

Abstract
Objective: To investigate whether high-mobility group box-1 induces cell proliferation, invasion and mediates inflammation in ectopic human endometrial stromal cells through Toll-like receptor 4. Methods: Ectopic endometrial specimens were retrieved from patients with ovarian endometrioma having laparoscopy. Ectopic HESCs were treated with H2O2 and recombinant HMGB-1 to induce oxidative stress. The effect of oxidative stress on cell proliferation and invasion was demonstrated. Receptors for HMGB-1 in NF-κB pathway (TLR4, RAGE), angiogenic molecule (VEGF), adhesion molecules (ICAM-1, E-cadherin), and inflammatory cytokines were measured simultaneously to the oxidative stress. Results: Ectopic HESCs showed markedly decreased cell viability with the increased release of HMGB-1 following treatment with H2O2. When ectopic HESCs were stressed by rHMGB-1, cell proliferation and cell migration numbers increased significantly in a dose-dependent manner. Increased TLR4 and RAGE mRNA and protein expression levels were noted to rHMGB-1 treatment in a dose-dependent manner. VEGF synthesis was also increased by rHMGB-1 treatment. The gene expression of ICAM-1 was upregulated, whereas that of E-cadherin was downregulated with rHMGB-1 treatment. Interleukin-6, IL-1β, tumor necrosis factor-alpha, and IL-10 were increased significantly by rHMGB-1 treatment. Inversely, after transfection of small interfering RNA against TLR4, rHMGB treatment resulted in decreased cell proliferation and invasion. Conclusion: HMGB-1 activates the NF-κB pathway via TLR4 to increase cell proliferation, invasion, and the production of various inflammatory markers in HESCs. Thus, HMGB-1, TLR4, and NF-κB may represent potential therapeutic targets for the treatment of endometriosis.
Keywords: Endometriosis, oxidative stress, HMGB-1, TLR4
Introduction
Endometriosis is a chronic, multifactorial disease that typically develops in women in their 30 s and 40 s, with negative effects on reproductive function. Since the pathogenesis of endometriosis is still unclear and has only been hypothesized, current clinical management of the condition is limited to conservative treatment such as pain control and fertility treatments as necessary. A study in the early 1990s demonstrated increased levels of inflammatory cytokines in the peritoneal cavity in endometriosis patients [1], suggesting a pathophysiological mechanism related to the immune system to trigger the establishment of endometriosis.
Oxidative stress and damage-associated molecular pattern (DAMP) are stimuli which leads activation of innate immunity, leading to an inflammatory response induced by non-infectious cause in humans. High-mobility group box-1 (HMGB-1) exists as an intranuclear DNA-binding protein in mammalian cells, resulting in the stabilization of DNA and control of transcriptional activity [2]. Extracellularly released HMGB-1 via cell death acts as an endogenous ligand activating inflammatory pathways, which is known a representative DAMP [3]. We previously reported the increased release of HMGB-1 in human endometrial stromal cells (HESCs) by oxidative stress-induced cell death. Recombinant HMGB-1 (rHMGB-1) treatment to HESCs showed a dose-dependent correlation with expression of its receptor Toll-like receptor 4 (TLR4), and changes were consistently correlated according to TLR4 antagonist and nuclear factor-kappa B (NF-κB) inhibitor treatment doses. Thus, the innate immune response via HMGB-1 and TLR4/NF-κB was suggested as a possible mechanism contributing to the development of endometriosis by inducing alterations in the naïve, eutopic endometrium. Accordingly, we hypothesized that HMGB-1 may play an important role in the early development of endometriosis.
To test this hypothesis, in the present study, we sought to elucidate whether HMGB-1 could induce the proliferation and invasion, and mediate inflammation in ectopic HESCs in vitro via TLR4, contributing to the progression of endometriosis. In addition to helping to elucidate the detailed pathogenic mechanism, these findings can offer new treatment targets for endometriosis.
Materials and methods
Participants
From October 2014 to March 2015, ectopic endometrial specimens were obtained from 10 patients who were surgically diagnosed endometriosis. The ectopic endometrium was collected from the endometrioma of the ovary. All 10 patients were of reproductive age and none had had hormonal medication for at least 3 months preoperatively. The study was approved by the institutional review board of Severance Hospital, Yonsei University College of Medicine (4-2014-0560). All participants have been provided written consent after agreement.
Sample collection
After the endometriotic cyst was enucleated, a 1.0 × 1.0 × 1.0-cm3 section was sampled at the cyst wall. Each specimen was placed in a buffered saline solution after extraction and sent to the laboratory, right after.
Cell culture
Isolated ectopic HESCs were cultured as described previously [4]. After washing and sectioning in phosphate-buffered saline, the cells were treated with 0.25% trypsin/ethylenediaminetetraacetic acid (Gibco, Invitrogen) at 37°C for 30 min. During incubation, the cells were tapped every 5 min to enhance detachment. Supernatant was isolated and then ectopic HESCs were unfolded in Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12, HyClone, Logan, UT, USA) with 10% fetal bovine serum (FBS; Gibco, Invitrogen). The stromal-enriched fraction was cultured in 75-mL culture flasks at 37°C, in humidified atmosphere of 95% air and 5% CO2. Passage 2-6 HESCs were used in the experiments.
Cell treatment and & study design
To induce oxidative stress in ectopic HESCs, the endometrial cells were incubated with 0, 0.01, 0.1 or 1 nM H2O2 for 48 h. H2O2 was used to induce passive release of extracellular HMGB-1 resulted from cell necrosis. To confirm oxidative stress-induced necrosis, the cell proliferation of ectopic HESCs and passive HMGB-1 release were examined with an MTT assay. To evaluate the influence of DAMP on ectopic HESCs, they were incubated with 0, 5, and 10 μM recombinant HMGB-1 (rHMGB-1; Sino, Beijing, China) for 48 h. TLR4, receptor for advanced glycation end products (RAGE), and vessel endothelial growth factor (VEGF) were examined for mRNA and protein expression. Likewise, mRNA expression levels for intracellular adhesion molecule-1 (ICAM-1) and E-cadherin, and for inflammatory cytokines were measured after 48 hr of rHMGB-1 treatment. Further, TLR4 was inhibited by transfection of small interfering RNA (siRNA) and treated with rHMGB-1 thereafter; the cell migration was evaluated to examine the changes in cell invasion.
Cell proliferation assays (CCK-8)
In 6-well tissue culture plates, ectopic HESCs were seeded at a density of 1 × 105 cells/well. After 24 h, the culture medium was exchanged to DMEM/F12 with 2% FBS. followed by treatment with 0, 0.01, 0.1, or 1 nM H2O2 for 48 h to induce oxidative stress. To confirm oxidative stress-induced necrosis, cell proliferation assays were performed by dropping 100 μL of Cell Counting Kit-8 (CCK-8; Dojindo, Japan) into each well, and incubating at 37°C for 1 h. The supernatants were then plated to 96-well and measured at an optical density (OD) of 450 nm using a VersaMax reader (Molecular Devices, Sunnyvale, CA, USA).
Examination of HMGB-1 release following cell death
To measure the amount of passive HMGB-1 released following cell death, 3 × 105 HESCs were seeded in 6-well plates and incubated at 37°C. After 24 h, the medium was changed to serum-free DMEM/F12. HESCs were incubated for 1 h with 0, 50, or 100 μM H2O2 and then with 100 μL of MTT solution (Sigma), which were incubated at 37°C in a 5% CO2 incubator for 4 h further. After removal of the medium, each well was treated with 500 μL dimethyl sulfoxide (DMSO; Sigma), and incubated on a shaker for 10 min. Lastly, the OD was measured with a VersaMax reader at 562 nm to determine the cell viability rates. The supernatants were harvested for subsequent western blotting to examine the extent of HMGB-1 release.
siRNA transfection
siTLR4 RNA was purchased from Thermo Fisher Scientific (CA, USA). HESCs were transfected with siRNA oligonucleotides using Lipofectamine RNAiMAX (Thermo Fisher Scientific, CA, USA) according to the manufacturer’s instruction. After 6 h, the cells were washed and incubated overnight in complete medium.
Cell invasion and migration assay
Transwell matrigel invasion chambers (Corning Inc., Lowell, MA, USA) were used for migration and invasion assays. After 24 h post-transfection, cells were spreaded to a single cell suspension, washed in PBS and resuspended in DMEM/F12 without FBS. For the migration assay, 2 × 105 cells in 100 μL DMEM/F12 without FBS were seeded in the upper chamber of 24-well plate. The lower chamber was filled with DMEM supplemented with 20% FBS and 0, 5, and 10 ng/mL of rHMGB-1. Cells were cultured in standard condition. After 48 h, cells on the upper surface of the plate were removed by wiping with a wet cotton swab and washed in PBS several times. The migrated cells on the lower surface of the upper chamber were fixed in methanol for 10 minutes, dried at room temperature, stained with Diff-Quik solution. The migrated cells were counted, and photographed under a Carl Zeiss microscope (LSM 710, Germany). Examinations were performed 3 rounds totally, and the cells in five random fields on each well were chosen for count. For the invasion assay, 5 × 104 ectopic HESCs were seeded onto the upper matrigel chamber with serum-free DMEM. After 48 h, the cells that invaded the lower surface were collected, stained, and then counted under the microscope (LSM 710, Carl Zeiss, Germany).
Real-time reverse transcription-polymerase chain reaction (RT-PCR)
Cell-free total RNA was pulled out from the cell lysates with a RNeasy kit (Qiagen, Hilden, Germany). cDNA was produced with an oligo-dT primers (Invitrogen) using 1 μg of total RNA. RT-PCR for TLR4, vascular endothelial growth factor (VEGF), and E-cadherin with ABI StepOnePlus system (Applied Biosystems, Foster City, CA, USA) and SYBR green RT-PCR master mix (Toyobo, Osaka, Japan) was utilized for each. The primers used were TLR4, 5’-CAGAGTTTCCTGCAATGGATCA-3’ and 5’-GCTTATCTGAAGGTGTTGCACAT-3’; VEGF, 5’-GGA GGAGGGCAGAATCATCAC-3’ and 5’-ATTGGATGGCAGTAGCTGCCT-3’; E-Cadherin, 5’-TCATGAGTGTCCCCCGGTAT-3’ and 5’-TCTTGAAGCGATTGCCCCAT-3’; ICAM-1, 5’-TCT TCC TCG GCC TTC CCA TA-3’ and 5’-AGG TAC CAT GGC CCC AAA TG-3’. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) 5’-TCGACAGTCAGCCGCATCTTCTTT-3’ and 5’-ACCAAATCCGTTGACTCCGACCTT-3’ was used as endogenous reference gene. The PCR conditions were 10 min at 95°C, 15 sec at 95°C, 1 min at 6°C, and 15 sec at 9°C, repeated 40 times in a row. The data were corrected for GAPDH expression and the PCR results were extended to a 1.5% agarose gel containing tidum for bromide and quantified by Image J (National Institute of Health).
Western blotting
Cell lysis was induced in a radioimmunoprecipitation analysis buffer (RIPA buffer; Intron, iNtRON Biotechnology, Seongnam, Korea) with protease inhibitor cocktail (Cell Signaling Technology, Beverly, MA, USA). Subsequently, after mixing the lysed cells, 40 μL was distributed in each. Then the divided cells were collected, centrifuged at 4°C for 30 min at 13,000 rpm, and the amount of protein in the supernatant was quantified with a BCA protein analysis kit (Thermo Scientific, Hudson, NH, USA). Western blotting was carried out in the following order. 30 micrograms of each lysate were placed in 5X buffer and boiled. The centrifuged supernatant was filtered through sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on an 8% gel and then moved to polyvinylidene fluoride membranes (Millipore, Eschborn, Germany). BSA treatment was then performed at room temperature for 1 hour and the membranes were each cultured overnight at 4°C with the primary antibody. The antibodies used were TLR4 (polyclonal anti-rabbit, 0.3 μg/mL; GeneTex, Irvine, CA, USA), VEGF (polyclonal anti-rabbit, 1 μg/mL; Abcam, Cambridge, UK), E-Cadherin (Monocle, private section--Rabbit; Cell Signaling Technology, Beverly, MA, USA) and GAPDH (monoclonal antibody--mouse, 0.1 μg/mL; Millipore). Secondary antibodies include anti-mouse antibody (IgG, 0.27 μg/mL; Jackson, West Grove, PA, USA) and anti-rabbit antibody (IgG, 0.27 μg/mL; Jackson) bound to horseradish peroxidase were used. Using enhanced chemiluminescence solution (Advansta, San Francisco, CA, USA), bands were detected and quantified with Image J.
Enzyme-linked immunosorbent assay (ELISA)
After ectopic HESC treatment with 0, 5, and 10 ng/mL rHMGB-1 for 48 h, the supernatant was collected. Supernatants was analyzed for interleukin (IL)-6 (BD bioscience, San Jose, CA, USA), tumor necrosis factor (TNF)-α (GE healthcare, Buckinghamshire, UK), IL-1β (Abcam, Cambridge, UK), and IL-10 (Abcam, Cambridge, UK) using respective ELISA kits. The OD was measured at 450 nm.
Statistical analysis
Kruskal-Wallis tests with Dunn’s procedure was performed to determine the differences in the levels of mRNA and protein. SPSS 23.0 (IBM, NY, USA) was used. P values of less than 0.05 were considered showing difference with significance, statistically.
Results
Cell proliferation and HMGB-1 release after H2O2 treatment
To present extracellular release of HMGB-1 caused by necrosis, ectopic HESCs were treated with H2O2. We intended to induce sustained oxidative stress by increasing extracellular HMGB-1. There was a gradual increase in ectopic HESC proliferation with increasing concentration of H2O2 to induce oxidative stress (Figure 1A). The MTT assay results showed that cell death was significantly induced with increasing H2O2 concentration (Figure 1B), and HMGB-1 release was increased due to cell death (Figure 1C).
Figure 1.
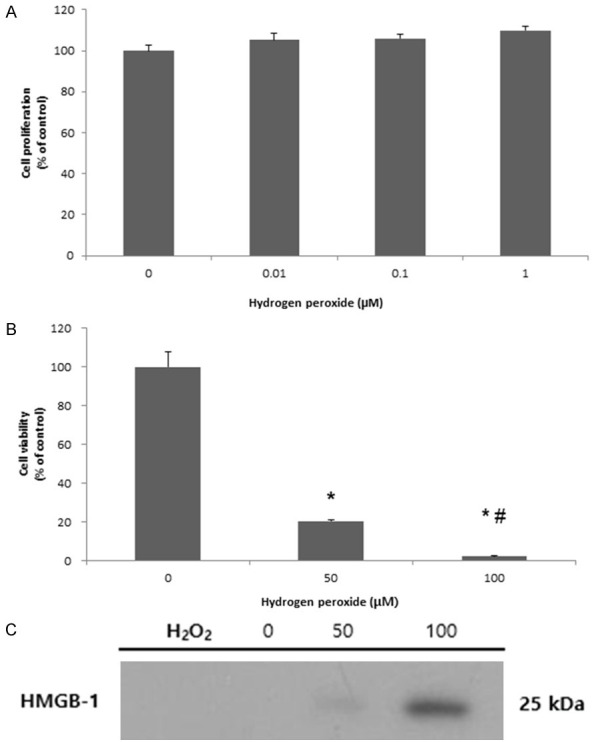
Cell changes and passive release of HMGB-1 after inducing necrosis by H2O2 treatment in ectopic HESCs. Oxidative stress caused cell proliferation, although not statistically significant (A), decrease of cell viability (B), and increase of HMGB-1 release (C) in ectopic HESCs. *P < 0.05 compared to 0 μM H2O2 treatment. #P < 0.05 compared to 50 μM H2O2 treatment.
Changes in ectopic HESCs after rHMGB-1 treatment
After H2O2, the cells were treated with rHMGB-1. The experiment was conducted to mimic the environment wherein passive release of HMGB-1 increases due to oxidative stress-induced cell death. Cell proliferation and invasion increased after rHMGB-1 treatment in a dose- and time-dependent manner (Figure 2A, 2B). Moreover, with increasing rHMGB-1 dosage, the expression of TLR4 and RAGE showed a marked increase at the mRNA (data not shown) and protein (Figure 3) levels.
Figure 2.
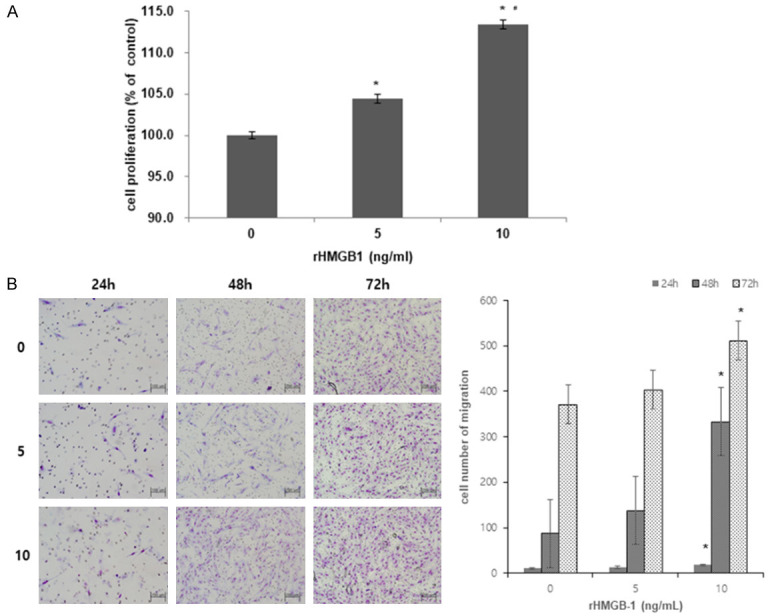
Increased cell proliferation and migration after rHMGB-1 treatment. Cell proliferation (A) and invasion assay (B) after inducing oxidative stress in ectopic HESCs with rHMGB-1 treatment. The migrating cell number was significantly increased in each culture group: 24 h, 48 h, 72 h compared between 0 ng/mL and 10 ng/mL of rHMGB-1 treatment. Treatment with 5 ng/mL, in contrast to 0 ng/mL of rHMGB-1, led to an increased migration of cells, however, the difference was not statistically significant. *P < 0.05 compared to 0 ng/mL rHMGB-1 treatment; #P < 0.05 compared to 5 ng/mL rHMGB-1 treatment.
Figure 3.
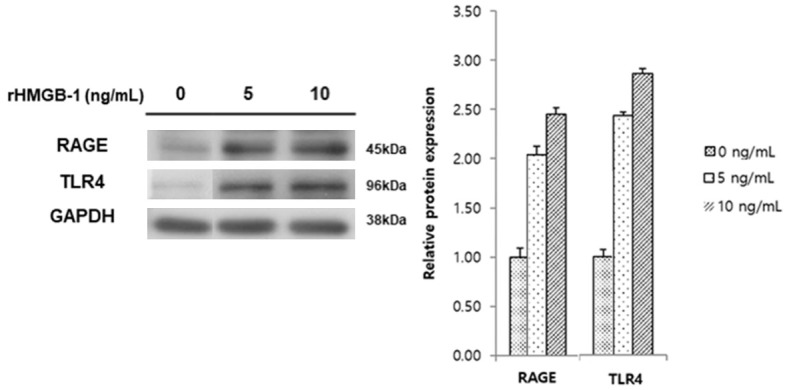
RAGE and TLR4 mRNA and protein expression after rHMGB-1 treatment. Protein expression levels of the receptors, RAGE and TLR4 were significantly increased after HMGB-1 treatment for 48 h in a dose-dependent manner.
Changes of adhesion and angiogenic factors after rHMGB-1 treatment
After applying rHMGB-1 at different doses for 48 h, expression of the angiogenic marker VEGF markedly increased at the mRNA and protein levels (Figure 4A, 4B). The mRNA level of another angiogenic marker, ICAM-1, also significantly increased at 10 ng/mL treatment of rHMGB-1 compared to the naïve control group (Figure 4A). By contrast, expression of the adhesion marker E-cadherin was significantly inhibited with rHMGB-1 treatment (Figure 4A).
Figure 4.
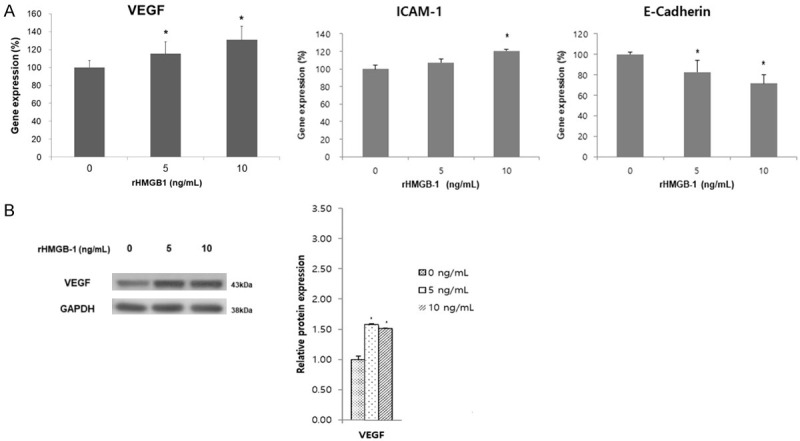
mRNA expression of VEGF, ICAM-1 and E-Cadherin (A) after treatment with rHMGB-1 by dosage (48 h) and protein expression of VEGF (B). (A) VEGF mRNA expression was increased significantly according to the dose increase of rHMGB-1. ICAM-1 mRNA expression was increased only at 10 ng/mL of rHMGB-1, whereas E-Cadherin showed a significant decrease by rHMGB-1 treatment in a dose-dependent manner. (B) VEGF protein synthesis was significantly increased by rHMGB-1 treatment; however, there was no differences between rHMGB-1 treatment doses. *P < 0.05 compared to 0 ng/mL rHMGB-1 treatment.
Inflammatory cytokine secretion in the HESC supernatant after rHMGB-1 treatment
After applying oxidative stress with rHMGB-1 at different doses for 48 h, the levels of inflammatory cytokines IL-6 and IL-10 significantly increased in the supernatants of the HESCs in a dose-dependent manner, and the levels of TNF-α and IL-1β increased significantly at the higher dose of rHMGB-1 (Figure 5).
Figure 5.
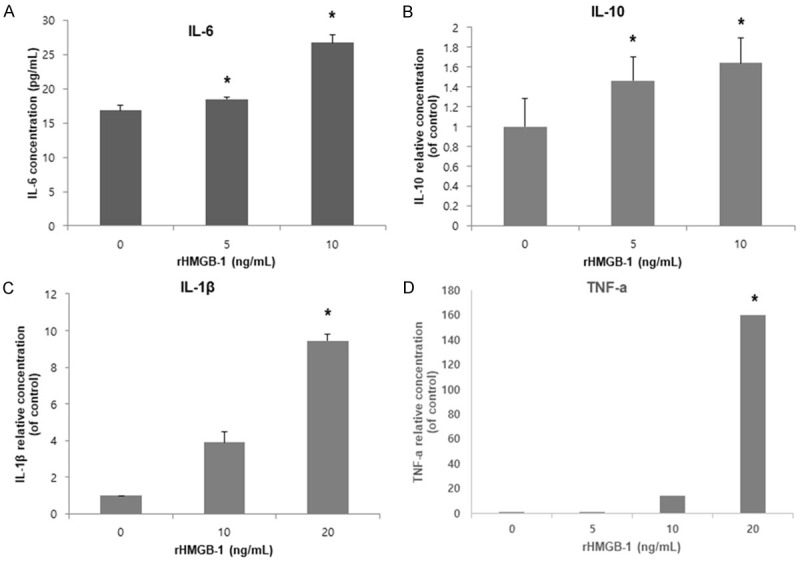
Cytokine secretion in ectopic HESCs after rHMGB-1 treatment. Secretion of pro-inflammatory cytokines including IL-6, TNF-α and IL-1β by ectopic HESCs was significantly increased with increased doses of rHMGB-1. Secretion of anti-inflammatory cytokine IL-6 was increased in a dose-dependent manner. *P < 0.05 compared to 0 ng/mL rHMGB-1 treatment.
Cell invasion after TLR4 inhibition
To examine signal transduction via TLR4 in inflammatory pathway in HESC, TLR4 was suppressed by transfection of small interfering RNA (siRNA) targeting TLR4 (siTLR4) and the cells were treated with different doses of rHMGB-1. Suppression of TLR4 led to significant decrease of HESC migration (Figure 6).
Figure 6.

Cell invasion and migration assay after treating rHMGB-1 to siTLR4-transfected HESCs. Cell invasion (A), migration (B) were significantly decreased with treatment of 5 ng/mL and 10 ng/mL rHMGB-1 in siTLR4-transfected HESCs compared to the untreated control. *P < 0.05 compared to 0 ng/mL rHMGB-1 treatment.
Discussion
In the present study, we obtained evidence for scheme of inflammatory pathway activated via TLR4 in ectopic HESC. The endogenous danger signal HMGB-1 released after cell death induced cell proliferation, increased expression of angiogenesis, adhesion and inflammation via TLR4. In our previous study [5], we demonstrated a scheme of altered human eutopic endometrial stromal cell by HMGB-1, suggesting initiation of inflammation by oxidative stress. Extracellular release of HMGB-1 from eutopic endometrial stromal cell induced by H2O2 was confirmed by RT-PCR and western blot. In the current study, we tried to demonstrate TLR4-mediated mechanism of inflammation induced by HMGB-1 in already altered endometrial stromal cells (ectopic HESC). HMGB-1 increased TLR4 expression in ectopic HESC, which showed a cascade of increased angiogenesis, adhesion, and inflammation probably due to activation of the NF-κB signaling pathway [6]. Inversely, after inhibiting TLR4 by siRNA transfection, cell proliferation and migration significantly decreased, further suggesting that the DAMP exerts its actions via TLR4 in ectopic HESCs.
As a possible trigger that activates the NF-κB pathway, chronic inflammation related to innate immunity has been suggested to play a pathophysiological role in endometriosis development [7]. NF-κB has been extensively studied in the field of endometriosis research [8,9], as well as in other inflammation-related diseases [10]. NF-κB mediates the induction of pro-inflammatory genes, including cytokines, chemokines, and other inflammatory mediators, in various immune cells such as macrophages, dendritic cells, and T and B lymphocytes. In endometriosis, an inflammatory signature representing a shift toward a relative dominance of Th2-type cytokines from Th1 dominant cell population has been suggested [11], and aberrantly regulated immune factors have been found, such as increased cytokines (TNF-α, IL-1β, IL-8, IL-6, IL-10) [12-14] and decreased natural killer cells in the peritoneal fluid [15]. Accordingly, there has been extensive research devoted to the role of the NF-κB pathway in endometriosis, with some investigations revealing increased expression of NF-κB subunits in the endometrium [8,16] and its activation in macrophages [17]. In addition to endometriosis, the NF-κB pathway has been widely implicated in carcinogenesis, autoimmune diseases, and inflammatory diseases, and these studies have provided clues into the possible stimuli that could activate the NF-κB pathway in endometriosis.
Stimuli such as hypoxia, oxidative stress, DAMP, microbium, iron burden, inflammasomes, and cytokines have been suggested to lead to NF-κB pathway activation, resulting in an increase of cell proliferation, decreased apoptosis, and inflammation modulation [7,9,18,19]. In the present study, we focused on the potential role of innate immunity involving DAMP and the NF-κB pathway in endometriosis. Previous studies demonstrated different proliferation, decidualization, and survival rates of stromal cells isolated from the eutopic endometrium, ovarian endometrioma, and deep-infiltrating endometriosis tissues, suggesting that different origins may reflect their different characteristics in endometriosis [20,21]. Here, we demonstrated eutopic endometrial stromal cells as a naïve cell type before the pathologic modifications occurring in endometriosis, and ectopic endometrial stromal cells as a post-modified cell due to pathologic changes. In our study, we demonstrated a possible mechanism regarding oxidative stress and HMGB-1 providing viscious, endogenous stimuli for chronic inflammation in pelvic cavity. Oxidative stress and HMGB-1 showed altering ability in eutopic endometrium, which leaded changes closer to ectopic endometrium in our previous work [5]. Serially, in current study, the danger signal has shown aggravating cell invasion, proliferation and changes of molecular level in ectopic HESCs. We suggest that oxidative stress and HMGB-1 may affect progression of endometriosis, not only initiation of the pathologic changes in eutopic endometrial cells.
HMGB-1 is stored in the nucleus where it acts as a DNA-binding protein that participates in DNA replication, recombination, transcription, and repair [3,22]. However, when HMGB-1 is released for any reason, extracellular HMGB-1 functions as a signaling molecule in the inflammation pathway, immune system, and tissue regeneration, including in cell differentiation and migration [2]. Extracellular HMGB-1 can be actively secreted by inflammatory cells stimulated by DAMP, cytokines, or chemokines, or can be passively released by dead, dying, or injured cells with several different mechanisms proposed [23]. Extracellular HMGB-1 acts as a DAMP molecule, and its activity depends on the redox state, receptors, and their interactive partners. HMGB-1 has been suggested as a key molecule inducing chronic inflammatory processes in rheumatic arthritis [24], retinopathy [25], sepsis [26], atherosclerosis [27,28], and liver diseases [23]. In particular, atherosclerosis and liver disease have another common feature with endometriosis in causing an increase of stromal fibrosis besides chronic inflammation. For example, a “two-hit” hypothesis has been proposed to explain the pathogenesis of non-alcoholic fatty liver disease (NAFLD) [29]. When the liver becomes sensitized to the injuries caused by steatosis, second hits by cytokines, chemokines, and oxidative stress result in de novo lipogenesis and increased lipid deposition in the liver. HMGB-1-TLR4-MyD88 signaling has been considered to be an important key mechanism in the early stage of progression to NAFLD [30]. In NAFLD, the HMGB-1 released by injured hepatocytes activates the NF-κB pathway via TLR4 and MyD88, leading to hepatic stellate cell activation and the increased expression and synthesis of alpha-smooth muscle actin and collagen, which collectively result in liver fibrosis [23].
Accordingly, our results suggest a similar pathogenic mechanism of endometriosis given the possible passive release and active secretion of HMGB-1 from ectopic endometriotic cells, which may activate the NF-κB pathway via TLR4. Activation of NF-κB resulted in decreased expression of E-cadherin, and increased expression of ICAM-1 and VEGF, along with the secretion of concomitant proinflammatory (IL-6, TNF-α, and IL-1β) and anti-inflammatory (IL-10) cytokines. The increased expression levels of VEGF [31], ICAM-1 [32], and cytokines is in line with previous results. The adhesion marker E-cadherin is important for maintaining cell-to-cell adhesion and cell polarity, and decreased expression of E-cadherin has been reported in various stages of endometriosis [33].
Our study has a limitation in that these findings are based only on in vitro experiments. Surely, suggested role of the HMGB-1-TLR4-NF-κB axis in early endometriosis development needs to be evaluated and validated through in vivo experiments. However, endometrial stromal cells have been suggested to reflect specific characteristics of ectopic endometrial cells [6]. Since endometriotic tissue is composed of glands, extracellular matrix and stroma mainly, many studies applied the stromal cells as a representative in vitro model to assess treatment of endometriosis [34]. Another possible limitation is the knockdown with siRNA for TLR4. Although siRNA transfection is a popularly used method in endometrial cell experiments, there may exist an off-target signature on an unexpected target of the pathway. Nevertheless, the aim of these experiments was to demonstrate a pathway stimulated by TLR4, and the results were consistent in terms of the changes observed pre- and post-downregulation. However, our study has the main advantage in terms of a rare serial demonstration of a possible pathogenic mechanism in endometriosis. Specifically, our results suggest that HMGB-1 via TLR4 may activate the NF-κB pathway to result in inflammatory changes in ectopic HESCs. Thus, the innate immune response may play an important role in the development of endometriosis, suggesting new targets for research and treatment.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (NRF-2012R1A1A1013167). We are thankful for the support of the Department of Obstetrics and Gynecology, Yonsei University College of Medicine.
Disclosure of conflict of interest
None.
References
- 1.Haney AF, Jenkins S, Weinberg JB. The stimulus responsible for the peritoneal fluid inflammation observed in infertile women with endometriosis. Fertil Steril. 1991;56:408–413. doi: 10.1016/s0015-0282(16)54532-4. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi ME, Beltrame M, Paonessa G. Specific recognition of cruciform DNA by nuclear protein HMG1. Science. 1989;243:1056–1059. doi: 10.1126/science.2922595. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 4.Seo SK, Nam A, Jeon YE, Cho S, Choi YS, Lee BS. Expression and possible role of non-steroidal anti-inflammatory drug-activated gene-1 (NAG-1) in the human endometrium and endometriosis. Hum Reprod. 2010;25:3043–3049. doi: 10.1093/humrep/deq277. [DOI] [PubMed] [Google Scholar]
- 5.Yun BH, Chon SJ, Choi YS, Cho S, Lee BS, Seo SK. Pathophysiology of endometriosis: role of high mobility group box-1 and toll-like receptor 4 developing inflammation in endometrium. PLoS One. 2016;11:e0148165. doi: 10.1371/journal.pone.0148165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meigs JV. Endometrial hematomas of the ovary. Boston Med Surg J. 1922;187:1–13. [Google Scholar]
- 7.Kajihara H, Yamada Y, Kanayama S, Furukawa N, Noguchi T, Haruta S, Yoshida S, Sado T, Oi H, Kobayashi H. New insights into the pathophysiology of endometriosis: from chronic inflammation to danger signal. Gynecol Endocrinol. 2011;27:73–79. doi: 10.3109/09513590.2010.507292. [DOI] [PubMed] [Google Scholar]
- 8.Guo SW. Nuclear factor-kappab (NF-kappaB): an unsuspected major culprit in the pathogenesis of endometriosis that is still at large? Gynecol Obstet Invest. 2007;63:71–97. doi: 10.1159/000096047. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Ramos R, Defrere S, Devoto L. Nuclear factor-kappaB: a main regulator of inflammation and cell survival in endometriosis pathophysiology. Fertil Steril. 2012;98:520–528. doi: 10.1016/j.fertnstert.2012.06.021. [DOI] [PubMed] [Google Scholar]
- 10.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2:e17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Podgaec S, Abrao MS, Dias JA Jr, Rizzo LV, de Oliveira RM, Baracat EC. Endometriosis: an inflammatory disease with a Th2 immune response component. Hum Reprod. 2007;22:1373–1379. doi: 10.1093/humrep/del516. [DOI] [PubMed] [Google Scholar]
- 12.Gilabert-Estelles J, Estelles A, Gilabert J, Castello R, Espana F, Falco C, Romeu A, Chirivella M, Zorio E, Aznar J. Expression of several components of the plasminogen activator and matrix metalloproteinase systems in endometriosis. Hum Reprod. 2003;18:1516–1522. doi: 10.1093/humrep/deg300. [DOI] [PubMed] [Google Scholar]
- 13.Sikora J, Mielczarek-Palacz A, Kondera-Anasz Z. Imbalance in cytokines from interleukin-1 family - role in pathogenesis of endometriosis. Am J Reprod Immunol. 2012;68:138–145. doi: 10.1111/j.1600-0897.2012.01147.x. [DOI] [PubMed] [Google Scholar]
- 14.Cameron MJ, Kelvin DJ. Cytokines and chemokines--their receptors and their genes: an overview. Adv Exp Med Biol. 2003;520:8–32. doi: 10.1007/978-1-4615-0171-8_2. [DOI] [PubMed] [Google Scholar]
- 15.Sikora J, Mielczarek-Palacz A, Kondera-Anasz Z. Role of natural killer cell activity in the pathogenesis of endometriosis. Curr Med Chem. 2011;18:200–208. doi: 10.2174/092986711794088416. [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, Ihm HJ, Oh YS, Chae HD, Kim CH, Kang BM. Increased nuclear expression of nuclear factor kappa-B p65 subunit in the eutopic endometrium and ovarian endometrioma of women with advanced stage endometriosis. Am J Reprod Immunol. 2013;70:497–508. doi: 10.1111/aji.12161. [DOI] [PubMed] [Google Scholar]
- 17.Lousse JC, Van Langendonckt A, Gonzalez-Ramos R, Defrere S, Renkin E, Donnez J. Increased activation of nuclear factor-kappa B (NF-kappaB) in isolated peritoneal macrophages of patients with endometriosis. Fertil Steril. 2008;90:217–220. doi: 10.1016/j.fertnstert.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 18.Kyama CM, Debrock S, Mwenda JM, D’Hooghe TM. Potential involvement of the immune system in the development of endometriosis. Reprod Biol Endocrinol. 2003;1:123. doi: 10.1186/1477-7827-1-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi H, Higashiura Y, Shigetomi H, Kajihara H. Pathogenesis of endometriosis: the role of initial infection and subsequent sterile inflammation (Review) Mol Med Rep. 2014;9:9–15. doi: 10.3892/mmr.2013.1755. [DOI] [PubMed] [Google Scholar]
- 20.Klemmt PA, Carver JG, Kennedy SH, Koninckx PR, Mardon HJ. Stromal cells from endometriotic lesions and endometrium from women with endometriosis have reduced decidualization capacity. Fertil Steril. 2006;85:564–572. doi: 10.1016/j.fertnstert.2005.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koster F, Jin L, Shen Y, Schally AV, Cai RZ, Block NL, Hornung D, Marschner G, Rody A, Engel JB, Finas D. Effects of an antagonistic analog of growth hormone-releasing hormone on endometriosis in a mouse model and in vitro. Reprod Sci. 2017;24:1503–1511. doi: 10.1177/1933719117691140. [DOI] [PubMed] [Google Scholar]
- 22.Giese K, Cox J, Grosschedl R. The HMG domain of lymphoid enhancer factor 1 bends DNA and facilitates assembly of functional nucleoprotein structures. Cell. 1992;69:185–195. doi: 10.1016/0092-8674(92)90129-z. [DOI] [PubMed] [Google Scholar]
- 23.Chen R, Hou W, Zhang Q, Kang R, Fan XG, Tang D. Emerging role of high-mobility group box 1 (HMGB1) in liver diseases. Mol Med. 2013;19:357–366. doi: 10.2119/molmed.2013.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersson U, Harris HE. The role of HMGB1 in the pathogenesis of rheumatic disease. Biochim Biophys Acta. 2010;1799:141–148. doi: 10.1016/j.bbagrm.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 25.Yang H, Hirooka K, Liu Y, Fujita T, Fukuda K, Nakamutra T, Itano T, Zhang J, Nishibori M, Shiraga F. Deleterious role of anti-high mobility group box 1 monoclonal antibody in retinal ischemia-reperfusion injury. Curr Eye Res. 2011;36:1037–1046. doi: 10.3109/02713683.2011.594201. [DOI] [PubMed] [Google Scholar]
- 26.Hu YM, Pai MH, Yeh CL, Hou YC, Yeh SL. Glutamine administration ameliorates sepsis-induced kidney injury by downregulating the high-mobility group box protein-1-mediated pathway in mice. Am J Physiol Renal Physiol. 2012;302:F150–158. doi: 10.1152/ajprenal.00246.2011. [DOI] [PubMed] [Google Scholar]
- 27.Sugimoto K, Ohkawara H, Nakamura Y, Takuwa Y, Ishibashi T, Takeishi Y. Receptor for advanced glycation end products - membrane type1 matrix metalloproteinase axis regulates tissue factor expression via RhoA and Rac1 activation in high-mobility group box-1 stimulated endothelial cells. PLoS One. 2014;9:e114429. doi: 10.1371/journal.pone.0114429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jialal I, Rajamani U, Adams-Huet B, Kaur H. Circulating pathogen-associated molecular pattern - binding proteins and High Mobility Group Box protein 1 in nascent metabolic syndrome: implications for cellular Toll-like receptor activity. Atherosclerosis. 2014;236:182–187. doi: 10.1016/j.atherosclerosis.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 29.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, Hou YJ, Chang YX, Tu QQ, Feng GS, Shen F, Wu MC, Wang HY. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54:1620–1630. doi: 10.1002/hep.24552. [DOI] [PubMed] [Google Scholar]
- 31.McLaren J. Vascular endothelial growth factor and endometriotic angiogenesis. Hum Reprod Update. 2000;6:45–55. doi: 10.1093/humupd/6.1.45. [DOI] [PubMed] [Google Scholar]
- 32.Vigano P, Gaffuri B, Somigliana E, Busacca M, Di Blasio AM, Vignali M. Expression of intercellular adhesion molecule (ICAM)-1 mRNA and protein is enhanced in endometriosis versus endometrial stromal cells in culture. Mol Hum Reprod. 1998;4:1150–1156. doi: 10.1093/molehr/4.12.1150. [DOI] [PubMed] [Google Scholar]
- 33.Saliminejad K, Edalatkhah H, Kamali K, Memariani T, Nasiri M, Saket M, Khorram Khorshid HR. Association of common variations of the E-cadherin with endometriosis. Gynecol Endocrinol. 2015;31:899–902. doi: 10.3109/09513590.2015.1101436. [DOI] [PubMed] [Google Scholar]
- 34.Ji M, Liu Y, Yang S, Zhai D, Zhang D, Bai L, Wang Z, Yu J, Yu C, Cai Z. Puerarin suppresses proliferation of endometriotic stromal cells in part via differential recruitment of nuclear receptor coregulators to estrogen receptor-alpha. J Steroid Biochem Mol Biol. 2013;138:421–426. doi: 10.1016/j.jsbmb.2013.07.006. [DOI] [PubMed] [Google Scholar]