

Abstract
The analysis of genetic diversity in SARS‐CoV‐2 is the focus of several studies, providing insights into how the virus emerged and evolves. Most common changes in SARS‐CoV‐2 are single or point nucleotide substitutions; meanwhile, insertions and deletions (indels) have been identified as a less frequent source of viral genetic variability. Here, we report the emergence of a 12‐nucleotide deletion in ORF7a, resulting in a 4‐amino acid in‐frame deletion. The Δ12 variant was identified in viruses from patients of a single outbreak and represents the first report of this deletion in South American isolates. Phylogenetic analysis revealed that Δ12 strains belong to the lineage B.1.1 and clustered separated from the remaining Uruguayan strains. The ∆12 variant was detected in 14 patients of this outbreak by NGS sequencing and/or two rapid and economic methodologies: Sanger amplicon sequencing and capillary electrophoresis. The presence of strong molecular markers as the deletion described here are useful for tracking outbreaks and reveal a significant aspect of the SARS‐CoV‐2 evolution on the robustness of the virus to keep its functionality regardless loss of genetic material.
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is responsible for the global pandemic of coronavirus disease 2019 (COVID‐19), the most serious threat to human health in the 21st century with a huge social and economic impact worldwide. SARS‐CoV‐2 (order Nidovirales, family Coronaviridae) is an enveloped, positive‐sense single‐stranded RNA virus with the largest known RNA genome (~ 30 kb) (Gorbalenya et al., 2006). SARS‐CoV‐2 exhibits a significant genetic diversity (Khailany et al., 2020) which is likely consequence of its rapid spreading in the naïve human hosts. To provide insights into how the virus emerged and evolves, a great research effort has been done to describe and understand SARS‐CoV‐2 genetic variants.
Tracing virus variability through genome sequencing is an analytical tool to assist epidemiological studies, including the identification of emergent variants with potential impact on virus control and vaccine development. With more than 200.000 complete genomes already available, several nucleotide changes are described in open reading frames (ORFs) coding for non‐structural (nsp1‐16), structural (spike, matrix, small envelope and nucleocapsid) and accessory (3a, 6, 7a, 7b, 8 and 10) proteins (Wu et al., 2020).
Most common changes in SARS‐CoV‐2 are single or point nucleotide substitutions (SNPs) (Badua et al., 2020; Franco‐Muñoz et al., 2020). Insertions and deletions (indels) constitute another relevant source of viral genetic variability. Indels reveal an additional aspect of genomic evolution and provide information about the robustness of the virus to maintain its functionality regardless gain and loss of genetic material. Indels are also useful and stable markers to use in conjunction with SNPs to trace origin and spreading of the SARS‐CoV‐2 outbreaks.
Short indels are unevenly distributed in the SARS‐CoV‐2 genome and are particularly common in the S gene that codes for S1 and S2 domain of the spike glycoprotein (Liu, Jiang, et al., 2020). Beside a role in intraspecific variability, indels have also been involved in the emergence of SARS‐CoV‐2. There is a unique 12‐nucleotide insertion, immediately upstream of the S1/S2 boundary that leads to a predictively solvent‐exposed PRRAR|SV sequence, which corresponds to a canonical furin‐like cleavage site that might affect the tissue tropism and/or transmissibility (Walls et al., 2020). Besides, deletion mutants involving this cleavage site are stable and replicative but it seems to be attenuated in the hamster infection model (Lau et al., 2020; Liu, Jiang, et al., 2020).
Indels occur in other structural and non‐structural coding regions, including nucleocapsid (N) and, less frequently, envelope (E) gene and in the ORF1ab (nsp1, 2 and 3) (Bal et al., 2020; Benedetti et al., 2020; Kumar et al., 2020). Accessory genes also harbour indels, particularly in the ORFs 3a, 7a and 8 (Gong et al., 2020; Holland et al., 2020; Su et al., 2020). Accessory genes of SARS‐CoV‐2 are dispensable for replication in cell culture but may have regulatory roles in several steps of the viral cycle and thus contribute to the virus fitness (Narayanan et al., 2008). Tracking indels in accessory genes is interesting because it could provide some clues about the functionality and evolution of these regions (Zinzula, 2020). Indels are also relevant because they may represent evidences of the ongoing viral adaptation of the virus to humans through natural attenuation (Lau et al., 2020).
As far as we know, there are not reports in South America that describe and characterize SARS‐CoV‐2 strains containing deletions in accessory genes. Here, we report the emergence of a 12‐nucleotide deletion in ORF7a that is exclusively shared by all the viruses coming from a single outbreak in Montevideo, Uruguay.
2. MATERIALS AND METHODS
Fourteen COVID‐19 case patients from a single outbreak were diagnosed as part of the ongoing laboratory surveillance at the Centro Nacional de Referencia de Influenza y otros Virus Respiratorios, Departamento de Laboratorios de Salud Pública del MSP (DLSP‐MSP) in Montevideo, Uruguay. Combined nasopharyngeal and oropharyngeal swab samples were obtained, and diagnosis was performed by RNA extraction (Qiamp Viral RNA minikit, Qiagen USA) followed by real‐time reverse transcription–polymerase chain reaction (RT‐qPCR) using the protocol recommended by the Panamerican Health Organization (PAHO‐WHO) (Corman et al., 2020). SARS‐CoV‐2 RNA samples were then submitted to the Genomic Platform at the Facultad de Ciencias, Universidad de la República Uruguay (UdelaR), for genome sequencing and molecular characterization.
Copy DNA (cDNA) was obtained by retro‐transcription with 12 µl of total RNA, Superscript II® reverse transcriptase (Thermo Fisher, USA) and random primers. On seven samples, multiplex PCR was performed using ARTIC primer scheme version 3 which produces 406 bp amplicons covering ~ 98% of SARS‐CoV‐2 genome; information on the primer sequences and protocols are available at the ARTIC network repository (https://artic.network/ncov‐2019). The Nextera DNA Flex Library Preparation kit (Illumina, USA) with dual indexing was used for library preparation. The libraries were purified with AMPure XP beads (Beckman Coulter, USA). All quantifications were carried out using a Qubit DNA High‐Sensitivity kit (Invitrogen, USA). Control quality and length of the library were assessed on a Fragment Analyzer 5,200 system (Agilent Technologies, USA) using the Standard Sensitivity NGS Analysis Kit (Agilent Technologies, USA). Whole genome sequencing was performed on an Illumina MiniSeq (Illumina, USA) platform using MiniSeqTM Mid Output Reagent Cartridge (300 cycles, paired‐end reads). Adapter/quality trimming and filtering of raw data were performed with BBDuk, and clean reads were mapped to the consensus genome using Geneious Prime 2020.1.2 (https://www.geneious.com).
The deletion was confirmed on the fourteen samples using RT‐PCR with Artic primers flanking the deleted region (forward: TCACTACCAAGAGTGTGTTAGAGGT, reverse: TTCAAGTGAGAACCAAAAGATAATAAGCA) and further Sanger sequencing of the generated amplicon (~ 406 bp) in Macrogen (Korea). Amplicons were also subjected to capillary electrophoresis on a Fragment Analyzer 5,200 system using the High Sensitivity NGS Analysis Kit (Agilent Technologies, USA) to detect size differences between deleted and wild‐type viruses.
The online web application CoV‐GLUE (http://cov‐glue.cvr.gla.ac.uk/#/deletion) was used to assess changes in SARS‐CoV‐2 genomes (Singer et al., 2020) (Table S1). Lineage was assigned according to the lineage nomenclature system proposed by Rambaut et al. (2020).
For DNA alignments, the best‐fit model of nucleotide substitution (GTR+G+I) was selected under the Akaike and the Bayesian formation criteria in jModelTest (Posada, 2008). Maximum‐likelihood trees, with 1000‐replicate bootstrap for the support of internal nodes, were inferred in Geneious using PhyML 3.0 plugin (Guindon et al., 2010). Phylogenetic trees were visualized and edited with FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Dataset was retrieved from the GISAID EpiCoV database (Shu & McCauley, 2017) and the GenBank database (Table S2). The structure of the ORF7a protein was obtained from the protein database (PDB ID: 6W37) and I‐TASSER server (https://zhanglab.ccmb.med.umich.edu/COVID‐19/).
3. RESULTS AND DISCUSSION
Uruguay is one of the less COVID‐19‐affected countries in South America, mainly due to its small density population and an effective quarantine‐free strategy that included contact tracing and isolation, distancing measures and mask‐wearing. The epidemic in the country is mainly occurring through sporadic outbreaks associated with social events, healthcare centres or residential care centres (DEVISA, 2020). In the present study, we obtained seven complete SARS‐CoV‐2 genomes from one of these outbreaks using NGS sequencing. Genomes were sequenced with high average coverage (>1,000) and submitted to the GenBank. The analysis evidences the presence of a 12‐nucleotide deletion (Δ12) at 27,629–27,640 position in ORF7a, which was found in 100% of the reads in all seven clinical samples. The Δ12 variant deletion was identified in the fourteen patients of this outbreak by Sanger sequencing and capillary electrophoresis without the detection of co‐infection with the wild‐type virus (Figure 1).
FIGURE 1.
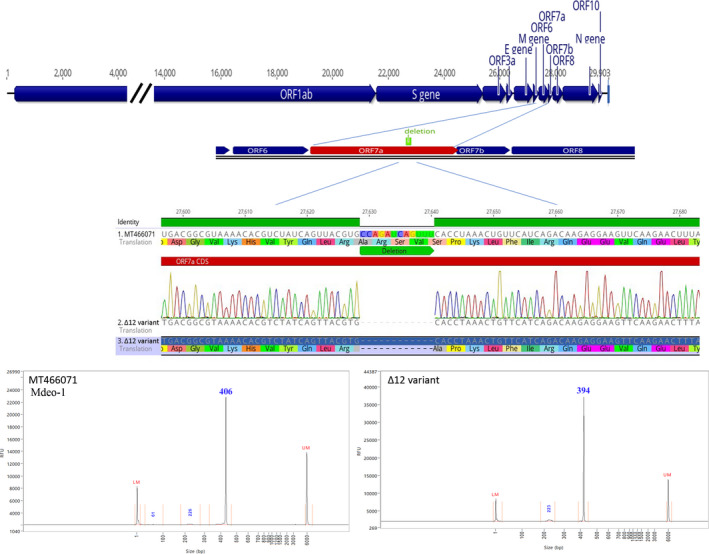
Top: complete SARS‐CoV‐2 genome organization and zoomed diagram showing location of the deletions in the ORF7a. Middle: zoomed diagram details of the nucleotide and amino acid deletion validated by Sanger sequencing; the Mdeo‐1 sequence (MT466071) was used as a reference. Bottom: amplicon capillary electrophoresis analysis. Chromatogram peaks using Mdeo‐1 reference sequence (MT466071) and Δ12 variant, left and right, respectively
The Δ12 mutation results in a 4‐amino acid in‐frame deletion (RSVS) in the accessory 121‐aa protein 7a. In SARS‐CoV, the 7a protein, also denoted as X4 or U122, is a viral antagonist of host restriction factor BST‐2/Tetherin and induces apoptosis via the caspase pathway in various cell types (Schaecher et al., 2007; Tan et al., 2004; Taylor et al., 2015; Yuan et al., 2006). The SARS‐CoV 7a protein is a type I 122‐aa membrane protein containing a 15‐aa signal peptide, a 81‐aa lumenal hydrophilic domain oriented inside the endoplasmic reticulum–Golgi apparatus, a 21‐aa transmembrane domain and a 5‐aa C‐terminal cytoplasmic tail (Nelson et al., 2005). The lumenal domain (ectodomain) has a 7‐stranded β‐sandwich fold typical of the immunoglobulin superfamily and a flexible and unstructured region (Hänel et al., 2006; Nelson et al., 2005). The ORF7a protein of SARS‐CoV‐2 has similar three‐dimensional structure, and the deletion here observed (residues 80‒83) spans the terminal residue of the last β‐strands and three residues of the beginning of the unstructured region of the 80‐aa lumenal domain (Figure 2).
FIGURE 2.
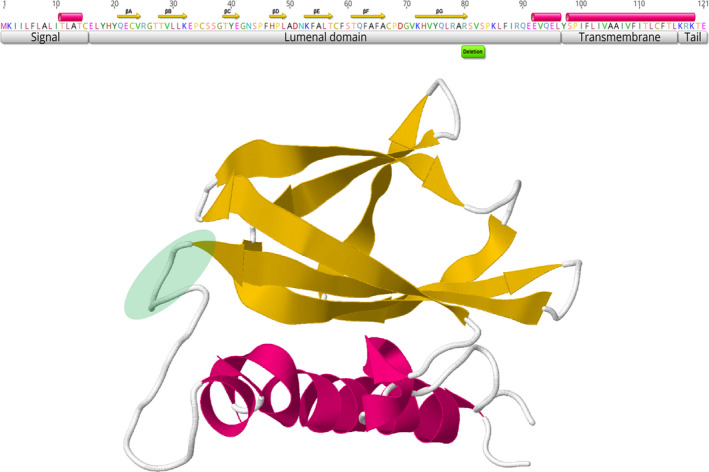
Structure of the 121‐aa ORF7a protein of SARS‐CoV‐2. Top: amino acid sequence and functional protein domains and folds. Cylinders represent α‐helices, and arrows denote ß‐strands. Bottom: three‐dimensional model. Green circle denotes the deletion region detected in Uruguayan samples
Previous studies reported deletions in ORF7a, including an 81nt deletion (Holland et al., 2020) and two large deletions of 392 nt and 227 nt that result in an in‐frame gene fusion of ORF7a with ORF8 and ORF7b, respectively (Addetia et al., 2020). We also observed the occurrence of several ORF7a deletions, particularly outside the β‐sandwich fold, of SARS‐CoV‐2 sequences from Europe, Asia, Africa, Australia and North America (Figure S1). The repeated emergence of SARS‐CoV‐2 viruses with deletions in ORF7a suggests that it is structurally tolerant to deletions. It is possible that most deletions were neutral or quasi‐neutral but the uneven deletion distribution and the fact that they were mostly in frame to keep the amino acid sequence of the remaining peptide, suggest that this gene is important for viral fitness. It has been recently established that the ORF7a protein binds to human monocytes leading to decreasing antigen‐presenting ability and inducing dramatic expression of pro‐inflammatory cytokines (Zhou et al., 2020). The larger 4‐stranded (A, C, F and G) β‐sheet in the lumenal domain is the putative functional interface, suggesting that variability in these regions affect binding capability. The deletion variant here identified spans the last residue of the βG strand and three residues of the following flexible region (Figure 2), and consequently, a clear effect of viral fitness or in the clinical course of COVID‐19 is not expected. However, it remains a possibility that variations in those regions may provide regulatory modulation and selective advantages without disorganizing the active domain.
Large deletions of 62 nt to 382 nt in the other accessory (ORF7b and/or ORF8) have also been described in circulating viruses from Singapore, Asia, Australia and Spain (Gong et al., 2020; Su et al., 2020). In contrast with these deletions, the Δ12 variant retained the presumptive single peptide and most of the β‐sheet of the Ig‐like fold of ORF7a (Holland et al., 2020) (Figure S1).
Genetic variations have been very useful to classify the virus in different lineages and to perform phylodynamic analyses to understand viral evolution and epidemiology, identifying patterns of spread (Deng et al., 2020) and sources of zoonotic infections (Sit et al., 2020). The Δ12 variant complete genomes were classified as belonging to the SARS‐CoV‐2 phylogenetic lineages B.1.1 and were clearly unrelated with A.5 or B.5 lineages. Phylogenetic analysis revealed that the Δ12 variants clustered in a separate subclade from the rest of the Uruguayan strains belonging to the B.1.1 lineage, also present in Uruguay (Figure 3). Sequences of the Uruguayan Δ12 variant were nearly identical across the genome, with only one difference among the strains, a C‐T non‐synonymous transition in one sample (GenBank accession number MW298639) that produces a Thr1100Ile substitution in the S protein. These findings confirm that the Δ12 variant arose through shared ancestry and was restricted in Uruguay to a unique local transmission cluster of at least fourteen patients. In addition, this deletion might have similar fitness than the wild type and is capable to be transmitted. In this sense, the deletion described here is a strong molecular marker or signature that could be useful for tracking outbreaks and nosocomial‐associated infections (Wong et al., 2020). Other reports evidenced ORF7a deletions that arose independently in the strains and suggest that deletions are not likely restricted to specific SARS‐CoV‐2 lineages (Addetia et al., 2020). This deletion was not detected in the 75 Uruguayan strains nor in any of the more than 2000 South American sequences (including neighbouring countries Brazil and Argentina) available in the GISAID database by November 2020. However, we found four sequences reported in CoV‐GLUE, one from the USA and three from New Zealand, that have the same deletion and belong to the B.1 lineage but have 13‒14 nucleotide differences with the Uruguayan Δ12 strains. Phylogenetic analysis shows that the USA, New Zealand and Uruguayan Δ12 variants are unrelated, suggesting an independent origin (Figure 3). Taking together, this result is an indicative that the Δ12 variant likely emerged and spread locally.
FIGURE 3.
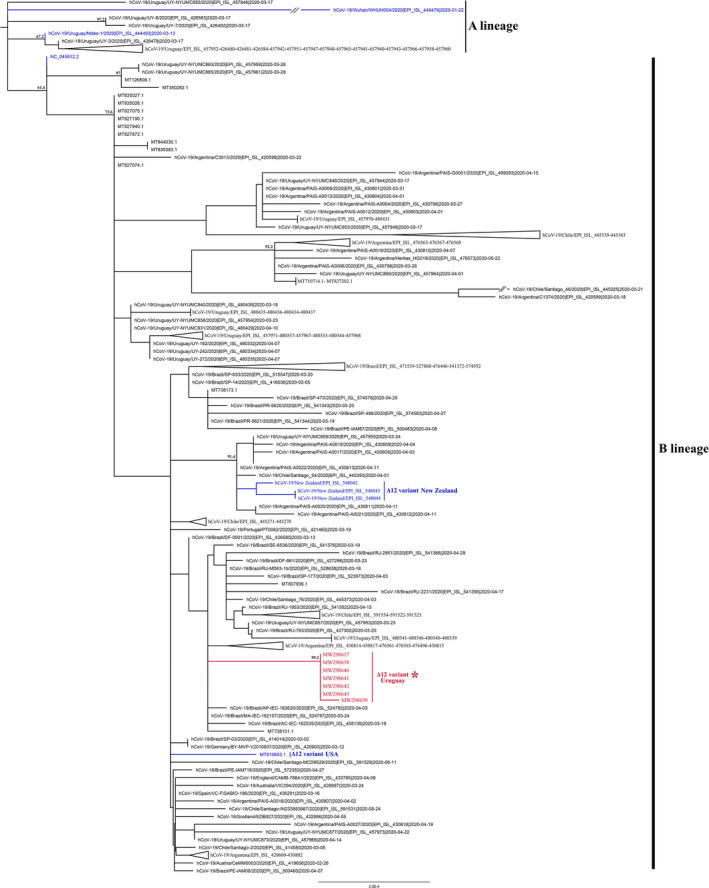
Maximum‐likelihood tree based on 158 SARS‐CoV‐2 sequences (Table S2). The seven Uruguayan Δ12 variant complete genomes characterized in this study are indicated in red. The reference sequence of the lineages B and A (Wuhan‐Hu‐1 and Wuhan‐Hu‐4), the Mdeo‐1 sequence (MT466071) and sequences with the same Δ12 deletion, three from New Zealand and one from the USA, are indicated by blue branches
The Δ12 variant has the Asp to Gly replacement at position 614 of the S gene (D614G), which has been related to a higher virus transmissibility in late outbreak sequences (Korber et al., 2020). There is not significant evidence of a higher transmissibility of this variant, but the D614G change is widespread in Uruguay and apparently substituted strains lacking the 614G residue. It is worth to note that finding several samples with the Δ12 deletion and the 614G change is not evidence that these viruses have any transmission advantage. Genome changes are more likely due to stochastic epidemiological phenomena, including founder effects and biased sampling that occur frequently in populations with fewer infected hosts (MacLean et al., 2020).
The question remaining is the emergence and spreading of the South American ∆12 variant. Future studies will have to determine its de novo occurrence in Uruguay and if it has a different evolutionary path (e.g. different substitution rates) compared with wild‐type viruses. The molecular mechanisms associated with indels in SARS‐CoV‐2 also remain to be determined. Indels occur preferentially at homo‐polymeric tracts and also at short, repeated, sequences which are prone to misalignment mutagenesis (Domingo, 2020). There is no evidence of homo‐polymers or repetitive sequences in the SARS‐CoV‐2 genome that could induce deletions as a result of slippage mutagenesis, suggesting that deletions are differentially retained in less constrained protein regions. It would be also interesting to establish whether the ∆12 variant goes extinct or increase its frequency in the Uruguayan population. In this sense, methodologies here described for rapid and economic detection of the ∆12 variant, Sanger sequences and capillary electrophoresis, can become powerful analytical tools for resolving potentially related outbreaks. Our findings may serve as a reference for study, describe novel deletions in infection clusters in South America and reinforce the need for continuous surveillance in different geographic areas of all continents to understand the worldwide dissemination of new SARS‐CoV‐2 variants.
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTIONS
All authors revised and approved the manuscript. YP and NR conceived the study. YP, NR, SF, LC and AD did the experiments. YP and RP analysed the data. AM, CT, SG and EF participated in protocol optimization. NG, VR, LC, CS and HC carried out the diagnostic. CM is Head of the DLSP. JA and RP got the financial support. RP, YP, AD, NR, SF, JA and LC wrote and revised the manuscript.
ETHICAL STATEMENT
The ‘Centro Nacional de Referencia de Influenza y otros Virus Respiratorios, Departamento de Laboratorios de Salud Pública. Ministerio de Salud Pública’, where the sample was diagnosed, belong to the WHO Global Influenza Surveillance and Response System (GISRS) for global surveillance of influenza and other respiratory viruses. Neither written informed consent nor explicit ethical approval was sought, as this study was only observational and carried out as part of the routine virologic surveillance (anonymously, without identification of patients).
Supporting information
Fig S1
Table S1
Table S2
ACKNOWLEDGEMENTS
We would like to thank all the authors who have kindly deposited and shared genome data on GISAID (Table S2).
Panzera Y, Ramos N, Frabasile S, et al. A deletion in SARS‐CoV‐2 ORF7 identified in COVID‐19 outbreak in Uruguay. Transbound Emerg Dis. 2021;68:3075–3082. 10.1111/tbed.14002
Yanina Panzera and Natalia Ramos contributed equally to this work.
Funding information
Facultad de Ciencias and Comisión Sectorial de Investigación Científica (CSIC), Fundación Manuel Pérez, UdelaR.
[Correction added on 4 May 2021, after first online publication: Adriana Delfraro was added as co‐corresponding author]
Contributor Information
Adriana Delfraro, Email: adriana@fcien.edu.uy.
Ruben Pérez, Email: rperez@fcien.edu.uy.
DATA AVAILABILITY STATEMENT
Viral sequences were deposited in the GenBank (accession numbers: MW298637 to MW298643).
REFERENCES
- Addetia, A. , Xie, H. , Roychoudhury, P. , Shrestha, L. , Loprieno, M. , Huang, M.‐L. , Jerome, K. R. , & Greninger, A. L. (2020). Identification of multiple large deletions in ORF7a resulting in in‐frame gene fusions in clinical SARS‐CoV‐2 isolates. Journal of Clinical Virology, 129, 104523. 10.1016/j.jcv.2020.104523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badua, C. L. D. C. , Baldo, K. A. T. , & Medina, P. M. B. (2020). Genomic and proteomic mutation landscapes of SARS‐CoV‐2. Journal of Medical Virology, 93(3), 1702–1721. 10.1002/jmv.26548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal, A. , Destras, G. , Gaymard, A. , Bouscambert‐Duchamp, M. , Valette, M. , Escuret, V. , Frobert, E. , Billaud, G. , Trouillet‐Assant, S. , Cheynet, V. , Brengel‐Pesce, K. , Morfin, F. , Lina, B. , & Josset, L. (2020). Molecular characterization of SARS‐CoV‐2 in the first COVID‐19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del). Clinical Microbiology & Infection, 26, 960–962. 10.1016/j.cmi.2020.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti, F. , Snyder, G. A. , Giovanetti, M. , Angeletti, S. , Gallo, R. C. , Ciccozzi, M. , & Zella, D. (2020). Emerging of a SARS‐CoV‐2 viral strain with a deletion in nsp1. Journal of Translational Medicine, 18, 329. 10.1186/s12967-020-02507-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman, V. M. , Landt, O. , Kaiser, M. , Molenkamp, R. , Meijer, A. , Chu, D. K. W. , Bleicker, T. , Brünink, S. , Schneider, J. , Schmidt, M. L. , Mulders, D. G. J. C. , Haagmans, B. L. , Van Der Veer, B. , Van Den Brink, S. , Wijsman, L. , Goderski, G. , Romette, J. L. , Ellis, J. , Zambon, M. , … Drosten, C. (2020). Detection of 2019 novel coronavirus (2019‐nCoV) by real‐time RT‐PCR. Eurosurveillance, 25(3), 23–30. 10.2807/1560-7917.ES.2020.25.3.2000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, X. , Gu, W. , Federman, S. , du Plessis, L. , Pybus, O. G. , Faria, N. , Wang, C. , Yu, G. , Pan, C.‐Y. , Guevara, H. , Sotomayor‐Gonzalez, A. , Zorn, K. , Gopez, A. , Servellita, V. , Hsu, E. , Miller, S. , Bedford, T. , Greninger, A. L. , Roychoudhury, P. , … Chiu, C. Y. (2020). A Genomic Survey of SARS‐CoV‐2 Reveals Multiple Introductions into Northern California without a Predominant Lineage. Science, 369, 582–587. 10.1101/2020.03.27.20044925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEVISA (2020). Informe epidemiológico, actualización 21.8 . DEVISA, Div. Epidemiología, MSP. 1‐26. Retrieved from https://www.gub.uy/ministerio‐salud‐publica/comunicacion/noticias/informe‐epidemiologico‐21‐agosto‐2020.
- Domingo, E. (2020). Molecular basis of genetic variation of viruses: error‐prone replication. In Virus as Populations (pp 35–71). Elsevier. Retrieved from https://www.sciencedirect.com/book/9780128163313/virus‐as‐populations. [Google Scholar]
- Franco‐Muñoz, C. , Álvarez‐Díaz, D. A. , Laiton‐Donato, K. , Wiesner, M. , Escandón, P. , Usme‐Ciro, J. A. , Franco‐Sierra, N. D. , Flórez‐Sánchez, A. C. , Gómez‐Rangel, S. , Rodríguez‐Calderon, L. D. , Barbosa‐Ramirez, J. , Ospitia‐Baez, E. , Walteros, D. M. , Ospina‐Martinez, M. L. , & Mercado‐Reyes, M. (2020). Substitutions in Spike and Nucleocapsid proteins of SARS‐CoV‐2 circulating in South America. Infection, Genetics and Evolution, 85, 104557. 10.1016/j.meegid.2020.104557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, Y.‐N. , Tsao, K.‐C. , Hsiao, M.‐J. , Huang, C.‐G. , Huang, P.‐N. , Huang, P.‐W. , Lee, K.‐M. , Liu, Y.‐C. , Yang, S.‐L. , Kuo, R.‐L. , Chen, K.‐F. , Liu, Y.‐C. , Huang, S.‐Y. , Huang, H.‐I. , Liu, M.‐T. , Yang, J.‐R. , Chiu, C.‐H. , Yang, C.‐T. , Chen, G.‐W. , & Shih, S.‐R. (2020). SARS‐CoV‐2 genomic surveillance in Taiwan revealed novel ORF8‐deletion mutant and clade possibly associated with infections in Middle East. Emerging Microbes & Infections, 9, 1457–1466. 10.1080/22221751.2020.1782271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya, A. E. , Enjuanes, L. , Ziebuhr, J. , & Snijder, E. J. (2006). Nidovirales: Evolving the largest RNA virus genome. Virus Research, 117, 17–37. 10.1016/j.virusres.2006.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J.‐F. , Lefort, V. , Anisimova, M. , Hordijk, W. , & Gascuel, O. (2010). New Algorithms and Methods to Estimate Maximum‐Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology, 59, 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Hänel, K. , Stangler, T. , Stoldt, M. , & Willbold, D. (2006). Solution structure of the X4 protein coded by the SARS related coronavirus reveals an immunoglobulin like fold and suggests a binding activity to integrin I domains. Journal of Biomedical Science, 13, 281–293. 10.1007/s11373-005-9043-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, L. A. , Kaelin, E. A. , Maqsood, R. , Estifanos, B. , Wu, L. I. , Varsani, A. , Halden, R. U. , Hogue, B. G. , Scotch, M. , & Lim, E. S. (2020). An 81 nucleotide deletion in SARS‐CoV‐2 ORF7a identified from sentinel surveillance in Arizona (Jan‐Mar 2020). Journal of Virology, 94, e00711‐20, 10.1128/jvi.00711-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khailany, R. A. , Safdar, M. , & Ozaslan, M. (2020). Genomic characterization of a novel SARS‐CoV‐2. Gene Reports, 19, 1–6. 10.1016/j.genrep.2020.100682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber, B. , Fischer, W. M. , Gnanakaran, S. , Yoon, H. , Theiler, J. , Abfalterer, W. , Hengartner, N. , Giorgi, E. E. , Bhattacharya, T. , Foley, B. , Hastie, K. M. , Parker, M. D. , Partridge, D. G. , Evans, C. M. , Freeman, T. M. , de Silva, T. I. , McDanal, C. , Perez, L. G. , Tang, H. , … Wyles, M. D. (2020). Tracking Changes in SARS‐CoV‐2 Spike: Evidence that D614G Increases Infectivity of the COVID‐19 Virus. Cell, 182, 812–827.e19. 10.1016/j.cell.2020.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, B. K. , Rohit, A. , Prithvisagar, K. S. , Rai, P. , Karunasagar, I. , & Karunasagar, I. (2020). Deletion in the C‐terminal region of the envelope glycoprotein in some of the Indian SARS‐CoV‐2 genome. Virus Research, 291, 198222. 10.1016/j.virusres.2020.198222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, S.‐Y. , Wang, P. , Mok, B.‐W.‐Y. , Zhang, A. J. , Chu, H. , Lee, A.‐C.‐Y. , Deng, S. , Chen, P. , Chan, K.‐H. , Song, W. , Chen, Z. , To, K.‐K.‐W. , Chan, J.‐F.‐W. , Yuen, K.‐Y. , & Chen, H. (2020). Attenuated SARS‐CoV‐2 variants with deletions at the S1/S2 junction. Emerging Microbes & Infections, 9, 837–842. 10.1080/22221751.2020.1756700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P. , Jiang, J.‐Z. , Wan, X.‐F. , Hua, Y. , Li, L. , Zhou, J. , Wang, X. , Hou, F. , Chen, J. , Zou, J. , & Chen, J. (2020). Are pangolins the intermediate host of the 2019 novel coronavirus (SARS‐CoV‐2)? PLoS Path, 16, e1008421. 10.1371/journal.ppat.1008421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean, O. A. , Orton, R. J. , Singer, J. B. , & Robertson, D. L. (2020). No evidence for distinct types in the evolution of SARS‐CoV‐2. Virus Evolution, 6, 1–6. 10.1093/ve/veaa034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan, K. , Huang, C. , & Makino, S. (2008). SARS coronavirus accessory proteins. Virus Research, 133, 113–121. 10.1016/j.virusres.2007.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, C. A. , Pekosz, A. , Lee, C. A. , Diamond, M. S. , & Fremont, D. H. (2005). Structure and Intracellular Targeting of the SARS‐Coronavirus Orf7a Accessory Protein. Structure, 13, 75–85. 10.1016/j.str.2004.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada, D. (2008). jModelTest: Phylogenetic Model Averaging. Molecular Biology and Evolution, 25, 1253–1256. 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- Rambaut, A. , Holmes, E. C. , O’Toole, Á. , Hill, V. , McCrone, J. T. , Ruis, C. , du Plessis, L. , & Pybus, O. G. (2020). A dynamic nomenclature proposal for SARS‐CoV‐2 lineages to assist genomic epidemiology. Nature Microbiology, 5, 1403–1407, DOI: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaecher, S. R. , Touchette, E. , Schriewer, J. , Buller, R. M. , & Pekosz, A. (2007). Severe Acute Respiratory Syndrome Coronavirus Gene 7 Products Contribute to Virus‐Induced Apoptosis. Journal of Virology, 81, 11054–11068. 10.1128/JVI.01266-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, Y. , & McCauley, J. (2017). GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance, 22, 30494. 10.2807/1560-7917.ES.2017.22.13.30494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, J. , Gifford, R. , Cotten, M. , & Robertson, D. (2020). CoV‐GLUE: A Web Application for Tracking SARS‐CoV‐2 Genomic Variation. Preprint. 10.20944/preprints202006.0225.v1 [DOI]
- Sit, T. H. C. , Brackman, C. J. , Ip, S. M. , Tam, K. W. S. , Law, P. Y. T. , To, E. M. W. , Yu, V. Y. T. , Sims, L. D. , Tsang, D. N. C. , Chu, D. K. W. , Perera, R. A. P. M. , Poon, L. L. M. , & Peiris, M. (2020). Infection of dogs with SARS‐CoV‐2. Nature, 586, 776–778. 10.1038/s41586-020-2334-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, Y. C. F. , Anderson, D. E. , Young, B. E. , Linster, M. , Zhu, F. , Jayakumar, J. , Zhuang, Y. , Kalimuddin, S. , Low, J. G. H. , Tan, C. W. , Chia, W. N. , Mak, T. M. , Octavia, S. , Chavatte, J.‐M. , Lee, R. T. C. , Pada, S. , Tan, S. Y. , Sun, L. , Yan, G. Z. , … Smith, G. J. D. (2020). Discovery and Genomic Characterization of a 382‐Nucleotide Deletion in ORF7b and ORF8 during the Early Evolution of SARS‐CoV‐2. Mbio, 11(4), 10.1128/mBio.01610-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, Y.‐J. , Fielding, B. C. , Goh, P.‐Y. , Shen, S. , Tan, T. H. P. , Lim, S. G. , & Hong, W. (2004). Overexpression of 7a, a Protein Specifically Encoded by the Severe Acute Respiratory Syndrome Coronavirus, Induces Apoptosis via a Caspase‐Dependent Pathway. Journal of Virology, 78, 14043–14047. 10.1128/JVI.78.24.14043-14047.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, J. K. , Coleman, C. M. , Postel, S. , Sisk, J. M. , Bernbaum, J. G. , Venkataraman, T. , Sundberg, E. J. , & Frieman, M. B. (2015). Severe Acute Respiratory Syndrome Coronavirus ORF7a Inhibits Bone Marrow Stromal Antigen 2 Virion Tethering through a Novel Mechanism of Glycosylation Interference. (A. García‐Sastre, Ed.). Journal of Virology, 89, 11820–11833. 10.1128/jvi.02274-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls, A. C. , Park, Y. J. , Tortorici, M. A. , Wall, A. , McGuire, A. T. , & Veesler, D. (2020). Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell, 181, 281–292.e6, DOI: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, S. C. Y. , Kwong, R. T.‐S. , Wu, T. C. , Chan, J. W. M. , Chu, M. Y. , Lee, S. Y. , Wong, H. Y. , & Lung, D. C. (2020). Risk of nosocomial transmission of coronavirus disease 2019: an experience in a general ward setting in Hong Kong. Journal of Hospital Infection, 105, 119–127. 10.1016/j.jhin.2020.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. , Zhao, S. , Yu, B. , Chen, Y. M. , Wang, W. , Song, Z. G. , Hu, Y. , Tao, Z. W. , Tian, J. H. , Pei, Y. Y. , Yuan, M. L. , Zhang, Y. L. , Dai, F. H. , Liu, Y. , Wang, Q. M. , Zheng, J. J. , Xu, L. , Holmes, E. C. , & Zhang, Y. Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579, 265–269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, X. , Wu, J. , Shan, Y. , Yao, Z. , Dong, B. , Chen, B. , Zhao, Z. , Wang, S. , Chen, J. , & Cong, Y. (2006). SARS coronavirus 7a protein blocks cell cycle progression at G0/G1 phase via the cyclin D3/pRb pathway. Virology, 346, 74–85. 10.1016/j.virol.2005.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z. , Huang, C. , Zhou, Z. , Huang, Z. , Su, L. , Kang, S. , Chen, X. , Chen, Q. , He, S. , Rong, X. , Xiao, F. , Chen, J. , & Chen, S. (2020). Structural Insight Reveals SARS‐CoV‐2 Orf7a as an Immunomodulating Factor for Human CD14+Monocytes. SSRN Electronic Journal. 10.2139/ssrn.3699795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzula, L. (2020). Lost in deletion: The enigmatic ORF8 protein of SARS‐CoV‐2. Biochemical and Biophysical Research Communications, 54, 251–255. 10.1016/j.bbrc.2020.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1
Table S2
Data Availability Statement
Viral sequences were deposited in the GenBank (accession numbers: MW298637 to MW298643).