

Abstract
Background
Traumatic injury is associated with increased concentrations of cell-free DNA (cfDNA) in the circulation, which contribute to post-injury complications. The endonuclease deoxyribonuclease 1 (DNase-1) is responsible for removing 90% of circulating cfDNA. Recently, DNase activity was reported to be significantly reduced following major non-traumatic brain injury (TBI), but the processes responsible were not investigated. Moreover, it is not known how quickly following injury DNase activity is reduced and whether this also occurs after TBI.
Methods
At 3 post-injury time points (≤1, 4–12 and 48–72 hours), blood samples were obtained from 155 adult trauma patients that had sustained an isolated TBI (n = 21), TBI with accompanying extracranial injury (TBI+) (n = 53) or an extracranial injury only (ECI) (n = 81). In addition to measuring cfDNA levels and the activity and expression of DNase, circulating concentrations of monomeric globular action (G-actin), an inhibitor of DNase-1, and the actin scavenging proteins gelsolin (GSN) and vitamin D binding protein (VDBP) were determined and values compared to a cohort of healthy controls.
Results
Significantly elevated concentrations of plasma cfDNA were seen in TBI, TBI+ and ECI patients at all study time points when compared to healthy controls. cfDNA levels were significantly higher at ≤1 hour post-injury in ECI patients who subsequently developed multiple organ dysfunction syndrome when compared to those who did not. Plasma DNase-1 protein was significantly elevated in all patient groups at all sampling time points. In contrast, DNase enzyme activity was significantly reduced, with this impaired function evident in TBI+ patients within minutes of injury. Circulating concentrations of G-actin were elevated in all patient cohorts in the immediate aftermath of injury and this was accompanied by a significant reduction in the levels of GSN and VDBP.
Conclusions
The post-traumatic increase in circulating cfDNA that occurs following extracranial trauma and TBI is accompanied by reduced DNase activity. We propose that, secondary to reduced GSN and VDBP levels, elevated circulating concentrations of G-actin underlie the post-injury reduction in DNase activity. Reducing circulating cfDNA levels via therapeutic restoration of DNase-1 activity may improve clinical outcomes post-injury.
Keywords: Cell-free DNA, Deoxyribonuclease, Extracellular actin scavenging system, Pre-hospital, Trauma
Highlights.
Via the analysis of blood samples acquired from major trauma patients within 1 hour of injury, this study is the first to examine the immediate impact of trauma on the activity of circulating deoxyribonuclease.
Following isolated traumatic brain injury, we detected a very rapid post-injury reduction in deoxyribonuclease activity that was accompanied by elevated concentrations of circulating cell-free DNA.
The post-injury impairment in deoxyribonuclease activity occurred secondary to elevated levels of monomeric globular actin, an inhibitor of deoxyribonuclease, and reduced concentrations of the actin scavengers gelsolin and vitamin D binding protein.
Significantly higher levels of cell-free DNA were detected in plasma samples acquired within 1 hour of injury from extracranial trauma patients who subsequently developed multiple organ dysfunction syndrome when compared to those with a non-eventful outcome.
Reducing circulating cell-free DNA levels via therapeutic restoration of deoxyribonuclease activity may improve clinical outcomes post-injury.
Background
Traumatic injury results in raised levels of cell-free DNA (cfDNA) in the circulation. Released from damaged/necrotic tissue, and as a by-product of immune cell activation [1–3], significantly increased concentrations of mitochondrial-derived DNA (mtDNA) [3–8] and nuclear-derived DNA (nDNA) [4–6, 9–14] have been measured in blood samples acquired from trauma patients with different mechanisms of injury, such as traumatic brain injury (TBI) [6, 9, 10, 13, 15], blunt trauma [4, 5, 12] and burns [11, 16, 17]. Attributed, in part, to its pro-thrombotic and cytotoxic nature [18–22], elevated cfDNA levels post trauma have been linked to a number of adverse clinical outcomes, which include the development of acute respiratory distress syndrome (ARDS) [23], multiple organ dysfunction syndrome (MODS) [8, 14, 24], multiple organ failure [14] and death [4, 7, 9, 10, 12, 13]. However, in spite of its potential impact on patient outcomes, the mechanisms behind the post-trauma elevation in circulating cfDNA are still uncertain.
Prospective observational cohort studies of trauma patients have shown that, relative to healthy controls (HCs), circulating cfDNA concentrations remain significantly elevated in the days, weeks and months following injury [5, 11, 13, 14, 25, 26]. With the half-life of DNA in circulation estimated to be less than 2 hours [27, 28], the persistent elevation in cfDNA levels post-injury could be a consequence of sustained production, triggered by secondary events such as surgical intervention [5, 29, 30] and/or its impaired breakdown and clearance. As the predominant endonuclease in bodily fluids, deoxyribonuclease 1 (DNase-1) is responsible for the breakdown of 90% of circulating cfDNA [31]. With the exception of a significant increase on the day of injury, major trauma has no effect on the circulating concentration of DNase-1 protein [25]. However, a persistent post-injury impairment in DNase activity has been described. Analysing plasma samples from 103 trauma patients with major extracranial injuries, McIlroy et al. reported elevated mtDNA levels and significantly reduced DNase activity in the circulation [26]. Although the first study to report a trauma-induced reduction in the activity of this endonuclease, the authors did not investigate the mechanism(s) involved, nor did they determine whether this reduction in DNase activity occurred in cohorts of trauma patients with different injuries, such as TBI.
DNase-1 activity is negatively regulated by actin, a cytoskeletal protein that exists in 2 forms: monomeric globular actin (G-actin) and filamentous actin (F-actin). Released into the circulation as a result of cell turnover, damage or death, G-actin forms a stable 1:1 stoichiometric complex with DNase-1, inhibiting its nuclease activity [32–34]. Facilitating the removal of actin from the circulation is the extracellular actin scavenger system (EASS), which comprises 2 plasma proteins: vitamin D binding protein (VDBP) and gelsolin (GSN). Mechanistically, GSN binds to and drives the depolymerization of F-actin, generating monomeric G-actin subunits that are bound by VDBP to form complexes that are cleared by the liver, spleen and kidneys [35–37]. The importance of the EASS for the breakdown of cfDNA is demonstrated by reports that GSN-mediated dissociation of actin from DNase-1 enhances its endonuclease activity [38].
Traumatic injury is associated with dysregulation of the EASS. Demonstrating the immediate post-injury release of actin into circulation, increased frequencies of actin–VDBP complexes have been detected in hospital admission blood samples of trauma patients [39], who also present with immediate and sustained reductions in circulating concentrations of VDBP [39, 40] and GSN [41–44]. However, to date, no single study has measured cfDNA, DNase activity/expression and components of the EASS in trauma patients. Moreover, it is currently unknown whether DNase activity is impaired in the immediate aftermath of trauma, and whether injuries other than extracranial trauma result in reduced activity of this endonuclease.
The aim of this prospective observational cohort study was to examine cfDNA levels, the expression and activity of DNase and the circulating concentrations of GSN and VDBP in blood samples acquired during the immediate (≤1 hour) and acute (4–72 hours) post-injury phase from patients that had sustained an isolated TBI, TBI with accompanying extracranial injury (TBI+) or an extracranial injury (ECI) only.
Methods
Study design
This article reports data acquired from subjects enrolled in the Brain Biomarkers after Trauma Study between 15 May 2014 and 26 August 2018. This study is an ongoing prospective longitudinal observational study of adult trauma patients conducted at a single major trauma centre in the UK (University Hospitals Birmingham National Health Service (NHS) Foundation Trust, Birmingham). Ethical approval for the study was granted by the North Wales Research Ethics Committee—West (REC reference: 13/WA/0399, protocol number: RG_13–164). Due to the nature of their injuries, patients were unlikely to be able to provide informed consent to enroll in the study. As such, patient recruitment into the study was performed under the guidance of the Mental Health Capacity Act for research in emergency situations, in accordance with the Declaration of Helsinki. Details on patient selection, study enrolment and the consenting procedure for patients who lacked capacity have been described previously [2]. A total of 75 adults (mean age, 41 years; range, 18–78 years; 47 male) were enrolled as a cohort of HCs (Table 1).
Table 1.
Patient Demographics
| Variables | HCs (n = 75) | TBI (n = 21) | TBI + (n = 53) | ECI (n = 81) |
|---|---|---|---|---|
| Age, years | 41 (18-78) | 41 (19-81) | 42 (18-87) | 40 (18-95) |
| Male, n (%) | 47 (63) | 18 (86) | 43 (81) | 73 (90) |
| Time to pre-hospital | - | 44 (26-60) | 42 (13-60) | 40 (18-60) |
| sample, minutes ISS |
- |
25 (9-45) |
34 (9-66) |
18 (9-75) |
| NISS | - | 48 (19-75) | 49 (12-75) | 25 (9-75) |
| GCS | - | 7 (3-15) | - | - |
| Mechanism of Injury | ||||
| Fall, n (%) | - | 9 (43) | 10 (19) | 10 (12) |
| A/P, n (%) | - | 3 (14) | 0 | 33 (41) |
| Blunt, n (%) | - | 1 (5) | 0 | 5 (6) |
| RTC, n (%) | - | 8 (38) | 43 (81) | 33 (41) |
| Outcomes | ||||
| ICU-free days | - | 21 (0-30) | 15 (0-30) | 25 (0-30) |
| Hospital free days | - | 12 (0-29) | 4 (0-26) | 15 (0-29) |
| MODS, n (%) | - | 11 (52) | 35 (66) | 17 (21) |
| Mortality, n (%) | - | 4 (19) | 15 (28) | 5 (6) |
Data are expressed as mean (range) unless otherwise stated
For TBI, TBI+ and ECI patient groups, the number of data points for the clinical variables ISS and NISS are 19, 52 and 73 respectively
ICU-free days and hospital-free days were calculated as 30 minus the number of days the patient stayed in the respective settings. Patients who died in ICU within 30 days of hospital admission were assigned a hospital and ICU-free day score of 0
HCs healthy controls, TBI traumatic brain injury, TBI+ TBI with accompanying extracranial injury, ECI extracranial injury, A/P assault/penetrating, GCS Glasgow coma scale, ICU intensive care unit, ISS Injury Severity Score, MODS multiple organ dysfunction syndrome, NISS new injury severity score, RTC road traffic collision
Clinical outcomes
The patient outcomes assessed in this study were MODS (defined as a Sequential Organ Failure Assessment (SOFA) score of ≥6 on 2 or more consecutive days, at least 48 hours post admission) and hospital-free and intensive care unit (ICU)-free days, which were calculated as 30 minus the number of days the patient stayed in hospital and ICU, respectively. Patients who died in the hospital or ICU setting within 30 days of admission were assigned a score of 0.
Clinical data collection
Patient and injury details were collected prospectively from contemporaneous electronic and physical medical records for included patients. These included age, gender, mechanism of injury, severity of injury (Injury Severity Score (ISS) and New ISS (NISS)), and Glasgow Coma Scale (GCS). Data required for SOFA (and MODS) assessment included ventilation status, PaO2, FiO2, mean arterial pressure and bilirubin, platelet and creatinine concentrations. The assigned SOFA scores for each day were taken as the highest score for that day.
Blood sampling
The blood samples were collected in BD Vacutainers® (BD Biosciences, UK) containing z-serum clotting activator or a 1/10 volume of 3.2% trisodium citrate at 3 post-injury time points pre-hospital (≤1-hour), 4–12 and 48–72 hours. For samples acquired in the pre-hospital setting, Vacutainers were stored at room temperature (RT) during transportation to hospital where, upon arrival, they were stored at 4°C and collected for analysis within 1 hour by a single laboratory researcher on a 24-hour basis.
Injury classification
Patients were divided into groups within the study based on their initial clinical and radiological assessments. In brief, radiological assessment consisted of a non-contrast head CT followed by a contrast-enhanced CT (with biphasic injection of contrast) from the circle of Willis to the level of the greater trochanters. These results, along with secondary and tertiary patient examinations, were used by a minimum of 2 independent clinicians to determine whether patients were classified as having suffered an isolated TBI, a TBI with accompanying ECI or an ECI only.
Preparation of platelet-free plasma (PFP) and serum
PFP was generated by a two-step centrifugation process. Citrate anticoagulated blood was centrifuged at 2000 × g for 20 minutes at 4°C, after which the top two-thirds of platelet-poor plasma was carefully removed. Following centrifugation of platelet-poor plasma at 13,000 × g for 2 minutes at 4°C, PFP was collected and stored at −80°C until analysed. To prepare serum, blood samples collected into Vacutainers containing z-serum clotting activator were incubated at RT for 30 minutes before centrifugation at 1620 × g for 10 minutes at RT. Serum aliquots were stored at −80°C prior to analysis.
Fluorometric analysis of cfDNA
cfDNA levels in PFP were measured using a fluorometric-based assay. A sample of 10 μL of PFP was incubated for 10 minutes in the dark at RT with 1 μM SYTOX green dye (Life Technologies, UK), after which fluorescence was measured using a BioTek Synergy 2 fluorometric plate reader (NorthStar Scientific Ltd, UK). with excitation and emission set at 485 nm and 528 nm, respectively. For quantification, a λ-DNA standard curve (Fisher Scientific, UK) was used. PFP samples were run in duplicate, with cfDNA concentrations calculated from the average value via extrapolation from the standard curve.
DNase activity
Neutrophil extracellular traps (NETs) served as the DNA substrate for our studies of DNase activity in serum [45]. To generate NETs, neutrophils (5 × 105) isolated from ethylenediaminetetraacetic acid anticoagulated blood by Percoll density gradient centrifugation were dispensed into wells of a 96-well flat-bottomed plate (BD Biosciences, UK) and stimulated for 3 hours (37°C/5% CO2) with 25 nM phorbol 12-myristate 13-acetate (Sigma-Aldrich, UK). Post stimulation, NETs were incubated for 6 hours (37°C/5% CO2) with 5% patient sera diluted in Hank’s balanced salt solution supplemented with calcium and magnesium (Gibco, Life Technologies, UK), or in Hank’s balanced salt solution supplemented with calcium and magnesium alone. Following a 30-minute fixation with 5% paraformaldehyde (Sigma-Aldrich, UK) (37°C/5% CO2), samples were washed 3 times with phosphate-buffered saline (Sigma-Aldrich, UK) prior to a 10-minute incubation at RT in the dark with 1 μM SYTOX green dye. Fluorescence was measured using a BioTek Synergy 2 fluorometric plate reader with excitation and emission set at 485 nm and 528 nm, respectively. NET degradation by 5% serum pooled from 10 HCs was used to define 100% DNase activity.
Enzyme-linked immunosorbent assay (ELISA)
Performed in accordance with the instructions of the manufacturers, serum concentrations of GSN (LifeSpan BioSciences Inc., UK) and G-actin (MyBioSource, USA) were quantified using ELISAs. PFP samples were analysed to determine the concentrations of circulating DNase-1 (LifeSpan BioSciences Inc, UK) and VDBP (Abcam, UK).
Lactate dehydrogenase (LDH) activity assay
LDH activity in 5-μL serum samples was quantified using the LDH activity assay kit (Sigma-Aldrich, UK) according to the manufacturer’s instructions.
Data analysis
Statistical analyses were performed using GraphPad Prism software version 5 (GraphPad Software Ltd, USA) and R (v3.6.1, https://www.R-project.org). Data distributions were examined using the Shapiro–Wilk normality test. For the comparison of HC and patient data, either the Mann–Whitney U test or the unpaired t-test with a Bonferroni correction for multiple comparisons was performed. Differences in cfDNA concentrations at each sampling time point between patients who did or did not develop MODS were examined using the Mann–Whitney U test. Spearman correlations were calculated to examine the relationship between cfDNA concentration and hospital- or ICU-free days. Differences in cfDNA concentrations and LDH activity between patient groups were compared using a two-way analysis of variance. Area under the receiver operating characteristic (AUROC) analysis was performed to estimate the predictability of MODS development based on the pre-hospital concentrations of cfDNA. The scaled (Z-transformed) values were considered for sensitivity, specificity and corresponding AUROC values. For Mann–Whitney U tests and unpaired t-tests, where a Bonferroni correction for multiple comparisons was performed, statistical significance was set at an adjusted p value of <0.016. For all other analyses, statistical significance was accepted at p < 0.05. Data are presented as Tukey-style box and whisker plots, where the line inside the box represents the median value and outliers are indicated by individual dots.
Results
Patient characteristics
Of the 155 trauma patients included in this study, 21, 53 and 81 were classified as having sustained an isolated TBI, TBI+ or ECI, respectively (Table 1). Isolated TBI patients, from whom pre-hospital blood samples were acquired within a mean time of 44 minutes (range, 26–60 minutes) post-injury, were a predominantly male cohort (86%) with severe head trauma, evidenced by a mean GCS score of 7 (range, 3–15) and an ISS of 25 (range, 9–45) (Table 1). The primary mechanism of injury was a fall from height, which accounted for 43% of injuries sustained (Table 1). For the TBI+ cohort, of which 81% were male, pre-hospital blood samples were acquired within a mean time of 42 minutes (range, 13–60 minutes) post-injury (Table 1). The mean ISS and NISS were 34 (range, 9–66) and 49 (range, 12–75), respectively, with road traffic collisions the predominant mechanism of injury (Table 1). Road traffic collisions and assault/penetrating injuries were the primary mechanisms of injury amongst our cohort of ECI patients, of whom 90% were male (Table 1). ECI patients presented with mean ISS and NISS scores of 18 (range, 9–75) and 25 (range, 9–75), respectively, with pre-hospital blood samples obtained within a mean time of 40 minutes (range, 18–60 minutes) post-injury (Table 1).
Increased plasma cfDNA levels after traumatic injury are associated with poor patient outcomes
At all post-injury time points, significantly higher concentrations of cfDNA were measured in plasma from isolated TBI, TBI+ and ECI patients when compared to HCs (Figure 1a). A comparison of cfDNA concentrations between patient groups revealed significantly higher levels of cfDNA in plasma samples acquired within 1 hour of injury from TBI+ patients when compared to individuals that had sustained either an isolated TBI or ECI (Figure 1b). For TBI+ and ECI patients, no relationship was found between the concentration of cfDNA in samples obtained ≤1 hour post-injury and either ISS (TBI+, r(n = 47) = 0.184, p = 0.21; ECI, r(n = 60) = 0.07, p = 0.56) or NISS (TBI+, r(n = 47) = −0.004, p = 0.976; ECI, r(n = 60) = 0.126, p = 0.33). Similarly, in our cohort of isolated TBI patients, cfDNA concentrations in plasma samples obtained ≤1 hour post-injury did not correlate with GCS scores (r(n = 18) = −0.338, p = 0.17).
Figure 1.
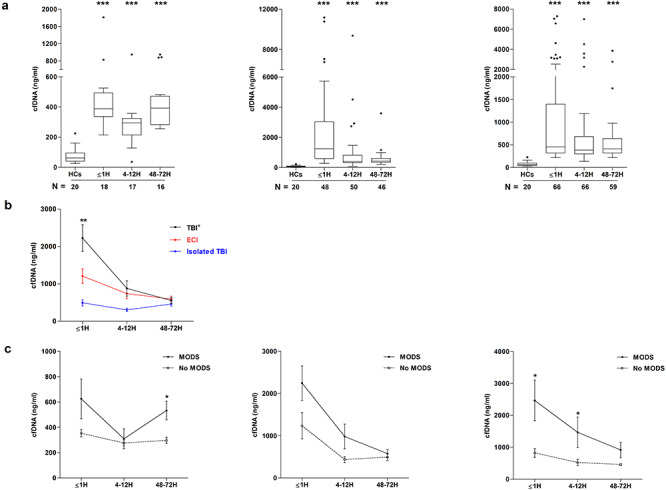
Trauma-induced changes in plasma cell-free DNA (cfDNA) levels and their relationship with patient outcome. (a) Comparison of cfDNA concentrations in plasma samples acquired from healthy controls (HCs) and patients that had sustained an isolated traumatic brain injury (TBI) (left panel), TBI with an accompanying extracranial injury (TBI+) (middle panel) or an extracranial injury (ECI) only (right panel). Patient samples were acquired at 3 post-injury time points (≤1 hour, 4–12 hours and 48–72 hours), with the number of samples analysed indicated below each time point. ***p < 0.0001 vs HCs. (b) Comparison of cfDNA concentrations in plasma samples from TBI, TBI+ and ECI patients at 3 post-injury time points. Number of samples analysed match those reported in (a). **p < 0.001 vs ECI and isolated TBI. (c) Comparison of cfDNA concentrations in plasma samples obtained from TBI, TBI+ and ECI patients who did or did not develop multiple organ dysfunction syndrome (MODS) during their hospital stay (TBI: MODS ≤1 hour (H) n = 9, 4–12H n = 10, 48–72H n = 11; no MODS ≤1H n = 7, 4–12H n = 5, 48–72H n = 5; TBI+: MODS ≤1H n = 32, 4–12H n = 34, 48–72H n = 35; no MODS ≤1H n = 11, 4–12H n = 13, 48–72H n = 11; ECI: MODS ≤1H n = 16, 4–12H n = 15, 48–72H n = 17; no MODS ≤1H n = 43, 4–12H n = 43, 48–72H n = 41). *p < 0.05 vs no MODS. H hour
To investigate whether cfDNA levels were associated with clinical outcomes, we first compared the concentration of cfDNA in plasma samples between patients who did or did not develop MODS during their hospital stay. In our isolated TBI cohort, plasma cfDNA levels were higher in samples obtained ≤1 hour post-injury from patients who developed MODS, although this was not statistically significant (p = 0.054) (Figure 1c). By 48–72 hours post-injury, cfDNA concentrations were significantly increased in isolated TBI patients who subsequently developed MODS (Figure 1c). At no sampling time point were cfDNA levels different between TBI+ patients who did or did not develop MODS (Figure 1c). In our ECI cohort, significantly elevated cfDNA concentrations were measured in the ≤1 hour and 4–12 hour post-injury plasma samples of subjects who developed MODS (Figure 1c). Focusing on ECI patients, who represented our largest cohort, we examined the ability of cfDNA to discriminate between the MODS and no MODS cohorts using AUROC analysis. We found that cfDNA concentrations in samples acquired within an hour of injury moderately discriminated between the 2 groups (AUROC 0.729, 95% CI 0.564–0.866; sensitivity 0.73, 95% CI 0.63–0.85; specificity 0.62, 95% CI 0.51–0.70).
An examination of whether cfDNA levels were associated with the number of hospital or ICU-free days revealed a significant negative relationship between cfDNA and both outcome measures at all sampling time points for patients that had sustained an ECI (Table 2). Within our isolated TBI cohort, cfDNA concentrations in ≤1 hour post-injury plasma samples were negatively associated with both ICU- and hospital-free days (Table 2). For TBI+ patients, a significant negative relationship was observed between cfDNA concentrations at the 4–12 hour sampling time point and the number of hospital-free days (Table 2).
Table 2.
Correlations between cfDNA concentrations at pre-hospital (≤1H), 4-12H and 48-72H post-injury time-points and length of stay outcomes
| Hospital-free days | ICU-free days | |||||
|---|---|---|---|---|---|---|
| ≤1H | 4-12H | 48-72H | ≤1H | 4-12H | 48-72H | |
| TBI | -0.522 (-0.800– -0.058) | -0.013 (-0.503–0.481) | -0.378 (-0.743–0.160) | -0.490 (-0.784– -0.015) | -0.118 (-0.577 – 0.397) | -0.242 (-0.668–0.302) |
| TBI+ | -0.131 (-0.407–0.167) | -0.319 (-0.555– -0.037) | -0.198 (-0.469–0.105) | -0.056 (-0.342–0.239) | -0.248 (-0.498–0.041) | -0.128 (-0.410–0.177) |
| ECI | -0.470 (-0.643– -0.250) | -0.289 (-0.502– -0.043) | -0.279 (-0.505– -0.017) | -0.482 (-0.652– -0.265) | -0.356 (-0.555– -0.117) | -0.342 (-0.555– -0.087) |
Data are presented as r, with 95% confidence intervals in parentheses
Significant associations according to Spearman’s rank correlation coefficient are indicated in bold font.
Number of pairs for hospital/ICU-free days and cfDNA correlations: ≤1H, TBI (n=18), TBI+ (n=48), ECI (n=66); 4-12H, TBI (n=17), TBI+ (n=49), ECI (n=66); 48-72H, TBI (n=16), TBI+ (n=46), ECI (n=59).
cfDNA cell-free DNA, ECI extracranial injury, ICU intensive care unit, TBI traumatic brain injury, TBI+ TBI with accompanying extracranial injury, H hour
Effect of traumatic injury on the activity and expression of circulating DNase
Reduced breakdown is a potential explanation for the persistent elevation in circulating cfDNA post trauma. To examine this possibility, we measured DNase activity. Compared to HCs, isolated TBI patients presented with significantly reduced DNase activity at the 4–12 and 48–72 hour post-injury time points (Figure 2a). In our TBI+ cohort, a significant impairment in DNase activity was detected in both pre-hospital and 48–72 hour post-injury samples (Figure 2a). At the 4–12 hour and 48–72 hour post-injury time points, ECI patients presented with significantly reduced DNase activity (Figure 2a). DNase activity was also lower in samples acquired from ECI patients within 1 hour of injury, although this was not statistically significant (p = 0.016) (Figure 2a).
Figure 2.
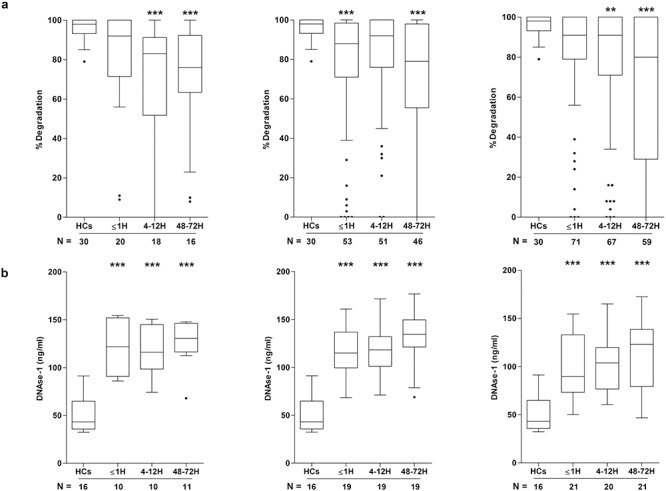
Effect of traumatic injury on deoxyribonuclease (DNase) activity and antigen levels. Comparison of DNase activity (a) and antigen levels (b) in peripheral blood samples acquired from healthy controls (HCs) and patients that had sustained an isolated traumatic brain injury (TBI) (left panel), TBI with an accompanying extracranial injury (middle panel) or an extracranial injury only (right panel). Patient samples were acquired at 3 post-injury time points (≤1 hour (H), 4–12 hours and 48–72 hours) and the number of samples analysed is indicated below each time point. **p < 0.01, ***p < 0.005 vs HCs. H hour
To investigate whether the post-trauma impairment in DNase activity was a consequence of reduced DNase protein levels, we quantified DNase-1 by ELISA. As shown in Figure 2b, compared to HCs, significantly elevated levels of DNase-1 were measured in blood samples acquired ≤1, 4–12 and 48–72 hours post-injury from all 3 patient groups.
Serum LDH activity and G-actin concentration are increased following traumatic injury
At all sampling time points, serum LDH activity, a marker of cell and tissue damage, was significantly higher in each of our patient groups when compared to HCs (Figure 3a). In serum samples acquired from TBI+ patients ≤1 hour and 4–12 hours post-injury, and from ECI patients 4–12 hours post-injury, LDH activity was significantly greater than that recorded in samples obtained at the corresponding time points from isolated TBI patients (Figure 3b). For both TBI+ and ECI patients, LDH activity and cfDNA levels in samples acquired within 1 hour of injury were positively associated (TBI+, r(n = 35) = 0.696, p = <0.0001; ECI, r(n = 29) = 0.524, p = 0.003).
Figure 3.
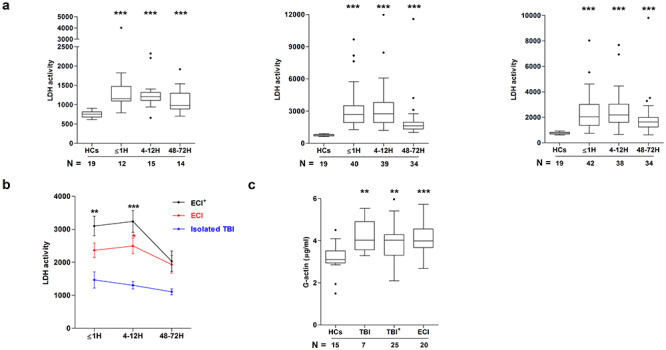
Trauma-induced elevation in circulating lactate dehydrogenase (LDH) activity and globular actin (G-actin) concentration. (a) Comparison of LDH activity in serum samples obtained from healthy controls (HCs) and patients that had sustained an isolated traumatic brain injury (TBI) (left panel), TBI with an accompanying extracranial injury (TBI+) (middle panel) or an extracranial injury (ECI) only (right panel). Patient samples were acquired at 3 post-injury time points (≤1 hour (H), 4–12 hours and 48–72 hours) and the number of samples analysed is indicated below each time point. LDH activity is presented as milliunits/ml. ***p < 0.005 vs HCs. (b) Comparison of LDH activity in serum samples from TBI, TBI+ and ECI patients at 3 post-injury time points. Number of samples analysed match those reported in (a). *p < 0.05, **p < 0.01, **p < 0.001 vs Isolated TBI. (c) Comparison of the concentration of G-actin in pre-hospital blood samples acquired from TBI, TBI+ and ECI patients within 1 hour of injury and HCs. The number of samples analysed are indicated below each time point. **p < 0.01, ***p < 0.005 vs HCs. H hour
Released from injured tissue, G-actin inhibits the nuclease activity of DNase-1 [32–34]. Focusing on samples collected in the immediate aftermath of injury, we detected a significant increase in the concentration of G-actin in pre-hospital blood samples acquired from all 3 patient groups when compared to HCs (Figure 3c).
Impact of trauma on the circulating levels of GSN and VDBP
As part of the EASS, the plasma protein GSN drives the depolymerization of F-actin [35]. At all 3 sampling time points, GSN concentrations, relative to HCs, were significantly reduced in samples acquired from patients that had sustained either an isolated TBI or ECI (Figure 4a). In the TBI+ cohort, a significant trauma-induced reduction in GSN levels was detected in samples obtained 4–12 hours and 48–72 hours post-injury (Figure 4a). Subunits of monomeric G-actin are bound by VDBP to form complexes that are cleared by the liver, spleen and kidneys [36, 37]. At all sampling time points, concentrations of VDBP were significantly reduced in plasma samples from ECI and TBI+ patients when compared to the levels recorded in samples from HCs (Figure 4b). In isolated TBI patients, VDBP levels were significantly lower ≤1 hour and 4–12 hours post-injury (Figure 4b).
Figure 4.
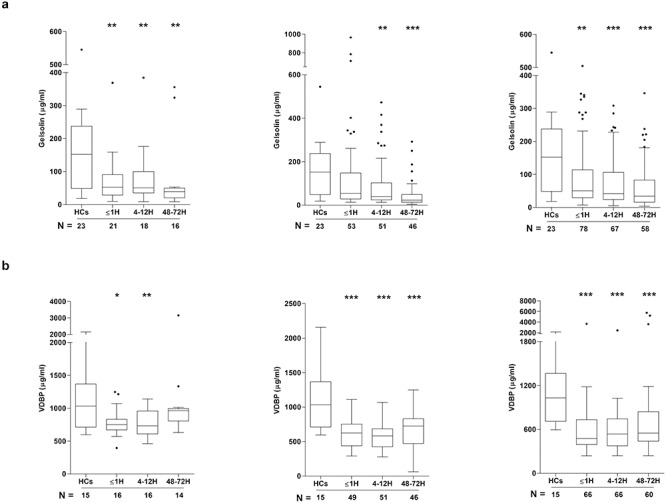
Effect of traumatic injury on the circulating extracellular actin scavenging system. Comparison of the concentrations of gelsolin (a) and vitamin D binding protein (VDBP) (b) in peripheral blood samples acquired from healthy controls (HCs) and patients that had sustained an isolated traumatic brain injury (TBI) (left panel), TBI with an accompanying extracranial injury (middle panel) or an extracranial injury only (right panel). Patient samples were acquired at 3 post-injury time points (≤1 hour (H), 4–12 hours and 48–72 hours) and the number of samples analysed is indicated below each time point. *p < 0.05, **p < 0.01, ***p < 0.005 vs HCs. H hour
Discussion
In this prospective observational cohort study of 155 trauma patients, from whom blood samples were acquired during the ultra-early (≤1 hour) and acute (4–72 hours) post-injury phase, we have shown for the first time that major trauma results in an immediate impairment in DNase activity. Accompanied by significantly elevated circulating concentrations of cfDNA, this reduction in DNase activity was observed in patients that had sustained extracranial and brain injuries. Identifying a potential underlying mechanism for this trauma-associated decline in DNase activity, circulating concentrations of G-actin, an inhibitor of DNase-1, were significantly increased within minutes of injury, whilst plasma levels of the actin scavengers GSN and VDBP were significantly reduced. With our data suggesting that elevated circulating concentrations of cfDNA are associated with the development of MODS in ECI and isolated TBI patients, therapeutic restoration of DNase activity may represent a strategy by which to reduce poor clinical outcome after a broad range of injuries.
In agreement with some, but not all, previous studies that have examined the relationship between injury burden and circulating cfDNA levels [46], we found no association between the concentration of cfDNA in pre-hospital plasma samples and injury severity. However, we did find that elevated cfDNA levels during the ultra-early (≤1 hour) and acute (4–72 hours) post-injury phases were associated with poorer clinical outcome. Mirroring the findings of Shaked et al. [15], we observed, in all 3 of our patient groups, a significant negative association between plasma cfDNA concentrations and the number of hospital- and/or ICU-free days. However, only in our cohort of ECI patients was this relationship between raised cfDNA levels and a longer hospital/ICU length of stay (LOS) evident at all 3 sampling time points, suggesting that cfDNA may be a better indicator of outcome in patients with extracranial injuries when compared to subjects with head trauma. Recently, it has been proposed that the release of cfDNA post trauma is more likely to be representative of injury burden and the degree of tissue damage sustained by a patient rather than a marker of early physiological derangement [47]. In line with this theory, Ren and colleagues found that the percentage of plasma samples positive for cfDNA was significantly higher in a cohort of trauma patients with penetrative injuries when compared to individuals that had sustained blunt trauma [48]. If, as these data suggest, soft tissue is the predominant source of cfDNA, and cfDNA is a marker of overall tissue damage and injury burden [47], then the higher incidence of assault/penetrative injuries and the greater degree of soft-tissue damage sustained by our ECI cohort may explain why, in these patients, cfDNA was positively associated with both hospital and ICU LOS at all 3 sampling time points. Indeed, when compared to isolated TBI patients, it is reasonable to think that in patients with polytrauma, longer hospital LOS is related to higher cfDNA levels based on the fact that they probably have a greater burden of injury. In addition to LOS, significantly higher cfDNA concentrations were measured in plasma samples obtained ≤1 hour and 4–12 hours post-injury from ECI patients who subsequently developed MODS, an observation that matches the results of previous prospective studies of hospitalized trauma patients [14, 49]. Of note, AUROC analyses revealed that a measurement of cfDNA in samples acquired within minutes of injury could moderately (AUROC 0.729, 95% CI 0.564–0.866) discriminate between ECI patients who did or did not develop MODS. Thus, taken together, these data suggest that an immediate assessment of cfDNA in patients that have sustained an ECI may help in the early identification of subjects at risk of poor outcome.
A number of groups have reported the presence of both nDNA and mtDNA in the circulation of trauma patients [4, 5, 26, 47, 50]. Derived from an organelle of bacterial origin, mtDNA shares many structural similarities with bacterial DNA, which includes a circular genome and the presence of unmethylated CpG motifs. Through activation of the pathogen recognition receptor toll-like receptor 9 and the DNA sensor cyclic GMP-AMP synthase, mtDNA triggers various pro-inflammatory responses [51, 52], which in the setting of traumatic injury has been shown to include the activation of neutrophils [3, 53, 54]. As such, mtDNA-driven inflammatory responses are thought to contribute to poor clinical outcomes post-injury. In line with this theory, significantly higher mtDNA levels have been detected in the post-hospital admission blood samples of non-survivors versus survivors of traumatic injury [4, 7], as well as in individuals who developed post-traumatic MODS [49, 55], ARDS [23] and a systemic inflammatory response [8, 26, 49]. In a previous study, in which we examined the origins of the cfDNA present within our patient blood samples, we found no difference in the concentration of mtDNA between patient samples and those of HCs [2]. However, we did observe a significant post-trauma elevation in the circulating levels of nDNA, suggesting that the associations we have reported in the current study between elevated cfDNA and poor clinical outcome are attributable to the actions of nDNA. On this note, in a recent prospective cohort study of 104 patients with severe blunt trauma, Stortz et al. found circulating concentrations of nDNA, but not mtDNA, were significantly higher ≤12 hours and 24 hours post-injury in patients who developed chronic critical illness when compared to those who experienced rapid recovery [47]. Furthermore, significantly elevated plasma concentrations of nDNA have been reported in non-survivors of major traumatic injury [4, 9] and in patients who develop post-traumatic MODS [14] and nosocomial infections [50]. Whilst appearing to be devoid of pro-inflammatory/immunogenic activity [18], nDNA and/or its associated proteins (e.g. histones) have been shown to trigger platelet activation [18, 20], inhibit fibrinolysis [56], perturb blood flow through capillary plexi [57] and induce endothelial/epithelial cell death [19, 21, 22]. Thus, these pro-thrombotic and cytotoxic properties of nDNA/histones may represent a potential mechanistic explanation for the associations that we and others have reported between raised circulating nDNA and poor clinical outcome post-injury [4, 9, 14].
In each of our patient cohorts, we detected, at all sampling time points, significantly elevated circulating concentrations of DNase-1. Expressed in a variety of tissues, DNase-1 localizes along the secretory pathway in Paneth cells of the small intestine and the hormone secreting cells of the pituitary gland [58, 59]. As blunt/penetrative trauma and TBI results in increased intestinal permeability and activation of the hypothalamic–pituitary–adrenal axis [60–62], the secretion/passive release of DNase-1 from these tissues could potentially explain the immediate and sustained elevation in the circulating levels of this endonuclease post trauma. The combination of raised DNase-1 antigen levels and cfDNA concentrations post trauma led us to investigate the effect of injury on the enzymatic activity of DNase. Confirming the findings of McIlroy et al., who had previously reported impaired DNase-1 activity in a cohort of major orthopaedic blunt trauma patients [26], we found DNase activity was significantly reduced in serum samples obtained from ECI patients 4–12 hours and 48–72 hours post-injury. Furthermore, we have shown for the first time that serum DNase activity is significantly reduced in patients who have sustained an isolated TBI or TBI with an accompanying ECI. This impairment in enzymatic activity was detectable within minutes of injury and persisted for up to 72 hours post trauma.
Our observation of reduced DNase activity in the background of elevated protein levels led us to investigate whether a circulating inhibitor was contributing to the trauma-induced decline in DNase activity. Released by tissue injury, G-actin binds to and inhibits the nuclease activity of DNase-1 [32–34]. Screening of pre-hospital blood samples obtained from all patient groups revealed an immediate significant increase in the circulating concentration of G-actin. Whilst elevated, the differences we detected in G-actin levels between HCs and injured patients were not as marked as we had expected. We attribute this to the fact that our methodology is likely to have underestimated the exact concentration of G-actin present in the circulation post-injury, as the ELISA we used only measures free G-actin. Thus, the assay would not have detected G-actin bound to DNase-1 or VDBP. Given that actin–VDBP complexes are formed within minutes of injury [39], the total circulating pool of G-actin in the immediate aftermath of injury is likely to be much greater than that we have reported here.
Responsible for the clearance of actin from the circulation is the EASS, which is comprised of the plasma-residing proteins GSN and VDBP [35–37]. Mechanistically, GSN drives the depolymerization of F-actin into monomeric G-actin subunits that are subsequently sequestered by VDBP and cleared via a receptor-dependent mechanism in the liver, lung and/or spleen [63, 64]. As reported by others [39–44], we detected, across our 3 patient cohorts, significantly reduced concentrations of GSN and VDBP in pre- and/or post-hospital admission blood samples. When complexed to actin, the circulating half-lives of GSN and VDBP are markedly decreased [65]. Although not investigated in this study, previous analyses of hospital admission blood samples of polytrauma patients have revealed an increased frequency of VDBP–actin complexes [39]. Thus, we suggest that the deficiency we observed in VDBP and GSN reflects, at least in part, their consumption by circulating actin. In light of these data, we propose a post-injury scenario in which actin release from damaged tissue reduces plasma GSN and VDBP levels. This dysregulation in the EASS results in elevated concentrations of circulating G-actin, the subsequent binding of which to DNase-1 significantly reduces its enzymatic activity [32–34]. Impaired DNase-1–mediated clearance of cfDNA, combined with the release of DNA from damaged tissue and/or activated immune cells following both the initial act of trauma and subsequent surgical interventions, results in an immediate and persistent elevation in circulating cfDNA [5]. Through activation of the clotting cascade [18, 20], promotion of endothelial dysfunction [19, 21, 22] and reduction of capillary blood flow [57], raised cfDNA may contribute to the development of such secondary complications as MODS and ARDS that would result in an increased ICU/hospital LOS.
In rodent models of haemorrhagic shock and sepsis, clearance of circulating DNA via systemic administration of DNase-1 has been shown to protect against organ damage and promote host survival [66, 67]. As human-based trauma studies have shown, raised cfDNA levels are associated with the development of MODS [8, 14, 24], multiple organ failure [14] and an increased risk of mortality [4, 7, 9, 10, 12, 13], could DNase-1 treatment reduce the incidence of secondary complications amongst hospitalized trauma patients? Although feasible, this approach is faced with a number of obstacles. For instance, not only does the timing of DNase-1 treatment appear to be critical for its success [67], but, based on the data presented in this manuscript, exogenous DNase-1 would be administered into an environment rich in circulating G-actin. Combined with the post-trauma dysregulation in the EASS that persists in the days and weeks following injury [39–44], raised concentrations of G-actin would promote the formation of G-actin–DNase complexes, an association that negatively impacts upon the activity of DNase-1 [32–34]. Interestingly, in a small pilot study of polytrauma patients, we measured significantly increased GSN concentrations and DNase activity in blood samples acquired from individuals that had received fresh frozen plasma (FFP) prior to hospital admission [68]. Although the underlying mechanism was not directly demonstrated, we speculated that GSN-mediated scavenging of circulating actin was responsible for this FFP-induced enhancement in DNase activity [68]. Recently, a multicentre phase 3 clinical trial demonstrated that pre-hospital administration of thawed plasma to trauma patients at risk of haemorrhagic shock was safe and associated with a reduced risk of mortality when compared to standard care [69]. As we have provided evidence of elevated cfDNA levels and reduced DNase activity within minutes of injury, a large-scale pre-hospital based trial of trauma patients that investigates whether administration of FFP or recombinant GSN enhances DNase-mediated clearance of cfDNA, via restoration of the EASS, would build upon our previous pilot study [68]. This would help determine whether increasing DNase activity is a potential therapeutic strategy by which to improve the clinical outcomes of severely injured patients.
In addition to targeting DNase, direct scavenging of DNA is currently being investigated as a means of preventing the onset of secondary complications associated with raised cfDNA. In a rodent model of combined tissue injury and haemorrhagic shock, clearance of circulating DNA via systemic administration of a nucleic acid scavenging polymer was found to attenuate pro-inflammatory responses and protect against organ injury [55]. Highlighting how such a strategy could translate into the treatment of trauma patients, Gocho et al. recently demonstrated that direct haemoperfusion through filters containing immobilized polymyxin B significantly reduced circulating DNA levels in a cohort of septic patients, an approach that the authors suggested could be an effective means by which to reduce unwarranted inflammation and thrombogenesis [70].
Whilst in this article we have focused upon how trauma-induced alterations in the circulating concentrations of G-actin and the EASS may, via their regulation of DNase-1 activity, contribute to the post-injury persistence in circulating cfDNA, a review of the literature suggests that these changes may have additional implications for the host. For example, VDBP and GSN have been shown to: (1) augment C5a-induced chemotaxis of monocytes and neutrophils [71, 72], (2) increase both the phagocytic and microbicidal activity of alveolar macrophages [73] and (3) induce nitric oxide synthesis [73]. Furthermore, GSN has been proposed to exhibit immune regulatory properties. Referred to as the ‘GSN mediator buffer hypothesis’, this plasma protein has been shown to bind to and inhibit the agonistic activity of a range of inflammatory mediators that includes lysophosphatidic acid [74], platelet-activating factor [74], lipopolysaccharide [75] and lipoteichoic acid [76]. Based in part on data from animal models of infection/injury that have demonstrated reduced inflammatory complications and increased survival following GSN administration [77–79], it is believed that GSN helps prevent excessive systemic inflammatory responses. Thus, depletion of GSN and VDBP may be a contributory factor in both the impaired immune responses and elevated systemic inflammation observed in traumatically injured patients. Interestingly, this scenario may be exacerbated by the trauma-induced elevation in circulating actin, a protein that is both immune suppressive, as demonstrated by its ability to impair macrophage bacterial defences [80] and immune stimulatory, with its recognition by cross-presenting dendritic cells potentially contributing to the activation of CD8+ T cells and the induction of pro-inflammatory immune responses [81, 82]. Thus, via their ability to modulate host immune responses, the consequences of a dysregulated EASS and elevated levels of circulating actin may extend beyond a post-trauma accumulation of cfDNA.
Conclusions
We have demonstrated a very rapid post-injury elevation in circulating cfDNA levels and a reduction in DNase activity in patients with both extracranial injuries and TBI. This impairment in endonuclease activity occurred secondary to reduced levels of the actin scavengers GSN and VDBP and elevated levels of G-actin. Impaired DNase activity offers a potential common mechanistic explanation for the persistently elevated concentrations of cfDNA associated with the development of secondary complications and/or death. Moreover, restoration of DNase-1 activity, which can be achieved via administration of FFP or GSN, may represent a therapeutic strategy to improve the clinical outcomes of hospitalized trauma patients.
Acknowledgments
The authors wish to thank all the research, nursing and administrative staff at the National Institute for Health Research Surgical Reconstruction Microbiology Research Centre for their assistance in data collection and management. We also thank all the emergency healthcare professionals in the West Midlands Ambulance Service NHS Foundation Trust, the Midlands Air Ambulance Charity and the University Hospitals Birmingham NHS Foundation Trust for their participation in the identification, enrolment and follow-up of patients in this study.
Abbreviations
ARDS: acute respiratory distress syndrome; AUROC: area under the receiver operating characteristics; cfDNA: cell-free deoxyribonucleic acid; DNase: Deoxyribonuclease; DNase-1: deoxyribonuclease-1; EASS: extracellular actin scavenger system; ECI: extracranial injury; ELISA: enzyme-linked immunosorbent assay; F-actin: filamentous actin; FFP: fresh frozen plasma; G-actin: monomeric globular actin; GCS: Glasgow Coma Scale; GSN: gelsolin; HCs: healthy controls; ICU: intensive care unit; ISS: Injury Severity Score; LDH: lactate dehydrogenase; LOS: length of stay; MODS: multiple organ dysfunction syndrome; mtDNA: mitochondrial-derived DNA; nDNA: nuclear-derived DNA; NETs: neutrophil extracellular traps; NHS: National Health Service; NISS: New Injury Severity Score; PFP: platelet-free plasma; RT: room temperature; SOFA: Sequential Organ Failure Assessment; TBI: traumatic brain injury; TBI+: traumatic brain injury with accompanying extracranial injury; VDBP: vitamin D binding protein.
Authors’ contributions
JH was involved in study design, sample processing, data curation, data analysis and drafted the manuscript. RJD was involved in study design, data collection, data analysis and critically reviewed and edited the manuscript. DNN contributed to data curation and critically reviewed and edited the manuscript. AA performed statistical analysis and critically reviewed the manuscript. JRBB performed statistical analysis and critically reviewed the manuscript. JML was involved in conceptualization of the study and critically reviewed and edited the manuscript. PH was involved in conceptualization of the study, study design and critically reviewed and edited the manuscript. All authors read and approved the final manuscript.
Funding
This work was funded by the National Institute for Health Research (NIHR) Surgical Reconstruction and Microbiology Research Centre and the Scar Free Foundation. JML is supported by the NIHR Birmingham Biomedical Research Centre and the Medical Research Council (MRC) Versus Arthritis Centre for Musculoskeletal Ageing Research. The funding bodies had no role in the design of the study, the collection, analysis or interpretation of the data or in the writing of the manuscript. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Conflicts of interest
The authors declare no conflicts of interest.
Availability of data and materials
All data upon which the conclusions of the paper rely are presented in the main manuscript.
Ethics approval and consent to participate
This study reports data acquired from subjects enrolled in the Brain Biomarkers after Trauma Study. Ethical approval for the study was granted by the North Wales Research Ethics Committee—West (REC reference: 13/WA/0399, protocol number: RG_13–164). Due to the nature of their injuries, patients were unlikely to be able to provide informed consent to enroll in the study. As such, patient recruitment into the study was performed under the guidance of the Mental Health Capacity Act for research in emergency situations, in accordance with the Declaration of Helsinki. If the patient lacked capacity, a written agreement for study participation was sought from a legal consultee, with written consent obtained from the patient after they regained capacity. In cases in which the patient did not regain capacity to consent, data were retained in accordance with the legal consultee’s assent.
Consent for publication
Not applicable.
References
- 1. Hazeldine J, Dinsdale RJ, Harrison P, Lord JM. Traumatic injury and exposure to mitochondrial-derived damage associated molecular patterns suppresses neutrophil extracellular trap formation. Front Immunol. 2019. 10.3389/fimmu.2019.00685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hazeldine J, Naumann DN, Toman E, Davies D, Bishop JRB, Su Z, et al. Prehospital immune responses and development of multiple organ dysfunction syndrome following traumatic injury: a prospective cohort study. PLoS Med. 2017. 10.1371/journal.pmed.1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Itagaki K, Kaczmarek E, Lee YT, Tang IT, Isal B, Adibnia Y, et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS One. 2015. 10.1371/journal.pone.0120549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lam NYL, Rainer TH, Chiu RWK, Joynt GM, Lo YMD. Plasma mitochondrial DNA concentrations after trauma. Clin Chem. 2004;50:213–6. [DOI] [PubMed] [Google Scholar]
- 5. McIlroy DJ, Bigland M, White AE, Hardy BM, Lott N, Smith DW, et al. Cell necrosis-independent sustained mitochondrial and nuclear DNA release following trauma surgery. J Trauma Acute Care Surg. 2015;78:282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang H-C, Lin Y-J, Tsai N-W, Su BY-J, Kung C-T, Chen W-F, et al. Serial plasma deoxyribonucleic acid levels as predictors of outcome in acute traumatic brain injury. J Neurotrauma. 2014;31:1039–45. [DOI] [PubMed] [Google Scholar]
- 7. Yamanouchi S, Kudo D, Yamada M, Miyagawa N, Furukawa H, Kushimoto S. Plasma mitochondrial DNA levels in patients with trauma and severe sepsis: time course and the association with clinical status. J Crit Care. 2013;28:1027–31. [DOI] [PubMed] [Google Scholar]
- 8. Gu X, Yao Y, Wu G, Lv T, Luo L, Song Y. The plasma mitochondrial DNA is an independent predictor for post-traumatic systemic inflammatory response syndrome. PLoS One. 2013. 10.1371/journal.pone.0072834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rodrigues Filho EM, Simon D, Ikuta N, Klovan C, Dannebrock FA, Oliveira de Oliveira C, et al. Elevated cell-free plasma DNA level as an independent predictor of mortality in patients with severe traumatic brain injury. J Neurotrauma. 2014;31:1639–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campello Yurgel V, Ikuta N, Brondani da Rocha A, Lunge VR, Fett Schneider R, Kazantzi Fonseca AS, et al. Role of plasma DNA as a predictive marker of fatal outcome following severe head injury in males. J Neurotrauma. 2007;24:1172–81. [DOI] [PubMed] [Google Scholar]
- 11. Hampson P, Dinsdale RJ, Wearn CM, Bamford AL, Bishop JRB, Hazeldine J, et al. Neutrophil dysfunction, immature granulocytes, and cell-free DNA are early biomarkers of sepsis in burn-injured patients: a prospective observational cohort study. Ann Surg. 2017;265:1241–9. [DOI] [PubMed] [Google Scholar]
- 12. Lo YMD, Rainer TH, Chan LYS, Hjelm NM, Cocks RA. Plasma DNA as a prognostic marker in trauma patients. Clin Chem. 2000;46:319–23. [PubMed] [Google Scholar]
- 13. Macher H, Egea-Guerrero JJ, Revuelto-Rey J, Gordillo-Escobar E, Enamorado-Enamorado J, Boza A, et al. Role of early cell-free DNA levels decrease as a predictive marker of fatal outcome after severe traumatic brain injury. Clin Chim Acta. 2012;414:12–7. [DOI] [PubMed] [Google Scholar]
- 14. Lam NYL, Rainer TH, Chan LYS, Joynt GM, Lo YMD. Time course of early and late changes in plasma DNA in trauma patients. Clin Chem. 2003;49:1286–91. [DOI] [PubMed] [Google Scholar]
- 15. Shaked G, Douvdevani A, Yair S, Zlotnik A, Czeiger D. The role of cell-free DNA measured by a fluorescent test in the management of isolated traumatic head injuries. Scand J Trauma Resusc Emerg Med. 2014. 10.1186/1757-7241-22-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fox A, Gal S, Fisher N, Smythe J, Wainscoat J, Tyler MPH, et al. Quantification of circulating cell-free plasma DNA and endothelial gene RNA in patients with burns and relation to acute thermal injury. Burns. 2008;34:809–16. [DOI] [PubMed] [Google Scholar]
- 17. Shoham Y, Krieger Y, Perry ZH, Shaked G, Bogdanov-Berezovsky A, Silberstein E, et al. Admission cell free DNA as a prognostic factor in burns: quantification by use of a direct rapid fluorometric technique. Biomed Res Int. 2014. 10.1155/2014/306580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhagirath VC, Dwivedi DJ, Liaw PC. Comparison of the proinflammatory and procoagulant properties of nuclear, mitochondrial, and bacterial DNA. Shock. 2015;44:265–71. [DOI] [PubMed] [Google Scholar]
- 19. Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584:3193–7. [DOI] [PubMed] [Google Scholar]
- 20. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NLet al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118:1952–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012. 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Faust HE, Reilly JP, Anderson BJ, Ittner CAG, Forker CM, Zhang P, et al. Plasma mitochondrial DNA levels are associated with ARDS in trauma and sepsis patients. Chest. 2020;157:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Margraf S, Lögters T, Reipen J, Altrichter J, Scholz M, Windolf J. Neutrophil-derived circulating free DNA (cf-DNA/NETs): a potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock. 2008;30:352–8. [DOI] [PubMed] [Google Scholar]
- 25. Meng W, Paunel-Görgülü A, Flohé S, Witte I, Schädel-Höpfner M, Windolf J, et al. Deoxyribonuclease is a potential counter regulator of aberrant neutrophil extracellular traps formation after major trauma. Mediators Inflamm. 2012. 10.1155/2012/149560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McIlroy DJ, Minahan K, Keely S, Lott N, Hansbro P, Smith DW, et al. Reduced deoxyribonuclease enzyme activity in response to high postinjury mitochondrial DNA concentration provides a therapeutic target for systemic inflammatory response syndrome. J Trauma Acute Care Surg. 2018;85:354–8. [DOI] [PubMed] [Google Scholar]
- 27. Lo YM, Zhang J, Leung TN, Lau TK, Chang AMZ, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet. 1999;64:218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Henriksen TV, Reinert T, Christensen E, Sethi H, Birkenkamp-Demtröder K, Gögenur Met al. The effect of surgical trauma on circulating free DNA levels in cancer patients-implications for studies of circulating tumor DNA. Mol Oncol. 2020;14:1670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. J Orthop Trauma. 2010;24:534–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nadano D, Yasuda T, Kishi K. Measurement of deoxyribonuclease I activity in human tissues and body fluids by a single radial enzyme-diffusion method. Clin Chem. 1993;39:448–52. [PubMed] [Google Scholar]
- 32. Lazarides E, Lindberg U. Actin is the naturally occurring inhibitor of deoxyribonuclease I. Proc Natl Acad Sci U S A. 1974;71:4742–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mannherz HG, Goody RS, Konrad M, Nowak E. The interaction of bovine pancreatic deoxyribonuclease I and skeletal muscle actin. Eur J Biochem. 1980;104:367–79. [DOI] [PubMed] [Google Scholar]
- 34. Eulitz D, Mannherz HG. Inhibition of deoxyribonuclease I by actin is to protect cells from premature cell death. Apoptosis. 2007;12:1511–21. [DOI] [PubMed] [Google Scholar]
- 35. Chaponnier C, Janmey PA, Yin HL. The actin filament-severing domain of plasma gelsolin. J Cell Biol. 1986;103:1473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dahl B. The extracellular actin scavenger system in trauma and major surgery. Clinical and experimental studies. Acta Orthop Suppl. 2005;76(317):2. [PubMed] [Google Scholar]
- 37. Lee WM, Galbraith RM. The extracellular actin-scavenger system and actin toxicity. N Engl J Med. 1992;326:1335–41. [DOI] [PubMed] [Google Scholar]
- 38. Chhabra D, Nosworthy NJ, Remedios CG. The N-terminal fragment of gelsolin inhibits the interaction of DNase I with isolated actin, but not with the cofilin-actin complex. Proteomics. 2005;5:3131–6. [DOI] [PubMed] [Google Scholar]
- 39. Dahl B, Schiødt FV, Kiaer T, Ott P, Bondesen S, Tygstrup N. Serum Gc-globulin in the early course of multiple trauma. Crit Care Med. 1998;26:285–9. [DOI] [PubMed] [Google Scholar]
- 40. Dahl B, Schiødt FV, Nielsen M, Kiaer T, Williams JG, Ott P. Admission level of Gc-globulin predicts outcome after multiple trauma. Injury. 1999;30:275–81. [DOI] [PubMed] [Google Scholar]
- 41. Xu J-F, Liu W-G, Dong X-Q, Yang S-B, Fan J. Change in plasma gelsolin level after traumatic brain injury. J Trauma Acute Care Surg. 2012;72:491–6. [DOI] [PubMed] [Google Scholar]
- 42. Jin Y, Li B-Y, Qiu L-L, Ling Y-R, Bai Z-Q. Decreased plasma gelsolin is associated with 1-year outcome in patients with traumatic brain injury. J Crit Care. 2012;27:527.e1–6. [DOI] [PubMed] [Google Scholar]
- 43. Mounzer KC, Moncure M, Smith YR, Dinubile MJ. Relationship of admission plasma gelsolin levels to clinical outcomes in patients after major trauma. Am J Respir Crit Care Med. 1999;160:1673–81. [DOI] [PubMed] [Google Scholar]
- 44. Dahl B, Schiødt FV, Ott P, Gvozdenovic R, Yin HL, Lee WM. Plasma gelsolin is reduced in trauma patients. Shock. 1999;12:102–4. [DOI] [PubMed] [Google Scholar]
- 45. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann Vet al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107:9813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gögenur M, Burcharth J, Gögenur I. The role of total cell-free DNA in predicting outcomes among trauma patients in the intensive care unit: a systematic review. Crit Care. 2017. 10.1186/s13054-016-1578-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stortz JA, Hawkins RB, Holden DC, Raymond SL, Wang Z, Brakenridge SC, et al. Cell-free nuclear, but not mitochondrial, DNA concentrations correlate with the early host inflammatory response after severe trauma. Sci Rep. 2019. 10.1038/s41598-019-50044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ren B, Liu F, Xu F, He J, Zhu H, Zou G. Is plasma cell-free DNA really a useful marker for diagnosis and treatment of trauma patients? Clin Chim Acta. 2013;424:109–13. [DOI] [PubMed] [Google Scholar]
- 49. Simmons JD, Lee Y-L, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg. 2013;258:591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Timmermans K, Kox M, Vaneker M, Berg M, John A, Laarhoven A, et al. Plasma levels of danger-associated molecular patterns are associated with immune suppression in trauma patients. Intensive Care Med. 2016;42:551–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020. 10.15252/embr.201949799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meyer A, Laverny G, Bernardi L, Charles AL, Alsaleh G, Pottecher J, et al. Mitochondria: an organelle of bacterial origin controlling inflammation. Front Immunol. 2018. 10.3389/fimmu.2018.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock. 2010;34:55–9. [DOI] [PubMed] [Google Scholar]
- 54. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aswani A, Manson J, Itagaki K, Chiazza F, Collino M, Wupeng WL, et al. Scavenging circulating mitochondrial DNA as a potential therapeutic option for multiple organ dysfunction in trauma hemorrhage. Front Immunol. 2018. 10.3389/fimmu.2018.00891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gould TJ, Vu TT, Stafford AR, Dwivedi DJ, Kim PY, Fox-Robichaud AE, et al. Cell-free DNA modulates clot structure and impairs fibrinolysis in sepsis. Arterioscler Thromb Vasc Biol. 2015;35:2544–53. [DOI] [PubMed] [Google Scholar]
- 57. Boneschansker L, Inoue Y, Oklu R, Irimia D. Capillary plexuses are vulnerable to neutrophil extracellular traps. Integr Biol (Camb). 2016;8:149–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shimada O, Ishikawa H, Tosaka-Shimada H, Yasuda T, Kishi K, Suzuki S. Detection of deoxyribonuclease I along the secretory pathway in Paneth cells of human small intestine. J Histochem Cytochem. 1998;46:833–40. [DOI] [PubMed] [Google Scholar]
- 59. Shimada O, Suzuki S, Tosaka-Shimada H, Ishikawa H. Detection of deoxyribonuclease I in a hormone-secretory pathway of pituitary cells in humans and rats. Cell Struct Funct. 1998;23:49–56. [DOI] [PubMed] [Google Scholar]
- 60. Ma EL, Smith AD, Desai N, Cheung L, Hanscom M, Stoica BA, et al. Bidirectional brain-gut interactions and chronic pathological changes after traumatic brain injury in mice. Brain Behav Immun. 2017;66:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Langkamp-Henken B, Donovan TB, Pate LM, Maull CD, Kudsk KA. Increased intestinal permeability following blunt and penetrating trauma. Crit Care Med. 1995;23:660–4. [DOI] [PubMed] [Google Scholar]
- 62. Faries PL, Simon RJ, Martella AT, Lee MJ, Machiedo GW. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44:1031–5. [DOI] [PubMed] [Google Scholar]
- 63. Herrmannsdoerfer AJ, Heeb GT, Feustel PJ, Estes JE, Keenan CJ, Minnear FL, et al. Vascular clearance and organ uptake of G- and F-actin in the rat. Am J Physiol. 1993;265:G1071–81. [DOI] [PubMed] [Google Scholar]
- 64. Dueland S, Nenseter MS, Drevon CA. Uptake and degradation of filamentous actin and vitamin D-binding protein in the rat. Biochem J. 1991;274:237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lind SE, Smith DB, Janmey PA, Stossel TP. Role of plasma gelsolin and the vitamin D-binding protein in clearing actin from the circulation. J Clin Invest. 1986;78:736–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cagliani J, Yang W-L, Brenner M, Wang P. Deoxyribonuclease reduces tissue injury and improves survival after hemorrhagic shock. J Surg Res. 2020;249:104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mai SHC, Khan M, Dwivedi DJ, Ross CA, Zhou J, Gould TJ, et al. Delayed but not early treatment with DNase reduces organ damage and improves outcome in a murine model of sepsis. Shock. 2015;44:166–72. [DOI] [PubMed] [Google Scholar]
- 68. Dinsdale RJ, Hazeldine J, Al Tarrah K, Hampson P, Devi A, Ermogenous C, et al. Dysregulation of the actin scavenging system and inhibition of DNase activity following severe thermal injury. Br J Surg. 2020;107:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sperry JL, Guyette FX, Brown JB, Yazer MH, Triulzi DJ, Early-Young BJ, et al. Prehospital plasma during air medical transport in trauma patients at risk for hemorrhagic shock. N Engl J Med. 2018;379:315–26. [DOI] [PubMed] [Google Scholar]
- 70. Gocho T, Mori H, Islam MM, Maruchi Y, Takenaka N, Tomino A, et al. Removal of circulating neutrophil extracellular trap components with an immobilized Polymyxin B filter: a preliminary study. Shock. 2020;54:44–9. [DOI] [PubMed] [Google Scholar]
- 71. Kew RR, Webster RO. Gc-globulin (vitamin D-binding protein) enhances the neutrophil chemotactic activity of C5a and C5a des Arg. J Clin Invest. 1988;82:364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Piquette CA, Robinson-Hill R, Webster RO. Human monocyte chemotaxis to complement-derived chemotaxins is enhanced by Gc-globulin. J Leukoc Biol. 1994;55:349–54. [DOI] [PubMed] [Google Scholar]
- 73. Yang Z, Chiou TT-Y, Stossel TP, Kobzik L. Plasma gelsolin improves lung host defense against pneumonia by enhancing macrophage NOS3 function. Am J Physiol Lung Cell Mol Physiol. 2015;309:L11–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Osborn TM, Dahlgren C, Hartwig JH, Stossel TP. Modifications of cellular responses to lysophosphatidic acid and platelet-activating factor by plasma gelsolin. Am J Physiol Cell Physiol. 2007;292:C1323–30. [DOI] [PubMed] [Google Scholar]
- 75. Bucki R, Georges PC, Espinassous Q, Funaki M, Pastore JJ, Chaby R, et al. Inactivation of endotoxin by human plasma gelsolin. Biochemistry. 2005;44:9590–7. [DOI] [PubMed] [Google Scholar]
- 76. Bucki R, Byfield FJ, Kulakowska A, McCormick ME, Drozdowski W, Namiot Z, et al. Extracellular gelsolin binds lipoteichoic acid and modulates cellular response to proinflammatory bacterial wall components. J Immunol. 2008;181:4936–44. [DOI] [PubMed] [Google Scholar]
- 77. Rothenbach PA, Dahl B, Schwartz JJ, O’Keefe GE, Yamamoto M, Lee WM, et al. Recombinant plasma gelsolin infusion attenuates burn-induced pulmonary microvascular dysfunction. J Appl Physiol. 2004;96:25–31. [DOI] [PubMed] [Google Scholar]
- 78. Christofidou-Solomidou M, Scherpereel A, Solomides CC, Christie JD, Stossel TP, Goelz S, et al. Recombinant plasma gelsolin diminishes the acute inflammatory response to hyperoxia in mice. J Invest Med. 2002;50:54–60. [DOI] [PubMed] [Google Scholar]
- 79. Lee P-S, Waxman AB, Cotich KL, Chung SW, Perrella MA, Stossel TP. Plasma gelsolin is a marker and therapeutic agent in animal sepsis. Crit Care Med. 2007;35:849–55. [DOI] [PubMed] [Google Scholar]
- 80. Ordija CM, Chiou TT-Y, Yang Z, Deloid GM, Oliveira VM, Wang Z, et al. Free actin impairs macrophage bacterial defenses via scavenger receptor MARCO interaction with reversal by plasma gelsolin. Am J Physiol Lung Cell Mol Physiol. 2017;312:L1018–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ahrens S, Zelenay S, Sancho D, Hanč P, Kjær S, Feest C, et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity. 2012;36:635–45. [DOI] [PubMed] [Google Scholar]
- 82. Zhang J-G, Czabotar PE, Policheni AN, Caminschi I, Wan SS, Kitsoulis S, et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity. 2012;36:646–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data upon which the conclusions of the paper rely are presented in the main manuscript.