

Abstract
The variety and widespread of coronavirus in natural reservoir animals is likely to cause epidemics via interspecific transmission, which has attracted much attention due to frequent coronavirus epidemics in recent decades. Birds are natural reservoir of various viruses, but the existence of coronaviruses in wild birds in central China has been barely studied. Some bird coronaviruses belong to the genus of Deltacoronavirus. To explore the diversity of bird deltacoronaviruses in central China, we tested faecal samples from 415 wild birds in Hunan Province, China. By RT‐PCR detection, we identified eight samples positive for deltacoronaviruses which were all from common magpies, and in four of them, we successfully amplified complete deltacoronavirus genomes distinct from currently known deltacoronavirus, indicating four novel deltacoronavirus stains (HNU1‐1, HNU1‐2, HNU2 and HNU3). Comparative analysis on the four genomic sequences showed that these novel magpie deltacoronaviruses shared three different S genes among which the S genes of HNU1‐1 and HNU1‐2 showed 93.8% amino acid (aa) identity to that of thrush coronavirus HKU12, HNU2 S showed 71.9% aa identity to that of White‐eye coronavirus HKU16, and HNU3 S showed 72.4% aa identity to that of sparrow coronavirus HKU17. Recombination analysis showed that frequent recombination events of the S genes occurred among these deltacoronavirus strains. Two novel putative cleavage sites separating the non‐structural proteins in the HNU coronaviruses were found. Bayesian phylogeographic analysis showed that the south coast of China might be a potential origin of bird deltacoronaviruses existing in inland China. In summary, these results suggest that common magpie in China carries diverse deltacoronaviruses with novel genomic features, indicating an important source of environmental coronaviruses closed to human communities, which may provide key information for prevention and control of future coronavirus epidemics.
Keywords: bird deltacoronavirus, common magpie, genome recombination, spike, viral genome
1. INTRODUCTION
Epidemics caused by viruses have been threatening the public health throughout the entire human history. Most human epidemic viruses originate from animal reservoirs and are transmitted to human by various animals serving as intermediate hosts. For instance, severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), the pathogenic virus of the recent COVID‐19 pandemic, is likely to originate from bats (Huang et al., 2020; Lu et al., 2020; Zhou, Yang, et al., 2020; Zhou, et al. 2020), but the infectious SARS‐CoV‐2 can be detected in a variety of animals, including cats (Sailleau et al., 2020), dogs (Sit et al., 2020) and other domesticated animals (Shi et al., 2020), which may act as the intermediate hosts bridging bats and humans. Similarly, MERS‐CoV and SARS‐CoV, the pathogenic viruses causing previous coronavirus (CoV) pandemics, were also reported to be transmitted from wild animals to humans through intermediate hosts (Forni et al., 2017; Shi & Hu, 2008; Su et al., 2016; Woo et al., 2006; Zhong et al., 2003;). Actually, the interspecies transmission of viruses from natural reservoir animals is a major cause for viral epidemics (Niederwerder & Hesse, 2018; Xu et al., 2018; Zhou et al., 2018). Therefore, monitoring the existence of potentially pathogenic viruses in animals is critical for predicting and preventing viral epidemics.
CoVs are enveloped, single‐stranded, positive‐sense ssRNA viruses belonging to the subfamily Orthocoronavirinae of the family Coronaviridae in the suborder Cornidovirineae of the order Nidovirales. CoVs are known to infect a wide variety of animals including humans, causing enteric, reproductive, respiratory and gastrointestinal diseases (Brian & Baric, 2005; Lin et al., 2016; Moreno et al., 2017). Currently, International Committee on Taxonomy of Viruses (ICTV) divides Orthocoronavirinae into four genera: Alpha‐, Beta‐, Gamma‐ and Delta‐CoV. The members of Alpha‐ and Beta‐CoV infect bats, humans and other mammals, whereas the ones of Gamma‐ and Delta‐CoV mainly infect birds but a few of them infect mammals (Dong et al., 2007; Lau et al., 2015; Wong et al., 2019; Woo et al., 2014).
Delta‐CoV is a novel genus of CoV discovered in recent years that can infect birds and mammals. In 2009, three novel avian coronaviruses were identified, including bulbul coronavirus HKU11 (BuCoV HKU11), thrush coronavirus HKU12 (ThCoV HKU12) and munia coronavirus HKU13 (MunCoV HKU13). According to their phylogenetic relationships and genomic structures, ICTV approved the classification of these three avian CoVs as a novel genus, Deltacoronavirus (Woo et al., 2009). Subsequently, in 2012, molecular epidemiological investigations in Hong Kong, China, revealed seven novel deltacoronaviruses, including six from birds and one from swine. Among them, porcine deltacoronavirus (PDCoV) HKU15 was the first deltacoronavirus found to infect non‐avian animals (Woo et al., 2012). Recently, four bird deltacoronaviruses, (falcon CoV UAE‐HKU27, houbara CoV UAE‐HKU28, pigeon CoV UAE‐HKU29 and quail CoV UAE‐HKU30), four novel sparrow CoVs and one quail CoV were found in the Middle East, the United States and Poland, respectively (Chen et al., 2018; Katarzyna et al., 2019; Lau et al., 2018). These findings suggested that deltacoronaviruses have the potential for Avian‐to‐Avian and Avian‐to‐Mammalian transmission, which suggest that the terrestrial birds may serve as the intermediate host for the transmission of CoV.
Birds are warm‐blooded vertebrates with high species biodiversity, featured behaviours (e.g. nesting and migration) and unique adaptive immune systems (Lee et al., 2014). Birds are the reservoir of major emerging viruses, including influenza virus, coronavirus and adenovirus (Chan et al., 2013, 2015). In this study, we tested samples from various avian species in the Hunan, a province located in central China. Based on the results of comparative genome and phylogenetic analyses, we propose four novel deltacoronavirus strains in common magpies.
2. MATERIALS AND METHODS
2.1. Sample collection
415 faecal samples of birds were collected in several areas of central China, including East Dongting Lake, Changde national wetland, Hengyang, in the Hunan Province over a 2‐month period (April 2018–June 2018). The samples were placed in virus transport medium (VTM) and kept in dry ice for transportation to the laboratory and stored at −80°C until use.
2.2. RNA extraction, PCR screening and sequencing
Viral RNA was extracted from the faecal samples using TIANamp Virus DNA/RNA Kit (Tiangen, China) following the manufacturer's instructions. RNA was eluted in 60 µl of RNase‐free water, aliquoted and stored at −80°C. For screening the presence of bird deltacoronaviruses, a set of primers to amplify a 440‐bp fragment of the RNA‐dependent RNA polymerase (RdRp) gene of CoVs using Deltacoronavirus conserved primers DCoVF (5′‐GTGGVTGTMTTAATGCACAGTC‐3′) and DCoVR (5′‐TACTGYCTGTTRGTCATRGTG‐3′) as described previously (Lau et al., 2018). PCR amplification was performed using the PrimeScript™ One Step RT‐PCR Kit Ver.2 (Takara). The PCR mixture (25 μL) contained 2 μL of extracted RNA, 12.5 μL 2 × 1 Step Buffer and 1 μL PrimeScript 1 Step Enzyme Mix. The mixtures were amplified by 60 cycles of 94°C for 1 min, 48°C for 1 min, and 72°C for 1 min and a final extension at 72°C for 10 min.
The PCR products were gel‐purified and sequenced with an ABI Prism 3,700 DNA analyzer (Applied Biosystems), using the PCR primers. The sequences of the PCR products were compared with known sequences of the RdRp genes of CoVs in the GenBank database.
2.3. Complete genome sequencing
Four complete genomes of common magpie CoV were amplified and sequenced using the RNAs extracted from birds faecal as templates. The RNA was amplified with degenerate primers designed by multiple alignments of the genomes of other CoVs with complete genomes available, using the PrimeScript™ One Step RT‐PCR Kit Ver.2. Additional primers were designed from the results of the first and subsequent rounds of sequencing. Sequences of the 5′ and 3′ genomic ends were obtained by 5′ and 3′ RACE (Roche), respectively. PCR products with expected size were gel‐purified and subjected directly to sequencing. Sequences were assembled to obtain the full‐length genome sequences.
The sequences obtained in this research have been deposited in GenBank with the accession numbers: MW345814(HNU1‐1), MW345815(HNU1‐2), MW 349841(HNU2), MW345816(HNU3), MW345810(CD1), MW345811(CD2), MW345812(CD3), MW345813(CD4).
2.4. Genome and phylogenetic analysis
Gene sequences and encoded putative protein amino acid (aa) sequences were compared to those of other CoVs using ORF finder (https://www.ncbi.nlm.nih.gov/orffifinder/). Sequence alignment was performed by using MAFFT v7.149 (Katoh & Standley, 2013). Phylogenetic trees were constructed using IQ‐tree v1.6.10, with 10,000 ultrafast bootstraps and the most appropriate model of aa substitution for each data sets was calculated by ModelFinder according to the corrected Akaike information criterion (Nguyen et al., 2015; Zhang et al., 2020).
2.5. Recombinant analysis
The preliminary identification of possible recombination events in our sample sequences using RDP v3.44 with seven different scanning methods, including RDP, GENECONV, BOOTSCAN, MAXCHI, CHIMAERA, SISCAN and 3SEQ (Martin et al., 2010). In order to minimize the existence of false positives, we used a Bonferroni correction and a highest acceptable p‐value = 0.05. At last, we used the Simplot v3.5.1 program to confirm the recombination events identified by RDP3 suite.
2.6. Discrete traits of Bayesian Phylogeographic analysis
We collected 20 bird deltacoronavirus sequences from GenBank, together with 4 samples that we amplified, a total of 24 bird deltacoronavirus sequences were from five different regions, Hong Kong (n = 10), United Arab Emirates (n = 5), USA (n = 4), Poland (n = 1) and Hunan (n = 4). To understand the spatial dynamics of bird deltacoronavirus, the above five regions were coded as discrete states and the phylogeographic analysis with RdRp gene to reconstruct ancestral geographical regions and migration patterns in BEAST v1.10.4 (Lemey et al., 2009). The marginal likelihood estimated by path sampling was used to compare the combinations of molecular clock models and coalescent models, and the results showed that the best‐fitting tree prior for our data set was the combination of strict clock and Bayesian skyline coalescent (Table S5) (Baele et al., 2012; Drummond et al., 2005). According to the latest report of the evolution rate on RdRp gene of deltacoronavirus, we used a uniformly distributions priori value (from 5 × 10–5 to 2 × 10–4 subs/site/year) of evolution rate in our analysis (Lau et al., 2018). Subsequently, phylogeographic analyses were performed by Bayesian Stochastic Search Variable Selection (BSSVS) combined with an asymmetric substitution model, running a markov chain of 40 million steps with sampling every 1,000 generations in BEAST. By discarding the first 10% of the samples, the remaining samples were used Tracer v1.7 to check the Effective Sample Size (ESS) of all estimated parameters, ensuring that there were enough ESS values (greater than 200) to achieve convergence (Rambaut et al., 2018). The Maximum Clade Credibility (MCC) tree was obtained by discarding the first 10% of trees in Tree Annotator package. The Bayes factors (BF) were calculated using SPREAD3 package to select significant migration pathways (BF > 3) among discrete locations.
3. RESULT
3.1. Prevalence of novel deltacoronaviruses in birds
The faecal samples for the deltacoronaviurs detection were from 415 birds collected in the Hunan province, China. These birds belonged to at least 26 species of 15 families. The presence of deltacoronaviruses was detected by RT‐PCR targeting a 440‐nt RNA‐dependent RNA polymerase (RdRp) fragment that is conserved among all known deltacoronaviruses. In total, eight samples were positive for deltacoronavirus. Host species identification by amplification of either Cytb or 12S rRNA gene suggested that all deltacoronavirus‐positive samples were exclusively from common magpie (family Corvidae) (Dinh et al., 2019; Wang et al., 2015) (Table 1). Preliminary analysis of the partial RdRp sequences suggested that all the eight sequences showed high similarity among themselves and with other reported deltacoronaviruses from swine and birds (Figure 1 and Figure S1). However, attempts to passage novel HNU CoV in cell cultures and embryonated chicken were failed, with no cytopathic effect or viral replication being detected.
TABLE 1.
Prevalence of deltacoronavirus in birds
| Animal type and scientific name | Common name | No. (%) of birds positive for deltacoronavirus | Sampling location |
|---|---|---|---|
| Ardeidae | |||
| Ardeola bacchus | Chinese pond heron | 0/13 | b |
| Columbidae | |||
| Streptopelia chinensis | Spotted dove | 0/1 | c |
| Streptopelia orientalis | Oriental turtle dove | 0/7 | b |
| Corvidae | |||
| Pica pica | Common magpie | 8/28 (0.28) | b *,c |
| Emberizidae | |||
| Emberiza sulphurata | Yellow bunting | 0/1 | a |
| Emberiza tristrami | Tristram's Bunting | 0/7 | a |
| Hirundinidae | |||
| Tachycineta euchrysea | Golden Swallow | 0/2 | a |
| Progne chalybea | Grey‐breasted Martin | 0/3 | a |
| Motacillidae | |||
| Anthus hodgsoni | Olive‐backed Pipit | 0/28 | a,b |
| Muscicapidae | |||
| Turdus hortulorum | Grey‐backed Thrush | 0/3 | a |
| Turdus kessleri | Kessler's Thrush | 0/2 | a |
| Paridae | |||
| Parus major | Great tit | 0/2 | a |
| Petroicidae | |||
| Petroica macrocephala | New zealand tit | 0/2 | a |
| Phasianidae | |||
| Bambusicola thoracica | Chinese Bamboo Partridge | 0/1 | a |
| Gallus gallus | Chicken | 0/5 | a |
| Pycnonotidae | |||
| Pycnonotus sinensis | Chinese bulbul | 0/10 | a |
| Pycnonotus taivanus | Tavaau bulbul | 0/2 | a |
| Pycnonotus xanthorrhous | Yellow‐vented Bulbul | 0/1 | a |
| Sturnidae | |||
| Acridotheres cristatellus | Crested myna | 0/7 | a |
| Sturnus sericeus | Red‐billed starling | 0/56 | a,b |
| Sturnus tristis | Common myna | 0/2 | a |
| Sylviida | |||
| Garrulax sannio | White‐checked Laughing Thrush | 0/1 | b |
| Timaliidae | |||
| Garrulax cineraceus | Moustached Laughing thrush | 0/1 | a |
| Turdidae | |||
| Turdus merula | Blackbird | 0/25 | b,c |
| Turdus migratorius | American robin | 0/3 | b |
| Zoothera princei | Grey Ground Thrush | 0/1 | c |
| Unknown | 0/201 | a,b,c |
a: Hengyang b: Changde c: Yueyang
Deltacoronavirus detected.
The bold values mean that deltaviruses have been detected in these samples, while abosultely no deltavirus has been detected in other samples.
FIGURE 1.
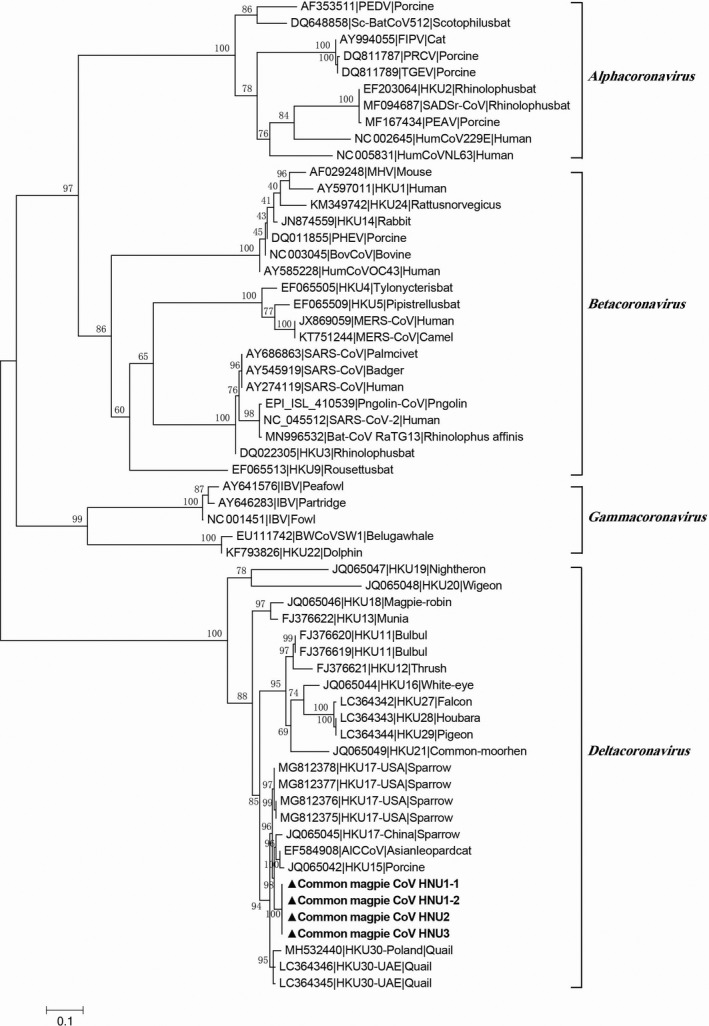
The maximum likelihood tree of C‐terminal domain fragment (261 amino acids) in RNA‐dependent RNA polymerase (RdRp). The tree was constructed using IQ‐treewith LG + I + G4 substitution model and 10,000 ultrafast bootstraps. The scale bar indicates amino acid substitutions per site. The four newly identified HNU CoVs are shown in triangle and bold
3.2. Genomic characterization of four novel common magpie deltacoronaviruses
To further understand the genetic diversity and structure of these bird deltacoronaviruses, complete genomic sequences of four novel common magpie CoVs (HNU1‐1, HNU1‐2, HNU2, HNU3) were successfully amplified with genome size ranging from 26,208 nt to 26,289 nt and G + C content ranging from 41.7% to 41.9%. The HNU1‐1 and HNU1‐2 genomes shared 99.8% nucleotide identity to each other but showed 93.2% and 92.7% identity to the stains HNU2 and HNU3, respectively. The identity between these CoVs and other avian CoVs was low (Tables S1 and S2). However, the genome organization of HNU CoVs was similar to other deltacoronaviruses and had the typical gene order of 5′‐UTR‐replicase ORF1ab, spike (S), envelope (E), membrane (M) and nucleocapsid (N)‐UTR‐3′. Moreover, additional ORFs coding for non‐structural (NS) proteins NS6, NS7a, NS7b and NS7c were identified. A putative transcription regulatory sequence (TRS) motif, 5′‐ACACCA‐3′ was detected, which is unique to deltacoronaviruses (Figure 2, Table S3).
FIGURE 2.
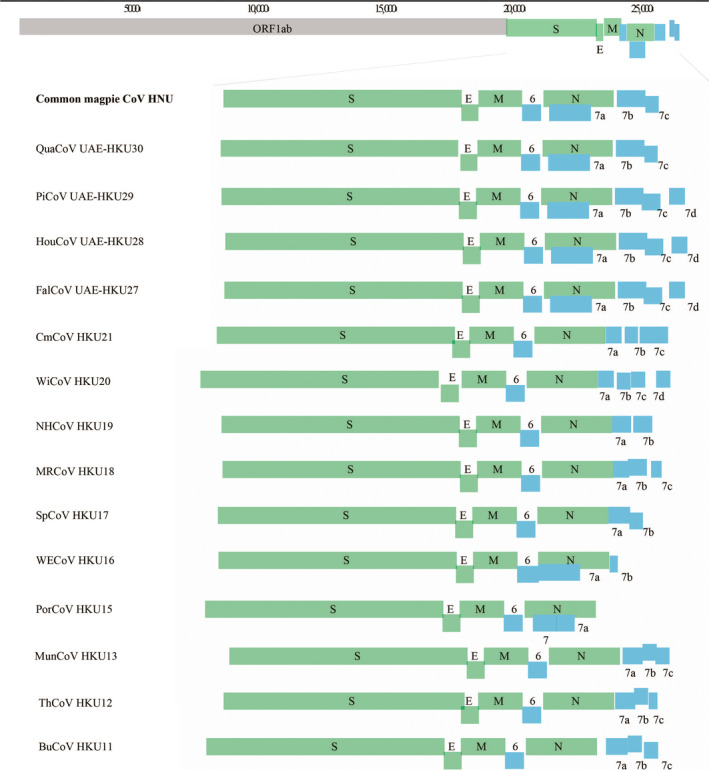
Genome organization of members of Deltacoronavirus. Open reading frames downstream of spike (S) gene are magnified to show the differences among the genomes of the 15 CoVs. S, envelope (E), membrane (M) and nucleocapsid (N) genes are represented by green boxes. Putative accessory proteins are represented by blue boxes. The novel HNU CoVs discovered in this study are shown in bold.
The coding potential and characteristics of putative non‐structural proteins (nsp's) coded by ORF1ab of HNU CoVs are shown in Table 2. The replicase ORF1ab polyprotein occupies 19.6 kb of the genome. This ORF encodes 16 putative non‐structural proteins, including nsp3 (putative papain‐like protease [PLpro]), nsp5 (putative chymotrypsin‐like protease [3CLpro]), nsp12 (putative RdRp), nsp13 (putative helicase) and other proteins with unclear functions. These proteins are produced via proteolytic cleavage of the large replicase polyprotein by PLpro and 3CLpro at specific sites. Especially, we found a putative cleavage site of AG/VP between nsp3 and nsp4 of HNU1‐1 and HNU1‐2 and another putative cleavage site of LQ/AG between nsp5 and nsp6 of all HNU CoVs.
TABLE 2.
Putative cleavage sites at the junctions between non‐structural proteins in four novel common magpie CoV compared with those in other deltacoronaviruses
| Cleavage site in | NSP pair | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NSP2/NSP3 | NSP3/NSP4 | NSP4/NSP5 | NSP5/NSP6 | NSP6/NSP7 | NSP7/NSP8 | NSP8/NSP9 | NSP9/NSP10 | NSP10/NSP11 | NSP12/NSP13 | NSP13/NSP14 | NSP14/NSP15 | NSP15/NSP16 | |
| JQ065042|HKU15|Porcine | AG/SD | AG/AP | LQ/AG | LQ/SG | VQ/NK | VQ/AV | LQ/NN | LQ/AS | LQ/NS | LQ/AS | LQ/SS | LQ/NL | LQ/SL |
| JQ065044|HKU16|White‐eye | AG/SD | AG/AR | LQ/AG | LQ/SN | VQ/NK | LQ/AV | IQ/NN | LQ/AN | LQ/GS | LQ/AS | LQ/SS | LQ/NL | VQ/SL |
| JQ065045|HKU17‐China|Sparrow | AG/SD | AG/AP | LQ/AG | LQ/SG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NS | LQ/AS | LQ/SS | LQ/NL | LQ/SL |
| JQ065046|HKU18|Magpie‐robin | AG/AD | AG/AM | LQ/AG | LQ/SG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NS | LQ/AS | LQ/AG | LQ/NL | LQ/SL |
| JQ065047|HKU19|Night heron | VG/GL | TG/GN | VQ/AG | LQ/GT | VQ/NK | LQ/VV | LQ/NN | LQ/SS | LQ/LG | LQ/AT | VQ/SL | LQ/TL | VQ/AL |
| JQ065048|HKU20|Wigeon | DG/VY | GG/SK | VQ/SG | LQ/AN | VQ/NR | LQ/VV | CQ/NN | LQ/AN | LQ/SN | LQ/AT | VQ/AE | LQ/TL | LQ/SL |
| JQ065049|HKU21|Common‐moorhen | AG/VS | AG/KF | LQ/AG | LQ/AS | VQ/NR | LQ/AV | IQ/NN | LQ/AT | LQ/NT | LQ/AS | VQ/CS | LQ/TI | VQ/SL |
| LC364342|HKU27|Falcon | AG/SD | AG/RK | LQ/AG | LQ/SG | VQ/NR | LQ/AV | LQ/NN | LQ/AN | LQ/GS | LQ/AS | LQ/SS | LQ/NL | VQ/AL |
| LC364343|HKU28|Houbara | AG/SD | AG/RK | LQ/AG | LQ/SG | VQ/NR | LQ/AV | LQ/NN | LQ/AN | LQ/GS | LQ/AS | LQ/SS | LQ/NL | VQ/AL |
| LC364344|HKU29|Pigeon | AG/SD | AG/RK | LQ/AG | LQ/SG | VQ/NR | LQ/AV | LQ/NN | LQ/AN | LQ/GS | LQ/AS | LQ/SS | LQ/NL | VQ/AL |
| LC364345|HKU30‐UAE|Quail | AG/SD | AG/AP | LQ/AG | LQ/SG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NS | LQ/AS | LQ/SG | LQ/NL | LQ/SL |
| MH532440|HKU30‐Poland|Quail | AG/SD | AG/VP | LQ/AG | LQ/SG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NA | LQ/AS | LQ/SG | LQ/NL | LQ/SL |
| Common magpie CoV HNU1−1 | AG/SD | AG/VP | LQ/AG | LQ/AG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NA | LQ/AS | LQ/SG | LQ/NL | LQ/SL |
| Common magpie CoV HNU1−2 | AG/SD | AG/VP | LQ/AG | LQ/AG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NA | LQ/AS | LQ/SG | LQ/NL | LQ/SL |
| Common magpie CoV HNU2 | AG/SD | AG/AP | LQ/AG | LQ/AG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NS | LQ/AS | LQ/SG | LQ/NL | LQ/SL |
| Common magpie CoV HNU3 | AG/SD | AG/AP | LQ/AG | LQ/AG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NS | LQ/AS | LQ/SG | LQ/NL | LQ/SL |
| MG812375|HKU17‐USA|Sparrow | AG/SD | AG/AP | LQ/AG | LQ/SG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NS | LQ/AS | LQ/SG | LQ/NL | LQ/SL |
| FJ376619|HKU11|Bulbul | AG/SD | AG/SD | LQ/AG | LQ/SG | VQ/NK | LQ/AV | IQ/NN | LQ/AN | LQ/NS | LQ/AS | LQ/SG | LQ/NL | VQ/SL |
| FJ376621|HKU12|Thrush | AG/SD | AG/SA | LQ/AG | LQ/SG | VQ/NK | LQ/AV | IQ/NN | LQ/AN | LQ/SS | LQ/AS | VQ/SS | LQ/NL | VQ/SL |
| FJ376622|HKU13|Munia | AG/AD | AG/AV | LQ/AG | LQ/SG | VQ/NK | VQ/AV | LQ/NN | LQ/AN | LQ/NS | LQ/AS | LQ/AG | LQ/NL | LQ/SL |
The bold values mean the sequences of the cleavage sites.
We further compared the aa identity of the seven conserved replicase domains for species demarcation (ADRP, nsp5 [3CLpro], nsp12 [RdRp], nsp13 [Hel], nsp14 [ExoN], nsp15 [NendoU] and nsp16 [O‐MT]) among the HNU CoVs. As shown in Table S4, in all the seven domains and overall, their aa sequences showed more than 90% identity to those of PDCoV HKU15, SpCoV HKU17 and QuaCoV HKU30, indicating that these CoVs should be subspecies of the same species.
3.3. Phylogenetic analyses of the novel bird deltacoronaviruses
The phylogenetic trees reconstructed using the amino acid sequences of ORF1ab, 3CLpro, Hel, S and N of the four novel HNU CoVs and other CoVs (Figure 3, Table S1). Except the S gene, the four novel HNU CoVs were clustered together. For ORF1ab, 3CLpro, Hel and N gene, the four HNU CoVs were clustered with PDCoV HKU15, QuaCoV UAE‐HKU30 and SpCoV HKU17, with amino acid identity higher than 85%. As for S gene, HNU1‐1 and HNU1‐2 were clustered with ThCoV HKU12, with 93.8% and 93.7% amino acid identity, respectively. HNU2 was clustered with WECoV HKU16, with 71.9% amino acid identity. And HNU3 was more closely related to SpCoV HKU17‐USA and PDCoV HKU15 than BuCoV HKU11.
FIGURE 3.
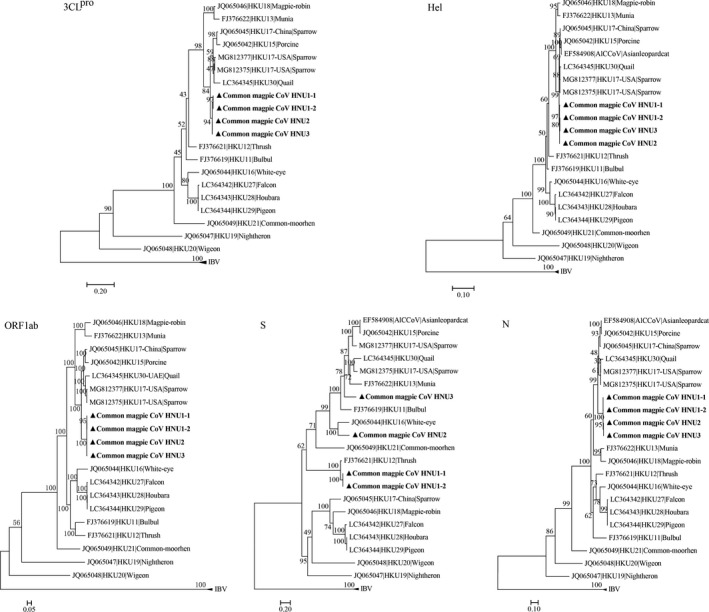
The maximum likelihood tree of amino acid sequences of chymotrypsin‐like protease (3CLpro), helicase (Hel), polyprotein 1ab (ORF1ab), spike (S) protein and nucleocapsid (N) protein. These trees were constructed using IQ‐tree with 10,000 ultrafast bootstraps, and their substitution models were as follows: LG + G4 (3CLpro), LG + F + R2 (Hel), LG + F + R5 (ORF1ab), WAG + I + G4 + F (S), LG + F + G4 (N). The scale bar indicates amino acid substitutions per site. The four newly identified HNU CoVs are shown in triangle and bold.
3.4. Recombinant events of HNU1 and HNU2 coronaviruses
Interestingly, switches of positions in phylogenetic trees based on different genes were observed for the four HNU CoVs, indicating that recombination events may have occurred frequently during the virus evolution. The full‐length genome sequences of all HNU CoVs in this study were screened for potential recombination events. The best possible recombination events in HNU1‐1 (or HNU1‐2) and HNU2 were identified by the GENECONV (p‐value: 2.263E‐307) and RDP (p‐value: 1.739E‐8) methods provided by RDP v3.44 program. These recombination events with strong p‐value were further confirmed in Simplot v3.5.1 program (Figure 4). The results suggested that HNU1‐1 and HNU1‐2 were likely to be recombinant strains from SpCoV HKU17‐USA and ThCoV HKU12, and the break points were identified at genome positions nt 21,017 and 25,056, which were closed to the start and end region of S protein (Figure 4a). Meanwhile, the newly identified QuaCoV UAE‐HKU30 and the previously reported WECoV HKU16 were suggested to be the major and minor parent of HNU2, and there are two break points nt 21,101 and 24,501 in HNU2, which covered a part of the S1 and the S2 of the S protein (Figure 4b). However, there is no obvious evidence showing that HNU3 was a recombinant strain (data were not show).
FIGURE 4.
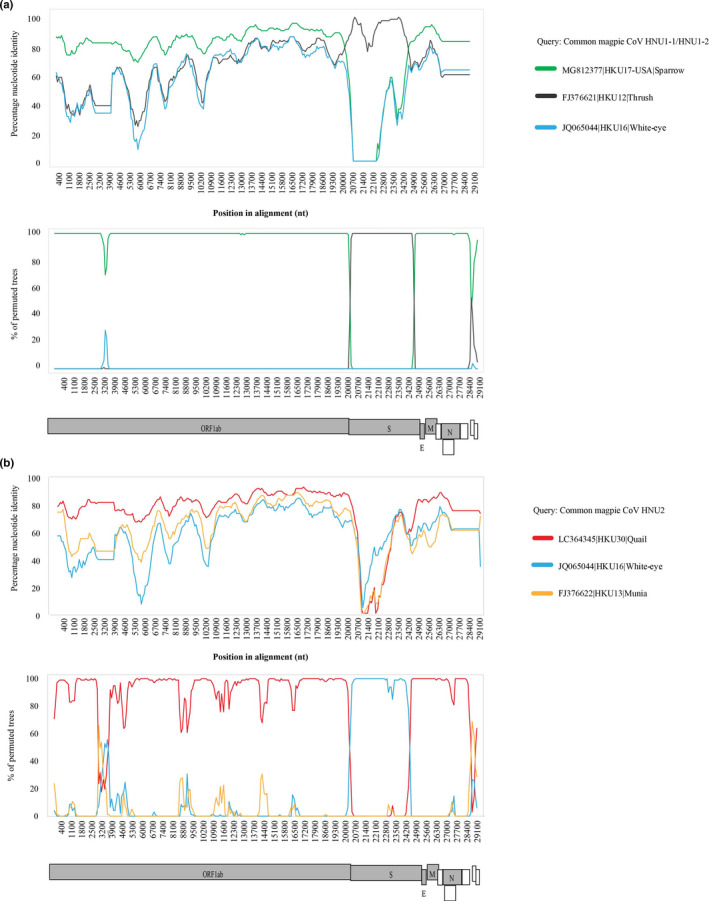
Detection of potential recombination events of novel HNU CoVs. (a) The possible recombination events in HNU1‐1 or HNU1‐2 strain. (b) The possible recombination events in HNU2. Both of Similarity Plot and Bootscan method in Simplot v3.5.1 were performed with an F84 distance model, a window size of 800 base pairs and a step size of 100 base pairs. The bootstrap replicates of Bootscan were set to 1,000.
3.5. Bayesian phylogeography of bird deltacoronaviruses
Molecular clock analysis was conducted to trace the temporal patterns of bird deltacoronaviruses evolution. The mean evolutionary rate was estimated as approximately 1.541 × 10−4/site/year (95% highest posterior density intervals or HPDs, 8.25 × 10–5–2.00 × 10–4) for the RdRp gene. Molecular clock analysis using the RdRp gene showed that the mean time to the most recent common ancestor (tMRCA) of bird deltacoronavirus was estimated to be 3,000 BC (95% HPDs, 8,119 BC to 1,414), and the root state probability distribution indicated that the USA was the probable original location with a posterior probability of 0.21652. The tMRCA of four novel magpie CoVs in this study was estimated to be 1,200 (95% HPDs, 590 to 1,504) and originated from Hong Kong (Figure 5). The BSSVS analysis filtered out three significant south‐to‐north migration links (BF > 3) in the diffusion processes of all bird deltacoronavirus worldwide, including the paths from Hong Kong to USA, from Hong Kong to Hunan and from UAE to Poland (Table 3). But it should be noted that the mean indicators of the three migration paths were lower than 0.5, which indicates that more bird coronavirus genome sequences needed to predict the more convincing migration paths.
FIGURE 5.
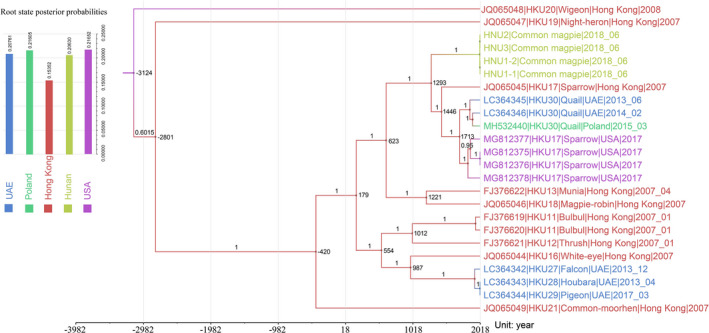
Bayesian phylogeographic analysis of 24 bird deltacoronaviruses strain sequences based on RdRp gene. Maximum clade credibility (MCC) tree with ancestral state reconstruction according to a discrete trait model, the substitution model was GTR + F + R3 which obtained by Model Finder under the BIC standard, the colours of branch and internal nodes indicated the most probable state reconstruction. The numbers on branch indicated posterior probability, and the time of divergence estimated was marked near the internal node. The root state posterior probabilities estimated for each regions of Bayesian phylogeographic MCC tree, which were inferred using an asymmetric substitution model.
TABLE 3.
Migration path and statistical of bird deltacoronaviruses
| Migration path | Mean migration rates | Mean indicators | Bayes factor |
|---|---|---|---|
| Hong Kong to Hunan | 0.8882 | 0.1656 | 3.713958182 |
| Hong Kong to Poland | 0.8268 | 0.265 | 2.161450775 |
| Hong Kong to UAE | 0.8727 | 0.3367 | 1.458047601 |
| Hong Kong to USA | 0.849 | 0.3077 | 3.65601402 |
| Hunan to Poland | 0.8672 | 0.3792 | −3.486333958 |
| Hunan to UAE | 0.9212 | 0.3709 | −5.206512576 |
| Hunan to USA | 0.9013 | 0.3707 | 0.061473914 |
| Hunan to Hong Kong | 0.8916 | 0.3089 | −3.586996146 |
| Poland to UAE | 1.428 | 0.5297 | 0.057528801 |
| Poland to USA | 1.3884 | 0.5285 | 0.054435634 |
| Poland to Hunan | 1.1547 | 0.5324 | 0.187082157 |
| Poland to Hong Kong | 1.1752 | 0.5394 | 0.169848657 |
| UAE to USA | 0.9557 | 0.4547 | 0.056965978 |
| UAE to Hong Kong | 1.0401 | 0.3986 | 0.07251349 |
| UAE to Hunan | 0.8937 | 0.3266 | −3.730958555 |
| UAE to Poland | 0.8739 | 0.4187 | 3.022568883 |
| USA to Hong Kong | 1.0466 | 0.4232 | 0.059406257 |
| USA to Hunan | 0.9111 | 0.3761 | 2.505014699 |
| USA to Poland | 0.9218 | 0.4344 | 2.349057809 |
| USA to UAE | 1.0719 | 0.481 | 1.965893096 |
4. DISCUSSION
Monitoring existence of viruses in wild animals is important for tracing viral transmission and controlling epidemics. Before transmitting to humans, the viruses mainly survive, transmit and evolve in animals, including their natural reservoirs and intermediate hosts. Birds are not only the reservoir animals for various viruses but also likely to be intermediate hosts of viruses, since birds are widely distributed around the world and capable of long‐distance migration, leading to frequent contact with human and domestic animals. In this study, we identified and characterized four common magpie CoVs belonging to deltacoronavirus genus from birds in central China. Deltacoronaviruses are widely distributed worldwide (Lee & Lee, 2014; Marthaler et al., 2014; Wang et al., 2014; Wang et al., 2015). The Coronaviridae Study Group of ICTV has established the following genus and species demarcation criteria in the family Coronaviridae: coronaviruses that cluster together and share more than 90% amino acid sequence identity in seven conserved replicase domains (ADRP, nsp5 [3CLpro], nsp12 [RdRp], nsp13 [Hel], nsp14 [ExoN], nsp15 [NendoU] and nsp16 [O‐MT]) are considered to belong to the same species (Lau et al., 2018). Hence, the four novel HNU CoVs belong to the same species as PDCoV HKU15, SpCoV HKU17 and QuaCoV HKU30. Notably, common magpies, sparrows and quails are small terrestrial birds with global distribution. Genome comparison showed that deltacoronaviruses from terrestrial birds share higher similarity with PDCoV, suggesting terrestrial birds may serve as the intermediate host for CoVs. These data suggested that birds are natural reservoirs which provide viral genes for evolution and interspecies transmission of viruses. Furthermore, our study provides evidence for genetic recombination among bird deltacoronaviruses. More specifically, evidence for at least two recombination events was observed in the S genes of the four novel HNU CoVs, including a recombination event between viruses in the HNU1‐1 (or HNU1‐2) and HKU12 that allowed the two viruses to acquire S from HKU12, and another recombination between viruses in the HNU2 and HKU16 clusters that allowed the two viruses to acquire partial S1 and an S2 subunit from HKU16 (Figure 4). These recombination events likely occurred in bird hosts. Similar to the case for birds in Hong Kong and Middle East, birds in mainland China are also important hosts for a diversity of deltacoronaviruses. Continuous surveillance studies on birds in other city of China will help to better understand the viral and host diversity of deltacoronaviruses and their potential for emergence in mammals.
At present, the genus Deltacoronavirus is subdivided into four subgenera and seven species (http://ictv.global/report). These virus species have been sampled from swine, as well as a variety of birds, including 14 bird species. In all, based on the known data of 24 deltacoronavirus isolates and their hosts from five different regions around the world, we investigated the molecular epidemiology of bird deltacoronavirus using Bayesian phylogeographic inference. Our phylogeographic analysis revealed a general south‐to‐north dispersal of bird deltacoronavirus. In particular, Hong Kong is a possible origin of bird deltacoronaviruses in central China. However, the three significant migration links (BF > 3) are in a lower value of mean indicators, suggesting that the dispersal pattern might be associated with the number of bird deltacoronavirus. More CoVs strains are needed to predict the root cause of avian coronavirus before more convincing evidence can be obtained.
In summary, our study indicates the widespread existence and high diversity of environmental deltacoronaviruses in birds, and some of them are classified into the same species as PDCoV. In order to understand the evolutionary history of bird deltacoronavirus and to prevent future emerging infectious diseases, it is critical to take extensive and long‐term surveillance on the environmental deltacoronaviruses.
CONFLICT OF INTEREST
The authors declare no competing interests.
ETHICAL APPROVAL
The ethical statement is not available since no sample collection or questionnaires from animals/human has been gathered.
Supporting information
Fig S1
Table S1‐S5
ACKNOWLEDGEMENTS
This work was jointly funded by the National Key Research and Development Program of China (grant number 2017YFD0500104), National Natural Science Foundation of China (grant number 32041001 and 81902070) and the Provincial Natural Science Foundation of Hunan Province (grant number 2019JJ20004, 2019JJ50035).
Wang Q, Zhou Z‐J, You Z, et al. Epidemiology and evolution of novel deltacoronaviruses in birds in central China. Transbound Emerg Dis.2022;69:632–644. 10.1111/tbed.14029
Qiong Wang and Zhi‐Jian Zhou contributed equally.
Contributor Information
Ye Qiu, Email: qiuye@hnu.edu.cn.
Xing‐Yi Ge, Email: xyge@hnu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Baele, G. , Lemey, P. , Bedford, T. , Rambaut, A. , Suchard, M. A. , & Alekseyenko, A. V. (2012). Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Molecular Biology and Evolution, 29(9), 2157–2167. 10.1093/molbev/mss084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brian, D. A. , & Baric, R. S. (2005). Coronavirus genome structure and replication. Current Topics in Microbiology and Immunology, 287, 1–30. 10.1007/3-540-26765-4_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, J. F. , To, K. K. , Chen, H. , & Yuen, K. Y. (2015). Cross‐species transmission and emergence of novel viruses from birds. Current Opinion in Virology, 10, 63–69. 10.1016/j.coviro.2015.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, J. F. , To, K. K. , Tse, H. , Jin, D. Y. , & Yuen, K. Y. (2013). Interspecies transmission and emergence of novel viruses: Lessons from bats and birds. Trends in Microbiology, 21(10), 544–555. 10.1016/j.tim.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q. I. , Wang, L. , Yang, C. , Zheng, Y. , Gauger, P. C. , Anderson, T. , Harmon, K. M. , Zhang, J. , Yoon, K.‐J. , Main, R. G. , & Li, G. (2018). The emergence of novel sparrow deltacoronaviruses in the United States more closely related to porcine deltacoronaviruses than sparrow deltacoronavirus HKU17. Emerging Microbes & Infections, 7(1), 105. 10.1038/s41426-018-0108-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh, T. D. , Ngatia, J. N. , Cui, L. Y. , Ma, Y. , Dhamer, T. D. , & Xu, Y. C. (2019). Influence of pairwise genetic distance computation and reference sample size on the reliability of species identification using Cyt b and COI gene fragments in a group of native passerines. Forensic Science International: Genetics, 40, 85–95. 10.1016/j.fsigen.2019.02.013 [DOI] [PubMed] [Google Scholar]
- Dong, B. Q. , Liu, W. , Fan, X. H. , Vijaykrishna, D. , Tang, X. C. , Gao, F. , Li, L. F. , Li, G. J. , Zhang, J. X. , Yang, L. Q. , Poon, L. L. M. , Zhang, S. Y. , Peiris, J. S. M. , Smith, G. J. D. , Chen, H. , & Guan, Y. (2007). Detection of a novel and highly divergent coronavirus from asian leopard cats and Chinese ferret badgers in Southern China. Journal of Virology, 81(13), 6920–6926. 10.1128/JVI.00299-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J. , Rambaut, A. , Shapiro, B. , & Pybus, O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular Biology and Evolution, 22(5), 1185–1192. 10.1093/molbev/msi103 [DOI] [PubMed] [Google Scholar]
- Forni, D. , Cagliani, R. , Clerici, M. , & Sironi, M. (2017). Molecular evolution of human coronavirus genomes. Trends in Microbiology, 25(1), 35–48. 10.1016/j.tim.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. , Wang, Y. , Li, X. , Ren, L. , Zhao, J. , Hu, Y. I. , Zhang, L. I. , Fan, G. , Xu, J. , Gu, X. , Cheng, Z. , Yu, T. , Xia, J. , Wei, Y. , Wu, W. , Xie, X. , Yin, W. , Li, H. , Liu, M. , … Cao, B. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet, 395, 497–506. 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katarzyna, D.‐B. , Kuczkowski, M. , & Sajewicz‐Krukowska, J. (2019). Whole genome characterisation of quail deltacoronavirus detected in Poland. Virus Genes, 55(2), 243–247. 10.1007/s11262-019-01639-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, S. K. P. , Wong, E. Y. M. , Tsang, C.‐C. , Ahmed, S. S. , Au‐Yeung, R. K. H. , Yuen, K.‐Y. , Wernery, U. , & Woo, P. C. Y. (2018). Discovery and sequence analysis of four deltacoronaviruses from birds in the middle east reveal interspecies jumping with recombination as a potential mechanism for avian‐to‐avian and avian‐to‐mammalian transmission. Journal of Virology, 92(15), 1–18. 10.1128/JVI.00265-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, S. K. P. , Woo, P. C. Y. , Li, K. S. M. , Tsang, A. K. L. , Fan, R. Y. Y. , Luk, H. K. H. , Cai, J.‐P. , Chan, K.‐H. , Zheng, B.‐J. , Wang, M. , & Yuen, K.‐Y. (2015). Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports the murine origin of Betacoronavirus 1 and has implications for the ancestor of Betacoronavirus lineage A. Journal of Virology, 89(6), 3076–3092. 10.1128/JVI.02420-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. S. , Cau, A. , Naish, D. , & Dyke, G. J. (2014). Dinosaur evolution. Sustained miniaturization and anatomical innovation in the dinosaurian ancestors of birds. Science, 345(6196), 562–566. 10.1126/science.1252243 [DOI] [PubMed] [Google Scholar]
- Lee, S. , & Lee, C. (2014). Complete genome characterization of korean porcine deltacoronavirus strain KOR/KNU14‐04/2014. Genome Announcements, 2(6), 10.1128/genomeA.01191-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey, P. , Rambaut, A. , Drummond, A. J. , & Suchard, M. A. (2009). Bayesian phylogeography finds its roots. PLoS Computational Biology, 5(9), e1000520. 10.1371/journal.pcbi.1000520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. M. , Saif, L. J. , Marthaler, D. , & Wang, Q. (2016). Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Research, 226, 20–39. 10.1016/j.virusres.2016.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, R. , Zhao, X. , Li, J. , Niu, P. , Yang, B. O. , Wu, H. , Wang, W. , Song, H. , Huang, B. , Zhu, N. A. , Bi, Y. , Ma, X. , Zhan, F. , Wang, L. , Hu, T. , Zhou, H. , Hu, Z. , Zhou, W. , Zhao, L. I. , … Tan, W. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet, 395(10224), 565–574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthaler, D. , Raymond, L. , Jiang, Y. , Collins, J. , Rossow, K. , & Rovira, A. (2014). Rapid detection, complete genome sequencing, and phylogenetic analysis of porcine deltacoronavirus. Emerging Infectious Diseases, 20(8), 1347–1350. 10.3201/eid2008.140526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. P. , Lemey, P. , Lott, M. , Moulton, V. , Posada, D. , & Lefeuvre, P. (2010). RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics, 26(19), 2462–2463. 10.1093/bioinformatics/btq467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno, A. , Franzo, G. , Massi, P. , Tosi, G. , Blanco, A. , Antilles, N. , Biarnes, M. , Majó, N. , Nofrarías, M. , Dolz, R. , Lelli, D. , Sozzi, E. , Lavazza, A. , & Cecchinato, M. (2017). A novel variant of the infectious bronchitis virus resulting from recombination events in Italy and Spain. Avian Pathology, 46(1), 28–35. 10.1080/03079457.2016.1200011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L. T. , Schmidt, H. A. , von Haeseler, A. , & Minh, B. Q. (2015). IQ‐TREE: A fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32(1), 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederwerder, M. C. , & Hesse, R. A. (2018). Swine enteric coronavirus disease: A review of 4 years with porcine epidemic diarrhoea virus and porcine deltacoronavirus in the United States and Canada. Transboundary and Emerging Diseases, 65(3), 660–675. 10.1111/tbed.12823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. , Drummond, A. J. , Xie, D. , Baele, G. , & Suchard, M. A. (2018). Posterior summarization in bayesian phylogenetics using tracer 1.7. Systematic Biology, 67(5), 901–904. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailleau, C. , Dumarest, M. , Vanhomwegen, J. , Delaplace, M. , Caro, V. , Kwasiborski, A. , Hourdel, V. , Chevaillier, P. , Barbarino, A. , Comtet, L. , Pourquier, P. , Klonjkowski, B. , Manuguerra, J.‐C. , Zientara, S. , & Le Poder, S. (2020). First detection and genome sequencing of SARS‐CoV‐2 in an infected cat in France. Transboundary and Emerging Diseases, 67, 2324–2328. 10.1111/tbed.13659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, J. , Wen, Z. , Zhong, G. , Yang, H. , Wang, C. , Huang, B. , Liu, R. , He, X. , Shuai, L. , Sun, Z. , Zhao, Y. , Liu, P. , Liang, L. , Cui, P. , Wang, J. , Zhang, X. , Guan, Y. , Tan, W. , Wu, G. , … Bu, Z. (2020). Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS‐coronavirus 2. Science, 368(6494), 1016–1020. 10.1126/science.abb7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Z. , & Hu, Z. (2008). A review of studies on animal reservoirs of the SARS coronavirus. Virus Research, 133(1), 74–87. 10.1016/j.virusres.2007.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit, T. H. C. , Brackman, C. J. , Ip, S. M. , Tam, K. W. S. , Law, P. Y. T. , To, E. M. W. , Yu, V. Y. T. , Sims, L. D. , Tsang, D. N. C. , Chu, D. K. W. , Perera, R. A. P. M. , Poon, L. L. M. , & Peiris, M. (2020). Infection of dogs with SARS‐CoV‐2. Nature, 586, 776–778. 10.1038/s41586-020-2334-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, S. , Wong, G. , Shi, W. , Liu, J. , Lai, A. C. K. , Zhou, J. , Liu, W. , Bi, Y. , & Gao, G. F. (2016). Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends in Microbiology, 24(6), 490–502. 10.1016/j.tim.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Byrum, B. , & Zhang, Y. (2014). Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerging Infectious Diseases, 20(7), 1227–1230. 10.3201/eid2007.140296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. P. , Geng, R. Q. , & Liu, Z. Q. (2015). An effective strategy for species identification of avian meats using the mitochondrial 12S rRNA gene fragment. Mitochondrial DNA, 26(2), 171–174. 10.3109/19401736.2013.845769 [DOI] [PubMed] [Google Scholar]
- Wang, Y. W. , Yue, H. , Fang, W. , & Huang, Y. W. (2015). Complete genome sequence of porcine deltacoronavirus strain CH/Sichuan/S27/2012 from mainland China. Genome Announcements, 3(5), 1–2. 10.1128/genomeA.00945-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, A. C. P. , Li, X. , Lau, S. K. P. , & Woo, P. C. Y. (2019). Global epidemiology of bat coronaviruses. Viruses, 11(2), 174. 10.3390/v11020174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. Y. , Lau, S. K. P. , Lam, C. S. F. , Lai, K. K. Y. , Huang, Y. I. , Lee, P. , Luk, G. S. M. , Dyrting, K. C. , Chan, K.‐H. , & Yuen, K.‐Y. (2009). Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. Journal of Virology, 83(2), 908–917. 10.1128/JVI.01977-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. Y. , Lau, S. K. P. , Lam, C. S. F. , Lau, C. C. Y. , Tsang, A. K. L. , Lau, J. H. N. , Bai, R. , Teng, J. L. L. , Tsang, C. C. C. , Wang, M. , Zheng, B.‐J. , Chan, K.‐H. , & Yuen, K.‐Y. (2012). Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. Journal of Virology, 86(7), 3995–4008. 10.1128/JVI.06540-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. Y. , Lau, S. K. P. , Lam, C. S. F. , Tsang, A. K. L. , Hui, S.‐W. , Fan, R. Y. Y. , Martelli, P. , & Yuen, K.‐Y. (2014). Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in Gammacoronavirus. Journal of Virology, 88(2), 1318–1331. 10.1128/JVI.02351-13 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Woo, P. C. , Lau, S. K. , Yip, C. C. , Huang, Y. , Tsoi, H. W. , Chan, K. H. , & Yuen, K. Y. (2006). Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. Journal of Virology, 80(14), 7136–7145. 10.1128/JVI.00509-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. , Zhong, H. , Zhou, Q. , Du, Y. , Chen, L. I. , Zhang, Y. , Xue, C. , & Cao, Y. (2018). A highly pathogenic strain of porcine deltacoronavirus caused watery diarrhea in newborn piglets. Virologica Sinica, 33(2), 131–141. 10.1007/s12250-018-0003-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. , Gao, F. , Jakovlić, I. , Zou, H. , Zhang, J. , Li, W. X. , & Wang, G. T. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Molecular Ecology Resources, 20(1), 348–355. 10.1111/1755-0998.13096 [DOI] [PubMed] [Google Scholar]
- Zhong, N. S. , Zheng, B. J. , Li, Y. M. , Poon, L. , Xie, Z. H. , Chan, K. H. , Li, P. H. , Tan, S. Y. , Chang, Q. , Xie, J. P. , Liu, X. Q. , Xu, J. , Li, D. X. , Yuen, K. Y. , Peiris, J. , & Guan, Y. (2003). Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet, 362(9393), 1353–1358. 10.1016/s0140-6736(03)14630-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Fan, H. , Lan, T. , Yang, X.‐L. , Shi, W.‐F. , Zhang, W. , Zhu, Y. , Zhang, Y.‐W. , Xie, Q.‐M. , Mani, S. , Zheng, X.‐S. , Li, B. , Li, J.‐M. , Guo, H. , Pei, G.‐Q. , An, X.‐P. , Chen, J.‐W. , Zhou, L. , Mai, K.‐J. , … Ma, J.‐Y. (2018). Fatal swine acute diarrhoea syndrome caused by an HKU2‐related coronavirus of bat origin. Nature, 556(7700), 255–258. 10.1038/s41586-018-0010-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Yang, X.‐L. , Wang, X.‐G. , Hu, B. , Zhang, L. , Zhang, W. , Si, H.‐R. , Zhu, Y. , Li, B. , Huang, C.‐L. , Chen, H.‐D. , Chen, J. , Luo, Y. , Guo, H. , Jiang, R.‐D. , Liu, M.‐Q. , Chen, Y. , Shen, X.‐R. , Wang, X. I. , … Shi, Z.‐L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z.‐J. , Qiu, Y. , Pu, Y. , Huang, X. , & Ge, X.‐Y. (2020). BioAider: An efficient tool for viral genome analysis and its application in tracing SARS‐CoV‐2 transmission. Sustainable Cities and Society, 63, 102466. 10.1016/j.scs.2020.102466 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1‐S5
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.