81), was identified as an inhibitor of the SARS‐CoV 3CLpro (IC50 = 1.5 μM) through high‐throughput screening of the NIH molecular libraries sample collection and was subsequently optimized to yield dipeptide‐like lead compounds (Figure 17A). 112 In a complex structure of the SARS‐CoV 3CLpro with ML188 (PDB ID: 3V3M), the compound adopted an orientation of an S enantiomer, similar to that of known covalent peptidomimetic inhibitors, with the tert‐butyl amide, tert‐butylanilido, 3‐pyridyl, and furyl amide groups occupying the S3, S2, S1, and S1ʹ subsites, respectively (Figure 17B). Particularly, the pyridyl ring formed a hydrogen bond with His163 side chain in the S1 subsite, which was recognized as the key interaction determining the inhibitory activity. 112 The recently released crystal structure of the SARS‐CoV‐2 3CLpro in complex of X77 (82), an analog of ML188, revealed a similar binding mode except that the imidazole group of 82 formed an additional water‐mediated hydrogen bond with the catalytic His41 (PDB ID: 6W63).
Figure 17.
Pyridyl‐based and other small molecule inhibitors: (A) chemical structures and (B–E) the binding modes of these small molecule inhibitors with various 3CLpros [Color figure can be viewed at wileyonlinelibrary.com]
5.2.3. Others
The structure‐based virtual screening and analog search observed two other classes of novel noncovalent small molecule inhibitors against the SARS‐CoV 3CLpro. 113 In one complex crystal structure of the SARS‐CoV 3CLpro (PDB ID: 2GZ7), 83 occupied the S3–S5 subsites where the phenyl ring made π–π interactions with the side chain of His41 and the 2‐nitro group was involved in a water‐mediated hydrogen bonding network connecting the side chains of His41 and Gln189 as well as the main chain of Met49 (Figure 17C). Moreover, the binding of 83 likely introduced electrostatic interactions with the His41 imidazole and thus led to a shifting of the latter to block the catalytic dyad function. Interestingly, this binding mode is actually different from the peptidomimetic inhibitors binding that experienced unshifted His41 side‐chain orientation, and also not observed in the structure of the SARS‐CoV 3CLpro in complex with 84 (Figure 17D). In the latter structure, the hydrogen bonding connection between 84 and Cys145 as well as Asn142 became the key interactions to stabilize the inhibitor binding.
We recently discovered baicalein (85), an aglycone of baicalin, to inhibit the SARS‐CoV‐2 3CLpro with an IC50 value of 0.94 μM. 114 As revealed by the crystal structure (PDB ID: 6M2N), 85 formed hydrogen bonds with the main chains of Leu141 and Gly143 and the side chains of Ser144 and Asn142 (belonging to the loop involved in the oxyanion hole) in the S1ʹ subsite and the main chain of Glu166 in the S1 subsite (Figure 17E). Jin et al. 89 identified carmofur (86), an FDA‐approved antineoplastic agent, as an inhibitor of the SARS‐CoV‐2 3CLpro in vitro (IC50 = 1.82 μM) by screening more than 10,000 compounds. 89 The resolved crystal structure of the SARS‐CoV‐2 3CLpro in complex with 86 (PDB ID: 7BUY) revealed that the carbonyl group of 86 formed the covalent bond with Cys145 to release the 5‐FU moiety and meanwhile, the carbonyl oxygen was connected to the main chain amides of Gly143 and Cys145 through hydrogen bonding, and the fatty acid was accommodated by the hydrophobic S2 subsite (Figure 17F). 115
6. INHIBITORS OF 3CLpro IN PRECLINICAL STUDIES
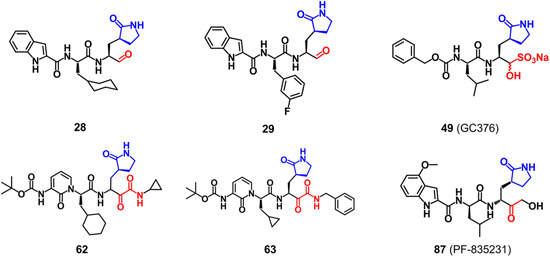
Up to now, no CoV 3CLpro inhibitors have entered the clinical trials. The preclinical inhibitors targeting CoV 3CLpros that can be searched from Cortellis Drug Discovery Intelligence are shown in Figure 18, and they are all classic peptidyl inhibitors. Among them, the inhibitors with a covalent warhead of aldehyde (28 and 29) or alpha‐ketoamide (62 and 63) inhibiting the SARS‐CoV‐2 3CLpro, or the aldehyde bisulfite adduct (49) inhibiting both the MERS‐CoV and SARS‐CoV‐2 3CLpros have been discussed in details in the above sections. Compound 49 displayed the highest IC50 value of 0.03 μM toward the SARS‐CoV‐2 3CLpro. Besides, compound 87 (Patent number: WO2005113580) with a covalent warhead of hydroxymethyl ketone that was invented by Hoffman et al. from Pfizer showed the best IC50 value of 0.004 μM against the SARS‐CoV 3CLpro by FRET assays.
Figure 18.
Chemical structures of 3CLpro inhibitors in preclinical studies [Color figure can be viewed at wileyonlinelibrary.com]
7. CONCLUSION AND PERSPECTIVES
Expert knowledge of the target structure and the relationship to its biological function is a priority for the structure‐based drug design. An amount of 3CLpro structures determined by X‐ray protein crystallography have been deposited in the PDB for a total of 12 CoVs, either in apo‐state or mostly bound with various substrates and inhibitors. The comparative analyses of these structures, in combination with the substrate specificity and inhibitory activities of the inhibitors, allow us to summarize the binding modes of known inhibitors as well as the structure–activity relationships that will provide a cumulative source for the development of new anti‐CoV chemotherapy.
Structurally, 3CLpro shares a significant similarity in structural features among CoVs, with the active site characterized by a large shallow binding pocket consisting of S4–S1ʹ subsites to accommodate substrates and an intact oxyanion hole to consolidate the carbonyl oxygen of the substrate scissile amide bond. Among these subsites, S1 and S2 are deeply buried and S4 is structurally shallow and physicochemically hydrophobic, whereas the remaining subsites are extensively solvent accessible. Residues involved in these subsites are highly conserved among 12 3CLpros and particularly, the S1 subsite most accounts for the high conservation.
The substrate specificity, accordingly, endows individual S4–S1ʹ subsites to recognize and interact with P4–P1ʹ positions of the substrate, and the catalytic Cys‐His dyad responds to the covalent linking with the substrate following a universe nucleophilic‐type catalytic mechanism (Figure 5). Generally, features of the substrate specificity among the CoV 3CLpros can be clearly seen on the specific residue accepted at the P1, P2, and P1ʹ positions (Figure 6). Among the three positions, Gln is highly specific for P1, various hydrophobic residues including Leu, Val, Met, Ile, and Phe are for P2, and small‐sized residues like Ser and Ala are for P1ʹ. In contrast, the substrate specificity at the P3 and P4 positions is less restricted, and it could be extremely low for the MERS‐CoV and HCoV‐HKU1 3CLpros or is even totally absent at the P3 position of the HCoV‐229E, PEDV, TGEV, and FIPV 3CLpros as compared to the other 3CLpros.
On the basis of the substrate specificity knowledge and the catalytic mechanism, peptidyl inhibitors, the majority of 3CLpro inhibitors, can be designed to imitate and compete with the native substrates. On the one hand, the chemical warhead in a peptidyl inhibitor that is utilized to form a covalent bond with the catalytic cysteine of 3CLpro could be recruited from chloromethyl ketones, epoxy ketones, Michael acceptors, aldehydes, ketones with leaving groups, aldehyde bisulfite adducts, nitriles, and alpha‐ketoamides. On the other hand, the individual P4–P1ʹ positions of the inhibitor make in contact with the corresponding S4–S1ʹ subsites to demonstrate their respective roles in determining the inhibitory activity. Using the SARS‐CoV‐2 3CLpro as an example, the pivotal protease‐inhibitor interactions include (1) the hydrogen bonding from the main chains of Gly143‐Cys145 to the P1–P1ʹ carbonyl groups of the inhibitor, from the main chain of Phe140 and side chain of His163/Glu166 to the P1‐position side chain, in which the hydrogen bonding with His163 is considered as the key interaction, from the main chains of His163/Glu166/Thr190 and the side chain of Gln189 to the main chain of the inhibitor, and (2) hydrophobic interactions of Met49, Met165, and Asp187 with the P2 side chain of the inhibitor.
The substitution at the P4–P1ʹ positions is supposed to have a direct impact on the inhibitory activity, particularly at the P1 and P2 positions. For instance, the cyclized Gln mimetic, (S) γ‐lactam, imidazole, and pyridine are often utilized for the side chain of the P1 position as it is favored by the highly Gln‐specific S1 subsite. While the flexible P2‐Leu is generally accepted by all 3CLpros, the bulkier group like a cyclohexyl, cyclopropyl, flurobenzyl, and phenyl ring at the P2 position could improve inhibitory activities, for example, against the SARS‐CoV and SAS‐CoV‐2 3CLpros. Additionally, various bulkier and hydrophobic groups are often placed at the P3 and P4 positions, for example, the addition of protective groups of Boc, Boc‐Ser, and benzoxy at the P4 position, to modify the inhibitory activity of compounds.
The overruling of the substrate specificity at the P1 and P2 positions can be, however, sometimes observed for the SARS‐CoV and SARS‐CoV‐2 3CLpros. For instance, the SARS‐CoV 3CLpro inhibitor 22 and the SARS‐CoV‐2 3CLpro inhibitor 26 have phenylalanine and methionine side chain at the P1 position, respectively. The pentapeptide aldehydes of Ac‐ESTLQ‐H, Ac‐NSFSQ‐H, Ac‐DSFDQ‐H, and Ac‐NSTSQ‐H that can bind with the SARS‐CoV 3CLpro with micromolar K i values contain P2‐Ser or P2‐Asp side chain. The nonclassic alpha‐ketoamide peptidyl inhibitors 65–68 that possessed inhibitory activities against the SARS‐CoV‐2 3CLpro emplace various hydrophobic side chains at the P1 positions. The corresponding crystal structures with the protease reveal that these “mismatched” P1 or P2 side chains are indeed accommodated by the S1 or S2 subsite except for 68.
Besides the caution in using the substrate specificity roles in designing inhibitors toward SARS‐CoV and SARS‐CoV‐2 3CLpros, it should be noticed that the trivial difference in the active site constituents among 3CLpros could influence the potency of inhibitors. For example, the rigid Pro189 in the HCoV‐NL63 3CLpro results in a relatively smaller and less plastic S2 subsite than the counterpart in the SARS‐CoV 3CLpro with Gln189. Accordingly, 58 inhibits the SARS‐CoV 3CLpro with an IC50 value of 1.95 μM but has no inhibitory effect on the HCoV‐NL63 3CLpro (IC50 > 50 μM), which is caused by the failure in fully accommodating the P2‐benzyl group by the HCoV‐NL63 3CLpro S2 subsite.
Different from the peptidyl inhibitors of which the substrate specificity generally regulated the binding modes, small molecule inhibitors display more diversity in their binding modes with the protease. The interactions from 3CLpro to these two kinds of inhibitors are mainly from the residues at the S1 and S2 subsites, either through noncovalent or covalent binding manner. In combination with the identified binding modes of the peptidyl and small molecules inhibitors with 3CLpro, the key residues of the protease involved in the ligand binding are the catalytic dyad, Phe140, Asn142, Gly143, His163, and Glu166 at the S1 subsite, and Met49 together with Gln189 at the S2 subsite in the SARS‐CoV‐2 3CLpro.
Among the reported inhibitors, the ones shown in Table 1 display distinguished inhibitory activities against specific 3CLpros. One might pay more attention to these covalent inhibitors in the anti‐CoV chemotherapy development. In addition, this review mainly focuses on the inhibitor design targeting the catalytic sites. It should be noticed that the dimer interface might be also a potential target since the formation and structure stabilization of the dimer of 3CLpro are the prerequisites for the performance of its biological function. To date, only a few studies have been reported on the dimerization inhibitors which reduced dimerization of the protease.52, 116, 117 Nevertheless, it may be still worth paying attention to the development of dimerization inhibitors as an alternative approach for the 3CLpro‐targeted drug design.
Table 1.
Selected potent 3CLpro inhibitors (EC50 characterizes the activity against virus cell, K i and IC50 respond to the activity against 3CLpro)
| CoV type | ||||
|---|---|---|---|---|
| Inhibitor type | Compound | SARS‐CoV | SARS‐CoV‐2 | MERS‐CoV |
| Aldehyde | 17 | K i = 0.053 μM | ||
| 26 | EC50 = 2.07 μM | |||
| 28 | IC50 = 0.053 μM | |||
| 29 | IC50 = 0.040 μM | |||
| Aldehyde bisulfite adduct | 49 | IC50 = 0.030 μM | EC50 = 0.9 μM | |
| 57 | IC50 = 0.042 μM | |||
| Alpha‐ketoamide | 68 | EC50 = 0.49 μM | ||
| 70 | IC50 = 0.045 μM | |||
| 71 | IC50 = 0.012 μM | |||
| Benzotriazole | 76 | IC50 = 0.051 μM | ||
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (Grant no. 21877122 and 32071248) and the Science and Technology Commission of Shanghai Municipality (Grant no. 20430780300).
Biographies
Muya Xiong received her BS (2019) from East China University of Science and Technology and is now pursuing her MS at Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences (CAS). She is interested in the molecular modeling of various drug target proteins and structure‐based drug design.
Haixia Su received her BS (2016) from East China University of Science and Technology and is now pursuing her PhD at SIMM, CAS. She is interested in the resolution of crystal structures of drug target proteins and structure‐based drug design.
Wenfeng Zhao received his BS (2014) and PhD (2020) from China Pharmaceutical University. He is interested in the resolution of crystal structures of drug target proteins, medicinal chemistry, and structure‐based drug design.
Hang Xie received her BS (2015) from Hubei University of Science and Technology and MS (2018) from China Pharmaceutical University. She then continued to work at SIMM, CAS. She is interested in the resolution of crystal structures of drug target proteins and pharmacology.
Qiang Shao received his BS (1999) from Nanjing University, MS from the College of William & Mary, and PhD (2009) from Texas A&M University. Since then, he worked as a postdoctoral researcher at Peking University and is currently a professor at SIMM, CAS. His research interest focuses on the molecular modeling of the structure and dynamics of drug target proteins, protein–ligand interactions, and structure‐based drug design.
Yechun Xu received her BS (1999) from East China Normal University and PhD (2004) at SIMM, CAS. Since then, she worked as a postdoctoral researcher at Weizmann Institute of Science and is currently a professor and principal investigator in SIMM, CAS. Her research group utilizes both computational and experimental approaches to clarify the nature as well as personalized characteristics of active compounds binding to targets, providing key information for rational drug design, and leading compound discovery. Recently, she has led the group devoted to structure‐based discovery and design of novel inhibitors targeting SARS‐CoV‐2 3CLpro, identified its crystal structure, and reported the multiple antiviral drug candidates.
Xiong M, Su H, Zhao W, Xie H, Shao Q, Xu Y. What coronavirus 3C‐like protease tells us: From structure, substrate selectivity, to inhibitor design. Med Res Rev. 2021;41:1965–1998. 10.1002/med.21783
Contributor Information
Qiang Shao, Email: qshao@simm.ac.cn.
Yechun Xu, Email: ycxu@simm.ac.cn.
REFERENCES
- 1. Bailey OT, Pappenheimer AM, Cheever FS, Daniels JB. A murine virus (JHM) causing disseminated encephalomyelitis with extensive destruction of myelin: II pathology. J Exp Med. 1949;90(3):195‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hamre D, Procknow JJ. A new virus isolated from the human respiratory tract. Proc Soc Exp Biol Med. 1966;121(1):190‐193. [DOI] [PubMed] [Google Scholar]
- 3. McIntosh K, Dees JH, Becker WB, Kapikian AZ, Chanock RM. Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc Natl Acad Sci USA. 1967;57(4):933‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bradburne AF, Bynoe ML, Tyrrell DA. Effects of a "new" human respiratory virus in volunteers. Br Med J. 1967;3(5568):767‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bradburne AF, Somerset BA. Coronative antibody tires in sera of healthy adults and experimentally infected volunteers. J Hyg. 1972;70(2):235‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. World Health Organization . Severe Acute Respiratory Syndrome (SARS) situation updates. https://www.who.int/csr/sars/archive/2003_05_07a/en/. Accessed September 4, 2020.
- 7. van der Hoek L, Pyrc K, Jebbink MF, et al. Identification of a new human coronavirus. Nat Med. 2004;10:368‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Woo PC, Lau SK, Chu CM, et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol. 2005;79(2):884‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. World Health Organization . Clinical Management of Severe Acute Respiratory Infection When Middle East Respiratory Syndrome Coronavirus (MERS‐CoV) Infection Is Suspected: Interim Guidance. https://apps.who.int/iris/bitstream/handle/10665/178529/WHO_MERS_Clinical_15.1_eng.pdf. Accessed September 4, 2020.
- 10. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. World Health Organization . Coronavirus Disease (COVID‐19) Weekly Epidemiological Update and Weekly Operational Update. https://www.who.int/publications/m/item/weekly-epidemiological-update---15-december-2020. Accessed December 16, 2020.
- 12. Woo PC, Lau SK, Lam CS, et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86(7):3995‐4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zumla A, Chan JF, Azhar EI, Hui DS, Yuen KY. Coronaviruses – drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15(5):327‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lau S, Wong E, Tsang CC, et al. Discovery and sequence analysis of four deltacoronaviruses from birds in the middle east reveal interspecies jumping with recombination as a potential mechanism for avian‐to‐avian and avian‐to‐mammalian transmission. J Virol. 2018;92(15):e00265‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pillaiyar T, Meenakshisundaram S, Manickam M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov Today. 2020;25(4):668‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lim YX, Ng YL, Tam JP, Liu DX. Human coronaviruses: a review of virus‐host interactions. Diseases. 2016;4(3):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti‐SARS drugs. Science. 2003;300(5626):1763‐1767. [DOI] [PubMed] [Google Scholar]
- 18. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu A, Peng Y, Huang B, et al. Genome composition and divergence of the novel Coronavirus (2019‐nCoV) originating in China. Cell Host Microbe. 2020;3(27):325‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang XC, Zhang JX, Zhang SY, et al. Prevalence and genetic diversity of coronaviruses in bats from China. J Virol. 2006;80(15):7481‐7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woo PCY, Wang M, Lau SKP, et al. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol. 2007;81(4):1574‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lau SKP, Li KSM, Tsang AKL, et al. Recent transmission of a novel alphacoronavirus, bat coronavirus HKU10, from Leschenault's Rousettes to pomona leaf‐nosed bats: first evidence of interspecies transmission of coronavirus between bats of different suborders. J Virol. 2012;86(21):11906‐11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galasiti KA, Kim Y, Damalanka VC, et al. Structure‐guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur J Med Chem. 2018;150:334‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang H, Bartlam M, Rao Z. Drug design targeting the main protease, the Achilles' heel of coronaviruses. Curr Pharm Des. 2006;12(35):4573‐4590. [DOI] [PubMed] [Google Scholar]
- 26. Liang PH. Characterization and inhibition of SARS‐coronavirus main protease. Curr Top Med Chem. 2006;6(4):361‐376. [DOI] [PubMed] [Google Scholar]
- 27. Xia B, Kang X. Activation and maturation of SARS‐CoV main protease. Protein Cell. 2011;2(4):282‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Q, Weber E, Yang H. Recent developments on coronavirus main protease/3C like protease inhibitors. Recent Pat Antiinfect Drug Discov. 2013;8(2):150‐156. [DOI] [PubMed] [Google Scholar]
- 29. Ullrich S, Nitsche C. The SARS‐CoV‐2 main protease as drug target. Bioorg Med Chem Lett. 2020;30(17):127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Y, Liang C, Xin L, et al. The development of coronavirus 3C‐like protease (3CLpro) inhibitors from 2010 to 2020. Eur J Med Chem. 2020;206:112711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Q, Kang C. Progress in developing inhibitors of SARS‐CoV‐2 3C‐like protease. Microorganisms. 2020;8(8):1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. An overview of Severe Acute Respiratory Syndrome‐Coronavirus (SARS‐CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59(14):6595‐6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang H, Yang M, Ding Y, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA. 2003;100(23):13190‐13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao Q, Li S, Xue F, et al. Structure of the main protease from a global infectious human coronavirus, HCoV‐HKU1. J Virol. 2008;82(17):8647‐8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang F, Chen C, Tan W, Yang K, Yang H. Structure of main protease from human Coronavirus NL63: insights for wide spectrum anti‐coronavirus drug design. Sci Rep. 2016;6:22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xue X, Yu H, Yang H, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82(5):2515‐2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang H, Xie W, Xue X, et al. Design of wide‐spectrum inhibitors targeting coronavirus main proteases. PLOS Biol. 2005;3(10):e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. John SES, Tomar S, Stauffer SR, Mesecar AD. Targeting zoonotic viruses: structure‐based inhibition of the 3C‐like protease from bat coronavirus HKU4—the likely reservoir host to the human coronavirus that causes Middle East Respiratory Syndrome (MERS). Bioorg Med Chem. 2015;23(17):6036‐6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang F, Chen C, Liu X, Yang K, Xu X, Yang H. Crystal structure of Feline Infectious Peritonitis Virus main protease in complex with synergetic dual inhibitors. J Virol. 2016;90(4):1910‐1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ye G, Deng F, Shen Z, et al. Structural basis for the dimerization and substrate recognition specificity of porcine epidemic diarrhea virus 3C‐like protease. Virology. 2016;494:225‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cui W, Cui S, Chen C, et al. The crystal structure of main protease from Mouse Hepatitis Virus A59 in complex with an inhibitor. Biochem Biophys Res Commun. 2019;511(4):794‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42(W1):320‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Diamond Light Source . Main Protease Structure and XChem Fragment Screen. https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem.html. Accessed September 4, 2020.
- 44. Wang H, He S, Deng W, et al. Comprehensive insights into the catalytic mechanism of Middle East Respiratory Syndrome 3C‐Like protease and Severe Acute Respiratory Syndrome 3C‐Like protease. ACS Catal. 2020;10:5871‐5890. [DOI] [PubMed] [Google Scholar]
- 45. Chen S, Hu T, Zhang J, et al. Mutation of Gly‐11 on the dimer interface results in the complete crystallographic dimer dissociation of severe acute respiratory syndrome coronavirus 3C‐like protease: crystal structure with molecular dynamics simulations. J Biol Chem. 2008;283(1):554‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu T, Zhang Y, Li L, et al. Two adjacent mutations on the dimer interface of SARS coronavirus 3C‐like protease cause different conformational changes in crystal structure. Virology. 2009;388(2):324‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Muramatsu T, Takemoto C, Kim YT, et al. SARS‐CoV 3CL protease cleaves its C‐terminal autoprocessing site by novel subsite cooperativity. Proc Natl Acad Sci USA. 2016;113(46):12997‐13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α‐helical domain. EMBO J. 2002;13(21):3213‐3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hsu WC, Chang HC, Chou CY, Tsai PJ, Lin PI, Chang GG. Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J Biol Chem. 2005;280(24):22741‐22748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen S, Chen L, Tan J, et al. Severe acute respiratory syndrome coronavirus 3C‐like proteinase N terminus is indispensable for proteolytic activity but not for enzyme dimerization. J Biol Chem. 2005;280(1):164‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuang WF, Chow LP, Wu MH, Hwang LH. Mutational and inhibitive analysis of SARS coronavirus 3C‐like protease by fluorescence resonance energy transfer‐based assays. Biochem Biophys Res Commun. 2005;331(4):1554‐1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wei P, Fan K, Chen H, et al. The N‐terminal octapeptide acts as a dimerization inhibitor of SARS coronavirus 3C‐like proteinase. Biochem Biophys Res Commun. 2006;339(3):865‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhong N, Zhang S, Zou P, et al. Without its N‐finger, the main protease of severe acute respiratory syndrome coronavirus can form a novel dimer through its C‐terminal domain. J Virol. 2008;82(9):4227‐4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xue X, Yang H, Shen W, et al. Production of authentic SARS‐CoV Mpro with enhanced activity: application as a novel tag‐cleaveage endopeptidacse for protein overproduciton. J Mol Biol. 2007;366(3):965‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang S, Zhong N, Xue F, et al. Three‐dimensional domain swapping as a mechanism to lock the active conformation in a super‐active octamer of SARS‐CoV main protease. Protein Cell. 2010;1(4):371‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hsu MF, Kuo CJ, Chang KT, et al. Mechanism of the maturation process of SARS‐CoV 3CL protease. J Biol Chem. 2005;280(35):31257‐31266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chou CY, Chang HC, Hsu WC, Lin TZ, Lin CH, Chang GG. Quaternary structure of the Severe Acute Respiratory Syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43(47):14958‐14970. [DOI] [PubMed] [Google Scholar]
- 58. Fan K, Wei P, Feng Q, et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C‐like proteinase. J Biol Chem. 2004;279(3):1637‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen S, Zhang J, Hu T, Chen K, Jiang H, Shen X. Residues on the dimer interface of SARS coronavirus 3C‐like protease: dimer stability characterization and enzyme catalytic activity analysis. J Biochem. 2008;143(4):525‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Graziano V, McGrath WJ, Yang L, Mangel WF. SARS CoV main proteinase: the monomer‐dimer equilibrium dissociation constant. Biochemistry. 2006;45(49):14632‐14641. [DOI] [PubMed] [Google Scholar]
- 61. Grum‐Tokars V, Ratia K, Begaye A, et al. Evaluating the 3C‐like protease activity of SARS‐Coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 2008;133(1):63‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kuo CJ, Chi YH, Hsu JT, Liang PH. Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem Biophys Res Commun. 2004;318(4):862‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kneller DW, Phillips G, O'Neill HM, et al. Room‐temperature X‐ray crystallography reveals the oxidation and reactivity of cysteine residues in SARS‐CoV‐2 3CL Mpro: insights into enzyme mechanism and drug design. IUCrJ. 2020;7(6):1028‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wu CG, Cheng SC, Chen SC, et al. Mechanism for controlling the monomer‐dimer conversion of SARS coronavirus main protease. Acta Crystallogr D Biol Crystallogr. 2013;69(5):747‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cheng SC, Chang GG, Chou CY. Mutation of Glu‐166 blocks the substrate‐induced dimerization of SARS coronavirus main protease. Biophys J. 2010;98(7):1327‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen S, Jonas F, Shen C, Hilgenfeld R. Liberation of SARS‐CoV main protease from the viral polyprotein: N‐terminal autocleavage does not depend on the mature dimerization mode. Protein Cell. 2010;1(1):59‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li C, Qi Y, Teng X, et al. Maturation mechanism of Severe Acute Respiratory Syndrome (SARS) coronavirus 3C‐like proteinase. J Biol Chem. 2010;285(36):28134‐28140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tomar S, Johnston ML, St. John SE, et al. Ligand‐induced dimerization of Middle East Respiratory Syndrome (MERS) coronavirus nsp5 protease (3CLpro). J Biol Chem. 2015;290(32):19403‐19422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188‐1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18(20):6097‐6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen H, Wei P, Huang C, Tan L, Liu Y, Lai L. Only one protomer is active in the dimer of SARS 3C‐like proteinase. J Biol Chem. 2006;281(20):13894‐13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tan J, Verschueren KH, Anand K, et al. PH‐dependent conformational flexibility of the SARS‐CoV Main Proteinase (Mpro) dimer: molecular dynamics simulations and multiple X‐ray structure analyses. J Mol Biol. 2005;354:25‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved α‐ketoamide inhibitors. Science. 2020;368(6489):409‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huang C, Wei P, Fan K, Liu Y, Lai L. 3C‐like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43(15):4568‐4574. [DOI] [PubMed] [Google Scholar]
- 75. Paasche A, Zipper A, Schafer S, Ziebuhr J, Schirmeister T, Engels B. Evidence for substrate binding‐induced zwitterion formation in the catalytic Cys‐His dyad of the SARS‐CoV main protease. Biochemistry. 2014;53(37):5930‐5946. [DOI] [PubMed] [Google Scholar]
- 76. Solowiej J, Thomson JA, Ryan K, et al. Steady‐state and pre‐steady‐state kinetic evaluation of severe acute respiratory syndrome coronavirus (SARS‐CoV) 3CLpro cysteine protease: development of an ion‐pair model for catalysis. Biochemistry. 2008;47(8):2617‐2630. [DOI] [PubMed] [Google Scholar]
- 77. Kneller DW, Phillips G, Weiss KL, et al. Unusual zwitterionic catalytic site of SARS‐CoV‐2 main protease revealed by neutron crystallography. J Biol Chem. 2020;295:17365‐17373. 10.1074/jbc.AC120.016154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27(2):157‐162. [DOI] [PubMed] [Google Scholar]
- 79. Ren Z, Yan L, Zhang N, et al. The newly emerged SARS‐like coronavirus HCoV‐EMC also has an "Achilles' heel": current effective inhibitor targeting a 3C‐like protease. Protein Cell. 2013;4(4):248‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. De Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N. Covalent inhibitors design and discovery. Eur J Med Chem. 2017;138:96‐114. [DOI] [PubMed] [Google Scholar]
- 81. Gehringer M, Laufer SA. Emerging and re‐Emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J Med Chem. 2019;62(12):5673‐5724. [DOI] [PubMed] [Google Scholar]
- 82. Lonsdale R, Ward RA. Structure‐based design of targeted covalent inhibitors. Chem Soc Rev. 2018;47(11):3816‐3830. [DOI] [PubMed] [Google Scholar]
- 83. Lee TW, Cherney MM, Huitema C, et al. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate‐like aza‐peptide epoxide. J Mol Biol. 2005;353(5):1137‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Asgian JL, James KE, Li ZZ, et al. Aza‐peptide epoxides: a new class of inhibitors selective for Clan CD cysteine proteases. J Med Chem. 2002;45(23):4958‐4960. [DOI] [PubMed] [Google Scholar]
- 85. James KE, Asgian JL, Li ZZ, et al. Design, synthesis, and evaluation of aza‐peptide epoxides as selective and potent inhibitors of caspases‐1, ‐3, ‐6, and ‐8. J Med Chem. 2004;47(6):1553‐1574. [DOI] [PubMed] [Google Scholar]
- 86. Matthews DA, Dragovich PS, Webber SE, et al. Structure‐assisted design of mechanism‐based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc Natl Acad Sci USA. 1999;96(20):11000‐11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shie JJ, Fang JM, Kuo TH, et al. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic alpha, beta‐unsaturated esters. Bioorg Med Chem. 2005;13(17):5240‐5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wang F, Chen C, Yang K, et al. Michael acceptor‐based peptidomimetic inhibitor of main protease from Porcine Epidemic Diarrhea Virus. J Med Chem. 2017;60(7):3212‐3216. [DOI] [PubMed] [Google Scholar]
- 89. Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS‐CoV‐2 and discovery of its inhibitors. Nature. 2020;582:289‐293. [DOI] [PubMed] [Google Scholar]
- 90. Ghosh AK, Xi K, Grum‐Tokars V, et al. Structure‐based design, synthesis, and biological evaluation of peptidomimetic SARS‐CoV 3CLpro inhibitors. Bioorg Med Chem Lett. 2007;17(21):5876‐5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yang S, Chen SJ, Hsu MF, et al. Synthesis, crystal structure, structure‐activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J Med Chem. 2006;49(16):4971‐4980. [DOI] [PubMed] [Google Scholar]
- 92. Lee CC, Kuo CJ, Ko TP, et al. Structural basis of inhibition specificities of 3C and 3C‐like proteases by zinc‐coordinating and peptidomimetic compounds. J Biol Chem. 2009;284(12):7646‐7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. St. John SE, Therkelsen MD, Nyalapatla PR, Osswald HL, Ghosh AK, Mesecar AD. X‐ray structure and inhibition of the Feline Infectious Peritonitis Virus 3C‐like protease: structural implications for drug design. Bioorg Med Chem Lett. 2015;25(22):5072‐5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Akaji K, Konno H, Mitsui H, et al. Structure‐based design, synthesis, and evaluation of peptide‐mimetic SARS 3CL Protease Inhibitors. J Med Chem. 2011;54(23):7962‐7973. [DOI] [PubMed] [Google Scholar]
- 95. Zhu L, George S, Schmidt MF, Al‐Gharabli SI, Rademann J, Hilgenfeld R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS‐coronavirus main protease. Antivir Res. 2011;92(2):204‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sacco MD, Ma C, Lagarias P, et al. Structure and inhibition of the SARS‐CoV‐2 main protease reveals strategy for developing dual inhibitors against Mpro and cathepsin L. Sci Adv. 2020;6:eabe0751. 10.1126/sciadv.abe0751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dai W, Zhang B, Su H, et al. Structure‐based design of antiviral drug candidates targeting the SARS‐CoV‐2 main protease. Science. 2020;368(6497):1331‐1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jain RP, Pettersson HI, Zhang J, et al. Synthesis and evaluation of keto‐glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J Med Chem. 2004;47(25):6113‐6116. [DOI] [PubMed] [Google Scholar]
- 99. Yin J, Niu C, Cherney MM, et al. A mechanistic view of enzyme Iinhibition and peptide hydrolysis in the active site of the SARS‐CoV 3C‐like peptidase. J Mol Biol. 2007;371(4):1060‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yin J, Cherney MM, Bergmann EM, et al. An episulfide cation (thiiranium ring) trapped in the active site of HAV 3C proteinase inactivated by peptide‐based ketone inhibitors. J Mol Biol. 2006;361(4):673‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chuck CP, Chen C, Ke Z, Wan DC, Chow HF, Wong KB. Design, synthesis and crystallographic analysis of nitrile‐based broad‐spectrum peptidomimetic inhibitors for coronavirus 3C‐like proteases. Eur J Med Chem. 2013;59:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kim Y, Liu H, Galasiti KA, et al. Reversal of the progression of fatal coronavirus infection in cats by a broad‐spectrum coronavirus protease inhibitor. PLOS Pathog. 2016;12(3):e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Pedersen NC, Kim Y, Liu H, et al. Efficacy of a 3C‐like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J Feline Med Surg. 2017;20(4):378‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC‐376, and calpain inhibitors II, XII inhibit SARS‐CoV‐2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Vuong W, Khan MB, Fischer C, et al. Feline coronavirus drug inhibits the main protease of SARS‐CoV‐2 and blocks virus replication. Nat Commun. 2020;11;4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kim Y, Lovell S, Tiew KC, et al. Broad‐spectrum antivirals against 3C or 3C‐like proteases of picornaviruses, noroviruses, and coronaviruses. J Virol. 2012;86(21):11754‐11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ye G, Wang X, Tong X, Shi Y, Fu ZF, Peng G. Structural basis for inhibiting Porcine Epidemic Diarrhea Virus replication with the 3C‐Like protease inhibitor GC376. Viruses. 2020;12(2):240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhang L, Lin D, Kusov Y, et al. α‐ketoamides as broad‐spectrum inhibitors of coronavirus and enterovirus replication: structure‐based design, synthesis, and activity assessment. J Med Chem. 2020;63(9):4562‐4578. [DOI] [PubMed] [Google Scholar]
- 109. Wu CY, King KY, Kuo CJ, et al. Stable benzotriazole esters as mechanism‐based inactivators of the severe acute respiratory syndrome 3CL protease. Chem Biol. 2006;13(3):261‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Verschueren KHG, Pumpor K, Anemüller S, Chen S, Mesters JR, Hilgenfeld R. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem Biol. 2008;15(6):597‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Turlington M, Chun A, Tomar S, et al. Discovery of N‐(benzo[1,2,3]triazol‐1‐yl)‐N‐(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS‐CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced‐fit binding. Bioorg Med Chem Lett. 2013;23(22):6172‐6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Jacobs J, Grum‐Tokars V, Zhou Y, et al. Discovery, synthesis, and structure‐based optimization of a series of N‐(tert‐Butyl)‐2‐(N‐arylamido)‐2‐(pyridin‐3‐yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the Severe Acute Respiratory Syndrome Coronavirus (SARS‐CoV) 3CL protease. J Med Chem. 2013;56(2):534‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lu IL, Mahindroo N, Liang PH, et al. Structure‐based drug design and structural biology study of novel nonpeptide inhibitors of Severe Acute Respiratory Syndrome coronavirus main protease. J Med Chem. 2006;49(17):5154‐5161. [DOI] [PubMed] [Google Scholar]
- 114. Su HX, Yao S, Zhao WF, et al. Anti‐SARS‐CoV‐2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol Sin. 2020;41:1167‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jin Z, Zhao Y, Sun Y, et al. Structural basis for the inhibition of SARS‐CoV‐2 main protease by antineoplastic drug carmofur. Nat Struct Mol Biol. 2020;27:529‐532. [DOI] [PubMed] [Google Scholar]
- 116. Goyal B, Goyal D. Targeting the dimerization of the main protease of Coronaviruses: a potential broad‐spectrum therapeutic strategy. ACS Comb Sci. 2020;22(6):297‐305. [DOI] [PubMed] [Google Scholar]
- 117. Ding L, Zhang X, Wei P, Fan K, Lai L. The interaction between severe acute respiratory syndrome coronavirus 3C‐like proteinase and a dimeric inhibitor by capillary electrophoresis. Anal Biochem. 2005;343(1):159‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]