

Abstract
Atherosclerosis is a chronic inflammatory disease driven by lipids, which occurs preferentially in the branches or curved areas of the middle and large arteries, contributing to increased morbidity and mortality of cardiovascular disease. Recently, it has been reported that STAT5 and its regulated immune response are closely related to non-tumor diseases. However, the role of STAT5 in the development of atherosclerosis remains unknown. In this study, atherosclerosis was induced by high-fat diet (HFD) in ApoE-/- mice, and STAT5-IN-1, a STAT5 inhibitor, was orally given. Macrophages stimulated by oxLDL were used as cell models in vitro. The effects of STAT5-IN-1 in ApoE-/- mice induced by HFD were assessed, and the underlying mechanisms were investigated by siRNA-induced gene silencing. The results revealed that treatment with STAT5 inhibitor significantly attenuated atherosclerosis in ApoE-/- mice induced by HFD via decreasing inflammation. Furthermore, it was demonstrated that inhibiting STAT5 could decrease oxLDL-induced inflammation. In summary, STAT5-IN-1 may be a potential drug for the treatment of atherosclerosis, and targeting STAT5 has the ability to be a potential therapeutic strategy for reducing atherosclerosis.
Keywords: Signal transducer and activator of transcription 5, atherosclerosis, inflammation
Introduction
According to the American Heart Association’s 2018 Heart Disease and Stroke Statistics report, there are 17.9 million annual death cases related to cardiovascular diseases, including atherosclerosis (AS), cardiac failure, cardiac hypertrophy, hypertension, stroke, peripheral artery disease, valvular heart disease, etc. And it is estimated that this number will continue to increase and reach 23.8 million by the year of 2030 [1]. AS is a chronic inflammatory disease driven by lipids, which develops preferentially in the branches or curved areas of the middle and large arteries, contributing to increased morbidity and mortality of cardiovascular diseases [2]. At the beginning of AS, lipids are trapped in the arterial endothelium, causing endothelial cells to activate and release trend factors to collect monocytes [3]. In lesions, inflammatory cells (including macrophages, T cells and dendritic cells) release pro-inflammatory cytokines and arachidonic acid, thereby promoting inflammatory response [4,5]. The environment of persistent inflammatory stimulation leads to plaque necrosis and further expansion of inflammatory response [6-8]. Therefore, inflammatory response is an important pathological process of AS, and inhibition of inflammatory response has been considered as an important approach to treating AS.
Signal transducer and activators of transcription (STATs) family are capable of binding to specific peptides containing phosphorylated tyrosine. The phosphorylation of STAT is often succeeded by its polymerization into a transcription activator, which is activated as a homodimer or heterodimer. This enables STAT to enter the nucleus to bind to a specific site of the target gene promoter sequence to promote its transcription. STAT5, a member of the STATs family, can be activated by JAK (Janus kinase), transferred to the nucleus, stimulate the transcription of downstream IFNs genes, and mediate the immune response [9,10]. Phosphorylation and expression of STAT5 is closely related to inflammation and a variety of tumors [11-13], which has important clinical research significance. In addition, STAT5 is also involved in non-tumor diseases. It has been reported that the IL-7/STAT5 axis promotes the generation of Th cells that produce GM-CSF/IL-3, and that the loss of function mainly mediates autoimmune neuritis [9]. A mounting body of evidence suggests that the Akt/STAT5 signaling pathway can affect the immune function of T cells by regulating glycolysis and glycosylation phosphorylation [14]. Moreover, liver specific knockout STAT5 attenuated liver inflammation and fibrosis in lithogenic diet-induced mice without affecting gallstones [15]. These above studies showed that STAT5 and its regulated immune response are closely related to non-tumor diseases. However, it is not clear whether STAT5 and its regulated immune response have an important role to play in AS.
In the current study, we intend to ascertain the therapeutic effects of STAT5 inhibitor on atherosclerotic therapy for ApoE-/- mice fed with HFD and clarify the underlying mechanisms. Our results demonstrated that treatment with STAT5 inhibitor could contribute to the attenuation of atherosclerosis in ApoE-/- mice induced by HFD, thus suppressing inflammatory response. In addition, the results suggest a close link between the beneficial effects of STAT5 and its ability to suppress inflammatory response. In summary, this study indicates that STAT5 could be a potential therapeutic target for treating atherosclerosis.
Materials and methods
Reagents and cell culture
STAT5-IN-1 was obtained from Selleck Chemicals (Houston, Texas, USA) and oxLDL were obtained from Yiyuan biotechnology Co., Ltd. (Guangzhou, China). For the in vitro experiments, the STAT5-IN-1 was dissolved in dimethyl sulfoxide (DMSO), and for the in vivo experiments, it was dissolved in 1% sodium carboxyl methylcellulose (CMC-Na). p-STAT5, STAT5 and TNF-α primary antibody were obtained from Cell Signaling (Danvers, MA, USA). α-Tubulin and β-actin primary antibody were obtained from Proteintech Group (Rosemont, IL, USA). The mouse tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) ELISA kits were procured from eBioScience (San Diego, CA).
siRNA-induced gene silencing
Gene silencing of the cells was performed with specific siRNA sequences. Mouse specific STAT5 siRNA sequences (sense sequence: 5’-GCCAGAUGCAAGUGUUGUATT-3’; antisense sequence: 5’-UACAACAUUGCAUCUGGCTT-3’) were procured from Gene Pharma Co. LTD. (Shanghai, China). LipofectAMINE™ 3000 (Invitrogen, Carlsbad, CA) was utilized to transfect siRNA into macrophage, with the transfection procedure implemented in accordance with manufacturer’s operating procedures.
Real-time quantitative PCR
In accordance with manufacturer’s operating procedures, TRIZOL (Thermo Fisher, Carlsbad, CA) was utilized to extract total RNA from cells and artery tissues. The experimental process was referred to our previous study [16]. Table 1 displays information about the primer sequences purchased from Thermo Fisher (Shanghai, China).
Table 1.
Primers used for real-time qPCR assay
| Gene | Species | Primers (FW) | Primers (RW) |
|---|---|---|---|
| TNF-α | Mouse | TGATCCGCGACGTGGAA | ACCGCCTGGAGTTCTGGAA |
| IL-6 | Mouse | CCAAGAGGTGAGTGCTTCCC | CTGTTGTTCAGACTCTCTCCCT |
| ICAM-1 | Mouse | GCCTTGGTAGAGGTGACTGAG | GACCGGAGCTGAAAAGTTGTA |
| VCAM-1 | Mouse | TGCCGAGCTAAATTACACATTG | CCTTGTGGAGGGATGTACAGA |
| β-actin | Mouse | CCGTGAAAAGATGACCCAGA | TACGACCAGAGGCATACAG |
Western immunoblot analysis
Solutes were isolated from cells or arterial tissues using 10% SDS-PAGE, and then electro-transferred to a nitrocellulose membrane. The detailed experimental process was referred to our previously study [16]. Compared with their respective control groups, they were quantitatively analyzed and normalized by the software Image J.
Enzyme-linked immunosorbent assay (ELISA)
The supernatant of culture medium was collected after treatment. Then, in accordance with ELISA kits’ operating procedures, the secretion levels of TNF-α and IL-6 were detected.
Animals
ApoE-/- mice (male, 16-22 g, 10 weeks) were procured from GemPharmatech Co. Ltd (Nanjing, China). All animal work was performed at the Animal Experiment Center of Zhejiang Academy of Traditional Chinese Medicine. The mice were subjected to a 12:12 hour light-dark cycle, fed with a standard rodent diet, and maintained at a constant room temperature. Prior to the study, the mice were brought into the laboratory for domestication for a minimum of three days. Protocols for mouse experiments were approved by the institutional review boards of Zhejiang Academy of Traditional Chinese Medicine (approval ID: KTSC075), Hangzhou, China.
ApoE-/- mice were randomly categorized into two weight-matched groups, HFD group (n=12) and STAT5-IN-1+HFD group (HFD+IN-1, n=12). Referring to previous publication [17,18], diet with 0.2% cholesterol (HFD) was given for 12 weeks and STAT5-IN-1 (10 mg/kg every other day) was administrated by gavage for 12 weeks. After the treatment, mice body weight was recorded weekly. Under ether anesthesia, the mice were euthanized under sodium pentobarbital anesthesia, after which their blood was collected. To perform gene and protein expression analysis, freeze arterial tissue by liquid nitrogen, and for microscopic analysis, they were embedded in 4% paraformaldehyde.
Histological analysis of atherosclerotic lesions
To assess the development of atherosclerosis in mice, the plaque lesions in aortic root and the entire aorta were analyzed. The heart near the aorta were excised and frozen by optimum cutting temperature compound and aortic roots were cut into sections. From aortic root to the iliac artery, the entire aorta was cut open longitudinally. For analysis of the lipid accumulation, oil red O staining was used. The detailed experimental process was referred to our previously study [16].
Immunofluorescence
Immunofluorescence assay was performed using the frozen sections. Firstly, the sections were sealed with 5% donkey serum albumin for 45 minutes. Subsequently, the sections were subjected to incubation with p-STAT5 primary antibody (1:100) at 4°C. After incubation with p-STAT5 primary antibody for 12 h, sections were subjected to incubation with second antibody (1:500) combined with appropriate Alexa Fluor 488 at room temperature for 1 hour, and mounted with DAPI (P36935, Life Technologies).
Determination of the serum lipid levels
The appropriate kit (Nanjing Jiancheng, Jiangsu, China) was utilized to measure triglycerides (TG), total cholesterol (TCH), high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C).
Statistical analysis
Data were presented as means ± SEM. To compare group differences, student’s t test or analysis of variance (ANOVA) was performed using the software GraphPad Pro 7.0 (GraphPad, San Diego, CA). A statistically significant result was obtained at P < 0.05.
Results
Phosphorylation of STAT5 is upregulated in atherosclerosis
We first evaluated the expression of phosphorylation of STAT5 in atherosclerosis. The results showed a significant increase in the phosphorylation levels of STAT5 in the artery tissue (Figure 1A). Similar results were obtained in the fluorescent staining experiment of aortic root (Figure 1B, 1C). Furthermore, the activation of STAT5 was tested in oxLDL-induced macrophage. There was a significant increase in the phosphorylation of STAT5 in oxLDL-stimulated macrophage (Figure 1D). These results indicate that the activation of STAT5 may contribute to the development and progression of AS.
Figure 1.
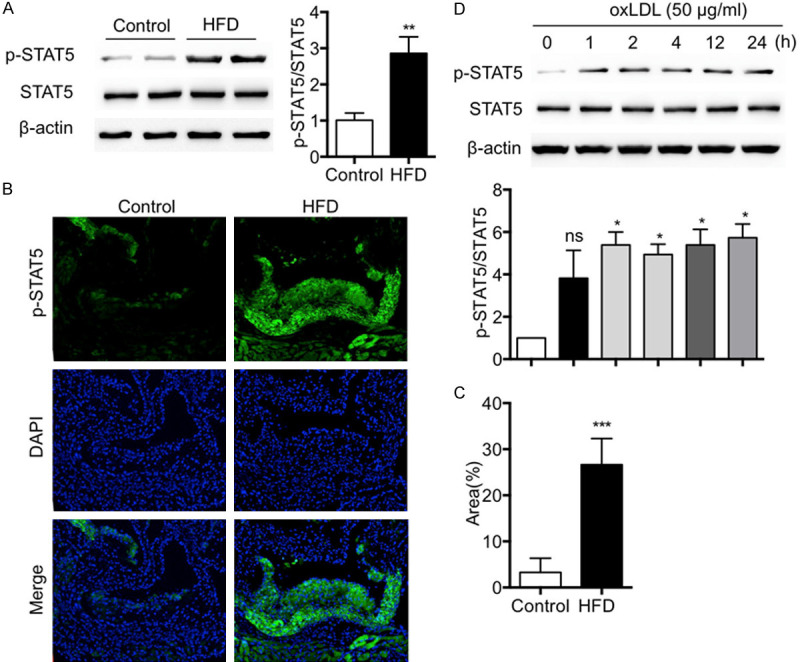
Phosphorylation of STAT5 is upregulated in atherosclerosis. A. Western blotting showed that p-STAT5 level in artery tissues of ApoE-/- mice was increased by HFD. B, C. Immunofluorescence staining showed that HFD increased p-STAT5 immunoreactivity in the aortic root of ApoE-/- mice. D. Western blotting showed that stimulation with oxLDL significantly induced p-STAT5. (n=6 in each group; *P < 0.05, **P < 0.01, ***P < 0.001, vs. Control).
Inhibiting STAT5 did not affect body weight and hyperlipidemia profile in HFD-fed ApoE-/- mice
To further study the role of STAT5 in AS, the anti-atherogenic effects of inhibiting the phosphorylation of STAT5 in HFD-induced ApoE-/- mice by STAT5-IN-1, which is a selective inhibitor of STAT5 and has no significant inhibitory effect on other members of STATs family validated by Judith Muller [19], were observed. The phosphorylation of STAT5 in arteries of ApoE-/- mice induced by HFD was inhibited by STAT5-IN-1, but interesingly, the expression of STAT5 was not changed (Figure 2A). Body weight increases and elevated levels of serum lipid were recorded in ApoE-/- mice induced by HFD, while no such change was recorded in HFD-fed ApoE-/- mice treated with STAT5 inhibitor for 12 weeks (Figure 2B) either in body weight, or in the levels of serum LDL-C (Figure 2C), HDL-C (Figure 2D), TCH (Figure 2E) and TG (Figure 2F), indicating that inhibition of STAT5 did not affect the hyperlipidemia profile.
Figure 2.
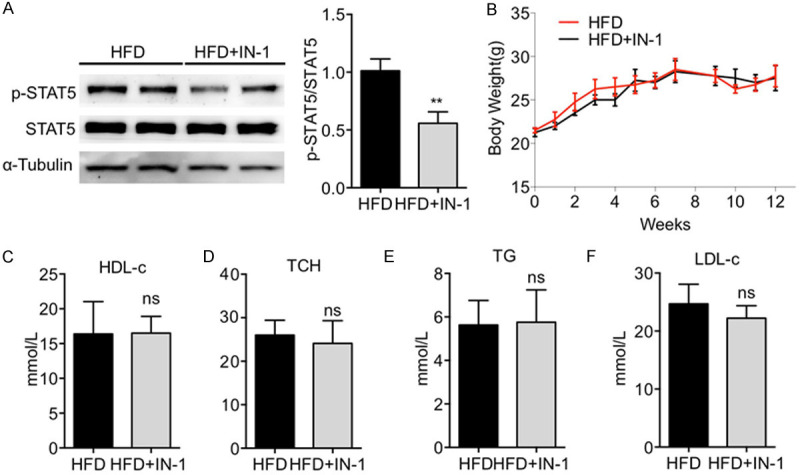
Inhibiting STAT5 did not affect body weight or hyperlipidemia profile in HFD-fed ApoE-/- mice. (A) Western blotting showed that p-STAT5 level in artery tissues of ApoE-/- mice induced by HFD was decreased by STAT5-IN-1. (B-E) STAT5-IN-1 failed to alter HFD-induced body weight changes (B) and serum lipid levels (C-F). (n=12 in each group; ns, no significance, vs. HFD).
Inhibition of STAT5 prevented the development of atherosclerosis in ApoE-/- mice induced by HFD
Treatment with STAT5 inhibitor contributed to the reduction of the atherosclerotic lesion area in aortic arch (Figure 3A). Oil red O staining of the en face showed that the atherosclerotic lesion area in the aorta of HFD-fed ApoE-/- mice was significantly increased, while that of HFD-fed ApoE-/- mice treated with STAT5 inhibitor is notably smaller (Figure 3B, 3C). In addition, oil red O staining of aortic root showed the plaques areas in aortic root of mice in HFD+IN-1 group were significantly smaller than those in the HFD group (Figure 3D-F). The above results suggested that inhibiting STAT5 protected against the plaque development of atherosclerosis in HFD-fed ApoE-/- mice.
Figure 3.
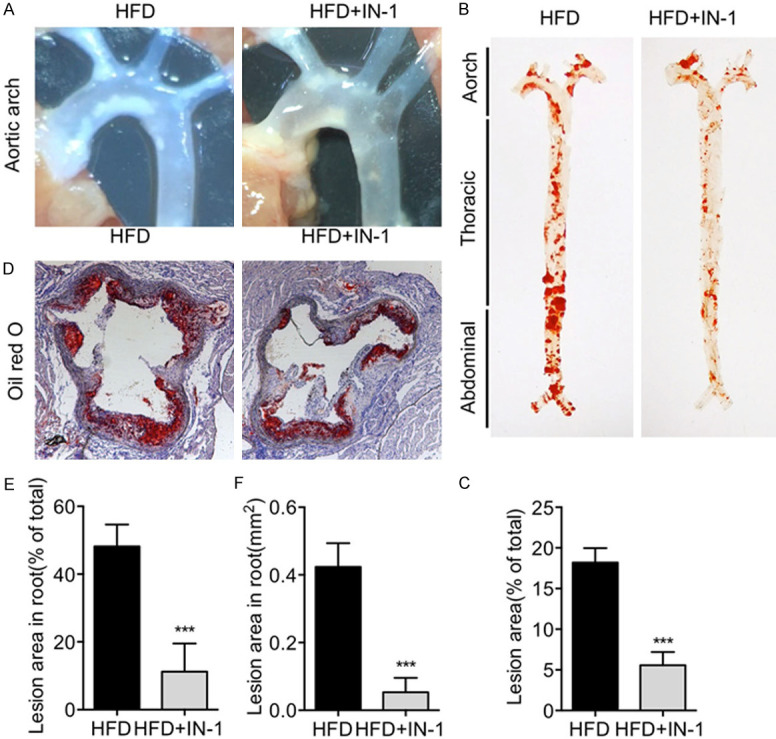
Inhibition of STAT5 prevented the plaque development of atherosclerosis in HFD-fed ApoE-/- mice. (A) Representative images of aortic arch showed that atherosclerotic lesion area was reduced by STAT5-IN-1. (B) Representative images of oil red O staining of en face showed that the atherosclerosis plaque was reduced by STAT5-IN-1. (C) Quantitative analysis of atherosclerotic plaque lesion area in the en face. (D-F) Representative images of oil red O staining in aortic root showed that the accumulation of lipid was reduced by STAT5-IN-1 (D) and quantitative analysis (E, F). (n=6 in each group; ***P < 0.001, vs. HFD).
STAT5 inhibitor inhibited inflammation in atherosclerotic aortas
Then we attempted to clarify the effect of STAT5 in inflammation of atherosclerotic aortas. As shown in Figure 4A-D, there was a marked decrease in the mRNA levels of ICAM-1, VCAM-1, TNF-α, and IL-6 in atherosclerotic aortas of HFD-fed mice treated with STAT5 inhibitor. In addition, the protein levels of TNF-α and IL-6 were reduced by STAT5 inhibitor (Figure 4E, 4F). These indicated that inhibiting STAT5 reduced inflammation in atherosclerotic aortas.
Figure 4.
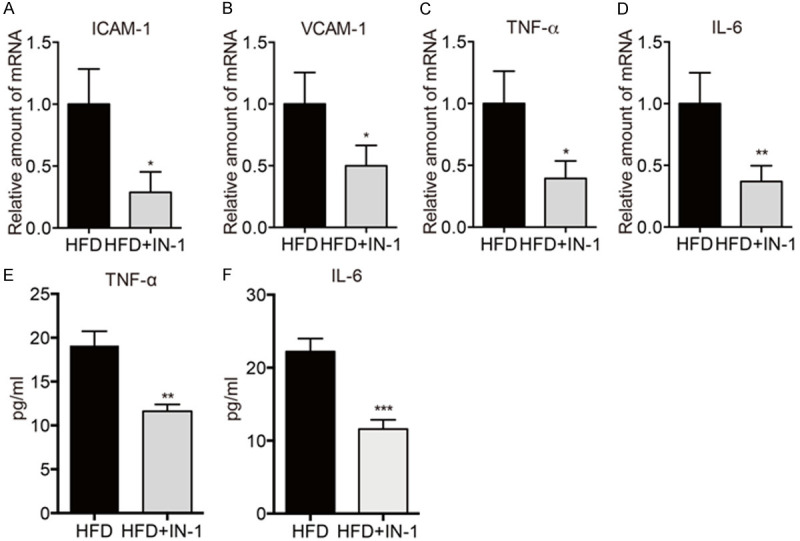
STAT5 inhibitor inhibited inflammation in atherosclerotic aortas. (A-D) The mRNA levels of ICAM-1 (A), VCAM-1 (B), IL-6 (C), and TNF-α (D) were detected by RT-qPCR. (E, F) The protein levels of TNF-α (E) and IL-6 (F) in arterial tissues were detected by ELISA. (n=6 in each group; *P < 0.05, **P < 0.01, ns, no significance, vs. HFD group).
Inhibiting STAT5 suppressed foam cell formatting and inflammation in oxLDL-stimulated macrophages
Based on the finding that inhibition of STAT5 could decrease inflammation in ApoE-/- mice induced by HFD and that there is a close relationship between inflammatory atherosclerosis and macrophages, the effects of STAT5 on oxLDL-stimulated inflammation of macrophages were investigated. Firstly, oxLDL induced the phosphosylation of STAT5 in macrophages, while the phosphosylation of STAT5 was decreased by STAT5 inhibitor (Figure 5A). As shown in Figure 5B, oxLDL induced macrophage to format foam cell, but treatment with STAT5 inhibitor suppressed oxLDL-induced foam cell formatting. Then, oxLDL led to an increase in the protein secretion and the mRNA levels of IL-6 and TNF-α, while these changes were reversed by STAT5 inhibitor (Figure 5C-F). Through real-time qPCR assay, the results further demonstrated that STAT5 inhibitor led to a decrease in the mRNA levels of VCAM-1 and ICAM-1 in oxLDL-stimulated macrophages (Figure 5G, 5H). Moreover, in oxLDL-stimulated macrophages, the protein level of TNF-α were reduced by STAT5-IN-1 (Figure 5I). Next, we knocked down STAT5 expression before exposing them to oxLDL. In contrast to the untreated group, the protein expression of STAT5 was reduced by over 70% by transfecting specific STAT5 siRNA (Figure 6A). In STAT5-knockdown macrophages, exposure to oxLDL did not induce foam cell formatting (Figure 6B), raise the secretion levels of IL-6 and TNF-α (Figure 6C, 6D) and the mRNA expression of ICAM-1, VCAM-1, IL-6 and TNF-α (Figure 6E-H). Furthermore, in oxLDL-stimulated macrophages, the protein level of TNF-α were reduced by knocking down STAT5 expression (Figure 6I). These results suggested that inhibiting STAT5 could serve as a suppressor of oxLDL-induced inflammation.
Figure 5.

STAT5 inhibitor suppressed foam cell formatting and inflammation in oxLDL-stimulated macrophages. (A) Macrophages were pretreated with STAT5-IN-1 (10 μM) or vehicle (DMSO, 1 μL) for 1 h and then stimulated with oxLDL (100 µg/mL) for 1 h. The foam cell formatting was observed by oil red O staining. The phosphorylation of STAT5 in macrophages was detected by western blotting. (B) Representative images of oil red O staining showed that STAT5-IN-1 inhibited oxLDL-induced foam cell formatting. Macrophages were pretreated with STAT5-IN-1 (10 μM) or vehicle (DMSO, 1 μL) for 1 h and then stimulated with oxLDL (100 µg/mL) for 24 h. The foam cell formatting was observed by oil red O staining. (C, D) STAT5-IN-1 decreased the secretion levels of inflammatory factors in oxLDL-induced macrophages. Pretreating with STAT5-IN-1 (10 μM) or vehicle (DMSO, 1 μL) for 1 h, Macrophages were stimulated with oxLDL (50 µg/mL) for 24 h. ELISA was used to measure the levels of IL-6 (C) and TNF-α (D). (E-H) STAT5-IN-1 reduced the mRNA levels of IL-6, TNF-α, VCAM-1 and ICAM-1 in oxLDL-induced macrophages. Pretreated with STAT5-IN-1 (10 μM) or vehicle (DMSO, 1 μL) for 1 h, Macrophages were stimulated with oxLDL (50 µg/mL) for 6 h. The mRNA expressions of IL-6 (E), TNF-α (F), VCAM-1 (G) and ICAM-1 (H) were detected by RT-qPCR. (I) STAT5-IN-1 reduced the protein expression of TNF-α in oxLDL-induced macrophages. Pretreated with STAT5-IN-1 (10 μM) or vehicle (DMSO, 1 μL) for 1 h, Macrophages were stimulated with oxLDL (50 µg/mL) for 24 h and then the total protein was collected. The expression of TNF-α was detected by western blot. (Bars represent the mean ± SEM of three independent experiments; #P < 0.05, ##P < 0.01, ###P < 0.001, vs. vehicle group; *P < 0.05, **P < 0.01, ***P < 0.001, vs. oxLDL group).
Figure 6.
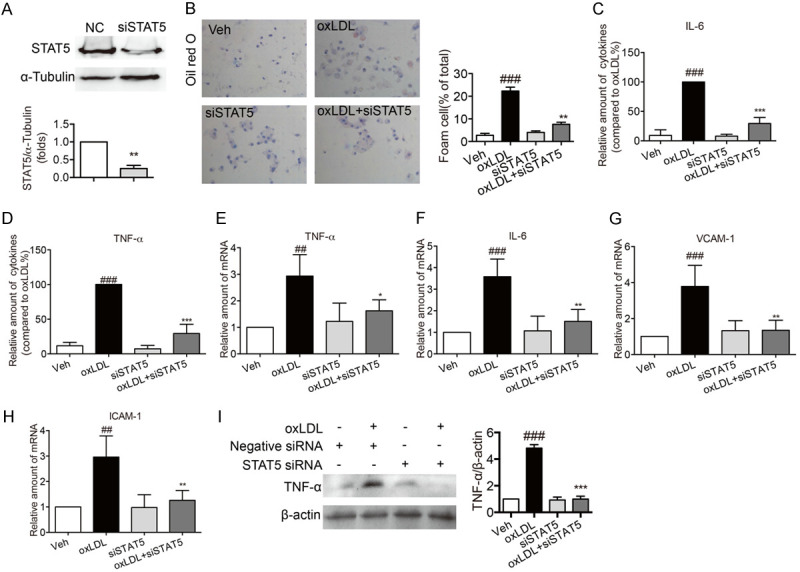
Knockdown of STAT5 by siRNA suppressed foam cell formatting and inflammation in oxLDL-stimulated macrophages. Primary macrophages were transfected with mouse specific STAT5 siRNA. (A) Western blotting for the expression of STAT5. (Bars represent the mean ± SEM of three independent experiments; **P < 0.01, vs. vehicle group). (B) Representative images of oil red O staining showed that STAT5 knockdown inhibited oxLDL-induced foam cell formatting. Macrophages were stimulated with oxLDL (100 µg/mL) for 24 h. The foam cell formatting was observed by oil red O staining. (C, D) STAT5 knockdown inhibited the decrease in the secretion levels of inflammatory factors in oxLDL-induced macrophages. oxLDL (50 µg/mL) stimulated macrophages for 24 h. ELISA was used to measure the levels of IL-6 (C) and TNF-α (D). (E-H) STAT5 knockdown reduced the mRNA levels of IL-6, TNF-α, VCAM-1 and ICAM-1 in oxLDL-induced macrophages. oxLDL (50 µg/mL) stimulated macrophages for 6 h. The mRNA levels of IL-6 (E), TNF-α (F), VCAM-1 (G) and ICAM-1 (H) were detected by RT-qPCR. (I) STAT5 knockdown inhibited the decrease in the protein expression of TNF-α in oxLDL-induced macrophages. oxLDL (50 µg/mL) stimulated macrophages for 24 h and then the total protein was collected. The expression of TNF-α was detected by western blot. (Bars represent the mean ± SEM of three independent experiments; ##P < 0.01, ###P < 0.001, vs. vehicle group; *P < 0.05, **P < 0.01, ***P < 0.001, vs. oxLDL group).
Discussion and conclusion
The pathogenesis of atherosclerosis is complex and diverse, and up to now, the molecular mechanism of atherosclerosis has not been clarified. Thus, it has become the top priority of clinical and basic research to find therapeutic targets and potential drugs for atherosclerosis. The current study investigated the potential role of STAT5 in the development and progression of atherosclerosis in ApoE-/- mice induced by HFD and our study found that inhibiting STAT5 attenuated the development of atherosclerosis and arterial inflammation.
STAT5 regulates the expression of cytokine target genes by binding to IFNγ-activated sequence motif. The transcriptional activation of the promoter and enhancer of the target gene requires contact with a co-activator and synergy with another transcription factor. The phosphorylation of STAT5 is considered to be induced by cytokine receptor-associated JAKs (Figure 1B). In the present study, we found that STAT5 was phosphorylated in oxLDL-stimulated macrophage. Interestingly, at 1 h after oxLDL stimulation, STAT5 was phosphorylated, suggesting that JAKs might not be the only upstream of STAT5 in oxLDL-induced activation. However, the molecular mechanism of its activation was not investigated in this study. Furthermore, the phosphorylation level of STAT5 in the aortas and aortic roots of ApoE-/- mice with HFD is higher than that in ApoE-/- mice fed with a normal diet (Figure 1A and 1C). The aforementioned findings indicated that STAT5 might be closely linked with the occurrence and progression of atherosclerosis. And the in vivo experiment results showed that the lesion area in aortic root and en face in STAT5 inhibitor-treated group was smaller than that in HFD group (Figure 3) and that STAT5 inhibitor did not induce any change either in mice body weight or in their serum lipid levels (Figure 2), confirming that STAT5 is implicated in the occurrence and progression of atherosclerosis. In the in vitro experiment, the results showed that inhibiting STAT5 by inhibitor or siRNA suppressed oxLDL-induced foam cell formatting, which is consistent with the in vivo results. The above findings provide compelling evidence that STAT5 might be the target for treating atherosclerosis.
Inflammation is present throughout every stage of the development of atherosclerosis [20]. The in vivo experiment results showed that the mRNA expressions of ICAM-1, VCAM-1, IL-6, and TNF-α and the protein levels of IL-6 and TNF-α in aorta were reduced by treatment with the STAT5 inhibitor (Figure 4). And in agreement with the in vivo experiment, the mRNA and protein secretion levels of IL-6 and TNF-α and the mRNA levels of ICAM-1 and VCAM-1 in oxLDL-stimulated macrophage were also reduced by treatment with the STAT5 inhibitor or genetic knockdown (Figures 5 and 6), suggesting that STAT5-regulated inflammation is implicated in the occurrence and progression of atherosclerosis.
Drug interventions, including statins, inhibitors, nicotinic acid, and calcium channel blockers, could prevent the development of atherosclerosis by targeting reactive oxygen species (ROS) and inflammation. However, the therapeutic effects of these drugs in the clinic are still limited, and targeted therapy offers a promising alternative in treating atherosclerosis. The results of this study suggest that STAT5 activation could contribute to the occurrence and progression of atherosclerosis. However, we failed to report how STAT5 is activated in atherosclerosis. It was found that STAT5-regulated inflammatory response could promote the development of atherosclerosis. To sum up, targeting STAT5 might be a potential therapeutic strategy to prevent atherosclerosis.
Acknowledgements
This research was supported by the Joint Funds of the Zhejiang Provincial Natural Science Foundation of China under Grant No. LQY20H300001; Zhejiang Provincial Natural Science Foundation of China under Grant No. LGF21H020002; Chinese Medical and Health Research Project of Zhejiang Province under Grant No. 2021ZA038.
Disclosure of conflict of interest
None.
References
- 1.Xu S, Kamato D, Little PJ, Nakagawa S, Pelisek J, Jin ZG. Targeting epigenetics and non-coding RNAs in atherosclerosis: from mechanisms to therapeutics. Pharmacol Ther. 2019;196:15–43. doi: 10.1016/j.pharmthera.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 5.Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38:1092–1104. doi: 10.1016/j.immuni.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolodgie FD, Virmani R, Burke AP, Farb A, Weber DK, Kutys R, Finn AV, Gold HK. Pathologic assessment of the vulnerable human coronary plaque. Heart. 2004;90:1385–1391. doi: 10.1136/hrt.2004.041798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ball RY, Stowers EC, Burton JH, Cary NR, Skepper JN, Mitchinson MJ. Evidence that the death of macrophage foam cells contributes to the lipid core of atheroma. Atherosclerosis. 1995;114:45–54. doi: 10.1016/0021-9150(94)05463-s. [DOI] [PubMed] [Google Scholar]
- 8.Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278:483–493. doi: 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM, Tan P, Moh A, Kaplan MH, Zhang Y, Fu XY. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014;24:1387–1402. doi: 10.1038/cr.2014.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herr F, Lemoine R, Gouilleux F, Meley D, Kazma I, Heraud A, Velge-Roussel F, Baron C, Lebranchu Y. IL-2 phosphorylates STAT5 to drive IFN-gamma production and activation of human dendritic cells. J Immunol. 2014;192:5660–5670. doi: 10.4049/jimmunol.1300422. [DOI] [PubMed] [Google Scholar]
- 11.Kaltenecker D, Themanns M, Mueller KM, Spirk K, Golob-Schwarzl N, Friedbichler K, Kenner L, Haybaeck J, Moriggl R. STAT5 deficiency in hepatocytes reduces diethylnitrosamine-induced liver tumorigenesis in mice. Cytokine. 2019;124:154573. doi: 10.1016/j.cyto.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 12.Luo XY, Zhou H, Wang SY, Xiong J, Mo CF, Guo HJ, Wang YT, Yang SX, Li LM, Zou Q, Liu Y. A benzoxazole derivative PO-296 inhibits T lymphocyte proliferation by the JAK3/STAT5 signal pathway. J Cell Biochem. 2019;120:9193–9202. doi: 10.1002/jcb.28195. [DOI] [PubMed] [Google Scholar]
- 13.Minieri V, De Dominici M, Porazzi P, Mariani SA, Spinelli O, Rambaldi A, Peterson LF, Porcu P, Nevalainen MT, Calabretta B. Targeting STAT5 or STAT5-regulated pathways suppresses leukemogenesis of Ph+ acute lymphoblastic leukemia. Cancer Res. 2018;78:5793–5807. doi: 10.1158/0008-5472.CAN-18-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones N, Vincent EE, Cronin JG, Panetti S, Chambers M, Holm SR, Owens SE, Francis NJ, Finlay DK, Thornton CA. Akt and STAT5 mediate naive human CD4+ T-cell early metabolic response to TCR stimulation. Nat Commun. 2019;10:2042. doi: 10.1038/s41467-019-10023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baik M, Kim J, Piao MY, Kang HJ, Park SJ, Na SW, Ahn SH, Lee JH. Deletion of liver-specific STAT5 gene alters the expression of bile acid metabolism genes and reduces liver damage in lithogenic diet-fed mice. J Nutr Biochem. 2017;39:59–67. doi: 10.1016/j.jnutbio.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 16.Ding X, Zheng L, Yang B, Wang X, Ying Y. Luteolin attenuates atherosclerosis via modulating signal transducer and activator of transcription 3-mediated inflammatory response. Drug Des Devel Ther. 2019;13:3899–3911. doi: 10.2147/DDDT.S207185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. 2015;125:4514–4528. doi: 10.1172/JCI82719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura A, Ishida Y, Furuta M, Nosaka M, Kuninaka Y, Taruya A, Mukaida N, Kondo T. Protective roles of interferon-gamma in cardiac hypertrophy induced by sustained pressure overload. J Am Heart Assoc. 2018;7:e008145. doi: 10.1161/JAHA.117.008145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller J, Sperl B, Reindl W, Kiessling A, Berg T. Discovery of chromone-based inhibitors of the transcription factor STAT5. Chembiochem. 2008;9:723–727. doi: 10.1002/cbic.200700701. [DOI] [PubMed] [Google Scholar]
- 20.Nahrendorf M, Swirski FK. Immunology. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science. 2015;349:237–238. doi: 10.1126/science.aac7801. [DOI] [PubMed] [Google Scholar]