

Abstract
Since its outbreak, the severe acute respiratory syndrome—coronavirus 2 (SARS‐CoV‐2) has impacted the quality of life and cost hundreds‐of‐thousands of lives worldwide. Based on its global spread and mortality, there is an urgent need for novel treatments which can combat this disease. To date, the 3‐chymotrypsin‐like protease (3CLpro), which is also known as the main protease, is considered among the most important pharmacological targets. The vast majority of investigated 3CLpro inhibitors are organic, non‐covalent binders. Herein, the use of inorganic, coordinate covalent binders is proposed that can attenuate the activity of the protease. ReI tricarbonyl complexes were identified that demonstrate coordinate covalent enzymatic inhibition of 3CLpro. Preliminary studies indicate the selective inhibition of 3CLpro over several human proteases. This study presents the first example of metal complexes as inhibitors for the 3CLpro cysteine protease.
Keywords: antiviral agents, bioinorganic chemistry, medicinal inorganic chemistry, protease inhibitor, SARS-CoV-2
The outbreak of the severe acute respiratory syndrome—coronavirus 2 has impacted the life quality and cost hundreds‐of‐thousands of lives worldwide. Herein, a novel approach using ReI tricarbonyl complexes as coordinate covalent inhibitors for the main protease is presented. These complexes target the active site cysteine residue and show selective inhibition of the main protease over several human proteases.
Introduction
Since its outbreak in December 2019 in Wuhan (Hubei, China), the severe acute respiratory syndrome—coronavirus 2 (SARS‐CoV‐2) has spread across the world. On 11th March, the World Health Organization (WHO) officially declared this pulmonary disease as a global pandemic. To date, more than 96 million cases have been reported with more than 2 million confirmed deaths, [1] creating an urgent need for the development of novel therapeutics.
The papain‐like protease (PLpro) and the 3‐chymotrypsin‐like protease (3CLpro), which is also known as the main protease, are considered among the most important viral targets for SARS‐CoV‐2. Clinical studies have indicated that infected patients treated with protease inhibitors have shown reduced symptoms and mortality. [2] The PLpro and 3CLpro proteases are responsible for the processing of the viral polyproteins pp. 1a and pp. 1b that mediate functions required for viral replication and transcription and are crucial for viral maturation and infectivity.[ 3 , 4 ] The inhibition of these proteases significantly disrupt the viral life cycle, presenting an important opportunity for therapeutic intervention. [5] The compound disulfiram, which is approved for chronic alcohol dependence, has been reported to inhibit the PLpro protease of SARS and is currently under investigation for SARS‐CoV‐2. [6] Recently, the use of Au complexes as potential therapeutics has been reported, including as PLpro inhibitors.[ 7 , 8 ] As inhibitors of the 3CLpro protease, the approved HIV therapeutics Lopinavir and Ritonavir are being studied in Phase III clinical trials for SARS‐CoV‐2. [9] In addition, a number of other organic compounds are under consideration in preclinical trials as inhibitors of these proteases.[ 10 , 11 , 12 , 13 ] Among these compounds, a handful of inhibitors have been identified that covalently bind to the catalytically active Cys145 amino acid.[ 13 , 14 , 15 ] Despite these contributions, irreversible Michael acceptors such as Rupintrivir have failed in clinical trials due to their low bioavailability, [16] in part due to reactions with off‐target biological thiols. To address this limitation, the generation of reversible Cys‐binding inhibitors has received increased attention in order to better target the Cys145 residue in the 3CLpro protease active site. [17]
Metal‐based coordination compounds can exhibit sophisticated 3‐dimensional (3D) shapes[ 18 , 19 , 20 , 21 , 22 , 23 , 24 ] and can be designed to have selective reactivity, providing a pathway to develop covalent enzyme inhibitors.[ 25 , 26 , 27 ] In this context, the use of 3D metal complexes as potential coordinate covalent inhibitors for the SARS‐CoV‐2 main protease 3CLpro is presented. A [Re(2,2′‐bipyridine)(CO)3]+ fragment was identified that could bind to the catalytically active Cys145 amino acid through a metal‐cysteine bond. The 2,2′‐bipyridine ligand of the fragment was derivatized with various functional groups and chloride and water axial capping ligands were examined to develop a rudimentary structure–activity relationship (SAR). The resulting complexes were synthesized, characterized, and their in vitro activity investigated. To the best of our knowledge, these are the first metal complexes reported as inhibitors for the SARS‐CoV‐2 main protease 3CLpro.
Results and Discussion
Rational Design
Previous studies by Fricker and co‐workers demonstrated that AuIII, PdII, and ReV complexes can inhibit the activity of the cysteine proteases Cathepsin B and K. These studies indicated that metal complexes can interact with catalytically active cysteine residues upon release of monodentate ligands to form coordinate covalent, but reversible, adducts.[ 28 , 29 ] These studies provided the motivation to examine a variety of metal complexes for activity against 3CLpro. Complexes of Pb, Bi, Te, Ni, Cu, Zn, Ru, Rh, Pd, Ag, Cd, Re, Pt, Au, and Hg were considered due to their high metal–sulfur bond enthalpy, especially in comparison to their metal‐oxygen or metal‐nitrogen bond enthalpy, which would be expected to produce high thiophilicity for Cys residues over other amino acid residues.[ 30 , 31 ] Coordination compounds of these metals that were generally associated with poorer biological compatibility were removed from further consideration, resulting in compounds based on Ru, Rh, Pd, Re, Pt, or Au as possible options. Subsequently, known classes of compounds (Figure S1) possessing one vacant coordination site to allow for a metal–Cys bond were then modelled inside the active site pocket of 3CLpro.
The molecular geometry of potential candidates (Figure S1) was determined using density‐functional theory (DFT) calculations; each candidate possessed a chlorine atom as a placeholder for the enzyme coordination site. The geometry of the calculated structures was verified by comparison with crystal structures of structurally related compounds from the Cambridge Crystallographic Data Centre (CCDC). For docking experiments, the chlorine atom was removed to yield the “active” fragment for enzyme binding which was further considered as a rigid body. The metal complex was then docked to the Cys residues found in 3CLpro. The enzyme possesses 12 Cys residues (Cys16, Cys22, Cys38, Cys44, Cys85, Cys117, Cys128, Cys145, Cys156, Cys160, Cys65, Cys300); however, the majority of these are buried inside the protein, with only three Cys (Cys85, Cys145, Cys156) surface accessible to fragments. After coordinating covalent docking of the complexes to these Cys residues, the binding pose was energetically minimized and scored using the GBVI/WSA dG force fields provided by MOE.
Among the modelled compounds, the fac‐Re(CO)3(NN) (NN=bidentate nitrogen‐donating ligand) fragment stood out as an attractive candidate (Table S1) that matched the protein architecture and could potentially react with the Cys145 active site residue (Figure 1, Figure S2). Among the docked binding poses, the fragment with the lowest energy was found to coordinate to Cys145 and not the other surface Cys residues. Previous studies have discussed the application of ReI tricarbonyl as anticancer agents, luminescent probes, or radio‐imaging agents, suggesting the biocompatibility of such compounds.[ 32 , 33 , 34 , 35 , 36 , 37 ] Additional docking studies were performed with the scaffold that possessed symmetric substituents in all positions of the 2,2′‐bipyridine ligand (vide infra). The docking pose of the ReI tricarbonyl complexes functionalized with polar groups demonstrated opportunities to interact with the protein active site by hydrogen bonding and hydrophobic interactions. With these docking studies in hand, a series of complexes with either chloride or water as a labile ligand were synthesized, characterized, and evaluated as 3CLpro inhibitors.
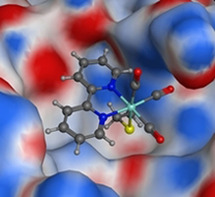
Figure 1.
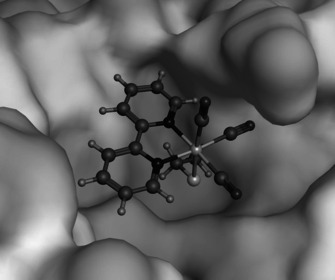
Docking pose of the [Re(2,2′‐bipyridine)(CO)3]+ fragment bound to the thiol of Cys145 in the active site of 3CLpro.
Synthesis and Characterization
The synthesis of complexes 1, 3–7, 10–12, 17–18, 20, 22, 27, and 32 have been previously described (see Supporting Information for details), while 2, 8–9, 13–16, 19, 21, 23–26, 28–31, and 33–42 have not been previously reported (Figure 2). The functionalized 2,2′‐bipyridine derivatives were prepared via literature procedures (see Supporting Information for details). The chloride capped ReI tricarbonyl complexes 1–21 were synthesized by complexation of pentacarbonylchlororhenium with the corresponding 2,2′‐bipyridine ligand. The aqua ReI tricarbonyl complexes 22–42 were prepared by treatment of the chloride coordinated complex with silver trifluoromethanesulfonate, followed by precipitation upon addition of water. The aqua complexes were found to generally have higher aqueous solubility then the corresponding chloride coordinated complexes. The identity of the resulting compounds was verified by NMR spectroscopy and high‐resolution mass spectrometry (HRMS). The purity of all compounds was confirmed by HPLC analysis (Figure S3–S7).
Figure 2.
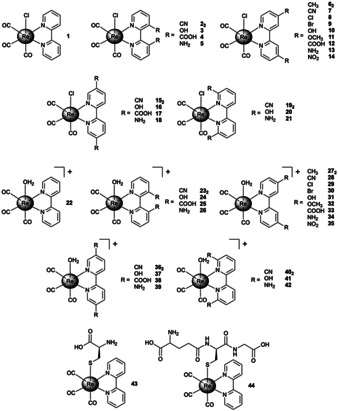
Chemical structure of ReI tricarbonyl complexes investigated in this study. The aqua coordinated complexes were isolated as triflate salts.
The molecular structure of several ReI tricarbonyl complexes was determined by single‐crystal X‐ray diffraction. The structures 1,[ 38 , 39 ] 6, [40] 7,[ 41 , 42 ] 10, [43] 11, [40] 17, [44] 18, [45] 20, [46] 27, [47] and 32 [47] have already been reported. The structures of compounds, 3, 8, 13, 14, 15, 19, 22, 37, and 42 are reported here (Figure 3, Table S2–S4). Although the structures of compounds 7, 11, and 20 have been reported elsewhere (as mentioned above), new structures of these compounds were obtained that possess different cell parameters reflective of solvent molecules used during the crystallization process. All of the structures show that a central ReI ion is bound to the bipyridine in a bidentate fashion, with the remaining ReI coordination sphere occupied by three carbonyl ligands and an axial chloride ion or water molecule. In the water bound complexes, triflate anions charge balance the overall monocationic charge of the complex. Interestingly, compound 3 is the first 3,3′‐disubstituted 2,2′‐bipyridine crystal structure using this ReI based scaffold and the bulkiness of the adjacent hydroxyl groups in the 3‐position resulted in distortion of the typically planar aromatic bipyridine rings to an out‐of‐plane motif. Moreover, the distorted planar angle resulting from steric bulk is also noteworthy in compounds 19 and 42 with 6,6′‐subsituted bipyridine ligands (Figure S8). In compound 19 the N1‐Re1‐Cl1 bond angle is 82.22(7)° and in compound 42 the N1‐Re1‐O1 bond angle is 79.52(12)° with deviations of 7.88° and 10.48° degrees from complete planarity (see Supporting Information for details). For several of the studies described below, the ReI tricarbonyl complexes with the unsubstituted 2,2′‐bipyridine ligand (chloride 1 and water 22 coordinated) were used as model compounds for each respective groups of compounds.
Figure 3.
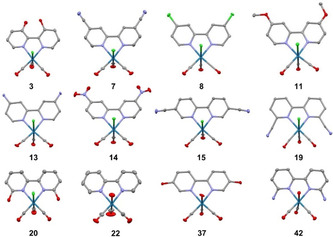
X‐ray crystal structures of ReI complexes 3, 7, 8, 11, 13, 14, 15, 19, 20, 22, 37, and 42 (50 % probability ellipsoids). Solvent, anions, and hydrogen atoms have been omitted for clarity. [57]
Stability
The stability of these compounds is an essential parameter for application as an enzyme inhibitor. [48] Representative complexes 1 and 22 were incubated in water or phosphate buffered saline (PBS) at 37 °C for 24 h in the dark and then analysed by HPLC. Compound 22 did not show any changes over the course of this experiment, while 1 showed slow hydrolysis of the chloride ligand, to yield complex 22 (Figures S9, S10). No other degradation products of these compounds were observed over this time period, suggesting that only the hydration of the chloride complex 1 needs to be considered in the context of the other experiments described herein.
Reactivity with Amino Acids
The interaction of the chloride (1) and aqua (22) ReI tricarbonyl complex with amino acids containing cationic (Nα‐p‐tosyl‐L‐arginine methyl ester, L‐histidine methyl ester, L‐lysine methyl ester), anionic (Nα‐tert‐butoxycarbonyl‐L‐aspartic acid tert‐butyl ester), polar (L‐serine methyl ester, L‐asparagine tert‐butyl ester), and sulphur (L‐cysteine methyl ester, L‐methionine methyl ester) side chains was investigated (Figure 4) using equimolar amounts of metal complex and amino acid. Previous studies have indicated that ReI tricarbonyl complexes are able to react with Cys derivatives[ 47 , 49 , 50 ] and a crystal structure has revealed that the Cys is covalently bound in axial position. [49]
Figure 4.
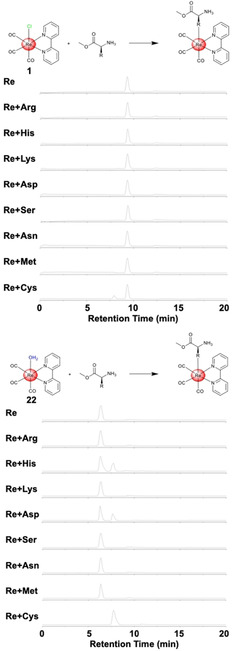
HPLC traces of the chloride 1 (top) and aqua 22 (bottom) ReI tricarbonyl complexes after exposure to amino acids for 24 h.
The ReI tricarbonyl complexes 1 and 22 were incubated in water at 37 °C with the corresponding amino acid for 24 h and analysed by HPLC. While 1 did not react with the majority of amino acids, it did react with L‐cysteine methyl ester, presumably due to the high thiophilicity of the ReI centre. In contrast, 22 showed reactivity toward the acidic Nα‐tert‐butoxycarbonyl‐l‐aspartic acid tert‐butyl ester and the basic L‐histidine methyl ester amino acids, but did not achieve full conversion over 24 h. Notably, the incubation of 22 with L‐cysteine methyl ester resulted in a single product peak with full conversion within 24 h. For additional insight into the kinetics of the capping group, the hydrolysis of the chloride ligand was investigated upon incubation of 1 in water and determination of the amount of hydrolysed product by HPLC in a time‐dependent manner. Based on this experiment, the difference in reactivity between 1 and 22 can be explained by the slow hydrolysis rate of the chloride ligand in 1 with a value of 0.355±0.051×10−3 s−1. Following this evaluation, the crude product of the incubation of 22 with L‐cysteine methyl ester was analysed by ESI‐MS confirming the generation of [Re(2,2′‐bipyridine)(L‐cysteine methyl ester)(CO)3] ([M+H]+ calcd for C17H17N3O5ReS: 562.0, found: 562.4). These results suggest that these compounds could serve as coordinate covalent inhibitors by binding to the catalytically active Cys145 in 3CLpro.
Following this assessment, the reaction of 22 with L‐cysteine methyl ester was further investigated. Using the carbonyl signals in the IR spectrum, the change of the coordination sphere of the ReI complex can be used to monitor its reactivity (Figure S11). As expected, the carbonyl signals from the starting material 22 (2047, 1935, 1905 cm−1) to [Re(2,2′‐bipyridine)(L‐cysteine methyl ester)(CO)3] (2050, 1930, 1900 cm−1) are shifted. Furthermore, additional peaks for the methyl ester (1719 cm−1) and the broad amine signal (2940 cm−1) stemming from the amino acid also appear. This interaction was also investigated by using UV/Vis absorption spectroscopy (Figure S11). Compound 22 and L‐cysteine methyl ester were mixed and the absorption spectrum was collected every 2 min for 120 min. The absorption spectrum shows immediate changes and within 30 min forms a new spectrum with the appearance of two isosbestic points, indicating a clean transformation of 22 to the Cys adduct. After 60 min, no further changes in the absorption spectrum was observed. The interaction of 22 with L‐cysteine methyl ester was also investigated by 1H NMR spectroscopy (Figure S11). Immediately after mixing of the two starting materials (≈5 min), a new set of signals was observed indicative of the formation of [Re(2,2′‐bipyridine)(L‐cysteine methyl ester)(CO)3]. While the aromatic protons of the bipyridine ligand were shifted high field, the β‐CH2 of the cysteine side chain and α‐CH protons were shifted slightly low field. Within 60 min, the signals for 22 completely disappeared, suggesting a full conversion to the amino acid metal complex.
The relatively rapid reactivity of 22 with thiol‐containing molecules could be potentially problematic due to the high concentration of glutathione in cells. The axial functionalized Cys (43) and glutathione (44) derivatives were synthesized by mixing of the corresponding thiol with 22 in water overnight (see Supporting Information for details). To study the reversibility of this reaction, 43 was incubated with equimolar amounts of glutathione and 44 with Cys at 37 °C. After 24 h, the generated products were analysed by HPLC. The bound thiol was exchanged in both cases, showing the reversibility of this reaction (Figure S12) and therefore the possibility of the ReI tricarbonyl complex to interact with Cys145 in 3CLpro despite the presence of other reactive biological thiols.
Biochemical Evaluation
After confirmation of the ability of the ReI tricarbonyl complexes to coordinate covalently bind to Cys derivatives, the binding to the SARS‐CoV‐2 3CLpro was investigated using ESI‐TOF MS. [51] While the native protein was found to have a deconvoluted mass of 33 797 (Figure S13), upon incubation with 22 for 2 h the mass shifted to 34 225 (Figure S14), corresponding to a single attached ReI tricarbonyl complex (m/z 427). To investigate if the metal complex was bound to the predicted cysteine residue, 3CLpro was first incubated with the well‐characterized inhibitor GC376, which covalently binds to Cys145 as confirmed by macromolecular X‐ray crystallography. [14] As expected, GC376 formed a new species (m/z 34 201) with a mass difference of m/z 404, corresponding to the correct mass for the GC376‐protein covalent adduct (Figure S15). Incubation of 3CLpro with GC376, followed by incubation with 22 resulted in only the formation of the GC376‐protein covalent adduct (m/z 34 201), with no mixture of adducts and no addition of the ReI tricarbonyl complex (Figure S16). These results strongly suggest that 22 is targeting the same residue as GC376, namely active site Cys145.
Following this initial assessment, the inhibition activity against the SARS‐CoV‐2 3CLpro was investigated. The 3CLpro protease was pre‐incubated with ReI compounds and enzyme inhibition was monitored by conversion of a non‐fluorescent substrate to a fluorescent product (see Supporting Information for details). Compounds 1–42 were screened against 3CLpro at a concentration of 200 μM (Figure 5). As expected, due to the slow release of the chloride atom, compounds 1–21 did not show significant inhibition activity. In contrast, all aqua compounds 22–42 were able to inhibit the activity of the protease, with remaining enzymatic activity reduced to ≈20–40 %. The aqua compounds were subsequently screened against 3CLpro at a lower concentration of 50 μM (Figure 5). Interestingly, the 4,4′‐ (27–35) and 5,5′‐ (36–39) substituted complexes generally displayed stronger inhibitory activity when compared to the 3,3′‐ (22–26) and 6,6′‐ (40–42) substituted complexes. However, none of the compounds displayed significantly greater activity than the unsubstituted parent compound 22. Overall, compounds 22, 31–34, 37–39 were identified as having the strongest inhibitory effect and were therefore studied further.
Figure 5.
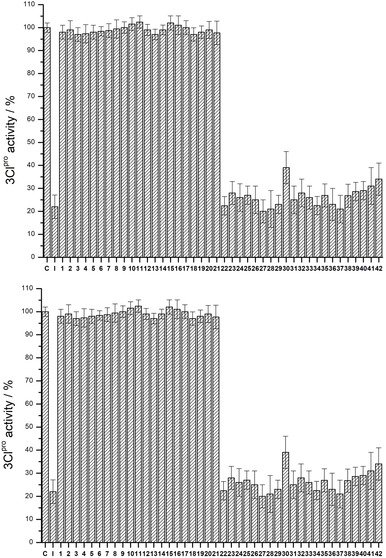
Enzyme activity assay of 3CLpro with 1–42 (C=control, no inhibitor; I=known covalent inhibitor GC376) at a concentration of: 200 μM (top) or 50 μM (bottom). Values and error bars are derived from three independent experiments.
The inhibition by the lead structures was quantified by determination of their IC50 values (Table 1). The complexes displayed IC50 values between 7.5–24.1 μM (Figure S17). The carboxylic acid functionalized compounds (33, 38) have the highest IC50 values while the amine functionalized compounds (34, 39) have the lowest IC50 values. This could be the result of these functional groups being directed at negatively charged surfaces inside the enzyme active site (Figure S18). Based on this hypothesis, the carboxylic acids on the ReI tricarbonyl complex would result in a repulsion with the protein surface, while the amine substituents would present attractive interactions, resulting in the observed differences in inhibition activity. Although only a preliminary SAR, the observation that preferred substitution patterns and functional groups on the bipyridine group can be identified suggests that more selective and active ReI compounds can be designed for inhibition of 3CLpro. Overall, 34 was identified as having the lowest IC50 value at 7.5±1.3 μM. To ensure that the intact coordination compound 34 is responsible for the inhibitory effect, the ligand 4,4′‐diamino‐2,2′‐bipyridine and the metal complex pentacarbonylchlororhenium were screened against 3CLpro. Both components showed no activity (IC50 value >100 μM), confirming that 34 is the active species.
Table 1.
Summary of the binding data of selected compounds against 3CLpro. Values and standard deviations are derived from three independent experiments.
|
Compound |
IC50 [μM] |
|---|---|
|
22 |
13.8±2.1 |
|
31 |
12.8±1.9 |
|
32 |
15.7±1.4 |
|
33 |
24.1±1.2 |
|
34 |
7.5±1.3 |
|
37 |
13.5±2.3 |
|
38 |
21.1±2.7 |
|
39 |
9.6±1.3 |
As glutathione and other biologically present thiols in cells could influence the activity of these compounds, the activity of the Cys 43 and glutathione 44 functionalized derivatives towards 3CLpro was tested. The complexes were found to display slightly mitigated activity compared to many of the aqua complexes (43, IC50=20.8±3.6 μM; 44, IC50=26.7±4.1 μM). A hallmark of covalent inhibitors is time‐dependent inhibition; therefore, 1 (axial Cl), 22 (axial H2O), 43 (axial Cys), and 44 (axial glutathione) derivatives (at a concentration of 20 μM) were pre‐incubated with 3CLpro for various time periods (30, 60, 120, and 240 min) and the remaining enzyme activity was measured (Figure S19). While 1 demonstrated no inhibition and 22 showed near complete inhibition over this time period, 43 and 44 showed a gradual reduction of 3CLpro activity as a function of time. Within 240 min, 43 and 44 had reduced enzymatic activity to the same level as compound 22, indicating that these axial thiols can be exchanged, acting as reversible, coordinate covalent inhibitors. Such thiol substituted ReI tricarbonyl complexes could even serve as a type of prodrug for this class of coordinate covalent inhibitors.
To further examine the effect of glutathione on inhibitory activity, an enzyme inhibition assay with compound 34 was performed with a 30 min pre‐incubation with 1 mM glutathione (which is comparable to levels of biological thiols), followed by a 30 min incubation with 3CLpro. As expected, the activity of the 34 was reduced, possessing an IC50 value of 13.6±2.8 μM; however, this represents only ≈2‐fold loss of activity. Following this, 34 was again pre‐incubated for 30 min with 1 mM glutathione followed by a 240 min incubation with 3CLpro. Interestingly, the inhibition by compound 34 with this extended incubation time showed an IC50 value of 9.1±1.8 μM, which is only slightly changed in comparison to the standard assay without addition of glutathione (Table 1). By comparison, the activity of the anticancer drug cisplatin is also reduced in the presence of glutathione, [52] and hence the reduced reactivity here is both expected and not an insurmountable impediment to further drug development.
Compound 34 was further investigated using a thermal shift assay as an orthogonal technique for evidence of enzyme binding. Compound 34 was incubated with 3CLpro for 30 min at 37 °C and the melting temperature of the enzyme measured. While 4,4′‐diamino‐2,2′‐bipyridine and the metal complex pentacarbonylchlororhenium showed only a negligible effect (ΔT=0.2±0.4), incubation with 34 gave a slight increase in the melting temperature of 3CLpro (ΔT=2.4±0.6), indicative of inhibitor binding. Interestingly, the change in melting temperature was found to be in the same range as for the known inhibitor GC376 (ΔT=3.1±0.5). [53] The binding to 3CLpro was further investigated by UV/Vis absorption spectroscopy (Figure S20). Compound 34 and 3CLpro were combined and their absorption properties monitored every 2 min for 120 min. While no changes in the absorption spectra for 34 and 3CLpro alone were observed under these conditions, the combined mixture of both showed changes. Within 30 min, an asymptotic absorption pattern was reached which showed no further changes after 60 min, suggestive of inhibitor binding (Figure S20).
As a means to characterize the binding affinity of the metal complex to the enzyme independently of intermediates, the ratio of the second reaction rate constant (k 2) in dependence of inhibition constant (K i) was determined according to previously published procedures. [54] Lead compound 34 was incubated with 3CLpro and the activity of the enzyme was monitored in a time‐dependent manner. The metal complex was found to slowly bind to 3CLpro with a ratio of k 2/K i of 127 M−1 s−1, which is two orders of magnitudes slower than the organic, covalent inhibitor GC376. [54] GC376 is a more potent inhibitor (IC50=0.19±0.04 μM) than 34, which is consistent with difference in aforementioned k 2/K i values. In addition, as found with compound 22, incubation of compound 34 with 3CLpro produced in an adduct that could be identified by mass spectrometry (see above). As expected, the deconvoluted mass peak of 3CLpro shifted to 34 255 (Figure S21) corresponding to a single attached ReI tricarbonyl complex 34 (m/z 457).
Covalent 3CLpro inhibitors could also bind and inhibit cathepsins, especially cathepsin B, resulting in undesired side reactions and off‐target effects. [55] Inhibition of cathepsins is particularly relevant because they are found in the respiratory system, where SARS‐CoV‐2 would infect. [56] To investigate this potential shortcoming, cathepsin B was incubated with 34 for 2 h followed by analysis by mass spectrometry. While matrix‐assisted laser ionization‐time of flight mass spectrometry (MALDI‐TOF‐MS) identified the covalent adduct of 34 with 3CLpro (Figure S22), no adduct formation was observed between 34 and cathepsin B (Figure S23), indicative of some selectivity of 34 towards 3CLpro over this cysteine protease. To further evaluate selectivity, the activity of 34 against the human serine protease dipeptidyl peptidase‐4 (DPP4), the aspartate protease beta‐secretase 1 (BACE1) and cysteine protease cathepsin B was measured in an inhibition assay (Figure 6). Encouragingly, compound 34 did not show measurable activity (IC50 value >100 μM) towards DPP4 and cathepsin B, and showed only weak inhibition of BACE1 (IC50 value=89.2±5.7 μM). These preliminary studies indicate that selective inhibition of 3CLpro can be achieved over several human proteases.
Figure 6.
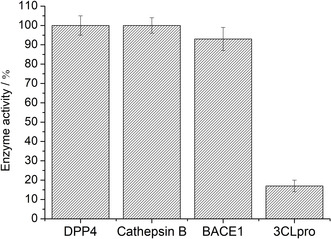
Enzymatic activity of the lead compound 34 at a concentration of 50 μM towards serine protease DPP4, aspartate protease BACE1, cysteine protease cathepsin B, and cysteine protease 3CLpro.
Conclusion
In summary, the synthesis and biophysical evaluation of ReI tricarbonyl complexes as inhibitors of the SARS‐CoV‐2 main protease 3CLpro has been achieved. A series of ReI complexes with chloride and water as capping ligands and differently substituted 2,2′‐bipyridine ligands were prepared. While the coordinated chloride was only slowly released, the aqua complex 22 showed reactivity towards amino acids within one hour. Mass spectrometry experiments verified the coordinate covalent binding of a single ReI tricarbonyl complex to 3CLpro. Using an enzymatic assay against 3CLpro, several complexes were found to be active, with IC50 values of <10 μM. Preliminary investigations show selectivity against human proteases. These results suggest that ReI tricarbonyl complexes can serve as a starting scaffold for the development of potent, selective SARS‐CoV‐2 inhibitors.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors acknowledge the help of Ryjul W. Stokes with the measurement of NMR spectra and Dr. Yongxuan Su (UCSD, Molecular Mass Spectrometry Facility) with mass spectrometry. M.K. is supported by the Department of Defense (DoD) through the National Defense Science and Engineering Graduate (NDSEG) Fellowship Program is the recipient of an Achievement Rewards for College Scientists (ARCS) Foundation Fellowship. This work was supported by a grant from the National Institutes of Health (R21 AI138934).
J. Karges, M. Kalaj, M. Gembicky, S. M. Cohen, Angew. Chem. Int. Ed. 2021, 60, 10716.
References
- 1. https://coronavirus.jhu.edu/map.html, last accessed 20.01.2021.
- 2. Chu C. M., Cheng V. C. C., Hung I. F. N., Wong M. M. L., Chan K. H., Chan K. S., Kao R. Y. T., Poon L. L. M., Wong C. L. P., Guan Y., Peiris J. S. M., Yuen K. Y., Thorax 2004, 59, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu I. L., Mahindroo N., Liang P.-H., Peng Y.-H., Kuo C.-J., Tsai K.-C., Hsieh H.-P., Chao Y.-S., Wu S.-Y., J. Med. Chem. 2006, 49, 5154–5161. [DOI] [PubMed] [Google Scholar]
- 4. Gorbalenya A. E., Donchenko A. P., Blinov V. M., Koonin E. V., FEBS Lett. 1989, 243, 103–114. [DOI] [PubMed] [Google Scholar]
- 5. Chen Y., Liu Q., Guo D., J. Med. Virol. 2020, 92, 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin M.-H., Moses D. C., Hsieh C.-H., Cheng S.-C., Chen Y.-H., Sun C.-Y., Chou C.-Y., Antiviral Res. 2018, 150, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gil-Moles M., Basu U., Büssing R., Hoffmeister H., Türck S., Varchmin A., Ott I., Chem. Eur. J. 2020, 26, 15140–15144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marzo T., Messori L., ACS Med. Chem. Lett. 2020, 11, 1067–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan J. F.-W., Yao Y., Yeung M.-L., Deng W., Bao L., Jia L., Li F., Xiao C., Gao H., Yu P., Cai J.-P., Chu H., Zhou J., Chen H., Qin C., Yuen K.-Y., J. Infect. Dis. 2015, 212, 1904–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Galasiti Kankanamalage A. C., Kim Y., Damalanka V. C., Rathnayake A. D., Fehr A. R., Mehzabeen N., Battaile K. P., Lovell S., Lushington G. H., Perlman S., Chang K.-O., Groutas W. C., Eur. J. Med. Chem. 2018, 150, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karypidou K., Ribone S. R., Quevedo M. A., Persoons L., Pannecouque C., Helsen C., Claessens F., Dehaen W., Bioorg. Med. Chem. Lett. 2018, 28, 3472–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu R.-J., Zhou K.-X., Yang H., Song G.-Q., Li Y.-H., Fu J.-X., Zhang X., Yu S.-J., Wang L.-Z., Xiong L.-X., Niu C.-W., Song F.-H., Yang H., Wang J.-G., Eur. J. Med. Chem. 2019, 167, 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R., Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vuong W., Khan M. B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H. A., McKay R. T., van Belkum M. J., Joyce M. A., Young H. S., Tyrrell D. L., Vederas J. C., Lemieux M. J., Nat. Commun. 2020, 11, 4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L. W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H., Nature 2020, 582, 289–293. [DOI] [PubMed] [Google Scholar]
- 16. Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H., J. Med. Chem. 2016, 59, 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malcolm B. A., Lowe C., Shechosky S., McKay R. T., Yang C. C., Shah V. J., Simon R. J., Vederas J. C., Smith D. V., Biochemistry 1995, 34, 8172–8179. [DOI] [PubMed] [Google Scholar]
- 18. Bregman H., Carroll P. J., Meggers E., J. Am. Chem. Soc. 2006, 128, 877–884. [DOI] [PubMed] [Google Scholar]
- 19. Feng L., Geisselbrecht Y., Blanck S., Wilbuer A., Atilla-Gokcumen G. E., Filippakopoulos P., Kräling K., Celik M. A., Harms K., Maksimoska J., Marmorstein R., Frenking G., Knapp S., Essen L.-O., Meggers E., J. Am. Chem. Soc. 2011, 133, 5976–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morrison C. N., Prosser K. E., Stokes R. W., Cordes A., Metzler-Nolte N., Cohen S. M., Chem. Sci. 2020, 11, 1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anthony E. J., Bolitho E. M., Bridgewater H. E., Carter O. W. L., Donnelly J. M., Imberti C., Lant E. C., Lermyte F., Needham R. J., Palau M., Sadler P. J., Shi H., Wang F.-X., Zhang W.-Y., Zhang Z., Chem. Sci. 2020, 10.1039/D1030SC04082G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karges J., ChemBioChem 2020, 21, 3044–3046. [DOI] [PubMed] [Google Scholar]
- 23. Romero-Canelón I., Sadler P. J., Proc. Natl. Acad. Sci. USA 2015, 112, 4187–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hartinger C. G., Metzler-Nolte N., Dyson P. J., Organometallics 2012, 31, 5677–5685. [Google Scholar]
- 25. Ghosh A. K., Samanta I., Mondal A., Liu W. R., ChemMedChem 2019, 14, 889–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh J., Petter R. C., Baillie T. A., Whitty A., Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- 27. Rausch M., Dyson P. J., Nowak-Sliwinska P., Adv. Ther. 2019, 2, 1900042. [Google Scholar]
- 28. Fricker S. P., Mosi R. M., Cameron B. R., Baird I., Zhu Y., Anastassov V., Cox J., Doyle P. S., Hansell E., Lau G., Langille J., Olsen M., Qin L., Skerlj R., Wong R. S. Y., Santucci Z., McKerrow J. H., J. Inorg. Biochem. 2008, 102, 1839–1845. [DOI] [PubMed] [Google Scholar]
- 29. Fricker S. P., Metallomics 2010, 2, 366–377. [DOI] [PubMed] [Google Scholar]
- 30. Kepp K. P., Inorg. Chem. 2016, 55, 9461–9470. [DOI] [PubMed] [Google Scholar]
- 31. Haynes W. M., CRC Handbook of Chemistry and Physics, CRC, Boca Raton, 2014. [Google Scholar]
- 32. Marker S. C., King A. P., Granja S., Vaughn B., Woods J. J., Boros E., Wilson J. J., Inorg. Chem. 2020, 59, 10285–10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Konkankit C. C., King A. P., Knopf K. M., Southard T. L., Wilson J. J., ACS Med. Chem. Lett. 2019, 10, 822–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Philippe C., Didier D., Veena V., Curr. Pharm. Des. 2019, 25, 3306–3322.31475892 [Google Scholar]
- 35. Capper M. S., Packman H., Rehkämper M., ChemBioChem 2020, 21, 2111–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Raszeja L. J., Siegmund D., Cordes A. L., Güldenhaupt J., Gerwert K., Hahn S., Metzler-Nolte N., Chem. Commun. 2017, 53, 905–908. [DOI] [PubMed] [Google Scholar]
- 37. Muñoz-Osses M., Godoy F., Fierro A., Gómez A., Metzler-Nolte N., Dalton Trans. 2018, 47, 1233–1242. [DOI] [PubMed] [Google Scholar]
- 38. Kurz P., Probst B., Spingler B., Alberto R., Eur. J. Inorg. Chem. 2006, 2966–2974. [Google Scholar]
- 39. Towrie M., Parker A. W., Ronayne K. L., Bowes K. F., Cole J. M., Raithby P. R., Warren J. E., Appl. Spectrosc. 2009, 63, 57–65. [DOI] [PubMed] [Google Scholar]
- 40. Smieja J. M., Kubiak C. P., Inorg. Chem. 2010, 49, 9283–9289. [DOI] [PubMed] [Google Scholar]
- 41. Coe B. J., Foxon S. P., Pilkington R. A., Sánchez S., Whittaker D., Clays K., Van Steerteghem N., Brunschwig B. S., Organometallics 2016, 35, 3014–3024. [Google Scholar]
- 42. Clark M. L., Rudshteyn B., Ge A., Chabolla S. A., Machan C. W., Psciuk B. T., Song J., Canzi G., Lian T., Batista V. S., Kubiak C. P., J. Phys. Chem. C 2016, 120, 1657–1665. [Google Scholar]
- 43. Manbeck G. F., Muckerman J. T., Szalda D. J., Himeda Y., Fujita E., J. Phys. Chem. B 2015, 119, 7457–7466. [DOI] [PubMed] [Google Scholar]
- 44. Bhat G. A., Rashad A. Z., Folsom T. M., Darensbourg D. J., Organometallics 2020, 39, 1612–1618. [Google Scholar]
- 45. Popov D. A., Luna J. M., Orchanian N. M., Haiges R., Downes C. A., Marinescu S. C., Dalton Trans. 2018, 47, 17450–17460. [DOI] [PubMed] [Google Scholar]
- 46. Lense S., Piro N. A., Kassel S. W., Wildish A., Jeffery B., Acta Crystallogr. Sect. E 2016, 72, 1201–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Knopf K. M., Murphy B. L., MacMillan S. N., Baskin J. M., Barr M. P., Boros E., Wilson J. J., J. Am. Chem. Soc. 2017, 139, 14302–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Keller S., Ong Y. C., Lin Y., Cariou K., Gasser G., J. Organomet. Chem. 2020, 906, 121059. [Google Scholar]
- 49. Capper M. S., Enriquez Garcia A., Macia N., Lai B., Lin J.-B., Nomura M., Alihosseinzadeh A., Ponnurangam S., Heyne B., Shemanko C. S., Jalilehvand F., J. Biol. Inorg. Chem. 2020, 25, 759–776. [DOI] [PubMed] [Google Scholar]
- 50. Lecina J., Palacios Ò., Atrian S., Capdevila M., Suades J., J. Biol. Inorg. Chem. 2015, 20, 465–474. [DOI] [PubMed] [Google Scholar]
- 51. Steel T. R., Hartinger C. G., Metallomics 2020, 12, 1627–1636. [DOI] [PubMed] [Google Scholar]
- 52. Chen H. H. W., Kuo M. T., Met.-Based Drugs 2010, 2010, 430939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim Y., Liu H., Galasiti Kankanamalage A. C., Weerasekara S., Hua D. H., Groutas W. C., Chang K.-O., Pedersen N. C., PLOS Pathog. 2016, 12, e1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma C., Sacco M. D., Hurst B., Townsend J. A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M. T., Chen Y., Wang J., Cell Res. 2020, 30, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Steuten K., Kim H., Widen J. C., Babin B. M., Onguka O., Lovell S., Bolgi O., Cerikan B., Cortese M., Muir R. K., Bennett J. M., Geiss-Friedlander R., Peters C., Bartenschlager R., Bogyo M., BioRxiv 2020, 10.1101/2020.11.21.392753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lukassen S., Chua R. L., Trefzer T., Kahn N. C., Schneider M. A., Muley T., Winter H., Meister M., Veith C., Boots A. W., Hennig B. P., Kreuter M., Conrad C., Eils R., EMBO J. 2020, 39, e105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Deposition Numbers 2026146, 2026147, 2026148, 2026149, 2026150, 2026151, 2026152, 2026153, 2026154, 2026155, 2026156 and 2026157 (for 3, 7, 8, 11, 13, 14, 15, 19, 20, 22, 37, and 42, respectively) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary