

Abstract
Background: Despite the essential functions of the intestinal microbiota in human physiology, little research was reported on gut microbiota alterations in intensive care patients. This investigation examined the dysbacteriosis of intestinal flora in critically ill patients and evaluated the prognostic performance of this dysbiosis to predict in-hospital mortality. Methods: A prospective cohort of patients were consecutively recruited in the Intensive Care Units (ICUs) in Guangdong Provincial People’s Hospital from March 2017 through October 2017. Acute Physiology and Chronic Health Evaluation (APACHE) II score and Sequential Organ Failure Assessment (SOFA) score were assessed, and fecal samples were taken for examination within 24 hours of ICU admission. The taxonomic composition of the intestinal microbiome was determined using 16S rDNA gene sequencing. Patients were divided into survival and death groups based on hospital outcomes. The two groups were statistically compared using the Wilcoxon test and Metastats analysis. The genera of bacteria showing significantly different abundance between groups were assessed as predictors of in-hospital death. The prognostic value of bacterial abundance alone and in combination with APACHE II or SOFA score was evaluated using the area under the receiver operating characteristic curve (AUROC). Results: Among the 61 patients examined, 12 patients (19.7%) died during their hospital stay. Bifidobacterium abundance was higher in the survival group than the death group (P = 0.031). The AUROC of Bifidobacterium abundance in identifying in-hospital death at a cut-off probability of 0.0041 was 0.718 (95% confidence interval [CI], 0.588-0.826). The panel of Bifidobacterium abundance plus SOFA (AUROC, 0.882; 95% CI, 0.774-0.950) outperformed SOFA (AUROC, 0.649; 95% CI, 0.516-0.767; P = 0.012) and Bifidobacterium abundance alone (P = 0.007). The panel of Bifidobacterium abundance plus APACHE II (AUROC, 0.876; 95% CI, 0.766-0.946) outperformed APACHE II (AUROC, 0.724; 95% CI, 0.595-0.831; P = 0.035) and Bifidobacterium abundance alone (P = 0.012). Conclusions: Dysbiosis of intestinal microbiota with variable degrees of reduction in Bifidobacterium abundance exhibited promising performance in the predicting of in-hospital mortality and provides incremental prognostic value to existing scoring systems in the adult intensive care unit (ICU) setting.
Keywords: Intestinal dysbiosis, bifidobacterium, in-hospital mortality, prognostic value, intensive care unit, scoring system
Introduction
Intestinal microbiota diversity plays a vital role in maintaining intestinal homeostasis, human nutrition and health [1]. However, this diversity is highly vulnerable to diseases, especially critical illnesses with high mortality rates [2,3]. Critical illnesses frequently cause gastrointestinal motility disorder, intestinal mucosal ischemia, and impaired intestinal barrier function [4,5], which lead to dramatically altered intestinal microbiota [3,6]. Dysbiosis of gut microbiota is widely viewed as a potential marker for many diseases [7]. Therefore, it may be a new mortality predictor in critically ill patients [5,7]. Xu et al. indicated that gut microbiota dysbiosis was associated with patients’ 180-day mortality in a neurological intensive care unit [6]. However, the prognostic ability of this potential marker in predicting in-hospital mortality in intensive care unit patients was not determined.
The present prospective observational cohort study investigated intestinal dysbiosis in critically ill patients at admission and detected that the altered abundance of some bacterial genera were associated with in-hospital mortality. Further assessments of the prognostic performance of this dysbiosis with existing scoring systems (APACHE II/SOFA score) were also performed to obtain an optimal clinical model to predict in-hospital mortality.
Materials and methods
Study design and population
A prospective cohort of patients was consecutively recruited in Intensive Care Units (ICUs) in Guangdong Provincial People’s Hospital from March 2017 through October 2017. Patients were admitted from general or emergency wards. Participants were excluded if any of the following criteria were met: (1) the participant was under the age of 18 years; (2) the participant received systematic antibiotic treatment within 48 hours before ICU admission; (3) the participant was admitted to the ICU for an end-stage chronic disease; or (4) the participant died within 24 hours of ICU admission. The local ethics committee approved the study. All patients provided informed consent form before participating in the study.
Data collection
Demographic, laboratory and clinical variables were recorded at admission to the ICU. SOFA and APACHE II scores were assessed twice within 24 hours of ICU admission, and the worst value was chosen. Stool samples or rectal swabs (if stool was unavailable) were collected at ICU admission and immediately frozen at -80°C. Taxonomic compositions of the intestinal microbiome were determined using 16S rDNA gene sequencing of the stool sample.
Intestinal microbiota analysis
Intestinal flora analysis was performed using the 16S rDNA amplicon sequencing method, which is commonly used for bacterial phylogeny and classification identification [8-10]. The genomic DNA of the sample was extracted using the sodium dodecyl sulphate (SDS) method. A sample without genomic DNA extraction was used as the blank control. The purity and concentration of the DNA were detected using 1% agarose gel electrophoresis. The DNA was diluted to 1 ng/ml with sterile water depending on the concentration. Specific primers (e.g., 16S V4: 515F-806R, 18S V4: 528F-706R, 18S V9: 1380F-1510R, et al.) with barcodes were used to amplify the 16S rDNA gene in different regions (16S V4/16S V3/16S V3-V4/16S V4-V5, 18S V4/18S V9, ITS1/ITS2, and Arc V4). All PCR reactions were performed using Phusion® High-Fidelity PCR Master Mix (New England Biolabs, Ipswich, MA, USA). The same volume of 1X loading buffer (containing SYB green) was mixed with the PCR product and electrophoresed on a 2% agarose gel. Samples with a bright master band between 400-450 base pairs were selected for further experiments. A sample without template was used as the PCR negative control. The mixed PCR products were purified using a Qiagen Gel Extraction Kit (Qiagen, Duesseldorf, Germany). The TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, San Diego, CA, USA) was used to generate sequencing libraries, according to the manufacturer’s recommendations, and index codes were added. A Qubit 2.0 fluorometer (Thermo Scientific, Waltham, MA, USA) and Agilent Bioanalyzer 2100 system (Agilent Technologies, Beijing, China) were used to evaluate the library quality [11]. The library was sequenced using an Illumina HiSeq2500 (Illumina, San Diego, CA, USA) platform, and 250 base-pair paired-end reads were generated [12].
Sequences analysis was performed using Uparse software (Uparse v7.0.1001, http://drive5.com/uparse/) [13]. By default, the sequences were clustered into operational taxonomic units (OTUs) with 97% identity. A representative sequence of OTUs was selected. According to the algorithm principle, the sequence with the highest frequency in OTUs was selected as the representative of OTUs. The Mothur method (with a cut-off value of 0.8) and Silva database (version 128, https://www.arb-silva.de) were used to annotate OTUS representative sequences and analyze species annotations to obtain classification information at each classification level: kingdom, phylum, taxa, order, family, genus and species [13-15]. Fast multi-sequence alignments were performed using MUSCLE (Version 3.8.31, http://www.drive5.com/muscle/) software to obtain phylogenetic relationships for all OTU representative sequences [15]. The data of each sample were homogenized, and the sample was homogenized according to the minimum amount of data in the sample.
Data analysis
Patients participating in the study were categorized into two groups according to their survival status in the hospital, i.e., survival and death groups. All baseline data between groups were assessed. Based on the OTU clustering results, the abundance analysis of OTUs was carried out to examine differences in the community structure between groups. The difference between the beta diversity index groups was analyzed. Metastats analysis was performed at each classification level (phylum, class, order, family, genus, and species). P values were obtained by performing permutation tests between groups, and abundance distribution boxes of different species between groups were plotted.
The prognosis prediction models for Bifidobacterium abundance, APACHE II score, and SOFA score was established using univariate logistic regression. The prognosis prediction models for APACHE II score plus Bifidobacterium abundance and SOFA score plus Bifidobacterium abundance were established using multivariate logistic regression. All prediction models were evaluated as the area under the receiver operating characteristic curves (AUROC). To avoid an overfitting effect due to relatively small sample size, the bootstrap method was used with 1000 resamples, and the bootstrap-corrected AUROCs and 95% CI are reported. The Youden index [16] was used to determine the optimal cut-off points. Comparisons between groups of AUROCs were performed using the DeLong [17] and bootstrapping methods [19].
Continuous variables are presented as means ± standard deviation (SD) and were compared using the Wilcoxon test. Categorical variables are presented as numbers (percentage) and were compared using Pearson’s χ2 test or Fisher’s exact test, where appropriate. The significance level was set at P<0.05. The analyses were performed using SPSS 24.0 software (SPSS, Chicago, IL, USA), MedCalc 12.5.0 software (MedCalc Software, Ostend, Belgium), and R 3.3.1 software (R Foundation for Statistical Computing, Vienna, Austria) using RStudio v1.0.136 (RStudio Inc., Boston, MA, USA).
Results
Sixty-one critically ill patients with a mean age 60.2 years were included in this study. Forty-nine patients (80.3%) survived, and 12 patients (19.7%) died in the hospital. The patients’ clinical and laboratory characteristics are shown in Table 1. Notably, the SOFA and APACHE II scores were markedly higher in the death group than the survival group (P = 0.001).
Table 1.
Clinical and demographic characteristics of patients
| Variables | Total (n = 61) | Death (n = 12) | Survival (n = 49) | P value |
|---|---|---|---|---|
| Age, y | 60.2 ± 19.0 | 61.1 ± 20.2 | 60.0 ± 18.9 | 0.871 |
| Male, n (%) | 42 (68.8) | 10 (83.3) | 32 (65.3) | 0.186 |
| Admission Type, n (%) | 0.541 | |||
| Neurologic | 25 (41.0) | 3 (25.0) | 22 (44.9) | 0.209 |
| Respiratory | 9 (14.8) | 2 (16.7) | 7 (14.3) | 0.836 |
| Digestive | 10 (16.4) | 3 (25.0) | 7 (14.3) | 0.369 |
| Sepsis | 5 (8.2) | 3 (25.0) | 2 (4.0) | 0.018 |
| Others | 12 (19.6) | 1 (8.3) | 11 (22.5) | 0.270 |
| CRP, mg/L | 67.3 ± 65.6 | 97.8 ± 81.9 | 59.4 ± 59.2 | 0.149 |
| PCT, ng/L | 10.5 ± 4.6 | 13.3 ± 5.1 | 8.5 ± 9.0 | 0.127 |
| ICU stay, days | 26.6 ± 43.7 | 14.7 ± 11.9 | 29.5 ± 48.1 | 0.057 |
| SOFA score | 9.8 ± 3.7 | 13.4 ± 3.4 | 8.9 ± 3.1 | 0.001 |
| APACHE II score | 20.4 ± 7.1 | 27.5 ± 6.6 | 18.7 ± 6.0 | 0.001 |
Continuous variables are presented as mean ± SD and categorical variables as number (percentage). APACHE II, Acute Physiology and Chronic Health Evaluation II; CRP, C reactive protein; ICU, intensive care unit; PCT, procalcitonin; SD, standard deviation; SOFA, sequential organ failure assessment.
After paired-end read merging and error correction of 16S rDNA sequencing, a total of 4,592,443 effective tags were obtained from 61 stool samples, for a mean of 75,286 tags. Based on 97% sequence identity, amplicons were clustered into 4954 OTUs. A total of 837 genera were identified in gut microbiomes in this study. OTU richness of the intestinal microbiota showed no significant difference between groups. According to the results of species annotation at the genus level, 10 genera exhibited a higher level of abundance in the two groups and accounted for greater than 50% of the total. The top 10 genera with the highest abundance at the genus classification level were enrolled to generate a columnar accumulation plot of relative abundance. The top 10 genera were Enterococcus, Clostridiuminnocuum, Subdoligranulum, Leuconostoc, Stenotrophomonas, Escherichia-Shigella, Streptococcus, Collinsella, Akkermansia, and Bifidobacterium, and did not include some of the most common organisms that are typically seen in the human gut, such as Bacteroides and Lachnospiraceae. Figure 1 shows the composition in each sample, and Figure 2 describes the relative abundance between groups. To identify different species in the 10 genera, we performed a t test to analyze the intestinal flora and identify species with a significant difference (P<0.05). Table 2 shows that only Bifidobacterium abundance exhibited a significant difference between groups, with a larger reduction in the death group than the survival group (P = 0.031). Notably, the abundance of Bifidobacterium in both groups was not relatively low, which may be related to the relative old age of the patients we included (the average age was approximately 60 years old). In general, Bifidobacterium, as a probiotic bacterium, decreases slowly with age. The abundance distribution of different species between groups was also mapped (Figure 3). Notably, there was a trend of a lower abundance of Escherichia-Shigella in the patients who died compared with patients who survived, which should be investigated in future studies using a larger sample size.
Figure 1.
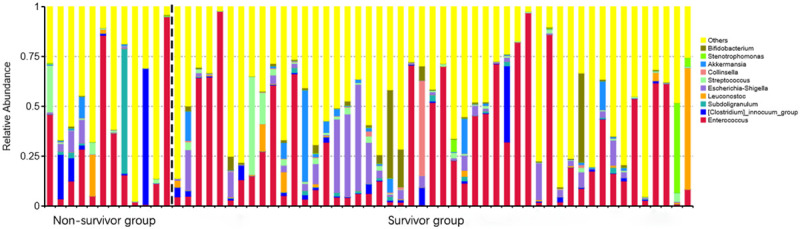
TOP 10 species relative abundance histogram at the genus level of the personal sample. The abscissa is the sample name; the ordinate is the relative abundance; the others represented the sum of the relative abundances of all the other genus except the 10 genera.
Figure 2.
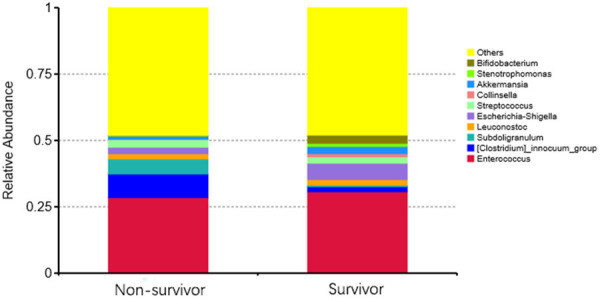
TOP 10 species relative abundance histogram at the genus level of the groups. The abscissa is the group name; the ordinate is the relative abundance; the others represent the sum of the relative abundances of all the other genus except the 10 genera.
Table 2.
Mean abundance of gut microbiome taxa in patients
| Taxa | Death | Survival | P value |
|---|---|---|---|
| Enterococcus | 0.1397 (0.0387-0.4347) | 0.1628 (0.0473-0.6092) | 0.830 |
| Clostridium innocuum | 0.0050 (0.0004-0.0924) | 0.0050 (0.0019-0.0119) | 0.253 |
| Subdoligranulum | 0.0007 (<0.0001-0.01451) | 0.0013 (0.0002-0.0059) | 0.342 |
| Leuconostoc | 0.0004 (<0.0001-0.0017) | 0.0010 (<0.0001-0.0055) | 0.933 |
| Escherichia-Shigella | 0.0023 (0.0007-0.0317) | 0.0170 (0.0035-0.0538) | 0.072 |
| Streptococcus | 0.0063 (0.0023-0.0188) | 0.0073 (0.0030-0.0205) | 0.810 |
| Collinsella | 0.0001 (<0.0001-0.0009) | 0.0010 (0.0001-0.0031) | 0.242 |
| Akkermansia | 0.0009 (<0.0001-0.0080) | 0.0021 (<0.0001-0.0128) | 0.268 |
| Stenotrophomonas | 0.0004 (<0.0001-0.0017) | 0.0002 (<0.0001-0.0013) | 0.261 |
| Bifidobacterium | 0.0009 (0.0002-0.0031) | 0.0047 (0.0009-0.0115) | 0.031 |
Abundance of gut microbiome are presented as median (IQR). IQR, interquartile range.
Figure 3.
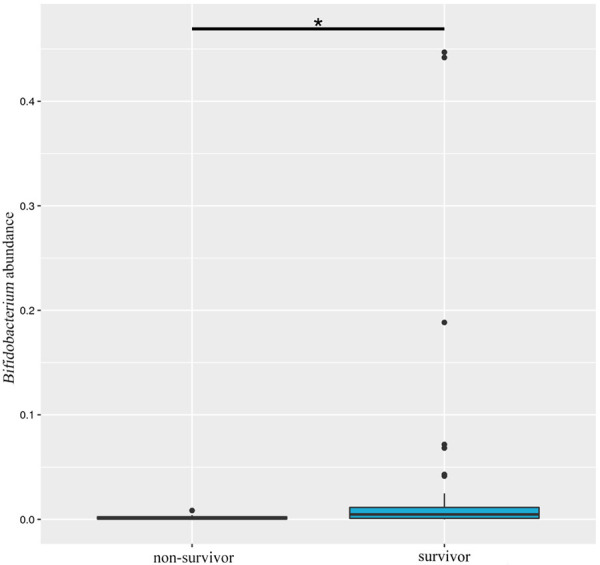
Box analysis of the abundance distribution of Bifidobacterium between groups. The horizontal axis is the sample grouping; the vertical direction is the relative abundance of Bifidobacterium; the horizontal lines represent two groups with significant differences. *P<0.05.
AUROC analysis was used to assess the predictive accuracy of APACHE II, SOFA, and Bifidobacterium abundance for the prognosis of hospital death in critically ill patients. The AUROC analysis of the three univariate models (APACHE II, SOFA, and Bifidobacterium abundance) indicated that APACHE II (AUROC, 0.724; 95% CI, 0.595-0.831) had a slightly better discrimination than SOFA (AUROC, 0.649; 95% CI, 0.516-0.767) and Bifidobacterium abundance (AUROC, 0.718; 95% CI, 0.588-0.826) (Figure 4A and Table 3). However, the pairwise comparative analysis showed no significant difference (Table 4).
Figure 4.
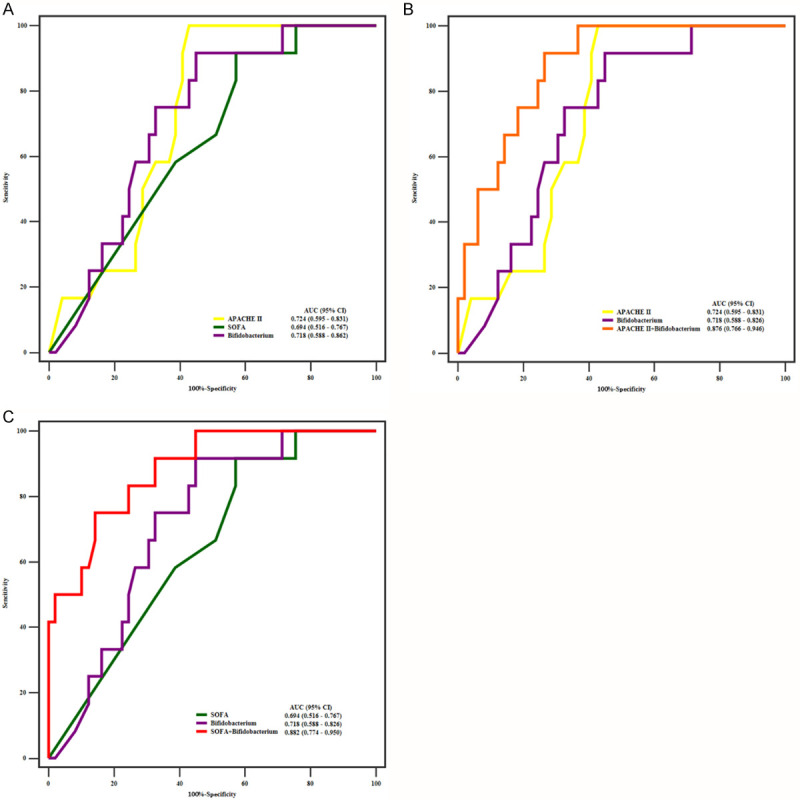
ROC curves for Univariate models and Multivariate models. The abscissa is the specificity, and the ordinate is the sensitivity. A. There is no significant difference among APACHE II, SOFA, and Bifidobacterium. B. The AUROC of APACHE II plus Bifidobacterium is larger than that of APACHE II and Bifidobacterium. C. The AUROC of SOFA plus Bifidobacterium is significantly larger than that of SOFA and Bifidobacterium.
Table 3.
Predictive characteristics of admission indicator and their combinations for ICU mortality
| Logistic regression model | AUROC (95% CI) | Youden index | Cut-off | OR (95% CI) | P |
|---|---|---|---|---|---|
| Univariate models | |||||
| APACHE II | 0.724 (0.595-0.831) | 0.5714 | 27.00 | 1.261 (1.091-1.458) | 0.002 |
| SOFA | 0.649 (0.516-0.767) | 0.3452 | 10.00 | 1.436 (1.159-1.777) | 0.001 |
| Bifidobacterium | 0.718 (0.588-0.826) | 0.4677 | 0.0041 | 0.800 (0.622-1.029) | 0.082 |
| Multivariate models | |||||
| APACHE II plus Bifidobacterium | 0.876 (0.766-0.946) | 0.6514 | 1.82 | 2.716 (1.435-5.141) | 0.002 |
| SOFA plus Bifidobacterium | 0.882 (0.774-0.950) | 0.6071 | 1.07 | 2.720 (1.488-4.973) | 0.001 |
Optimal cut-off value is determined by Youden’s index. Cut-off points of the multivariate indicator models were the predicted probability generated by the multiple logistic regression model. AUROC, area under the receiver operating characteristic curve. OR, odds ratio. CI, confidence interval.
Table 4.
Comparisons of AUROCs among prediction models
| Comparison pairs | P value |
|---|---|
| AUROC comparison among univariate models | |
| APACHE II vs SOFA | 0.419 |
| APACHE II vs Bifidobacterium | 0.927 |
| SOFA vs Bifidobacterium | 0.463 |
| AUROC comparison between multivariate and univariate model | |
| APACHE II plus Bifidobacterium vs APACHE II | 0.035 |
| APACHE II plus Bifidobacterium vs Bifidobacterium | 0.012 |
| SOFA plus Bifidobacterium vs SOFA | 0.012 |
| SOFA plus Bifidobacterium vs Bifidobacterium | 0.007 |
APACHE II, Acute Physiology and Chronic Health Evaluation II; AUROC, area under receiver operating characteristic curve; CI, Confidence Interval; SOFA, sequential organ failure assessment.
Go assess the performance of combinations of these predictors, we performed logistic regression using two panels of predictors (APACHE II score plus Bifidobacterium abundance and SOFA score plus Bifidobacterium abundance). The ROC curve analysis showed that the panel of Bifidobacterium abundance and APACHE II score had the largest AUROC of 0.876 (95% CI, 0.766-0.946) (Figure 4B and Table 3) with no significant difference (P = 0.9274). Further comparison of AUROCs between individual and combined predictors revealed that APACHE II score plus Bifidobacterium abundance significantly outperformed Bifidobacterium abundance (P = 0.018) and APACHE II score (P = 0.035) alone (Table 3). The AUROC of SOFA was 0.649 (95% CI, 0.516-0.767), and the AUROC of SOFA plus Bifidobacterium abundance was 0.882 (95% CI, 0.774-0.950). The combination of Bifidobacterium abundance and SOFA had higher specificity (85.71%) but lower sensitivity (75%) (Figure 4C). We compared the AUROC of SOFA versus SOFA plus Bifidobacterium abundance, and Bifidobacterium abundance versus SOFA plus Bifidobacterium abundance, and both comparisons showed significant differences with P values of 0.007 and 0.012, respectively (Table 3).
Discussion
The results of our study suggest that dysbiosis of intestinal microbiota with variable degrees of reduction in Bifidobacterium abundance is associated with critically ill patients’ risk of death in the hospital. The abundance of intestinal Bifidobacterium distribution was markedly lower in the death group than the survival group. Therefore, this abundance exhibited good prognostic values for in-hospital mortality. It also provided incremental prognostic value to existing scoring systems in the ICU.
Bifidobacterium is a genus of Gram-positive, non-motile, often branched anaerobic bacteria that were first discovered and isolated by Tissier et al. in 1899 from breastfeeding infant feces [18]. Bifidobacterium are a major genera of bacteria that make up the gut flora in humans, and its physiological functions include the promoting of nutrient absorption [19], maintaining intestinal micro-ecological balance [20], immune regulation [21], and anti-tumor activity [22-24]. However, bifidobacteria are susceptible to some adverse conditions, like aging and disease [25], and are likely reduced in intensive care patients [3,26]. The pathophysiological effects of critical illness (e.g., decreased oral intake, intestinal dysmotility, gut hypoperfusion, reperfusion injury, and impaired mucosal integrity) and the clinical interventions of intensive care (e.g., supine positioning, gastric-acid suppression, sedatives, opiates and neuromuscular blockade, and systemic antibiotics) cause ecological imbalances that substantially alter the gut microbiome.
The advent of culture-independent microbiology and high-throughput sequencing methods has enabled researchers to identify specific features of the microbiome that promote and disrupt homeostasis in critically ill patients. The present study used 16S rDNA amplicon sequencing for bacterial phylogeny and classification identification. Notably, our study found that Bifidobacterium abundance was associated the severity of illness. Patients with a lower Bifidobacterium content in gut had a higher risk for death in the hospital. Subsequent statistical analyses of logistic regression determined that Bifidobacterium abundance was a predictor for in-hospital mortality with an AUROC of 0.718. These findings are consistent with previous findings that the reduction of Bifidobacterium abundance is a sign of disorders [10,26,27].
Notably, the results of our study conflict with Xu et al. [6] who investigated the dysbiosis of gut microbiota in patients in a neurocritical care unit. Rather than Bifidobacterium, Xu et al. discovered that the gut genera with significantly decreased abundance longitudinally over time were Ruminococcaceae and Lachnospiraceae using the same 16S rRNA gene sequence analysis. This inconsistency may be attributed to several factors. First, participants in our study were consisted critically ill patients in a medical ICU rather than a neurosurgical ICU. Second, in contrast to healthy volunteers as the control cohort in the previous study, our control cohort was composed of patients who survived in the hospital. We also focused on the outcome of in-hospital death as a binomial variable for the logistic regression analysis rather than 180-day death as a time-dependent covariate for COX regression analysis. Despite these differences in designs and results, the two studies effectively enhance our common understanding that intestinal microbiota dysbiosis is associated with patients’ adverse outcomes and may be a predictor for death risk in critically ill patients.
As new diagnostic, therapeutic and prognostic techniques become available and ICU populations change, the scoring systems used in ICUs may not be very closely relevant to each patient population. This evolving situation calls for fresh biological or clinical markers to reflect current practice patterns and treatments for critical illness. Notably, we found that Bifidobacterium abundance was a promising prognosis factor for in-hospital mortality, and it added to the prognostic value of existing scoring systems, including SOFA and APACHE II scores. Bifidobacterium abundance exhibited a prognostic value that was slightly lower than the APACHE II score but higher than the SOFA score for in-hospital death in critically ill patients. When combined with Bifidobacterium abundance, the APACHE II score and SOFA score performed better in the prognosis prediction of critically ill patients. The reason for this result may be that indicators with different characteristics have different sensitivities and specificities, and a reasonable combination may improve the diagnostic performance. This result shares a similar mechanism with Deng [28], who combined a functional marker (sCysC) and a tubular injury marker (uNAG) to predict outcomes in patients with severe AKI, and the combination demonstrated superior discriminating performance compared to sCysC and uNAG alone.
To the best of our knowledge, there are few reports in the literature on gut microbiota dysbiosis in ICU patients. We demonstrated that the reduction of gut Bifidobacterium content was an independent risk factor for the prognosis of critically ill patients. Some Bifidobacterium strains [29] are important probiotics that show promise in the treatment of ulcerative colitis [30] and infection reduction, including ventilator-associated pneumonia (VAP), which is common in critical illness [31]. Therefore, we suggest that some pharmaceutical probiotic products containing Bifidobacterium may facilitate the maintenance or restoration of the homeostatic balance of infected intestine and improve the survival prognosis of patients in the ICU. However, further prospective clinical trials are required to confirm this proposal.
Our study has some limitations. First, the relatively small sample size as a result of the implementation of rigorous inclusion criteria and avoiding the use of too many resources (e.g., subjects, time, and financial costs), may cause an overestimation of the predictability of Bifidobacterium abundance. However, we used bootstrapping methods with 1000 resamples to avoid an overfitting effect in logistic regression, and we reported the bootstrap-corrected AUROC and 95% CI. Our findings are limited to a single-center study, and center-specific effects cannot be excluded. Further multicenter studies with larger sample sizes are required to verify the present results. The PCR and 16S rRNA amplicon sequencing methods are based on DNA and RNA extraction, respectively, and exhibit high accuracy but are cumbersome and time-consuming, and cannot quickly quantify the intestinal flora [8,32,33]. Therefore, it is especially important to develop a method that can quickly and accurately quantify the abundance of intestinal flora.
Conclusion
Dysbiosis of intestinal microbiota with variable degrees of reduction in Bifidobacterium abundance exhibited promising performance in the predicting of in-hospital mortality and provided incremental prognostic value to existing scoring systems in an intensive care unit (ICU) setting.
Acknowledgements
The authors would like to thank all the doctors, nurses, technicians, and patients for their dedication to the study. Ru Wei is currently receiving a grant (#A2018239) from the Medical Scientific Research Foundation of Guangdong Province; Chunbo Chen is currently receiving a grant (#201803010058) from the Guangzhou Livelihood Science and Technology Project; Weihong Sha is currently receiving a grant (#201803) from the Guangdong High-level Hospital Construction Project; Shixue Dai is currently receiving a grant (#81300370) from the National Natural Science Foundation of China; Linhui Hu is currently receiving a grant (#zx2020017) from the High-level Hospital Construction Research Project of Maoming People’s Hospital.
Informed consent was obtained from all individual participants included in the study.
Disclosure of conflict of interest
None.
Abbreviations
- APACHE-II
Acute Physiology and Chronic Health Evaluation-II
- AUROC
area under receiver operating characteristic curve
- CI
Confidence interval
- ICU
Intensive care unit
- IQR
Interquartile range
- OUT
Operational taxonomic unit
- PCR
polymerase chain reaction
- ROC
receiver operating characteristic curve
- SD
Standard deviation
- SOFA
Sequential Organ Failure Assessment
References
- 1.Thomas S, Izard J, Walsh E, Batich K, Chongsathidkiet P, Clarke G, Sela DA, Muller AJ, Mullin JM, Albert K, Gilligan JP, DiGuilio K, Dilbarova R, Alexander W, Prendergast GC. The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res. 2017;77:1783–1812. doi: 10.1158/0008-5472.CAN-16-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zaborin A, Smith D, Garfield K, Quensen J, Shakhsheer B, Kade M, Tirrell M, Tiedje J, Gilbert JA, Zaborina O, Alverdy JC. Membership and behavior of ultra-low-diversity pathogen communities present in the gut of humans during prolonged critical illness. mBio. 2014;5:e01361-14. doi: 10.1128/mBio.01361-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickson RP. The microbiome and critical illness. Lancet Respir Med. 2016;4:59–72. doi: 10.1016/S2213-2600(15)00427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carroll IM, Threadgill DW, Threadgill DS. The gastrointestinal microbiome: a malleable, third genome of mammals. Mamm Genome. 2009;20:395–403. doi: 10.1007/s00335-009-9204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H, Zhang D, Wang Y, Zhao S. Association between acute gastrointestinal injury grading system and disease severity and prognosis in critically ill patients: a multicenter, prospective, observational study in China. J Crit Care. 2016;36:24–28. doi: 10.1016/j.jcrc.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Xu R, Tan C, Zhu J, Zeng X, Gao X, Wu Q, Chen Q, Wang H, Zhou H, He Y, Pan S, Yin J. Dysbiosis of the intestinal microbiota in neurocritically ill patients and the risk for death. Crit Care. 2019;23:195. doi: 10.1186/s13054-019-2488-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freedberg DE, Zhou MJ, Cohen ME, Annavajhala MK, Khan S, Moscoso DI, Brooks C, Whittier S, Chong DH, Uhlemann AC, Abrams JA. Pathogen colonization of the gastrointestinal microbiome at intensive care unit admission and risk for subsequent death or infection. Intensive Care Med. 2018;44:1203–1211. doi: 10.1007/s00134-018-5268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G, Luo S, Clark DS, Chen F, Zhang T, Mackie RI, Pennacchio LA, Tringe SG, Visel A, Woyke T, Wang Z, Rubin EM. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 10.O’Mahony L, McCarthy J, Kelly P, Hurley G, Luo F, Chen K, O’Sullivan GC, Kiely B, Collins JK, Shanahan F, Quigley EM. Lactobacillus and bifidobacterium in irritable bowel syndrome: symptom responses and relationship to cytokine profiles. Gastroenterology. 2005;128:541–551. doi: 10.1053/j.gastro.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 11.Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 14.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3:32–35. doi: 10.1002/1097-0142(1950)3:1<32::aid-cncr2820030106>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 17.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 18.Tissier H. Recherches sur la flore intestinale normale et pathologique du nourisson. Thèse de Paris. 1900:1–253. [Google Scholar]
- 19.Plantinga TS, van Maren WW, van Bergenhenegouwen J, Hameetman M, Nierkens S, Jacobs C, de Jong DJ, Joosten LA, van’t Land B, Garssen J, Adema GJ, Netea MG. Differential Toll-like receptor recognition and induction of cytokine profile by Bifidobacterium breve and Lactobacillus strains of probiotics. Clin Vaccine Immunol. 2011;18:621–628. doi: 10.1128/CVI.00498-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469:543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- 21.Asahara T, Shimizu K, Nomoto K, Hamabata T, Ozawa A, Takeda Y. Probiotic bifidobacteria protect mice from lethal infection with Shiga toxin-producing Escherichia coli O157:H7. Infect Immun. 2004;72:2240–2247. doi: 10.1128/IAI.72.4.2240-2247.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, Chang EB, Gajewski TF. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu H, Li Z, Mao S, Ma B, Zhou S, Deng L, Liu T, Cui D, Zhao Y, He J, Yi C, Huang Y. Antitumor effect of sFlt-1 gene therapy system mediated by Bifidobacterium infantis on Lewis lung cancer in mice. Cancer Gene Ther. 2011;18:884–896. doi: 10.1038/cgt.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li ZJ, Zhu H, Ma BY, Zhao F, Mao SH, Liu TG, He JP, Deng LC, Yi C, Huang Y. Inhibitory effect of Bifidobacterium infantis-mediated sKDR prokaryotic expression system on angiogenesis and growth of Lewis lung cancer in mice. BMC Cancer. 2012;12:155. doi: 10.1186/1471-2407-12-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato K, Odamaki T, Mitsuyama E, Sugahara H, Xiao JZ, Osawa R. Age-related changes in the composition of gut bifidobacterium species. Curr Microbiol. 2017;74:987–995. doi: 10.1007/s00284-017-1272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimizu K, Ogura H, Goto M, Asahara T, Nomoto K, Morotomi M, Yoshiya K, Matsushima A, Sumi Y, Kuwagata Y, Tanaka H, Shimazu T, Sugimoto H. Altered gut flora and environment in patients with severe SIRS. J Trauma. 2006;60:126–133. doi: 10.1097/01.ta.0000197374.99755.fe. [DOI] [PubMed] [Google Scholar]
- 27.Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, Zwahlen MC, Desiere F, Bork P, Delley M, Pridmore RD, Arigoni F. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc Natl Acad Sci U S A. 2002;99:14422–14427. doi: 10.1073/pnas.212527599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng Y, Chi R, Chen S, Ye H, Yuan J, Wang L, Zhai Y, Gao L, Zhang D, Hu L, Lv B, Long Y, Sun C, Yang X, Zou X, Chen C. Evaluation of clinically available renal biomarkers in critically ill adults: a prospective multicenter observational study. Crit Care. 2017;21:46. doi: 10.1186/s13054-017-1626-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Florindo RN, Souza VP, Manzine LR, Camilo CM, Marana SR, Polikarpov I, Nascimento AS. Structural and biochemical characterization of a GH3 beta-glucosidase from the probiotic bacteria Bifidobacterium adolescentis. Biochimie. 2018;148:107–115. doi: 10.1016/j.biochi.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 30.Ghouri YA, Richards DM, Rahimi EF, Krill JT, Jelinek KA, DuPont AW. Systematic review of randomized controlled trials of probiotics, prebiotics, and synbiotics in inflammatory bowel disease. Clin Exp Gastroenterol. 2014;7:473–487. doi: 10.2147/CEG.S27530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manzanares W, Lemieux M, Langlois PL, Wischmeyer PE. Probiotic and synbiotic therapy in critical illness: a systematic review and meta-analysis. Crit Care. 2016;19:262. doi: 10.1186/s13054-016-1434-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barko PC, McMichael MA, Swanson KS, Williams DA. The gastrointestinal microbiome: a review. J Vet Intern Med. 2018;32:9–25. doi: 10.1111/jvim.14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng TY, Liu GH. PCR denaturing gradient gel electrophoresis as a useful method to identify of intestinal bacteria flora in Haemaphysalis flava ticks. Acta Parasitol. 2017;62:269–272. doi: 10.1515/ap-2017-0034. [DOI] [PubMed] [Google Scholar]